

Abstract
The semiempirical orthogonalization-corrected OMx methods (OM1, OM2, and OM3) go beyond the standard MNDO model by including additional interactions in the electronic structure calculation. When augmented with empirical dispersion corrections, the resulting OMx-Dn approaches offer a fast and robust treatment of noncovalent interactions. Here we evaluate the performance of the OMx and OMx-Dn methods for a variety of ground-state properties using a large and diverse collection of benchmark sets from the literature, with a total of 13035 original and derived reference data. Extensive comparisons are made with the results from established semiempirical methods (MNDO, AM1, PM3, PM6, and PM7) that also use the NDDO (neglect of diatomic differential overlap) integral approximation. Statistical evaluations show that the OMx and OMx-Dn methods outperform the other methods for most of the benchmark sets.
1. Introduction
Fast quantum-chemical methods are indispensable for computationally demanding calculations of electronic properties of large molecules. In the 1970s and 1980s, semiempirical quantum-chemical (SQC) methods were the workhorse in computational studies of ground-state properties. Since the 1990s, they have largely been replaced in such studies by ab initio and density functional theory (DFT) approaches, which are typically slower by at least 3 orders of magnitude but also generally more accurate and robust.
However, even nowadays, SQC methods often remain the only practical choice when treating huge molecules or very large numbers of molecules or when performing extensive molecular dynamics (MD) simulations, e.g., in the context of quantum mechanics/molecular mechanics (QM/MM) studies on large biomolecular systems.1,2 Examples of recent SQC applications include the calculation of electronic properties used in 3D-QSAR models (3D-quantitative structure–activity relationship),3 QM-based computer-aided drug design,4−7 the study of band gaps, UV/vis spectra, and charge-transfer processes in systems relevant for molecular nanoelectronics and organic photovoltaics,8,9 the investigation of local properties used to understand electron and hole transport mechanisms in transistors,10,11 gas-phase MD simulations of electron impact mass spectra,12−15 ground-state QM/MM MD simulations of enzymes,2,16 excited-state nonadiabatic dynamics simulations of organic chromophores,1 and studies of organic and enzymatic reactions in solution.17−20
Most of the widely used SQC methods are variants of the MNDO model21,22 which is based on the NDDO (neglect of diatomic differential overlap) integral approximation.1 These MNDO-type methods include AM1,23 PM3,24,25 MNDO/d,26−28 AM1*,29 RM1,30 PDDG/MNDO and PDDG/PM3,31,32 PM6,33 and PM734 (see the cited references for details and the meaning of the acronyms). A common feature of these approaches is that they attempt to improve the accuracy within the confines of the given electronic structure model, mainly by adding modifications to the core repulsion functions and by performing a more thorough and extensive parametrization. Prime examples of this strategy are the PMx methods (PM3, PM6, and PM7).
An alternative strategy is to improve the underlying model. All MNDO-based methods solve the Hartree–Fock secular equations as if the basis set were orthogonal, without explicitly accounting for the terms arising in the ab initio treatment when transforming the Fock matrix from the original nonorthogonal to an orthogonal basis.1 This neglect of orthogonalization terms gives rise to several qualitative deficiencies of standard SQC methods that cannot always be eliminated simply by parametrization.35 It has been shown that reintroduction of the overlap matrix into the secular equations can indeed significantly improve the accuracy of the MNDO method.36 In the orthogonalization models (OMx) developed in our group, orthogonalization corrections are included into the Fock matrix to different extent (along with other interactions of similar size), which leads to the following general-purpose SQC methods: OM1,37,38 OM2,39,40 and OM3.41 The theoretical formalism, the optimized parameters, and the initial validation of the OMx methods are described in detail in a companion Article.42
Both the MNDO-type and OMx methods formally neglect dispersion which causes their poor performance in systems where dispersion interactions play an important role.43 In recent years, a number of empirical dispersion corrections have been developed and successfully applied for DFT methods, e.g., the D2 and D3 corrections proposed by Grimme.44−46 The same types of dispersion corrections can also be combined with the OMx methods, with no change in the OMx parameters and only minor adjustment of the D2 and D3 parameters.42,47,48 The resulting OMx-Dn methods provide a much improved treatment of noncovalent interactions.42,47−49 Their formalism, parameters, and initial validation are described in detail in ref (42). Among the MNDO-type methods, the latest variant (PM7) incorporates explicit dispersion corrections in its definition, and unlike in the case of the OMx-Dn methods, its parameters were optimized with these corrections included.34
Given the severe approximations in the formalism of SQC methods and the presence of empirically determined parameters, it is essential to validate these methods carefully against reliable experimental data and/or accurate high-level theoretical results. Some validation studies are available for OMx methods, which cover both ground-state and excited-state properties.42,48,50−52 They indicate that the OMx methods outperform other SQC methods in most cases.42,50−52 Some of this validation work also offers comparisons between SQC methods and standard DFT approaches showing that the former may approach or sometimes even exceed the accuracy of DFT results for ground-state properties of organic molecules.1,8,31,48,52,53
Considering the extensive recent benchmark exercises for ab initio and DFT methods, it is obvious that a more comprehensive validation of the OMx and OMx-Dn methods is required to establish their reliability and to allow for a more detailed assessment of their accuracy compared with other SQC methods. The objective of this Article is to provide such validation for ground-state properties. We cover standard MNDO-type SQC methods (MNDO, AM1, PM3, PM6, and PM7), OMx methods (OM1, OM2, and OM3), and dispersion-corrected OMx-Dn methods (OM2 and OM3 with D2 and D3 corrections as well as D3T corrections with additional three-body terms). The OMx and OMx-Dn methods are fully specified in ref (42).
In addition to our own validation sets, we use benchmark sets mostly taken from the available ab initio and DFT literature. We include from these sets only those reference molecules that contain the elements H, C, N, O and/or F, for the simple reason that the OMx methods have up to now only been parametrized for these elements. We address the following ground-state properties: heats of formation, bond lengths, bond angles, dihedral angles, relative energies, reaction energies, dissociation energies, atomization energies, proton affinities, activation barriers, vertical and adiabatic ionization potentials (IPs), adiabatic electron affinities (EAs), dipole moments, noncovalent interaction energies, and geometries of noncovalent complexes. Overall, the benchmarks contain 13035 original or derived reference data. The results from the benchmarking will help to establish the accuracy of different SQC methods in different areas and can also serve as a guide for choosing the most appropriate SQC method in specific application projects.
2. Methods and Computational Details
All MNDO, AM1, PM3, OMx, and OMx-Dn calculations were carried out with our locally modified MNDO2005 program.54 The PM6, PM7, and RM1 calculations were done with the MOPAC2012 program.55,56 Molecules were visualized with Chemcraft 1.7.57
Standard conventions were used in all calculations.42 For the sake of documentation, we specify some of these standard options here. The convergence criterion for the SCF energy was set to 10–9 eV; in addition, the diagonal elements of the density matrix were required to be converged to 10–9 in MNDO2005. Doublet and triplet states were treated with the restricted open-shell half-electron approach,58 and higher spin states were described using unrestricted Hartree–Fock (UHF) calculations. We did not use molecular mechanics corrections for peptides in PM6 or PM7, and we did not apply any cutoffs for the three-center orthogonalization corrections in the OMx and OMx-Dn methods.
Geometry optimizations were considered converged when the gradient norm became smaller than 0.01 kcal/(mol·Å). In MNDO2005 the BFGS optimization algorithm was used by default; in difficult cases, we also applied eigenvector following, with the Hessian being computed numerically by one-sided finite differences of the gradient (at the first step and every 10 following steps) and with a minimum trust radius of 0.00001. In MOPAC the eigenvector following algorithm was used throughout; in most cases the full Hessian matrix was constructed and recalculated every 10 steps using numerical single-sided derivatives of the gradient. Frequently MOPAC stopped when the heat of formation remained essentially constant in subsequent cycles; in these cases the gradient norm was usually smaller than 0.1 kcal/(mol·Å).
It is well-known that SQC methods generally predict the heat of formation of the proton with very large errors.52 Therefore, in line with common semiempirical practice,59 we used the experimental value of 367.171 kcal/mol60 for the heat of formation of the proton at 298 K to calculate the proton affinities included in the GMTKN30-CHNOF and CE345-CHNOF databases (see later text).
The reference molecules in the various benchmark sets are generally still quite small by semiempirical standards. Therefore, they were computed using single-CPU serial versions of the MNDO2005 and MOPAC programs. For large-scale applications, parallel versions of MNDO2005 are available using shared-memory and distributed-memory message-passing parallelization,61,62 as well as a hybrid version using graphics processing units (GPUs).63 Similarly, there are shared-memory and GPU-parallelized versions of MOPAC.55,56 To illustrate the scope of SQC calculations, we note that the hybrid GPU version of MNDO2005 has been used to compute water clusters of up to 5400 atoms and to optimize the geometries of a large series of representative proteins.63,64
Calculations with D3 and D3T dispersion corrections were done using our interface of the MNDO2005 program to the DFT-D3 stand-alone program by Grimme (versions 3.0 Rev 2 and 3.1 Rev 0).65 Default cutoffs (95 au for two-body terms and 40 au for coordination numbers) were used with version 3.0 and increased cutoffs (95 au for two-body terms, coordination numbers, and three-body terms) with version 3.1.
3. Data Sets
In this section we define the benchmark sets used presently to evaluate different SQC methods. We divide these sets into two groups: those designed to benchmark ground-state properties in general (OVS7-CHNOF, G2G3-CHNOF, W4-11-CHNOF, GMTKN30-CHNOF, CE345-CHNOF, PDDG, PM7-CHNOF, and C7H10O2) and those specifically designed to benchmark noncovalent interactions (A24-CHNOF, S66, S66a8, JSCH-2005-CHNOF, S7L, S30L-CHNOF, and AF6). We note that this division is somewhat arbitrary as some reference molecules and complexes from the first group also feature noncovalent interactions and may thus appear again in the second group.
All data sets and subsets from the first and second groups are listed in Tables 1 and 2, respectively. Most of them are taken from the literature and described in the same terms as in the published work. Included are all molecules from the original data sets that contain only the elements C, H, N, O, and/or F, for which OMx parameters are available;42 all other molecules are ignored.
Table 1. Data Sets Used for Benchmarking General Ground-State Propertiesa.
| data set | N | description | ref values | ref |
|---|---|---|---|---|
| OVS7-CHNOF | 1131 | our own compilation of 7 validation sets | expt and theor | |
| radicals71 | 71 | heats of formation, relative energies and ionization potentials of radicals | expt and theor | (66) |
| anions24 | 24 | heats of formation of nonradical organic and inorganic anions | expt | (37) |
| cations41 | 41 | heats of formation and relative energies of organic and inorganic cations | expt and theor | (37) |
| BIGMOL20 | 20 | heats of formation of relatively large organic molecules | expt | (37, 67) |
| conformers30 | 30 | heats of formation and barriers of different organic conformers | expt and theor | (39) |
| isomers44 | 44 | heats of formation and relative energies of isomers of organic compounds | expt | (39) |
| fluorine91 | properties of 91 fluorine-containing compounds | theor | (68) | |
| 91 | heats of formation | G3 | ||
| 455 | bond lengths | MP2/6-31G(d) | ||
| 355 | bond angles | MP2/6-31G(d) | ||
| G2G3-CHNOF | 173 | |||
| G2 | 93 | heats of formation of small molecules from the G2 set | expt | (69) |
| G3 | 52 | heats of formation of midsize molecules from the G3 set | expt | (70) |
| alkanes28 | 28 | heats of formation and relative energies of alkanes C1–C16 | expt | (41, 71) |
| W4-11-CHNOF | 593 | high-confidence benchmark data set for computational thermochemistry | W4 or higher | (72) |
| TAE140 | 88 | atomization energies | W4 | (72) |
| TAE_nonMR124 | 43 | TAE140 without multireference cases | W4 | (72) |
| BDE99 | 79 | bond dissociation reaction energies | W4 | (72) |
| HAT707 | 394 | heavy-atom transfer energies | W4 | (72) |
| ISOMER20 | 19 | isomerization energies | W4 | (72) |
| SN13 | 13 | “nucleophilic substitution” energies | W4 | (72) |
| GMTKN30-CHNOF | 480 | general main group thermochemistry, kinetics, and noncovalent interactions | back-corr expt and theorb | (73) |
| MB08-165 | 25 | decomposition energies of artificial molecules | est CCSD(T)/CBS | (74) |
| W4-08 | 50 | atomization energies of small molecules | W4 | (75) |
| W4-08woMR | 43 | W4-08 without multireference cases | W4 | (75) |
| G21IP | 15 | adiabatic ionization potentials | expt | (76) |
| G21EA | 12 | adiabatic electron affinities | expt | (76) |
| PA | 8 | adiabatic proton affinities | est CCSD(T)/CBS and W1 | (77, 78) |
| SIE11 | 5 | self-interaction error related problems | est CCSD(T)/CBS | (79) |
| BHPERI | 22 | barrier heights of pericyclic reactions | W1 and CBS-QB3 | (75, 80−83) |
| BH76 | 54 | barrier heights of hydrogen and heavy-atom transfers, nucleophilic substitution, unimolecular, and association reactions | W1 and theor est | (84, 85) |
| BH76RC | 22 | reaction energies of the BH76 set | W1 and theor est | (84, 85) |
| RSE43 | 34 | radical stabilization energies | est CCSD(T)/CBS | (86) |
| O3ADD6 | 6 | reaction and association energies and barrier heights for addition of O3 to C2H4 and C2H2 | est CCSD(T)/CBS | (87) |
| G2RC | 15 | reaction energies of selected G2/97 systems | expt | (69) |
| ISO34 | 34 | isomerization energies of small and medium-sized organic molecules | expt | (88) |
| ISOL22 | 18 | isomerization energies of large organic molecules | SCS-MP3/CBS | (89) |
| DC9 | 7 | nine difficult cases for DFT | expt and theor | (79, 82, 90−95) |
| DARC | 14 | reaction energies of Diels–Alder reactions | est CCSDT/CBS | (96) |
| BSR36 | 36 | bond separation reactions of saturated hydrocarbons | est CCSD(T)/CBS | (97) |
| IDISP | 6 | intramolecular dispersion interactions | expt and theor | (88, 98, 99) |
| WATER27 | 27 | binding energies of water, H+(H2O)n and OH–(H2O)n clusters | est CCSD(T)/CBS; MP2/CBS | (100) |
| S22 | 22 | binding energies of noncovalently bound dimers | est CCSD(T)/CBS | (101, 102) |
| ADIM6 | 6 | interaction energies of n-alkane dimers | est CCSD(T)/CBS | (97, 103) |
| PCONF | 10 | relative energies of phenylalanyl–glycyl–glycine tripeptide conformers | est CCSD(T)/CBS | (104) |
| ACONF | 15 | relative energies of alkane conformers | W1h-val | (105) |
| SCONF | 17 | relative energies of sugar conformers | est CCSD(T)/CBS | (79, 106) |
| CE345-CHNOF | 187 | chemistry energetic database with 345 data | back-corr expt and theorb | (107, 108) |
| MGAE109/11 | 74 | main group atomization energies | expt, W4, W4.2, W4.3, W4.4 | (109, 110) |
| IsoL6/11 | 6 | isomerization energies of large molecules | CCSD(T)-F12a/aug-cc-pVDZ | (111) |
| IP21 | 4 | adiabatic ionization potentials | expt | (109, 112−116) |
| EA13/03 | 4 | adiabatic electron affinities | expt | (109, 112−114) |
| PA8/06 | 4 | proton affinities | expt | (78) |
| ABDE12 | 12 | alkyl bond dissociation energies | expt | (109, 117−119) |
| HC7/11 | 7 | hydrocarbon chemistry | expt and theor | (119) |
| πTC13 | 13 | thermochemistry of π systems | expt, CCSD(T)/cc-pVTZ, est CCSD(T)/CBS | (78, 112, 117) |
| HTBH38/08 | 26 | hydrogen transfer barrier heights | expt and theor | (85, 109, 120, 121) |
| NHTBH38/08 | 23 | non-hydrogen transfer barrier heights | Wn (n = 1, 4)c | (85, 109, 120, 121) |
| NCCE31/05 | 14 | noncovalent complexation energies | W1, est CCSD(T)/CBS | (113, 122) |
| PDDG | 979 | set used for training and validating PDDG/MNDO and PDDG/PM3 | expt | (31) |
| 622 | heats of formation | |||
| 153 | bond lengths | |||
| 54 | bond angles | |||
| 6 | dihedral angles | |||
| 97 | ionization potentials | |||
| 47 | dipole moments | |||
| PM7-CHNOF | 1595 | set used for training and validating PM7 | expt and theor | (34) |
| 1168 | heats of formation | |||
| 175 | bond lengths | |||
| 90 | bond angles | |||
| 104 | ionization potentials | |||
| 58 | dipole moments | |||
| C7H10O2 | 6095 | atomization enthalpies at 298 K of 6095 isomeric C7H10O2 molecules | G4MP2 | (123) |
Subsets are indented. Descriptions are taken from the cited literature.
Data in these databases were normally corrected to represent total energies without any zero-point vibrational energies and thermal corrections; this is also true for the “expt” and “theor” values given for the subsets; “theor” usually means “best theoretical estimate”.
Most of the data in this subset.
Table 2. Data Sets Used for Benchmarking Noncovalent Interactionsa.
| data set | N | description | ref values | ref |
|---|---|---|---|---|
| A24-CHNOF | 21 | very accurate interaction energies of small noncovalent complexes | est CCSDT(Q)/CBS + relativistic corrections | (124) |
| 23 | selected interatomic distancesb | est CCSD(T)/CBS | ||
| 40 | selected anglesb | est CCSD(T)/CBS | ||
| S66 | 66 | interaction energies of 66 noncovalent complexes | CCSD(T)/CBS | (125, 126) |
| 172 | selected interatomic distancesb | MP2/cc-pVTZ + est CCSD(T)/CBSc | ||
| 141 | selected anglesb | MP2/cc-pVTZ + est CCSD(T)/CBSc | ||
| S66a8 | 528 | sampling angular degrees of freedom in the S66 complexes | est CCSD(T)/CBS | (126) |
| JSCH-2005-CHNOF | 134 | interaction energies of base and amino acids pairs | CCSD(T) or MP2 with est CBS | (101) |
| S7L | 7 | energies of σ–σ and π–π interactions of 7 large complexes | est CCSD(T)/CBS | (127) |
| 28 | selected interatomic distancesb | B3LYP + CCSD(T)/ha-cc-pVDZd | ||
| S30L-CHNOF | 24 | interaction energies of very large complexes | back-corr expt | (49) |
| AF6 | 6 | folding energies of alkanes | CCSD(T)/CBS, CCSD+FNO(T)/cc-pVTZ | (128) |
| 6 | folding enthalpies at 298 K | CCSD+FNO(T)/cc-pVTZ+ZPE(MP2/cc-pVTZ)+temp.dep.shifts | ||
| 27 | selected interatomic distancesb | MP2/cc-pVTZ | ||
| 74 | selected anglesb | MP2/cc-pVTZ |
Descriptions are taken from the cited literature.
This work; based on geometries from the cited references.
Intermolecular distances were obtained from MP2/cc-pVTZ geometries by interpolating estimated CCSD(T)/CBS energies along dissociation curves.125
Reference geometries were obtained by optimizing intermolecular distances at the CCSD(T)/ha-cc-pVDZ level using monomer geometries optimized at the B3LYP level with large basis sets.127
Detailed numerical results on all individual reference molecules from all benchmark sets are compiled in the Supporting Information (SI) which also cites the origin of the published reference data. In the following two sections, we will focus on statistical evaluations of these results for all sets and subsets (with more than two molecules).
Following semiempirical tradition, all SQC methods tested presently were parametrized with regard to heats of formation. Their performance can thus be evaluated in a straightforward manner when reliable reference data for heats of formation are available either from experiment or from high-level ab initio calculations. This is the case for the CHNO and FLUOR data sets used during the parametrization of the OMx methods42 and for the OVS7-CHNOF, G2G3-CHNOF, PDDG, and PM7-CHNOF sets used in the present benchmarking. The corresponding reference data are mostly taken from experiment. Performance evaluation of atomization enthalpies at 298 K is also straightforward for the C7H10O2 set as reference values can be easily obtained from the reference ab initio enthalpies at 298 K.
Most of the entries in the other databases of Tables 1 and 2 represent relative energies, for example isomerization energies, interaction energies, binding energies, reaction energies, barrier heights, proton affinities, ionization potentials, and electron affinities. The corresponding ab initio reference data are generally obtained from energy differences, whereas the respective semiempirical values are differences of heats of formation (i.e., enthalpies at 298 K) that implicitly include zero-point vibrational energies (ZPVEs) and thermal enthalpic corrections (from 0 to 298 K). These corrections are normally quite similar for related molecules since they tend to be bond-specific and transferable,129 and hence they will cancel to a large extent when comparing related systems. As in many semiempirical studies over the past decades, we will thus mostly ignore the distinction between relative energies and heats of formation in the following, and consider the semiempirical values to be directly comparable with ab initio relative energies.
This line of reasoning obviously breaks down when it comes to atomization energies, because there is no longer any cancellation. For a realistic comparison between ab initio and semiempirical atomization energies, the ZPVEs and thermal enthalpic corrections have to be taken into account explicitly. This is computationally demanding at an accurate ab initio level,130 and therefore we computed both these corrections at the SQC level using the harmonic-oscillator and rigid-rotor approximations. To illustrate the necessity of including these corrections, we compare the OM2 results for propane with experimental and ab initio W4 reference data:75 the experimental heat of formation at 298 K (−25.0 kcal/mol) is well-reproduced by a straight OM2 calculation (−24.4 kcal/mol at the ab initio reference geometry), whereas the W4 atomization energy (1007.9 kcal/mol) is well-reproduced by OM2 (1005.6 kcal/mol) only after applying the corrections described previously, which are far from negligible (51.8 kcal/mol). The distinction between relative energies and heats of formation is also relevant for the reaction energies in other fragmentations, e.g., bond dissociation reactions: while the corresponding ZPVE and thermal enthalpic corrections are much smaller than for atomization energies, they are still non-negligible.
In the next section, we will apply these SQC corrections to all benchmarks for atomization energies (especially W4-11-CHNOF). We will also investigate their effect on the reaction energies covered by the four subsets of the W4-11-CHNOF set and by those subsets of the GMTKN30-CHNOF and CE345-CHNOF sets that address fragmentation reactions. The SQC corrections are combined with single-point SQC results obtained at the ab initio reference geometries. The conversion of semiempirical heats of formation at 298 K to ZPVE-exclusive relative energies at 0 K is described in detail in the Supporting Information.
4. General Benchmark of Ground-State Properties
The OMx methods have been parametrized using the relatively small CHNO and FLUOR training sets (140 and 48 molecules, respectively).42 For these sets, the OMx results are generally found to be superior to the results from other SQC methods, as documented in detail in a companion Article.42 These data are not presented here again.
4.1. OVS7-CHNOF
Various OMx parametrization runs starting from different initial values were often found to produce different parameter sets that gave results of similar quality for the molecules in the training set; therefore we performed further tests on several validation sets to help identify the most suitable parameters.42 The main validation sets are collected in the OVS7-CHNOF benchmark which consists of seven subsets. Detailed numerical results are given in the SI (Tables S9–S14), while statistical evaluations in terms of mean absolute errors (MAEs) are provided in Table 3.
Table 3. Mean Absolute Errors in Calculated Heats of Formation (kcal/mol), Relative Energies (kcal/mol), Ionization Potentials (eV), Barriers (kcal/mol), Bond Lengths (Å), and Bond Angles (deg) for the OVS7-CHNOF Benchmark Set: MNDO, AM1, PMx, and OMx.
| method |
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| subset | N | MNDO | AM1 | PM3 | PM6 | PM7 | OM1 | OM2 | OM3 |
| Heats of Formation | |||||||||
| radicals71 | 42 | 11.86 | 10.60 | 10.94 | 11.13 | 10.04 | 7.28 | 4.98 | 5.57 |
| anions24 | 24 | 14.41 | 11.29 | 9.59 | 11.43 | 10.65 | 11.55 | 8.37 | 9.56 |
| cations41 | 36 | 11.52 | 9.96 | 11.45 | 11.12 | 12.07 | 9.32 | 6.93 | 6.89 |
| BIGMOL20 | 20 | 9.20 | 12.11 | 8.25 | 9.43 | 8.57 | 10.45 | 4.85 | 5.05 |
| conformers30 | 11 | 3.14 | 6.48 | 2.72 | 3.40 | 2.40 | 1.83 | 2.95 | 3.05 |
| isomers44 | 27 | 6.41 | 4.40 | 2.92 | 2.92 | 2.05 | 3.32 | 1.05 | 1.81 |
| fluorine91 | 91 | 11.13 | 11.05 | 7.76 | 9.04 | 8.17 | 7.17 | 7.15 | 7.34 |
| Relative Energies | |||||||||
| radicals71 | 4 | 13.00 | 10.09 | 9.94 | 11.78 | 10.61 | 8.74 | 3.95 | 5.46 |
| cations41 | 5 | 13.20 | 9.30 | 9.02 | 20.97 | 10.96 | 5.65 | 3.68 | 3.53 |
| isomers44 | 17 | 8.04 | 5.59 | 3.22 | 1.81 | 1.70 | 5.67 | 0.80 | 2.07 |
| Ionization Potentials | |||||||||
| radicals71 | 25 | 0.88 | 0.73 | 0.84 | 0.79 | 0.80 | 0.38 | 0.37 | 0.53 |
| Barriers | |||||||||
| conformers30 | 19 | 2.38 | 2.00 | 2.17 | 2.65 | 2.46 | 1.50 | 1.26 | 1.34 |
| Bond Lengths | |||||||||
| fluorine91 | 455 | 0.027 | 0.022 | 0.015 | 0.017 | 0.021 | 0.015 | 0.016 | 0.022 |
| Bond Angles | |||||||||
| fluorine91 | 355 | 3.41 | 3.28 | 2.94 | 3.68 | 3.17 | 1.97 | 2.04 | 1.78 |
With regard to heats of formation, OM2 and OM3 outperform all other SQC methods for six of the seven subsets, namely, radicals71,66 anions24,37 cations41,37 BIGMOL20,37,67 isomers44,39 and fluorine91;68 the corresponding MAEs for OM2 and OM3 are 5.0–5.6, 8.4–9.6, ca. 6.9, ca. 5.0, 1.1–1.8, and 7.2–7.3 kcal/mol, respectively. In the conformers3039 subset, OM1 gives the lowest MAE (1.8 kcal/mol) followed by PM7 (2.4 kcal/mol) and OM2 and OM3 (ca. 3.0 kcal/mol). Considering relative energies, OM2 and OM3 again outperform all other SQC methods in the case of the radicals71 and cations41 subsets, and they also give very good results for the isomers44 subset (MAEs of 0.8–2.1 kcal/mol).
Ionization potentials for the radicals71 subset (from Koopmans’ theorem) are best predicted by the three OMx methods, with MAEs of 0.4–0.5 eV. This is also true for the barriers in the conformers30 subset, for which the MAEs for the OMx methods range between 1.3 and 1.5 kcal/mol. In the fluorine91 subset, bond lengths are well-described by OM1, OM2, PM3, and PM6, while bond angles are best reproduced by the OMx methods.
Finally, we note that the inclusion of dispersion corrections must deteriorate the accuracy of the OMx methods for heats of formation, because the OMx parameters were optimized without such corrections. Since dispersion effects are always attractive, their subsequent introduction will systematically lower the computed heats of formation and thus increase their errors compared with experiment. On the other hand, dispersion corrections have no or very little effect on ionization potentials and barriers; they improve the relative energies for the isomers44 subset while leaving those for the radicals71 and cations41 subsets almost unaffected (for further details see the SI Table S1).
4.2. G2G3-CHNOF
The G269 and G370 sets provide well-established accurate reference data that have often been used to validate the performance of ab initio and DFT methods. They have been published after the development of OM1 and OM2. In previous work50 we have reported an evaluation of the OMx methods for the G2 and G3 sets, and we can thus be brief. The statistical results are summarized in Table 4 (for detailed numerical results, see SI Tables S16–S18). Overall, OM2 and OM3 perform best closely followed by PM7. OM2 yields the lowest MAEs for the heats of formations in G2 and G3 (3.2–3.4 kcal/mol) and for the relative energies in the alkanes28 subset41,71 of the G3 set (0.6 kcal/mol), while OM3 gives the lowest MAE for the heats of formations in alkanes28 (0.7 kcal/mol).
Table 4. Mean Absolute Errors in Calculated Heats of Formation (kcal/mol) and Relative Energies (kcal/mol) for the G2G3-CHNOF Benchmark Set: MNDO, AM1, PMx, and OMx.
| method |
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| subset | N | MNDO | AM1 | PM3 | PM6 | PM7 | OM1 | OM2 | OM3 |
| Heats of Formation | |||||||||
| G2 | 93 | 7.71 | 7.44 | 6.86 | 5.69 | 5.14 | 4.64 | 3.37 | 3.83 |
| G3 | 52 | 7.51 | 6.73 | 5.01 | 3.86 | 3.40 | 4.25 | 3.18 | 3.71 |
| alkanes28 | 22 | 3.26 | 8.81 | 2.03 | 4.20 | 1.76 | 2.16 | 1.91 | 0.72 |
| Relative Energies | |||||||||
| alkanes28 | 6 | 6.16 | 4.35 | 1.76 | 1.05 | 1.51 | 4.68 | 0.61 | 1.48 |
As pointed out before,50 the performance of OM2 and OM3 for the G2 and G3 sets is respectable, even when compared with standard DFT approaches, and there is no systematic error for alkane chains of increasing length that plagues some standard DFT functionals (including B3LYP).
Again, as in the OVS7-CHNOF set and for the same reasons, dispersion corrections deteriorate the accuracy of the OMx methods for heats of formation in the G2G3-CHNOF set, while improving relative energies (SI Table S2).
4.3. W4-11-CHNOF
The W4-11 benchmark set72 includes the W4-08 subset of the GMTKN30 set (see later discussion) and, in addition, contains numerous further atomization energies (collected in the extended subset TAE140) as well as four other subsets with reaction energies (BDE99, HAT707, ISOMER20, and SN13; see Table 1). We reduced the W4-11 to the W4-11-CHNOF data set in the usual manner, by eliminating entries for species containing elements other than H, C, N, O, and F. The SQC results from single-point calculations at the reference geometries are compared with the reference data obtained from ab initio total energies without zero-point vibrational corrections. For reasons discussed above, we converted the semiempirical heats of formation to atomization energies at 0 K by applying ZPVE and thermal enthalpic corrections. We also compared the results for the reaction energies in the subsets calculated with and without such corrections (see Table 5 for the statistical evaluation).
Table 5. Mean Absolute Errors in Calculated Atomization and Reaction Energies (kcal/mol) for the W4-11-CHNOF Benchmark Set: MNDO, AM1, PMx, and OMxa.
| method |
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| subset | N | MNDO | AM1 | PM3 | PM6 | PM7 | OM1 | OM2 | OM3 |
| TAE140 (corr) | 88 | 11.90 | 9.09 | 8.00 | 7.73 | 6.51 | 7.02 | 4.81 | 6.47 |
| TAE_nonMR124 (corr) | 80 | 9.45 | 8.26 | 7.28 | 7.73 | 6.75 | 6.25 | 4.84 | 6.05 |
| BDE99 | 79 | 17.24 | 12.75 | 13.77 | 12.19 | 10.01 | 10.54 | 8.15 | 9.65 |
| BDE99 (corr) | 79 | 14.91 | 10.86 | 12.22 | 10.56 | 8.26 | 8.28 | 6.25 | 7.51 |
| HAT707 | 394 | 20.85 | 16.50 | 12.72 | 13.96 | 11.64 | 10.55 | 8.92 | 9.44 |
| HAT707 (corr) | 394 | 21.08 | 16.70 | 12.74 | 13.89 | 11.52 | 10.83 | 9.17 | 9.73 |
| ISOMER20 | 19 | 8.65 | 7.65 | 7.68 | 9.37 | 8.48 | 8.67 | 8.54 | 8.32 |
| ISOMER20 (corr) | 19 | 8.90 | 7.77 | 8.09 | 9.23 | 8.33 | 8.47 | 8.34 | 8.13 |
| SN13 | 13 | 8.23 | 7.70 | 4.98 | 5.43 | 3.14 | 4.02 | 5.55 | 4.31 |
| SN13 (corr) | 13 | 7.30 | 6.01 | 4.05 | 4.39 | 3.59 | 5.14 | 5.36 | 4.98 |
(corr) means that energies are obtained by removing ZPVE and thermal corrections from the SQC results (see text).
The atomization energies in the TAE140 subset are computed with errors somewhat larger than typical errors for heats of formation. These errors are of similar magnitude in the OMx and PM7 methods (MAEs of 4.81–7.02 kcal/mol, OM2 lowest). The MAEs remain roughly of similar size when multireference cases are excluded (TAE_nonMR124, MAEs of 4.84–6.75 kcal/mol, OM2 lowest).
Errors in the corrected and uncorrected reaction energies for heavy-atom transfer (HAT707 subset), for isomerization (ISOMER20 subset), and for transformations in the SN13 subset are very close to each other, without any systematic improvement due to the corrections. This provides further justification for the usual practice of directly comparing the semiempirical SCF energies with ab initio relative energies in such cases. Thus, we will discuss for these sets and similar benchmark sets of this kind only the noncorrected reaction energies calculated from the semiempirical SCF energies.
On the other hand, removing the ZPVE and thermal corrections from the semiempirical SCF energies systematically improves the computed bond dissociation energies (BDE99 subset, MAE lowered by ca. 2 kcal/mol). This indicates that these corrections are generally relevant for fragmentation reactions, and hence we will discuss them for such transformations in the following.
The reference energies for bond dissociation and heavy-atom transfer are best reproduced by OM2 (MAEs of 6.25 and 8.92 kcal/mol) followed by OM3 (MAEs of 7.51 and 9.44 kcal/mol). Isomerization energies are computed reasonably well by all SQC methods considered (MAEs of 7.65–9.37 kcal/mol, AM1 lowest). The reaction energies in the SN13 subset are also well-reproduced, particularly by PM7, OM1, and OM3 (MAEs of 3.14, 4.02, and 4.31 kcal/mol, respectively).
The largest outliers in the computed atomization energies are found for C2 (OM1), isocyanic acid HNCO (OM2), HNNN (OM3), and H2 and N2 (PM7), with errors ranging from 21 to 35 kcal/mol.
Dispersion corrections have only very little effect on any of the statistical results reported presently (see SI Table S19).
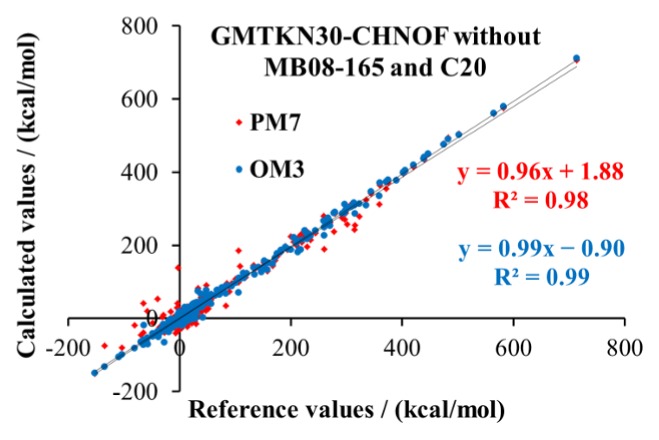
4.4. GMTKN30-CHNOF
The Grimme group published comprehensive collections of validation sets for general main group thermochemistry, kinetics, and noncovalent interactions,73,79,131 to allow for a thorough assessment of the performance of various DFT methods. In previous work,52 we extracted from the initial GMTKN24 database79 all species containing only the elements H, C, N, and O. For the resulting GMTKN24-hcno database, we performed calculations using the AM1, PM6, OM1, OM2, OM3, SCC-DFTB, B3LYP, and PBE methods and their dispersion-corrected counterparts to compare their accuracy in a comprehensive manner.52
Here we update our previous evaluation of SQC methods by starting from the more recent GMTKN30 database73,131 and extracting all species containing only the elements H, C, N, O, and F. The resulting GMTKN30-CHNOF database differs from the previous GMTKN24-hcno variant in the following aspects. (a) It incorporates fluorine-containing compounds. (b) It includes three new subsets ISOL22, BSR36, and ADIM6 from GMTKN30; the other three new subsets in GMTKN30 contain elements other than H, C, N, O, and F and are therefore disregarded. (c) It also includes the full W4-08 subset75 with multireference (MR) cases, in addition to the previously used W4-08woMR subset75 without MR cases. (d) It utilizes updated, more accurate reference data from the GMTKN30 database whenever available (in subsets IDISP, S22, BHPERI, and PA). Specifically, the GMTKN30 reference data for the BHPERI subset75,80−83 are now based on W1 rather than CBS-QB3 calculations;75 the latter had been employed in GMTKN24-hcno for some reaction barriers. The GMTKN30 reference values for the proton affinities of small molecules in the PA set are now taken from vibrationally back-corrected W1 data77 rather than from “best estimates“ from vibrationally back-corrected experimental values78 in GMTKN24-hcno.
Overall, the GMTKN30-CHNOF database consists of 24 subsets with 480 reference data that are determined from 806 single-point calculations at the reference geometries.73,131 Six subsets from the full GMTKN30 database are missing (AL2X, NBPRC, ALK6, RG6, HEAVY28, and CYCONF), because they consist of species containing elements not yet parametrized for the OMx methods.
Detailed numerical results for the GMTKN30-CHNOF database are documented in the Supporting Information. The corresponding statistical evaluations are given in Tables 6 and 7. Generally speaking, our present results support and generalize the conclusions on the performance of SQC methods obtained previously for the GMTKN24-hcno set.52
Table 6. Mean Absolute Errors (kcal/mol) for the GMTKN30-CHNOF Benchmark Set: MNDO, AM1, PMx, and OMxa.
| method |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| no. | subset | N | MNDO | AM1 | PM3 | PM6 | PM7 | OM1 | OM2 | OM3 |
| overall | 480 | 27.48 | 16.45 | 14.44 | 16.29 | 16.49 | 11.93 | 7.94 | 7.17 | |
| overall*b | 454 | 27.34 | 14.65 | 11.26 | 10.17 | 9.60 | 11.29 | 6.95 | 6.30 | |
| 1 | MB08-165 | 25 | 25.53 | 43.66 | 68.73 | 124.39 | 138.35 | 19.47 | 22.47 | 19.46 |
| 1 | MB08-165 (corr)c | 25 | 26.52 | 31.84 | 52.27 | 119.53 | 134.55 | 11.04 | 12.20 | 15.03 |
| 2 | W4-08 (corr) | 50 | 14.11 | 10.24 | 9.33 | 7.95 | 6.53 | 7.58 | 4.19 | 6.20 |
| 2a | W4-08woMRd (corr) | 43 | 10.32 | 8.90 | 7.66 | 8.00 | 6.51 | 6.00 | 4.12 | 5.37 |
| 3 | G21IP | 15 | 24.75 | 28.12 | 20.41 | 35.74 | 32.29 | 22.45 | 12.00 | 11.45 |
| 4 | G21EA | 12 | 27.44 | 25.84 | 14.23 | 20.08 | 18.94 | 24.81 | 11.39 | 9.31 |
| 5 | PA | 8 | 12.48 | 12.87 | 16.12 | 18.45 | 21.46 | 4.96 | 14.82 | 11.99 |
| 6 | SIE11 | 5 | 21.22 | 9.49 | 12.00 | 4.03 | 4.82 | 4.39 | 7.78 | 4.31 |
| 7 | BHPERI | 22 | 25.15 | 9.97 | 14.05 | 9.73 | 6.13 | 10.67 | 8.21 | 8.25 |
| 8 | BH76 | 54 | 23.84 | 13.06 | 13.36 | 13.39 | 13.68 | 10.39 | 9.72 | 10.66 |
| 9 | BH76RC | 22 | 11.88 | 13.49 | 11.57 | 15.72 | 16.28 | 5.28 | 4.29 | 5.37 |
| 10 | RSE43 | 34 | 5.00 | 3.55 | 4.02 | 6.10 | 5.83 | 3.74 | 4.31 | 5.24 |
| 11 | O3ADD6e | 6 | 14.90 | 10.57 | 9.87 | 2.03 | 26.84 | 4.01 | 12.24 | 10.97 |
| 12 | G2RC | 15 | 9.23 | 13.42 | 21.68 | 29.47 | 33.48 | 9.07 | 8.23 | 4.16 |
| 13 | ISO34 | 34 | 7.44 | 6.45 | 4.04 | 3.46 | 2.92 | 4.45 | 4.44 | 4.37 |
| 14 | ISOL22 | 18 | 16.76 | 10.25 | 8.27 | 7.41 | 6.55 | 7.99 | 5.31 | 6.05 |
| 15 | DC9 | 7 | 41.46 | 35.75 | 25.99 | 17.39 | 21.94 | 26.38 | 25.02 | 24.69 |
| 15a | DC9woC20f | 6 | 24.66 | 15.68 | 13.30 | 5.18 | 8.64 | 11.40 | 13.59 | 13.20 |
| 15b | C20g | 1 | 142.25 | 156.16 | 102.09 | 90.66 | 101.72 | 116.24 | 93.63 | 93.61 |
| 16 | DARC | 14 | 13.10 | 4.65 | 5.32 | 3.91 | 4.26 | 4.10 | 7.24 | 4.91 |
| 17 | BSR36 | 36 | 52.36 | 39.56 | 16.66 | 7.38 | 9.63 | 30.29 | 10.77 | 3.46 |
| 17 | BSR36 (corr)h | 36 | 56.86 | 44.24 | 18.53 | 14.33 | 17.40 | 35.01 | 7.08 | 1.90 |
| 18 | IDISP | 6 | 34.58 | 13.52 | 8.58 | 13.78 | 16.82 | 13.69 | 7.34 | 6.19 |
| 19 | WATER27 | 27 | 165.05 | 48.62 | 31.61 | 17.81 | 5.78 | 36.09 | 12.28 | 9.19 |
| 19 | WATER27 (upd)i | 27 | 164.26 | 47.83 | 30.82 | 17.02 | 6.51 | 35.30 | 11.49 | 8.40 |
| 20 | S22 | 22 | 16.74 | 6.78 | 5.91 | 3.37 | 0.76 | 5.10 | 3.05 | 3.54 |
| 21 | ADIM6 | 6 | 11.37 | 3.14 | 0.48 | 2.78 | 0.22 | 4.30 | 3.13 | 4.09 |
| 22 | PCONF | 10 | 10.08 | 5.35 | 3.68 | 2.27 | 2.97 | 3.60 | 1.28 | 1.33 |
| 23 | ACONF | 15 | 1.97 | 0.44 | 0.44 | 0.56 | 0.56 | 0.52 | 0.64 | 0.86 |
| 24 | SCONF | 17 | 17.52 | 2.39 | 3.05 | 2.61 | 2.38 | 5.87 | 1.67 | 1.32 |
(corr) means that energies are obtained by removing ZPVE and thermal corrections from the SQC results (see text).
Without MB08-165 and C20.
Upon geometry optimization, some of the artificial molecules adopted structures very different from the reference geometries so that the computed corrections may be less accurate in these cases (see text).
Subset W4-08 without multireference cases.
The adduct O3 + C2H2 is better described as an open-shell singlet at OMx-Dn.
Subset DC9 without C20 bowl/cage isomerization energy.
C20 bowl/cage isomerization energy.
For some SQC methods the corrections suffer from large accumulation errors (see text).
Reference dissociation energies of four (H2O)20 clusters were updated with more accurate values from ref (132) (see text).
Table 7. Mean Absolute Errors (kcal/mol) for the GMTKN30-CHNOF Benchmark Set: OMx-Dn Resultsa.
| method |
||||||||
|---|---|---|---|---|---|---|---|---|
| OM2 |
OM3 |
|||||||
| no. | subset | N | D2 | D3 | D3T | D2 | D3 | D3T |
| overall | 480 | 7.91 | 7.76 | 7.76 | 7.21 | 7.26 | 7.24 | |
| overall*b | 454 | 6.94 | 6.77 | 6.77 | 6.36 | 6.37 | 6.35 | |
| 1 | MB08-165 | 25 | 22.31 | 22.35 | 22.36 | 19.51 | 20.11 | 20.11 |
| 1 | MB08-165 (corr)c | 25 | 11.53 | 11.50 | 11.51 | 15.44 | 15.72 | 15.72 |
| 2 | W4-08 (corr) | 50 | 4.40 | 4.41 | 4.41 | 6.17 | 6.26 | 6.26 |
| 2a | W4-08woMRd (corr) | 43 | 4.39 | 4.41 | 4.41 | 5.34 | 5.51 | 5.51 |
| 3 | G21IP | 15 | 12.00 | 12.00 | 12.00 | 11.46 | 11.45 | 11.45 |
| 4 | G21EA | 12 | 11.40 | 11.39 | 11.39 | 9.33 | 9.31 | 9.31 |
| 5 | PA | 8 | 14.90 | 14.88 | 14.88 | 11.77 | 11.69 | 11.69 |
| 6 | SIE11 | 5 | 8.20 | 8.07 | 8.07 | 4.52 | 4.70 | 4.70 |
| 7 | BHPERI | 22 | 6.42 | 6.68 | 6.69 | 6.98 | 6.76 | 6.78 |
| 8 | BH76 | 54 | 9.69 | 9.71 | 9.71 | 10.83 | 10.93 | 10.93 |
| 9 | BH76RC | 22 | 4.22 | 4.22 | 4.22 | 5.43 | 5.48 | 5.48 |
| 10 | RSE43 | 34 | 4.16 | 4.24 | 4.24 | 5.03 | 5.12 | 5.12 |
| 11 | O3ADD6e | 6 | 12.70 | 12.61 | 12.61 | 11.12 | 11.39 | 11.38 |
| 12 | G2RC | 15 | 7.71 | 7.75 | 7.75 | 3.73 | 3.62 | 3.62 |
| 13 | ISO34 | 34 | 4.59 | 4.55 | 4.55 | 4.47 | 4.49 | 4.48 |
| 14 | ISOL22 | 18 | 5.04 | 4.96 | 4.95 | 6.28 | 6.23 | 6.17 |
| 15 | DC9 | 7 | 25.25 | 24.98 | 24.93 | 22.64 | 23.27 | 23.30 |
| 15a | DC9woC20f | 6 | 14.97 | 14.00 | 13.94 | 11.87 | 12.32 | 12.36 |
| 15b | C20g | 1 | 86.93 | 90.92 | 90.89 | 87.27 | 88.96 | 88.94 |
| 16 | DARC | 14 | 10.34 | 9.42 | 9.38 | 7.88 | 9.08 | 9.03 |
| 17 | BSR36 | 36 | 15.37 | 14.08 | 13.99 | 7.86 | 7.14 | 7.05 |
| 17 | BSR36 (corr)h | 36 | 11.71 | 10.37 | 10.28 | 4.35 | 3.48 | 3.40 |
| 18 | IDISP | 6 | 10.85 | 9.97 | 9.86 | 7.63 | 8.11 | 8.00 |
| 19 | WATER27 | 27 | 7.36 | 6.99 | 7.13 | 6.65 | 6.84 | 6.81 |
| 19 | WATER27 (upd)i | 27 | 6.57 | 6.20 | 6.34 | 6.42 | 7.52 | 7.38 |
| 20 | S22 | 22 | 1.14 | 0.91 | 0.94 | 1.21 | 0.97 | 0.95 |
| 21 | ADIM6 | 6 | 0.45 | 0.11 | 0.09 | 0.69 | 0.26 | 0.39 |
| 22 | PCONF | 10 | 0.94 | 1.11 | 1.02 | 1.25 | 1.54 | 1.39 |
| 23 | ACONF | 15 | 0.19 | 0.20 | 0.22 | 0.29 | 0.30 | 0.31 |
| 24 | SCONF | 17 | 1.57 | 1.63 | 1.62 | 1.35 | 1.35 | 1.34 |
(corr) means that energies are obtained by removing ZPVE and thermal corrections from the SQC results (see text).
Without MB08-165 and C20.
Upon geometry optimization, some of the artificial molecules adopted structures very different from the reference geometries so that the computed corrections may be less accurate in these cases (see text).
Subset W4-08 without multireference cases.
The adduct O3 + C2H2 is better described as an open-shell singlet at OMx-Dn.
Subset DC9 without C20 bowl/cage isomerization energy.
C20 bowl/cage isomerization energy.
For some SQC methods the corrections suffer from large accumulation errors (see text).
Reference dissociation energies of four (H2O)20 clusters were updated with more accurate values from ref (132) (see text).
Overall, the OMx and OMx-Dn methods provide the most accurate results. The MAEs for the entire GMTKN30-CHNOF set are lowest for OM3 (7.17 kcal/mol) followed by OM2 (7.94 kcal/mol), but are substantially higher for the PMx methods (14.44–16.49 kcal/mol) and AM1 (16.45 kcal/mol). Inclusion of dispersion corrections for OM2 and OM3 changes the overall MAEs only very slightly (OM3-Dn, 7.21–7.26 kcal/mol; OM2-Dn, 7.76–7.91 kcal/mol).
Compared to the previously reported uncorrected values in ref (52) (W4-08woMR subset), the semiempirical SCF atomization enthalpies at 298 K become much more accurate by correcting to ZPVE-exclusive atomization energies at 0 K (errors reduced by ca. 50%). The OMx methods remain the most accurate SQC methods in the W4-08woMR subset (MAEs of 4.12–6.00 kcal/mol). For the full W4-08 subset, the MAEs of the corrected atomization energies are somewhat higher at the OMx level (4.19–7.58 kcal/mol) and still slightly larger for PM6 and PM7 (6.53–7.95 kcal/mol). Dispersion corrections have only little effect on these atomization energies.
In the case of the MB08-165 subset,74 the ZPVE and thermal corrections reduce the errors for all of the methods except for MNDO. This subset is generally most challenging as it was especially designed to test the robustness of computational approaches. It consists of the dissociation energies of randomly generated species with unusual bonding situations to diatomics and hydrides. For some of the artificial molecules in this subset, the geometry optimization (needed to evaluate the ZPVE and thermal corrections) led to structures very different from the reference geometries (especially with MNDO), and hence to corrections that may be less accurate; the corrected values were included in the statistics also in these cases. The OMx methods have the smallest MAEs for both the uncorrected (19.46–22.47 kcal/mol, OM1 and OM3 lowest) and the corrected (11.04–15.03 kcal/mol, OM1 lowest) dissociation energies of the MB08-165 subset, while the MAEs for PM6 and PM7 exceed 100 kcal/mol.
In the case of the BSR36 subset, going from the SCF bond separation enthalpies at 298 K to the corresponding ZPVE-exclusive energies at 0 K reduces the errors only for the OM2, OM3, and OMx-Dn methods. The reason for the increased error with the other methods is the way in which this benchmark is constructed: bond separation energies refer to reactions of hydrocarbons with methane to produce ethane molecules; thus large numbers of CH4 and C2H6 molecules are involved (up to 22 and 18, respectively), and any errors in the computed ZPVE and thermal corrections for these two molecules will accumulate. This happens, for instance, in the case of PM7 where the MAE for the BSR36 subset drops from 17.40 to 9.08 kcal/mol (similar to the value of 9.63 kcal/mol without corrections) when using the ZPVE energies of methane and ethane calculated at the W4 level72 instead of the PM7 values. Such problems are not encountered with the OM2, OM3, and OMx-Dn methods which give realistic ZPVE and thermal corrections for methane and ethane. OM3 is most accurate for the BSR36 subset, both for the uncorrected (MAE of 3.46 kcal/mol) and the corrected (MAE of 1.90 kcal/mol) bond separation energies.
As noted previously,52 the cage/bowl isomerization of C20 fullerene is also a very challenging problem for SQC methods: the absolute error in the isomerization energy exceeds 85 kcal/mol for all SQC methods tested here. C20 is part of the DC9 subset with only seven members; to avoid misleading impressions because of the C20 outlier, we provide MAEs not only for the complete DC9 subset but also for DC9woC20 (DC9 without C20), as well as the errors for C20.
If we remove the most challenging test cases (MB08-165 and C20) from the overall statistics, the MAEs remain lowest for OM3 (6.30 kcal/mol) and OM2 (6.95 kcal/mol), whereas those for the PMx methods drop considerably (9.60–11.26 kcal/mol); AM1 benefits less (14.65 kcal/mol). Inclusion of dispersion corrections for OM2 and OM3 again affects their MAEs only slightly.
According to the statistical evaluations, the OMx and OMx-Dn methods are most accurate for barrier heights of hydrogen transfers, heavy-atom transfers, nucleophilic substitutions, unimolecular reactions, and association reactions (BH7684,85) and for the corresponding reaction energies (BH76RC84,85); and for selected reaction energies in the G2 set (G2RC69). The OM2, OM3, and OMx-Dn methods also perform best for adiabatic ionization potentials (G21IP76) and electron affinities (G21EA76); for isomerization energies of large organic molecules (ISOL2289); and for relative energies of phenylalanyl–glycyl–glycine tripeptide conformers (PCONF104) and of sugar conformers (SCONF79,106).
The lowest MAEs are provided for proton affinities (subset PA) by OM1 followed by OM3 and OM3-Dn; for systems with intramolecular dispersion interactions (IDISP88,98,99) by OM2, OM3, and OM3-Dn; for interaction energies between n-alkane dimers (ADIM697,103) by OMx-D3, OMx-D3T, and PM7; and for relative energies of alkane conformers (ACONF105) by OMx-Dn.
PM6 is statistically most accurate for systems with problems related to the DFT self-interaction error (SIE1179); for other molecules that are difficult cases for DFT (DC979,82,90−95 and DC9woC20); for reaction energies and barriers of ozone addition to C2H4 and C2H2 (O3ADD687); and for energies of Diels–Alder reactions (DARC96).
PM7 appears best suited for calculating barrier heights of pericyclic reactions (BHPERI); isomerization energies of small- and medium-sized organic molecules (ISO3488); and binding energies of noncovalently bound dimers (S22).101,102 In the latter case, the OMx-Dn methods are of similar accuracy (see section 5).
Benchmark energies for four (H2O)20 clusters used in the WATER27 subset (binding energies of water, H+(H2O)n, and OH–(H2O)n clusters)100 were taken from MP2/CBS calculations.133 Recently more accurate reference energies were obtained at the CCSD(T)/CBS(45) level of theory for those four complexes.132 When including these updated reference data, the PM7, OM2-Dn, and OM3-D2 methods perform similarly well (MAEs of 6.20–6.57 kcal/mol, OM2-D3 lowest).
Interestingly, radical stabilization energies (RSE4386) are best predicted by the venerable AM1 method closely followed by OM1. This demonstrates that the most recent SCQ models and parametrizations may not always be the best for a problem at hand. Hence, if possible, SQC methods should generally be calibrated against available experimental or high-level theoretical data for related systems before using them in actual applications.
As already discussed earlier the largest outliers (SI Figure S1 and Table S20) are often found for species in the MB08-165 subset; this is the case for PMx, OM2, and OM2-Dn, with especially large errors of more than 100 kcal/mol seen frequently with PMx. In the case of the OM3 and OM3-Dn methods, the largest errors are encountered for the isomerization energy of C20, followed by species in the MB08-165 subset. Other notable outliers (Figure 1) occur for OM1 in the WATER27 subset (four errors larger than 100 kcal/mol); for OM2 in the PA subset (error of −58 kcal/mol in the proton affinity of molecular hydrogen); and for OM3 in the O3ADD6 subset (error of 45 kcal/mol in the energy of adduct formation between O3 and C2H2).
Figure 1.

Error distribution of ground-state properties calculated at the OMx and PM7 levels for the GMTKN30-CHNOF benchmark set (excluding the MB08-165 subset and the isomerization energy of C20). The subsets are marked with alternating gray and white backgrounds, and their numbers correspond to those in Table 6.
According to the statistical evaluations, the OMx-Dn methods have essentially the same overall accuracy as the standard OMx methods. As expected, the dispersion corrections significantly improve the accuracy for systems, in which noncovalent interactions play a significant role. This is especially true for the WATER27, S22, and ADIM6 subsets. The barriers of pericyclic reactions (BHPERI subset) are also slightly improved when using the OMx-Dn methods, on average by more than 1 kcal/mol.
On the other hand, as already noted, inclusion of dispersion corrections (without any change in the standard OMx parameters) will lower the total energies systematically and thus tend to increase the errors in the calculated heats of formation. Interestingly, the dispersion corrections even deteriorate the results for the IDISP subset designed to capture the effects of intramolecular dispersion. Apparently, intramolecular noncovalent interactions of this kind are already largely taken into account (on average) by the general parametrization of OMx methods such that adding further dispersion interactions is detrimental.
Overall, the positive and negative effects of including empirical dispersion corrections (while retaining the standard OMx parameters) seem to cancel out in the case of the GMTKN30-CHNOF benchmark set so that the overall MAEs are similar for the OMx and OMx-Dn methods.
4.5. CE345-CHNOF
The chemistry energetic database with 345 entries (CE345)107,108 is a collection of accurate reference data used for the evaluation and parametrization of DFT functionals.108 We note that some of the reference data in the CE345 database are also present in the GMTKN30 database, although the actual reference values may differ slightly because of the use of different methodology for obtaining them.73
We reduced the CE345 database to the CE345-CHNOF set by accepting only species containing exclusively the elements H, C, N, O and/or F. Therefore, the SRMBE13 subset had to be excluded. The MRBE10 and DC9 subsets were also excluded from the error analysis because there are only two surviving species in each subset. Finally, we also disregarded the AE17 subset with atomic energies. The final CE345-CHNOF set consists of 11 subsets with a total of 187 entries.
The results from the statistical evaluations are summarized in Tables 8 and 9. Overall, the lowest MAEs for the CE345-CHNOF set are obtained from the OMx and OMx-Dn methods (6.17–7.91 kcal/mol). As expected, the inclusion of dispersion corrections improves the results for the NCCE31/05 subset targeting noncovalent complexation energies; this is also true for the subsets ABDE12, HC7/11, and πTC13 that contain systems in which dispersion plays a prominent role.
Table 8. Mean Absolute Errors (kcal/mol) for the CE345-CHNOF Benchmark Set: MNDO, AM1, PMx, and OMxa.
| method |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| no. | subset | N | MNDO | AM1 | PM3 | PM6 | PM7 | OM1 | OM2 | OM3 |
| overallb | 186 | 15.33 | 10.08 | 9.85 | 10.94 | 10.85 | 7.91 | 6.40 | 6.89 | |
| 1 | MGAE109/11 (corr) | 74 | 7.89 | 6.94 | 6.73 | 7.42 | 6.28 | 4.98 | 4.26 | 4.73 |
| 2 | IsoL6/11 | 6 | 9.25 | 4.63 | 3.74 | 2.41 | 2.05 | 7.68 | 1.99 | 3.22 |
| 3 | IP21 | 4 | 25.29 | 21.39 | 19.48 | 61.38 | 55.91 | 31.85 | 13.24 | 11.91 |
| 4 | EA13/03 | 4 | 28.56 | 21.36 | 11.75 | 16.67 | 18.20 | 31.16 | 9.80 | 9.18 |
| 5 | PA8/06 | 4 | 11.68 | 16.27 | 19.52 | 25.28 | 29.98 | 4.83 | 25.04 | 17.89 |
| 6 | ABDE12 (corr) | 12 | 28.94 | 17.92 | 21.64 | 18.39 | 19.92 | 8.19 | 8.98 | 10.52 |
| 7 | HC7/11 | 7 | 17.97 | 16.24 | 6.26 | 4.57 | 9.18 | 12.81 | 8.66 | 6.75 |
| 8 | πTC13 | 13 | 12.25 | 6.16 | 12.00 | 9.22 | 10.58 | 2.82 | 2.54 | 5.17 |
| 9 | HTBH38/08 | 26 | 25.36 | 10.79 | 9.52 | 10.50 | 11.77 | 11.29 | 4.96 | 5.99 |
| 10 | NHTBH38/08 | 23 | 22.16 | 15.16 | 15.66 | 16.44 | 15.53 | 9.60 | 13.64 | 14.18 |
| 11 | NCCE31/05b | 13 | 11.46 | 4.52 | 3.19 | 2.70 | 1.58 | 3.48 | 2.15 | 2.61 |
(corr) means that energies are obtained by removing ZPVE and thermal corrections from the SQC results (see text).
SCF calculations of the NH3···F2 complex in the NCCE31 subset could not be converged with OM2 and OM3; thus we excluded this complex from the statistics.
Table 9. Mean Absolute Errors (kcal/mol) for the CE345-CHNOF Benchmark Set: OMx-Dna.
| method |
||||||||
|---|---|---|---|---|---|---|---|---|
| OM2 |
OM3 |
|||||||
| no. | subset | N | D2 | D3 | D3T | D2 | D3 | D3T |
| overallb | 186 | 6.17 | 6.18 | 6.18 | 6.51 | 6.52 | 6.52 | |
| 1 | MGAE109/11 (corr) | 74 | 4.19 | 4.20 | 4.19 | 4.38 | 4.53 | 4.53 |
| 2 | IsoL6/11 | 6 | 2.26 | 2.19 | 2.16 | 3.15 | 3.28 | 3.28 |
| 3 | IP21 | 4 | 13.23 | 13.24 | 13.24 | 11.91 | 11.91 | 11.91 |
| 4 | EA13/03 | 4 | 9.80 | 9.80 | 9.80 | 9.18 | 9.18 | 9.18 |
| 5 | PA8/06 | 4 | 24.98 | 24.97 | 24.97 | 17.83 | 17.77 | 17.77 |
| 6 | ABDE12 (corr) | 12 | 7.32 | 7.77 | 7.78 | 8.97 | 8.90 | 8.91 |
| 7 | HC7/11 | 7 | 7.31 | 7.15 | 7.16 | 4.35 | 3.20 | 3.21 |
| 8 | πTC13 | 13 | 2.74 | 2.73 | 2.73 | 4.90 | 4.77 | 4.77 |
| 9 | HTBH38/08 | 26 | 4.95 | 4.96 | 4.96 | 6.44 | 6.61 | 6.61 |
| 10 | NHTBH38/08 | 23 | 13.55 | 13.58 | 13.58 | 14.05 | 14.07 | 14.07 |
| 11 | NCCE31/05b | 13 | 1.26 | 1.08 | 1.11 | 1.51 | 1.23 | 1.26 |
(corr) means that energies are obtained by removing ZPVE and thermal corrections from the SQC results (see text).
SCF calculations of the NH3···F2 complex in the NCCE31 subset could not be converged with OM2 and OM3; thus we excluded this complex from the statistics.
Looking at the statistical evaluation for the individual subsets, atomization energies (after correcting SCF atomization enthalpies at 298 K to ZPVE-exclusive energies at 0 K, MGAE109/11109,110) are found to be best reproduced by OMx and OMx-Dn methods (MAEs of 4.19–4.98 kcal/mol, OM2 and OM2-Dn lowest).
Isomerization energies (IsoL6/11111) are best reproduced by OM2 and PM7 (MAE of ca. 2.0 kcal/mol). We note that this subset contains the six smallest pairs of isomers from the ISOL22 subset of GMTKN30; their isomerization energies were calculated at a more accurate level of theory than those in ISOL22.
The subsets for adiabatic ionization potentials (IP21109,112−116) and electron affinities (EA13/03109,112−114) overlap with the corresponding GMTKN30-CHNOF subsets (G21IP and G21EA). In all cases, the OM2 and OM3 methods perform best (MAEs of 0.52–0.57 eV for IP21 and 0.40–0.42 eV for EA13/03), with substantially larger errors for the other SQC methods. As expected, dispersion corrections have essentially no effect on the OM2 and OM3 results for these properties.
All entries in the subset for proton affinities (PA8/0678) also appear in the PA subset of GMTKN30 (albeit with slightly different reference values). As in the PA case, OM1 has the lowest statistical error (MAE of 4.8 kcal/mol). Alkyl bond dissociation energies (ABDE12109,117−119) are best reproduced by the OMx-Dn and OMx methods (MAEs after accounting for ZPVE and thermal corrections of 7.32–10.52 kcal/mol and above 17 kcal/mol for the MNDO-type methods), while hydrocarbon chemistry (HC7/11119) is best described by OM3-Dn and PM6 (MAEs of 3.20–4.34 and 4.57 kcal/mol, respectively). The thermochemistry of π systems (πTC1378,112,117) is best treated by the OM2 and OM2-Dn methods (MAEs of 2.54 and 2.73–2.74 kcal/mol, respectively; the data set includes some proton affinities of polyenes that are part of the PA subset of GMTKN30).
The entries in the subsets for hydrogen and non-hydrogen transfer reactions (HTBH38/08 and NHTBH38/08)85,109,120,121 are also present in the BH76 subset of GMTKN30. OM2 and OM2-Dn perform best for the barrier heights to hydrogen transfer followed by OM3 and OM3-Dn (MAEs of 4.95–4.96 and 5.99–6.61 kcal/mol, respectively), while the barrier heights to non-hydrogen transfer are described less well (lowest MAE of OM1 9.60 kcal/mol).
As expected, noncovalent complexation energies (NCCE31/05113,122) are best reproduced by the OMx-Dn methods (MAEs of 1.08–1.51 kcal/mol) followed by PM7 (MAEs of 1.58 kcal/mol).
The error distributions of the OMx and PM7 methods with regard to the CE345-CHNOF set are shown in Figure 2. They appear similar overall; however, there are more outliers for PM7 compared with the OMx methods and especially with OM2.
Figure 2.

Error distribution of ground-state properties calculated at the OMx and PM7 levels for the CE345-CHNOF set. The subsets are marked with alternating gray and white backgrounds, and their numbers correspond to those in Table 8.
The largest outliers at the OMx and OMx-Dn levels have errors between 40 and 63 kcal/mol in the computed energies. These are the ionization energy of the carbon atom (OM1); the proton affinity of molecular hydrogen (OM2 and OM2-Dn); and the barrier of the HCN → HNC isomerization (OM3 and OM3-Dn); see the Supporting Information.
4.6. PDDG
The PDDG set was used in the development and validation of the PDDG/MNDO and PDDG/PM3 methods.31 It contains 979 experimental reference data for heats of formation, bond lengths, bond angles, dihedral angles, ionization potentials, and dipole moments of 622 closed-shell molecules (elements H, C, N, ond O). The PDDG set is divided in the original work into CH, CHN, CHO, and CHNO subsets. We use the same conventions for these subsets as in the original work. In addition, we introduce subsets for bond lengths, bond angles, and dihedral angles according to the elements involved.
In our evaluations, full geometry optimizations were performed for each molecule. Ionization potentials were calculated using Koopmans’ theorem. The resulting MAEs for the different properties are summarized in Table 10 for the MNDO, AM1, PMx, and OMx methods and in SI Table S3 for the OMx-Dn methods.
Table 10. Mean Absolute Errors in Calculated Heats of Formation (kcal/mol), Bond Lengths (Å), Bond Angles (deg), Dihedral Angles (deg), Ionization Potentials (eV), and Dipole Moments (D) for the PDDG Benchmark Set and Its Subsets: MNDO, AM1, PMx, and OMx.
| method |
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| subset | N | MNDO | AM1 | PM3 | PM6 | PM7 | OM1 | OM2 | OM3 |
| Heats of Formation | |||||||||
| overall | 622 | 8.04 | 6.72 | 4.24 | 4.05 | 3.34 | 5.51 | 3.55 | 3.68 |
| CH | 254 | 7.92 | 5.63 | 3.40 | 3.70 | 3.19 | 4.92 | 2.30 | 2.38 |
| CHN | 89 | 6.01 | 7.30 | 4.61 | 4.54 | 4.63 | 6.77 | 5.01 | 4.86 |
| CHO | 238 | 8.03 | 7.15 | 4.55 | 4.12 | 3.01 | 4.97 | 3.86 | 4.58 |
| CHNO | 41 | 13.25 | 9.70 | 6.93 | 4.76 | 3.41 | 9.53 | 6.24 | 3.96 |
| Bond Lengths | |||||||||
| overall | 153 | 0.013 | 0.016 | 0.011 | 0.012 | 0.011 | 0.012 | 0.014 | 0.018 |
| CH | 81 | 0.011 | 0.013 | 0.012 | 0.009 | 0.010 | 0.013 | 0.011 | 0.010 |
| CHN | 34 | 0.012 | 0.015 | 0.011 | 0.012 | 0.010 | 0.009 | 0.015 | 0.025 |
| CHO | 35 | 0.019 | 0.020 | 0.009 | 0.016 | 0.013 | 0.013 | 0.020 | 0.026 |
| CHNO | 3 | 0.024 | 0.038 | 0.009 | 0.026 | 0.018 | 0.010 | 0.027 | 0.044 |
| C–H | 38 | 0.008 | 0.011 | 0.008 | 0.010 | 0.012 | 0.009 | 0.010 | 0.013 |
| C–C | 52 | 0.012 | 0.015 | 0.012 | 0.011 | 0.010 | 0.013 | 0.010 | 0.010 |
| C=C | 15 | 0.012 | 0.011 | 0.010 | 0.008 | 0.007 | 0.013 | 0.009 | 0.009 |
| C≡C | 5 | 0.009 | 0.010 | 0.015 | 0.002 | 0.005 | 0.004 | 0.019 | 0.025 |
| N–C | 7 | 0.016 | 0.020 | 0.021 | 0.029 | 0.017 | 0.016 | 0.018 | 0.018 |
| N–H | 4 | 0.005 | 0.012 | 0.019 | 0.006 | 0.002 | 0.003 | 0.007 | 0.044 |
| N≡C | 5 | 0.009 | 0.007 | 0.006 | 0.002 | 0.004 | 0.005 | 0.017 | 0.046 |
| O–H | 6 | 0.012 | 0.013 | 0.010 | 0.032 | 0.025 | 0.011 | 0.037 | 0.058 |
| O–C | 7 | 0.018 | 0.015 | 0.015 | 0.024 | 0.011 | 0.015 | 0.018 | 0.022 |
| O=C | 9 | 0.014 | 0.021 | 0.006 | 0.006 | 0.007 | 0.011 | 0.015 | 0.019 |
| Bond Angles | |||||||||
| overall | 54 | 2.97 | 1.98 | 2.09 | 2.15 | 2.12 | 2.04 | 2.17 | 1.90 |
| CH | 20 | 1.57 | 0.73 | 0.89 | 0.88 | 1.12 | 1.14 | 1.37 | 0.94 |
| CHN | 12 | 1.94 | 1.42 | 1.88 | 1.74 | 1.80 | 1.90 | 1.30 | 1.33 |
| CHO | 21 | 4.46 | 3.21 | 3.12 | 3.33 | 3.04 | 2.66 | 3.08 | 2.76 |
| ∠CCH | 16 | 1.32 | 1.03 | 1.14 | 1.62 | 1.64 | 1.23 | 1.02 | 1.00 |
| ∠CCC | 13 | 2.11 | 0.96 | 1.17 | 0.78 | 1.06 | 1.27 | 1.78 | 1.19 |
| ∠OCH | 3 | 1.61 | 3.26 | 3.46 | 4.04 | 3.75 | 2.27 | 3.03 | 3.02 |
| ∠COH | 3 | 7.71 | 2.38 | 2.91 | 5.41 | 6.16 | 2.43 | 2.75 | 3.23 |
| ∠OCC | 5 | 2.18 | 2.06 | 2.38 | 2.13 | 2.01 | 1.62 | 2.59 | 1.81 |
| Dihedral Angles | |||||||||
| overall | 6 | 26.42 | 19.94 | 28.90 | 28.88 | 28.87 | 8.02 | 8.53 | 5.74 |
| CH (∠CCCC) | 3 | 3.40 | 5.16 | 5.83 | 5.39 | 5.83 | 5.20 | 6.00 | 5.43 |
| Ionization Potentials | |||||||||
| overall | 97 | 0.70 | 0.53 | 0.60 | 0.52 | 0.52 | 0.48 | 0.37 | 0.60 |
| CH | 41 | 0.74 | 0.49 | 0.61 | 0.48 | 0.54 | 0.50 | 0.35 | 0.57 |
| CHN | 21 | 0.61 | 0.46 | 0.51 | 0.52 | 0.55 | 0.40 | 0.28 | 0.44 |
| CHO | 31 | 0.67 | 0.60 | 0.60 | 0.53 | 0.45 | 0.46 | 0.42 | 0.75 |
| CHNO | 4 | 1.00 | 0.83 | 1.01 | 0.83 | 0.74 | 0.85 | 0.66 | 0.66 |
| Dipole Moments | |||||||||
| overall | 47 | 0.32 | 0.24 | 0.27 | 0.37 | 0.38 | 0.26 | 0.27 | 0.25 |
| CH | 10 | 0.24 | 0.15 | 0.15 | 0.26 | 0.26 | 0.10 | 0.11 | 0.11 |
| CHN | 14 | 0.52 | 0.46 | 0.38 | 0.52 | 0.55 | 0.43 | 0.25 | 0.30 |
| CHO | 20 | 0.20 | 0.12 | 0.22 | 0.34 | 0.34 | 0.23 | 0.34 | 0.27 |
| CHNO | 3 | 0.56 | 0.25 | 0.53 | 0.31 | 0.33 | 0.24 | 0.44 | 0.41 |
Considering heats of formation, the PDDG/PM3 method gives the lowest MAE for the PDDG set (3.2 kcal/mol), which is not too surprising since it was trained on this set.31 Slightly higher are the MAEs for PM7 (3.34 kcal/mol), OM2 (3.55 kcal/mol), and OM3 (3.68 kcal/mol), while those for the other SQC methods are above 4.0 kcal/mol. The OM2 and OM3 methods systematically overestimate the heats of formation in the PDDG set (mean signed errors (MSEs) of 0.92 and 1.08 kcal/mol, respectively), more so than PM7 (MSE of 0.49 kcal/mol). The error distribution is shown in Figure 3 for PM7 and the OMx methods. It is broadest for OM1 and rather similar for the other three methods. OM3 has the least number of outliers.
Figure 3.
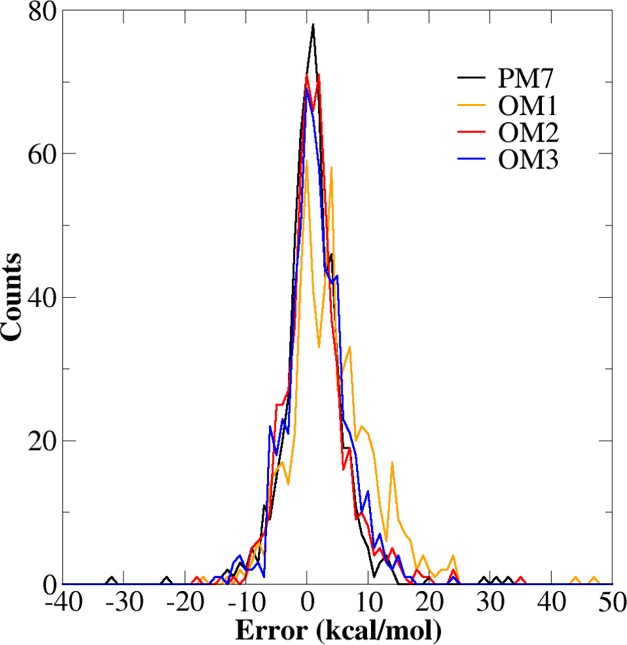
Error histogram of heats of formation calculated at the OMx and PM7 levels of theory for the PDDG benchmark set.
As noted previously, post-SCF dispersion corrections deteriorate the accuracy of heats of formation calculated by SQC methods parametrized at the SCF-MO level (compare Table 10 and SI Table S3). This shortcoming can be avoided by simultaneously fitting the parameters of the SQC method and of the dispersion corrections, as has been done in PM7;34 this may contribute to its good performance for the PDDG set.
Considering geometries, all tested SQC methods perform reasonably well for bond lengths (overall MAEs of 0.011–0.018 Å, lowest for PM3 and PM7). Except for MNDO, this is also true for bond angles (overall MAEs of 1.90–2.15°, lowest for OM3). Dihedral angles are described best by the OMx methods (overall MAEs of 5.7–8.5°, lowest for OM3).
Ionization potentials are reproduced best by OM2 (MAE of 0.37 eV); the other SQC methods have somewhat larger errors (0.48–0.70 eV). The MAEs in the calculated dipole moments are in the range between 0.24 and 0.38 D; the AM1, PM3, and OMx methods provide the best estimates (MAEs of 0.24–0.27 D).
4.7. PM7-CHNOF
The PM7-CHNOF benchmark set was assembled from the online database134 of reference data that have been used for the development and validation of the PM7 method.34 It includes all species of the database that consist only of the elements H, C, N, O, and F. It contains 1595 experimental and high-level ab initio reference values for heats of formation, bond lengths, bond angles, ionization potentials, and dipole moments of 1177 neutral and charged, open-shell and closed-shell species. It is divided into the DA, CH, CHN, CHO, CHF, and CHNO subsets. The diatomic DA subset consists of the H2, N2, O2 (triplet and singlet), and F2 molecules.
In our evaluations, full geometry optimizations were performed for each species. Ionization potentials were determined using Koopmans’ theorem. The resulting MAEs for the different properties are summarized in Table 11 for the MNDO, AM1, PMx, and OMx methods and in SI Table S4 for the OMx-Dn methods.
Table 11. Mean Absolute Errors in Calculated Heats of Formation (kcal/mol), Bond Lengths (Å), Bond Angles (deg), Dihedral Angles (deg), Ionization Potentials (eV), and Dipole Moments (D) for the PM7-CHNOF Benchmark Set and Its Subsets: MNDO, AM1, PMx, and OMx.
| method |
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| subset | N | MNDO | AM1 | PM3 | PM6 | PM7 | OM1 | OM2 | OM3 |
| Heats of Formation | |||||||||
| overall | 1168 | 11.26 | 9.30 | 5.51 | 4.43 | 3.78 | 8.25 | 4.85 | 4.83 |
| DA | 5 | 8.31 | 17.43 | 12.09 | 20.62 | 18.61 | 5.27 | 3.26 | 2.15 |
| CH | 310 | 13.67 | 9.85 | 5.23 | 4.72 | 4.18 | 9.23 | 4.16 | 3.63 |
| CHN | 214 | 7.63 | 7.19 | 5.32 | 3.73 | 3.42 | 7.90 | 5.43 | 5.69 |
| CHO | 373 | 9.26 | 8.03 | 5.42 | 4.33 | 3.40 | 6.43 | 4.22 | 5.18 |
| CHF | 32 | 6.73 | 8.15 | 6.25 | 3.77 | 4.54 | 2.42 | 3.50 | 2.44 |
| CHNO | 234 | 15.24 | 12.48 | 5.97 | 4.59 | 3.74 | 11.01 | 6.46 | 5.43 |
| Bond Lengths | |||||||||
| overall | 175 | 0.026 | 0.020 | 0.015 | 0.015 | 0.013 | 0.015 | 0.015 | 0.019 |
| DA | 4 | 0.079 | 0.056 | 0.039 | 0.043 | 0.033 | 0.023 | 0.036 | 0.053 |
| CH | 74 | 0.010 | 0.014 | 0.012 | 0.009 | 0.010 | 0.013 | 0.011 | 0.011 |
| CHN | 31 | 0.011 | 0.015 | 0.011 | 0.013 | 0.010 | 0.010 | 0.014 | 0.020 |
| CHO | 31 | 0.015 | 0.015 | 0.009 | 0.012 | 0.009 | 0.011 | 0.016 | 0.021 |
| CHF | 18 | 0.016 | 0.015 | 0.013 | 0.009 | 0.012 | 0.013 | 0.016 | 0.020 |
| CHNO | 17 | 0.146 | 0.061 | 0.037 | 0.043 | 0.032 | 0.043 | 0.028 | 0.040 |
| Bond Angles | |||||||||
| overall | 90 | 2.48 | 1.82 | 1.82 | 2.24 | 2.27 | 1.48 | 1.76 | 1.54 |
| CH | 32 | 1.61 | 1.01 | 0.76 | 2.14 | 2.33 | 0.64 | 0.90 | 0.49 |
| CHN | 20 | 1.62 | 1.19 | 1.53 | 0.96 | 1.08 | 1.39 | 1.44 | 1.35 |
| CHO | 20 | 3.47 | 2.08 | 2.33 | 2.31 | 2.12 | 1.79 | 2.33 | 1.98 |
| CHF | 11 | 2.59 | 2.38 | 1.43 | 2.35 | 2.18 | 1.45 | 1.47 | 1.11 |
| CHNO | 7 | 5.93 | 5.67 | 6.68 | 6.03 | 5.93 | 4.72 | 5.42 | 6.26 |
| Ionization Potentials | |||||||||
| overall | 104 | 0.59 | 0.49 | 0.50 | 0.42 | 0.42 | 0.40 | 0.33 | 0.53 |
| CH | 36 | 0.62 | 0.37 | 0.48 | 0.38 | 0.43 | 0.39 | 0.25 | 0.47 |
| CHN | 18 | 0.58 | 0.33 | 0.34 | 0.36 | 0.38 | 0.43 | 0.28 | 0.42 |
| CHO | 29 | 0.66 | 0.60 | 0.57 | 0.41 | 0.37 | 0.41 | 0.44 | 0.77 |
| CHF | 14 | 0.34 | 0.68 | 0.41 | 0.36 | 0.39 | 0.27 | 0.34 | 0.29 |
| CHNO | 5 | 0.70 | 0.67 | 0.78 | 0.64 | 0.59 | 0.59 | 0.47 | 0.55 |
| Dipole Moments | |||||||||
| overall | 58 | 0.34 | 0.25 | 0.27 | 0.35 | 0.33 | 0.26 | 0.24 | 0.22 |
| CH | 10 | 0.25 | 0.16 | 0.16 | 0.25 | 0.26 | 0.11 | 0.12 | 0.12 |
| CHN | 17 | 0.50 | 0.44 | 0.39 | 0.50 | 0.52 | 0.40 | 0.25 | 0.30 |
| CHO | 20 | 0.21 | 0.15 | 0.21 | 0.34 | 0.26 | 0.18 | 0.28 | 0.23 |
| CHF | 10 | 0.38 | 0.21 | 0.24 | 0.18 | 0.19 | 0.34 | 0.25 | 0.16 |
The PM7 method was trained and validated on this benchmark set, and it is thus not surprising that PM7 shows the best overall accuracy for the heats of formation (MAE of 3.78 kcal/mol, MSE of −0.18 kcal/mol); its error distribution is narrow and symmetric (Figure 4). The OMx methods tend to overestimate the heats of formation for this set, especially OM1 (Figure 4). OM2 and OM3 perform similarly (MAEs of 4.83–4.85 kcal/mol). They outperform PM7 in the DA and CHF subsets and have similar accuracy in the CH subset, while PM7 is superior in the CHN, CHO, and CHNO subsets. Again, for the same reasons as discussed before, post-SCF dispersion corrections deteriorate the accuracy of the heats of formation calculated by the OMx methods (compare Table 11 and SI Table S4).
Figure 4.
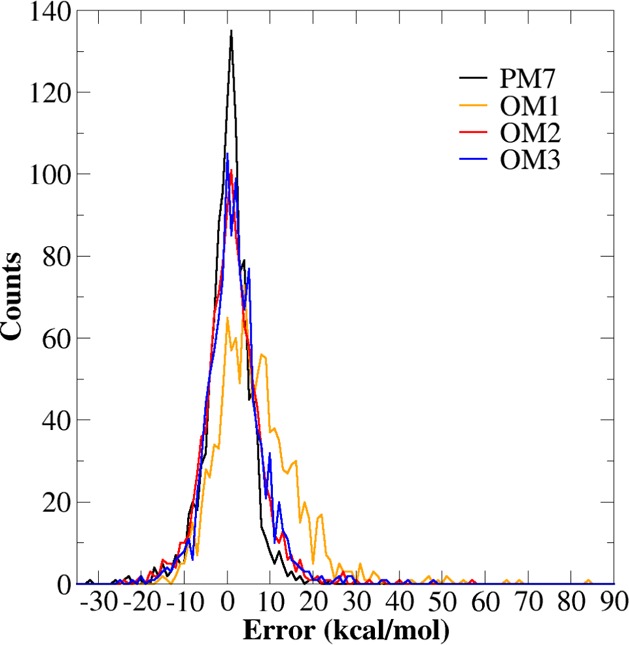
Error histogram for heats of formation calculated at the OMx and PM7 levels of theory for the PM7-CHNOF benchmark set.
Concerning the other properties, the conclusions for the PM7-CHNOF set are similar to those for the PDDG set (see preceding discussion). Bond lengths and bond angles are generally reproduced reasonably well by all SQC methods, and there are no great differences in the overall accuracy of the computed ionization potentials and dipole moments. For bond lengths, PM7 has the lowest overall MAE (0.013 Å) followed by PM3, PM6, OM1, and OM2 (0.015 Å in each case). The OMx methods have a slight overall advantage over the other SQC methods for bond angles (MAEs of 1.48–1.76°, OM1 lowest), ionization potentials (0.33–0.53 eV, OM2 lowest), and dipole moments (0.22–0.26 D, OM3 lowest).
4.8. C7H10O2
Accurate thermochemical calculations at the G4MP2135 level of theory were recently performed for a set of 6095 constitutional isomers C7H10O2.123 They were drawn from the chemical universe database GDB-17 that contains 166.4 billion molecules with up to 17 non-hydrogen atoms.136 Many of GDB-17 molecules belong to drug-like compounds.136
We derived atomization enthalpies at 298 K for these 6095 C7H10O2 isomers from the corresponding G4MP2 enthalpies reported in ref (123). The reported geometries were obtained at the B3LYP/6-31G(2df,p) level of theory,123 which is not considered accurate enough to be used as reference in our present benchmarking.
We performed full geometry optimizations for all these molecules. The statistical evaluation of the results is given in Table 12. The OMx and PMx methods perform similarly, with MAEs ranging between 6.30 and 8.92 kcal/mol (OM2 lowest, closely followed by PM7). Dispersion corrections have essentially no effect on the atomization enthalpies for the molecules in the C7H10O2 set (SI Table S26).
Table 12. Mean Absolute Errors in Calculated Atomization Enthalpies at 298 K (kcal/mol) for the C7H10O2 Benchmark Set.
| method |
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| subset | N | MNDO | AM1 | PM3 | PM6 | PM7 | OM1 | OM2 | OM3 |
| overall | 6095 | 9.27 | 13.43 | 7.92 | 7.26 | 6.44 | 8.92 | 6.30 | 7.67 |
4.9. RM1 Benchmark Results
Upon the request of a reviewer, we have carried out analogous RM1 calculations for all benchmark sets considered in preceding discussion. The statistical evaluations of the RM1 results are given in the Supporting Information (Tables S34–S41) along with a brief assessment. Overall RM1 tends to be generally more accurate than AM1, about as accurate as the PMx methods, and somewhat less accurate than the OMx methods.
5. Benchmarks of Noncovalent Interactions
Noncovalent interactions are difficult to describe for standard SQC methods which do not include dispersion in their formalism and often also have well-documented problems with hydrogen bonding. In this section, we will focus on the PM7 and OMx-Dn methods with explicit dispersion corrections, while presenting the results for the other (less accurate) SQC methods only in the Supporting Information.
We selected seven data sets from the literature to benchmark the accuracy of SQC methods for noncovalent interactions. As usual, we excluded in these sets the species containing elements for which OMx parameters are not yet available. Thus, three of the data sets were reduced: A24124 from 24 to 21 entries in A24-CHNOF, JSCH-2005101 from 143 to 134 entries in JSCH-2005-CHNOF, and S30L49 from 30 to 24 entries in S30L-CHNOF. The remaining four sets were not changed (S66, S66a8, S7L, and AF6). The resulting sets are briefly specified in Table 2.
In the following, we first report results from single-point calculations at the reference geometries for all seven data sets. Thereafter we present results from SQC geometry optimizations for the A24-CHNOF,124 S66,125 S7L,127 and AF6128 sets, for which high-level reference geometries are available. We note in this context that the S66a8 set is specifically designed for single-point calculations.125,126 The reference geometries in the JSCH-2005-CHNOF set are taken from different sources and include experimental, purely theoretical, and combined experimental/theoretical data,101,137 with purely theoretical geometries constituting only a minor part; many of these reference geometries are not minima in the gas phase, which would make it meaningless to compare them with optimized gas-phase SQC geometries.
In the validation studies with full geometry optimization, our focus is on the accuracy of the optimized SQC geometries. The reason is simple: if a method fails to predict the correct geometry for a noncovalent complex, the computed interaction energy will be meaningless. Hence, we primarily examine the most important intermolecular distances and angles for each complex (except for AF6 where we check the intramolecular geometry parameters in the folded conformation). The corresponding interaction energies are documented in the Supporting Information.
5.1. Single-Point Calculations
The statistical evaluation of the interaction energies obtained from single-point SQC calculations is given in Table 13 for the PM7 and OMx-Dn methods and in SI Table S5 for all other methods.
Table 13. Mean Absolute Errors of the Interaction Energies (kcal/mol) for the A24-CHNOF, S66, S66a8, JSCH-2005-CHNOF, S7L, and S30L-CHNOF Sets and of the Folding Enthalpies and Energies (kcal/mol) for the AF6 Set, Calculated at the Reference Geometries Using PM7 and OMx-Dn.
| method |
||||||||
|---|---|---|---|---|---|---|---|---|
| OM2 |
OM3 |
|||||||
| subset | N | PM7 | D2 | D3 | D3T | D2 | D3 | D3T |
| A24-CHNOF | ||||||||
| overall | 21 | 1.10 | 0.56 | 0.56 | 0.56 | 0.69 | 0.67 | 0.67 |
| overall*a | 20 | 0.64 | 0.43 | 0.43 | 0.43 | 0.49 | 0.47 | 0.46 |
| H-bonded | 5 | 2.66 | 1.39 | 1.40 | 1.40 | 1.36 | 1.32 | 1.32 |
| H-bonded*a | 4 | 0.73 | 0.97 | 0.95 | 0.95 | 0.52 | 0.46 | 0.46 |
| mixed | 10 | 0.49 | 0.19 | 0.17 | 0.17 | 0.41 | 0.34 | 0.34 |
| dispersion | 6 | 0.84 | 0.47 | 0.52 | 0.52 | 0.61 | 0.67 | 0.67 |
| S66 | ||||||||
| overall | 66 | 0.79 | 1.02 | 0.82 | 0.85 | 1.05 | 0.82 | 0.81 |
| electrostatic | 23 | 0.95 | 1.91 | 1.80 | 1.80 | 1.99 | 1.75 | 1.75 |
| mixed | 20 | 0.71 | 0.39 | 0.26 | 0.27 | 0.64 | 0.37 | 0.39 |
| dispersion | 23 | 0.70 | 0.68 | 0.32 | 0.39 | 0.45 | 0.27 | 0.23 |
| S66a8 | ||||||||
| overall | 528 | 0.78 | 0.77 | 0.60 | 0.61 | 0.80 | 0.62 | 0.60 |
| electrostatic | 184 | 0.84 | 1.44 | 1.34 | 1.34 | 1.52 | 1.33 | 1.33 |
| mixed | 160 | 0.60 | 0.32 | 0.22 | 0.22 | 0.48 | 0.26 | 0.26 |
| dispersion | 184 | 0.86 | 0.50 | 0.20 | 0.21 | 0.36 | 0.22 | 0.18 |
| JSCH-2005-CHNOF | ||||||||
| overall | 134 | 1.93 | 2.27 | 1.72 | 1.81 | 1.86 | 1.33 | 1.37 |
| overall*b | 128 | 1.83 | 2.07 | 1.52 | 1.61 | 1.62 | 1.10 | 1.13 |
| H-bonded base pairs | 31 | 2.62 | 3.50 | 3.11 | 3.05 | 3.12 | 2.53 | 2.47 |
| interstrand base pairs | 32 | 2.46 | 0.99 | 0.72 | 0.72 | 0.90 | 0.70 | 0.70 |
| stacked base pairs | 54 | 1.15 | 2.11 | 1.25 | 1.50 | 1.32 | 0.64 | 0.75 |
| amino acid pairs | 17 | 2.19 | 2.95 | 2.51 | 2.55 | 3.08 | 2.56 | 2.62 |
| amino acid pairs*b | 11 | 1.17 | 1.01 | 0.65 | 0.65 | 0.96 | 0.49 | 0.52 |
| S7L | ||||||||
| overall | 7 | 4.91 | 3.19 | 2.25 | 2.38 | 1.97 | 0.66 | 0.79 |
| π–π | 5 | 4.79 | 4.39 | 2.05 | 2.72 | 2.63 | 0.29 | 0.95 |
| S30L-CHNOF | ||||||||
| overall | 24 | 15.44 | 6.69 | 5.32 | 5.01 | 4.90 | 4.36 | 3.59 |
| π–π stacking | 7 | 18.54 | 6.13 | 3.58 | 3.04 | 4.20 | 5.08 | 3.17 |
| H-bondedc | 8 | 13.17 | 6.93 | 6.95 | 6.49 | 5.84 | 4.49 | 4.62 |
| charged complexesc | 8 | 21.76 | 9.55 | 9.46 | 8.41 | 5.71 | 6.38 | 4.56 |
| AF6 | ||||||||
| folding enthalpies | 6 | 1.18 | 0.20 | 0.45 | 0.34 | 0.79 | 0.76 | 1.00 |
| folding energies | 6 | 1.05 | 0.18 | 0.44 | 0.34 | 0.96 | 0.93 | 1.17 |
Without the HF···HF complex, see text.
Without the complexes of charged amino acids, see text.
Two complexes correspond to both H-bonded and charged complexes subsets; see ref (49).
The OMx-Dn methods perform best overall for the A24-CHNOF set (MAEs of 0.56–0.69 kcal/mol, OM2 lowest), while PM7 has somewhat larger errors (MAE of 1.10 kcal/mol). The most problematic case in A24-CHNOF is the HF···HF complex: at the nonoptimized reference geometry, the reference interaction energy is attractive (−4.57 kcal/mol); OM2 and OM2-Dn give only a rather weak attraction (−1.47 to −1.15 kcal/mol), whereas MNDO, PM6, PM7, OM1, OM3, and OM3-Dn yield a repulsive interaction energy (up to 5.81 kcal/mol in PM7); see the discussion in section 5.2. When this complex is disregarded in the statistics, the errors drop further (MAEs of OMx-Dn 0.43–0.49 kcal/mol and PM7 0.64 kcal/mol). Considering the subsets, the OMx-Dn methods outperform PM7 for complexes with dispersion-dominated and mixed electrostatic/dispersion interactions, and the OM3-Dn methods are best for H-bonded complexes.
In the S66 set, the PM7 and OMx-D3 methods are of similar accuracy (MAEs of 0.79 vs 0.82 kcal/mol), whereas the latter are superior in the S66a8 set (MAEs of 0.78 vs 0.60–0.62 kcal/mol); the OMx-D2 treatments are generally somewhat less accurate. In all three sets, the OMx-Dn methods outperform PM7 for the complexes with dispersion-dominated and mixed interactions.
In the JSCH-2005-CHNOF set (interaction energies of DNA base pairs and amino acid pairs), the OMx-D2 results are again slightly less accurate than the OMx-D3 results, and among the latter, OM3-D3 has generally the lowest or close-to-lowest errors. OM3-D3 also outperforms PM7 for the JSCH-2005-CHNOF set overall (MAEs of 1.33 vs 1.93 kcal/mol) as well as for two of the subsets (intrastrand and stacked base pairs), whereas the MAEs are similar in the case of H-bonded base pairs (MAEs of 2.53 vs 2.62 kcal/mol) and higher for OM3-D3 in the case of amino acid pairs (MAEs of 2.56 vs 2.19 kcal/mol). The largest outliers generally occur for pairs of oppositely charged amino acids: when these six complexes are excluded from the analysis, the errors drop substantially, as can be seen from the MAEs for OM3-D3 and PM7 for the neutral amino acid pairs (MAEs of 0.49 vs 1.17 kcal/mol) and for the overall set (MAEs of 1.10 vs 1.83 kcal/mol). Evidently, OM3-D3 performs best for the JSCH-2005-CHNOF set.
In a similar vein, OM3-D3 describes the σ–σ and π–π interactions for large complexes (S7L set) significantly better than any of the other SQC methods considered. The corresponding MAE value for OM3-D3 (0.66 kcal/mol) is small on an absolute scale and much lower than that for PM7 (4.91 kcal/mol).
The S30L set49 is an expansion of the S12L set48,138 and consists of very large noncovalent complexes, which are deemed useful to check ”against overfitting to too small molecular cases”.48 The OMx-Dn methods perform fairly well (MAEs of 3.59–6.69 kcal/mol, OM3-D3T lowest). They outperform other SQC methods investigated here and in ref (49), and especially PM7, which strongly overestimates most interaction energies (MAE of 15.44 kcal/mol). The OMx-Dn methods generally describe π–π stacking interactions better than H-bonding interactions; however, even in the latter case, they are more accurate than PM7, which appears to suffer from an accumulation of errors in the very large noncovalent complexes of the S30L-CHNOF set. The largest outliers in the OMx-Dn results are found for positively charged complexes, as previously reported for OM2-D3 and OM3-D3 in the case of the S12L set.48
The AF6 set provides reference folding energies and enthalpies for a series of alkanes.128 The PM7 values should be compared to the enthalpies, since PM7 was parametrized against heats of formation. In the case of the OMx-Dn methods, one may argue that the computed values can be considered as enthalpies or as energies, since the parameters for the underlying OMx methods and for the dispersion corrections were calibrated against heats of formation and interaction energies, respectively. To be unbiased, we provide data for both types of comparisons, and the results differ only very slightly (see Table 13). The PM7 and OMx-Dn methods have rather small errors both for the folding enthalpies (MAEs of 0.20–1.18 kcal/mol, OM2-D2 lowest, PM7 highest) and energies (MAEs of 0.18–1.05 kcal/mol, OM2-D2 lowest, PM7 highest). However, more important than these statistics is a qualitative finding: only OM2-D2, OM3-D2, and OM3-D3 correctly predict that the hairpin conformation of alkanes becomes more favorable than the linear conformation for 17 ± 1 carbon atoms, in agreement with experiment,139 while the hairpin conformation is more favorable already for alkanes with less than 16 carbon atoms in the case of PM7 and OM2-D3.
Finally, we briefly compare the effect of the different dispersion corrections applied in the OMx-Dn calculations. In the large majority of cases, the D3 correction turns out to be superior to the D2 correction, albeit often only by a small margin. The role of three-body corrections is negligible in small complexes (A24-CHNOF, S66, and S66a8 sets) and still quite small but not entirely negligible in hydrogen-bonded and stacked base pairs (JSCH-2005-CHNOF) and in folded alkanes (AF6). As expected, three-body corrections become significant in stacked systems modeling π–π interactions between graphene layers (S7L) and in very large noncovalent complexes (S30L-CHNOF). Since OM2-D3 and OM3-D3 underestimate the attractive π–π dispersion interactions, the repulsive three-body corrections increase the corresponding errors (on average by 0.7 kcal/mol). On the other hand, OM2-D3 and OM3-D3 tend to overestimate the binding energies in most of the very large noncovalent complexes, and hence the repulsive three-body corrections decrease the error in this case by 0.31–0.77 kcal/mol for the S30L set, as already reported earlier for the S12L set48 (OM2-D3 and OM3-D3) and for the S30L set49 (OM2-D3).
5.2. Geometry Optimizations
Full geometry optimizations were carried out for the A24-CHNOF, S66, S7L, and AF6 sets using all SQC methods examined presently.
The ab initio reference geometries were taken from published work at the following levels: CCSD(T)/CBS extrapolation for A24-CHNOF;124 counterpoise-corrected MP2/cc-pVTZ for S66 with intermolecular distances obtained by interpolating estimated CCSD(T)/CBS energies along dissociation curves;125,137 intermolecular distances optimized at the CCSD(T)/ha-cc-pVDZ level with monomer geometries optimized at the B3LYP level with large basis sets for S7L;127 and MP2/cc-pVTZ for AF6.128 In the original work on the A24 set, three π–π stacked complexes were not optimized but constrained artificially;124 therefore they are not considered here.
The statistical evaluation of selected optimized interatomic distances and bond angles is presented in Table 14 for the PM7 and OMx-Dn methods and in SI Table S6 for the other SQC methods. The corresponding statistical data for the associated interaction energies are documented in the Supporting Information (Tables S7 and S8). We note that some of the noncovalent complexes are present in several data sets, with slightly different reference geometries.
Table 14. Mean Absolute Errors in Selected Interatomic Distances (Å) and Angles (deg) for the A24-CHNOF, S66, S7L, and AF6 Benchmark Sets for Geometries Optimized with PM7 and OMx-Dn.
| method |
||||||||
|---|---|---|---|---|---|---|---|---|
| OM2 |
OM3 |
|||||||
| subset | N | PM7 | D2 | D3 | D3T | D2 | D3 | D3T |
| A24-CHNOF | ||||||||
| Selected Interatomic Distances | ||||||||
| overall | 23 | 0.375 | 0.472 | 0.500 | 0.500 | 0.322 | 0.261 | 0.262 |
| H-bonded | 5 | 0.365 | 0.202 | 0.197 | 0.198 | 0.333 | 0.335 | 0.335 |
| mixed | 13 | 0.168 | 0.421 | 0.413 | 0.413 | 0.258 | 0.324 | 0.326 |
| dispersion | 5 | 0.926 | 0.876 | 1.027 | 1.027 | 0.477 | 0.023 | 0.023 |
| Selected Angles | ||||||||
| overall | 40 | 12.86 | 14.83 | 9.86 | 9.97 | 12.79 | 8.69 | 8.80 |
| H-bonded | 13 | 14.83 | 11.15 | 11.39 | 11.39 | 11.08 | 11.07 | 11.08 |
| mixed | 21 | 7.31 | 6.57 | 6.46 | 6.45 | 13.01 | 9.35 | 9.56 |
| dispersion | 6 | 28.00 | 51.74 | 18.46 | 19.18 | 15.73 | 1.19 | 1.20 |
| S66 | ||||||||
| Selected Interatomic Distances | ||||||||
| overall | 172 | 0.407 | 0.381 | 0.348 | 0.348 | 0.290 | 0.320 | 0.317 |
| electrostatic | 28 | 0.083 | 0.181 | 0.173 | 0.173 | 0.308 | 0.333 | 0.283 |
| mixed | 63 | 0.449 | 0.433 | 0.415 | 0.415 | 0.303 | 0.344 | 0.361 |
| dispersion | 81 | 0.486 | 0.409 | 0.356 | 0.356 | 0.273 | 0.297 | 0.295 |
| Selected Angles | ||||||||
| overall | 141 | 15.83 | 11.68 | 12.27 | 12.32 | 11.32 | 13.15 | 12.99 |
| electrostatic | 28 | 6.29 | 14.41 | 13.37 | 13.30 | 11.83 | 12.37 | 9.97 |
| mixed | 52 | 25.14 | 14.28 | 14.50 | 14.54 | 15.74 | 18.95 | 19.73 |
| dispersion | 61 | 12.28 | 8.22 | 9.86 | 9.97 | 7.32 | 8.56 | 8.63 |
| S7L | ||||||||
| Selected Interatomic Distances | ||||||||
| overall | 28 | 0.325 | 0.510 | 0.428 | 0.420 | 0.390 | 0.389 | 0.393 |
| C···C | 20 | 0.310 | 0.547 | 0.433 | 0.421 | 0.408 | 0.467 | 0.468 |
| H···H | 8 | 0.361 | 0.419 | 0.415 | 0.416 | 0.345 | 0.193 | 0.207 |
| AF6 | ||||||||
| Selected Interatomic Distances | ||||||||
| overall | 27 | 0.090 | 0.140 | 0.169 | 0.168 | 0.121 | 0.168 | 0.165 |
| Selected Angles | ||||||||
| overall | 74 | 6.58 | 5.17 | 6.48 | 6.42 | 6.42 | 7.05 | 7.03 |
We start with some general remarks before discussing individual data sets and complexes. The selected distances and angles mostly refer to intermolecular geometrical parameters, e.g., distances defining the separation between the monomers or the length of hydrogen bonds and angles characterizing the relative orientation of the monomers in a complex. The associated minima on the potential energy surface are usually rather extended and flat, and hence the deviations of the SQC results for the geometrical parameters from the ab initio reference values are expected to be much larger than in the intramolecular case. Inspection of Table 14 confirms that this is indeed the case: the MAEs are typically of the order of 0.3 Å for the intermolecular distances and 10° for the angles.
The overall statistics in Table 14 for the OMx-Dn methods indicate that the OM3-based results are mostly superior to the OM2-based results, the D3 correction normally performs better than the D2 correction, and the three-center dispersion correction usually has no significant influence on the geometries of the rather small complexes considered here (D3 vs D3T). Hence, we will focus on the OM3-D3 results in the following.
In an overall assessment, OM3-D3 outperforms PM7 for the A24-CHNOF set (MAEs of 0.26 vs 0.38 Å and 8.7 vs 12.9°) and the S66 set (MAEs of 0.32 vs 0.41 Å and 13.2 vs 15.8°), while it shows somewhat larger errors for the S7L set (MAEs of 0.39 vs 0.33 Å) and the AF6 set (MAEs of 0.17 vs 0.09 Å and 7.1 vs 6.6°). OM3-D3 works especially well for the dispersion-dominated complexes in the A24-CHNOF set (MAEs of 0.02 Å and 1.2°).
We now address individual sets and complexes. In the case of the A24-CHNOF set, all SQC methods considered give qualitatively reasonable complex geometries in general (realistic shape and intermolecular distances within ±1 Å of the reference values), except for MNDO which often fails qualitatively and yields much too large intermolecular separations.
A problematic case is the HF dimer: the reference geometry has a linear hydrogen bond (F–H···F), which is also found with PM7 (but much too long) whereas the OMx and OMx-Dn methods give cyclic hydrogen-bonded structures (SI Figure S2). If the angle F–H···F is constrained to the reference value, the HF molecules drift apart upon SQC geometry optimization: the hydrogen bond is too long by 0.16–0.18 Å at OM2 and OM2-Dn, by 0.51–0.56 Å at OM3 and OM3-Dn, and by 1.04–1.05 Å at PM6 and PM7. Thus, when calculations are run on the reference geometry, the SQC methods either underestimate the binding energy (OM2 and OM2-Dn) or even predict a repulsive interaction (OM3, OM3-Dn, and especially PM6 and PM7; see section 5.1). As discussed previously,42 the methane dimer is also problematic: OM3-D3 gives a qualitatively correct geometry, while other SQC methods fail in this regard.
The reference interaction energies are derived in the original work on the A24 set124 without accounting for the deformation of the monomers (i.e., from single-point calculations on monomers at the geometry of the respective complex). However, for small monomers such as those in the A24 set, these deformation energies are negligible, and the interaction energies computed with and without them are essentially the same for all tested SQC methods (SI Table S27).
In the S66 set, the OMx-Dn methods again give the lowest MAEs for the distances and angles, but there are some notable outliers, in particular the benzene···AcOH (OH-π), H2O···MeOH, C6H6···MeNH2, peptide···peptide, and T-shaped pyridine dimer complexes (for their optimized reference and SQC structures see SI Figures S3–S7). The PM7 geometries are reasonable for some of these complexes, presumably due to the inclusion of special H-bond correction terms (see SI Figures S3–S7). Finally, a severe failure of the OM3 and OM3-Dn methods is that they give symmetric structures for carboxylic acid dimers, in which two hydrogen atoms are equidistant to the oxygen atoms in the corresponding hydrogen bonds. This is found for the acetic acid dimer in the S66 set and for the formic acid dimer in the validation of OMx and OMx-Dn methods.42 Since the monomer moieties in such dimers are strongly distorted, their interaction energies are strongly overestimated (see SI Table S28 for acetic acid dimer).
In the S7L set, PM7 gives overall the best intermolecular distances; OM3-D3 has slightly smaller errors for the H···H distances. A specific problem is that PM7 optimizes the parallel displaced naphthalene dimer to the sandwich conformation, contrary to OMx-Dn.
In the AF6 set, the overall statistical errors of the PM7 and OMx-Dn methods are rather similar, with slight advantages for PM7. All SQC methods without explicit dispersion corrections fail qualitatively since MNDO, AM1, PM6, and the OMx methods predict the most favorable conformation to be linear for all alkanes while PM3 predicts the opposite. Only the OM2-D2 and OM3-Dn methods give the correct result that the linear conformation dominates for short chain lengths and that the hairpin conformation becomes more favorable for alkanes with 17 ± 1 carbon atoms, in agreement with experiment.139
6. Conclusions
At the end of such a large and diverse benchmark study, it is impossible to provide a single unbiased “grand error estimate” for each SQC method. The chosen benchmark sets strongly differ in their size and in the quality of the reference data, and they address different properties. Thus, instead of attempting to provide such grand error estimates, we summarize the results for all subsets and identify the methods which are best for a given subset/property combination and which are most robust in general. This information can be used as a guide for selecting the SQC method that performs best for applications of the kind considered here.
Tables 15 and 16 present the benchmark results for ground-state properties and noncovalent interactions, respectively; they list the two top-performing methods with their MAEs for all data sets and subsets (as described in Tables 1 and 2).
Table 15. Methods with Lowest MAEs in Calculated Ground-State Propertiesa.
| data set | N | properties | first best | MAE | second best | MAE |
|---|---|---|---|---|---|---|
| OVS7-CHNOF | ||||||
| radicals71 | 42 | heats of formation | OM2 | 4.98 | OM3 | 5.57 |
| 4 | relative energies | OM2 | 3.95 | OM3 | 5.46 | |
| 25 | ionization potentialsd | OM1, OM2 | 0.37–0.38 | OM3 | 0.53 | |
| anions24 | 24 | heats of formation | OM2 | 4.98 | OM3 | 5.57 |
| cations41 | 36 | heats of formation | OM2 | 4.98 | OM3 | 5.57 |
| 5 | relative energies | OM3 | 3.53 | OM2 | 3.68 | |
| BIGMOL20 | 20 | heats of formation | OM2 | 4.85 | OM3 | 5.05 |
| conformers30 | 11 | heats of formation | OM1 | 1.83 | PM7 | 2.40 |
| 19 | barriers | OM2, OM3 | 1.26–1.34 | OM1 | 1.50 | |
| isomers44 | 27 | heats of formation | OM2 | 1.05 | OM3 | 1.81 |
| 17 | relative energies | OM2-D3, OM2-D3T | 0.69–0.70 | OM2, OM2-D2 | 0.80–0.82 | |
| fluorine91 | ||||||
| 91 | heats of formation | OM1, OM2 | 7.15–7.17 | OM3 | 7.34 | |
| 455 | bond lengths | OM1, OM2, PM3 | 0.015–0.016 | PM6 | 0.017 | |
| 355 | bond angles | OMx | 1.78–2.04 | MNDO, AM1, PMx | 2.94–3.68 | |
| G2G3-CHNOF | ||||||
| G2-CHNOF | 93 | heats of formation | OM2 | 3.37 | OM3 | 3.83 |
| G3-CHNOF | 52 | heats of formation | OM2 | 3.18 | PM7 | 3.40 |
| alkanes28 | 22 | heats of formation | OM3 | 0.72 | PM7 | 1.76 |
| 6 | relative energies | OM2-Dn | 0.21–0.24 | OM2 | 0.61 | |
| W4-11-CHNOF | ||||||
| TAE140 | 88 | atomization energies | OM2b | 4.81 | OM3b, PM7 | 6.47–6.51 |
| TAE_nonMR124 | 80 | atomization energies | OM2b | 4.84 | OM3b | 6.05 |
| BDE99 | 79 | reaction energies | OM2b | 6.25 | OM3b | 7.51 |
| HAT707 | 394 | reaction energies | OM2b | 8.92 | OM3b | 9.44 |
| ISOMER20 | 19 | reaction energies | OM3b | 8.32 | PM7, OM2b | 8.48–8.54 |
| SN13 | 13 | reaction energies | PM7 | 3.14 | OM1 | 4.02 |
| GMTKN30-CHNOF | 480 | energies | OM3b | 7.17 | OM2b | 7.94 |
| MB08-165 | 25 | reaction energies | OM3b, OM1 | 19.46–19.47 | OM2b | 22.47 |
| W4-08 | 50 | atomization energies | OM2b | 4.19 | OM3b | 6.20 |
| W4-08woMR | 43 | atomization energies | OM2b | 4.12 | OM3b | 5.37 |
| G21IP | 15 | adiabatic ionization potentials | OM3b | 11.45 | OM2b | 12.00 |
| G21EA | 12 | adiabatic electron affinities | OM3b | 9.31 | OM2b | 11.39 |
| PA | 8 | adiabatic proton affinities | OM1 | 4.96 | OM3b | 11.99 |
| SIE11 | 5 | reaction energies | PM6 | 4.03 | OM3b, OM1 | 4.31–4.39 |
| BHPERI | 22 | barriers | PM7 | 6.13 | OM2-D2 | 6.42 |
| BH76 | 54 | barriers | OM2b | 9.72 | OM1 | 10.39 |
| BH76RC | 22 | reaction energies | OM2b | 4.29 | OM1, OM3b | 5.28–5.37 |
| RSE43 | 34 | reaction energies | AM1 | 3.55 | OM1 | 3.74 |
| O3ADD6 | 6 | reaction energies and barriers | PM6 | 2.03 | OM1 | 4.01 |
| G2RC | 15 | reaction energies | OM3b | 4.16 | OM2b | 8.23 |
| ISO34 | 34 | relative energies | PM7 | 2.92 | PM6 | 3.46 |
| ISOL22 | 18 | relative energies | OM2b | 5.31 | OM3b | 6.05 |
| DC9woC20c | 6 | relative and reaction energies | PM6 | 5.18 | PM7 | 8.64 |
| DARC | 14 | reaction energies | PM6 | 3.91 | OM1 | 4.10 |
| BSR36 | 36 | reaction energies | OM3 | 3.46 | PM6 | 7.38 |
| IDISP | 6 | interaction and reaction energies | OM3 | 6.19 | OM2 | 7.34 |
| WATER27 | 27 | interaction energies | PM7 | 5.78 | OM3-D2 | 6.65 |
| S22 | 22 | interaction energies | PM7 | 0.76 | OMx-D3, OMx-D3T | 0.91–0.97 |
| ADIM6 | 6 | interaction energies | OM2-D3, OM2-D3T | 0.09–0.11 | PM7, OM3-D3 | 0.22–0.26 |
| PCONF | 10 | relative energies | OM2b, OM3b | 1.28–1.33 | PM6 | 3.27 |
| ACONF | 15 | relative energies | OMx-Dn | 0.19–0.31 | AM1, PM3, OM1 | 0.44–0.52 |
| SCONF | 17 | relative energies | OM3b | 1.32 | OM2b | 1.67 |
| CE345-CHNOF | 187 | energies | OM2b | 6.40 | OM3b | 6.89 |
| MGAE109/11 | 74 | atomization energies | OM2b | 4.26 | OM3b | 4.73 |
| IsoL6/11 | 6 | relative energies | OM2b, PM7 | 1.99–2.05 | PM6 | 2.41 |
| IP21 | 4 | adiabatic ionization potentials | OM3b | 11.91 | OM2b | 13.24 |
| EA13/03 | 4 | adiabatic electron affinities | OM3b | 9.18 | OM2b | 9.80 |
| PA8/06 | 4 | proton affinities | OM1 | 4.83 | MNDO | 11.68 |
| ABDE12 | 12 | dissociation energies | OM2-D2 | 7.32 | OM2-D3, OM3-D3T | 7.77–7.78 |
| HC7/11 | 7 | reaction energies | OM3-D3, OM3-D3T | 3.20–3.21 | OM3-D2 | 4.35 |
| πTC13 | 13 | relative energies and proton affinities | OM2b | 2.54 | OM1 | 2.82 |
| HTBH38/08 | 26 | barriers | OM2b | 4.96 | OM3b | 5.99 |
| NHTBH38/08 | 23 | barriers | OM1 | 9.60 | OM2b | 13.64 |
| NCCE31/05 | 14 | interaction energies | OM2-D3, OM2-D3T | 1.08–1.11 | OM3-D3, OM3-D3T, OM2-D2 | 1.23–1.26 |
| PDDG | 622 | heats of formation at 298 K | PM7 | 3.34 | OM2 | 3.55 |
| 153 | bond lengths | all | 0.011–0.018 | |||
| 54 | bond angles | all | 1.90–2.97 | |||
| 6 | dihedral angles | OM3b | 5.74 | OM1, OM2b | 8.02–8.53 | |
| 97 | ionization potentialsd | OM2b | 0.37 | OM1 | 0.48 | |
| 47 | dipole moments | all | 0.24–0.32 | |||
| PM7-CHNOF | 1168 | heats of formation at 298 K | PM7 | 3.78 | PM6 | 4.43 |
| 175 | bond lengths | all | 0.013–0.026 | |||
| 90 | bond angles | all | 1.48–2.48 | |||
| 104 | ionization potentialsd | OM2b | 0.33 | OM1 | 0.40 | |
| 58 | dipole moments | all | 0.22–0.34 | |||
| C7H10O2 | 6095 | atomization enthalpies at 298 K | OM2 | 6.30 | PM7 | 6.44 |
Subsets are indented. MAEs for energies in kcal/mol, for bond lengths in Å, for angles in degrees, for ionization potentials in eV, and for dipole moments in D. If MAEs differ by less than ca. 0.1 kcal/mol for energies or 0.01 Å for bond lengths, the corresponding methods are listed together.
MAE of dispersion-corrected counterpart (OM2-Dn or OM3-Dn) is very close (within 1.0 kcal/mol for energies and within 1.0° for angles) and may actually be slightly lower.
Without the isomerization energy of C20; see text.
Calculated using Koopmans’ theorem.
Table 16. Dispersion-Corrected Methods Predicting the Best Interaction Energies (kcal/mol) and Geometries (Distances, Å; Angles, deg) of Noncovalent Complexesa.
| data set | N | property | first best | MAE | second best | MAE |
|---|---|---|---|---|---|---|
| A24-CHNOF | 21 | interaction energiesbc | OMx-Dn | 0.43–0.49 | PM7 | 0.64 |
| 23 | selected interatomic distancesd | OM3-D3, OM3-D3T | 0.261–0.262 | OM3-D2 | 0.322 | |
| 40 | selected anglesd | OM3-D3, OM3-D3T | 8.69–8.80 | OM2-D3, OM2-D3T | 9.86–9.97 | |
| S66 | 66 | interaction energiesc | PM7, OMx-D3, OMx-D3T | 0.79–0.85 | OMx-D2 | 1.02–1.05 |
| 172 | selected interatomic distancesd | OM3-D2 | 0.290 | OM3-D3, OM3-D3T | 0.317–0.320 | |
| 141 | selected anglesd | OM3-D2, OM2-Dn | 11.32–12.32 | OM3-D3, OM3-D3T | 12.99–13.15 | |
| S66a8 | 528 | interaction energiesc | OMx-D3, OMx-D3T | 0.60–0.62 | all other | 0.77–0.80 |
| JSCH-2005-CHNOF | 134 | interaction energiesc | OM3-D3, OM3-D3T | 1.33–1.37 | OM2-D3, OM2-D3T | 1.72–1.81 |
| S7L | 7 | interaction energiesc | OM3-D3 | 0.66 | OM3-D3T | 0.79 |
| 28 | selected interatomic distancesd | PM7 | 0.325 | OM3-Dn | 0.389–0.393 | |
| S30L-CHNOF | 24 | interaction energiesc | OM3-D3T | 3.59 | OM3-D3 | 4.36 |
| AF6 | 6 | folding energiesc | OM2-D2 | 0.20 | OM2-D3T | 0.34 |
| 6 | folding enthalpiesc | OM2-D2 | 0.18 | OM2-D3, OM2-D3T | 0.34–0.44 | |
| 27 | selected interatomic distancesd | PM7 | 0.090 | OM3-D2 | 0.121 | |
| 74 | selected anglesd | OM2-D2 | 5.17 | all other | 6.42–7.05 |
Only the OMx-Dn and PM7 methods are considered here since the other SQC methods do not adequately describe noncovalent interactions. If MAEs differ by less than 0.1 kcal/mol for energies or 0.01 Å for distances or 1.0° for angles, the corresponding methods are listed together.
Without the HF···HF complex; see text.
Single-point calculations on reference geometries.
Deviations upon optimization with SQC methods.
Most of the entries in Table 15 involve one of the OMx or OMx-Dn methods, which appear to perform best overall for the large variety of ground-state molecules and properties considered presently. As a rule of thumb, the OMx and OMx-Dn methods can thus be recommended as default for ground-state SQC studies, especially when a specific validation against experimental data or high-level calculations is not feasible. Common organic molecules are also described with good accuracy by PM6 and PM7 because of their extensive parametrization. This indicates that SQC methods with orthogonalization corrections still have a large potential for improvement when larger data sets are used for their parametrization; this is one target of our current research.
The OMx-Dn and PM7 methods have similar overall accuracy for noncovalent binding energies and geometries of noncovalent complexes. The OMx-Dn methods generally outperform PM7 for dispersion-dominated complexes and mixed electrostatic/dispersion interactions, while PM7 is normally superior for hydrogen-bonded complexes; the latter can be attributed to special hydrogen-bond corrections that are present in PM7 but not in the OMx-Dn methods. While it has been shown for water clusters that the description of hydrogen bonds can be improved by a special parametrization of OMx methods,140 it would seem more promising to target a complete reparametrization at the OMx-Dn level that includes noncovalent interaction energies and geometries as reference data.
Acknowledgments
We are grateful to William L. Jorgensen, Martin Korth, and Tell Tuttle for providing their data sets.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jctc.5b01047.
Details on converting semiempirical heats of formation at 298 K to ZPVE-exclusive energies at 0 K, extensive tables with numerical results for individual molecules and properties in all data sets covered presently, tables with additional statistical evaluations not included in the main text, figures with error distributions and with reference and optimized SQC geometries of selected noncovalent complexes (309 pages) (PDF)
We thank the European Research Council for financial support within an ERC Advanced Grant (OMSQC).
The authors declare no competing financial interest.
This paper was published ASAP on January 29, 2016, with errors in Table 15. The corrected version was reposted on February 2, 2016.
Supplementary Material
References
- Thiel W. WIREs: Comput. Mol. Sci. 2014, 4, 145–157. (see also references therein) 10.1002/wcms.1161. [DOI] [Google Scholar]
- Senn H. M.; Thiel W. Angew. Chem., Int. Ed. 2009, 48, 1198–1229. 10.1002/anie.200802019. [DOI] [PubMed] [Google Scholar]
- El Kerdawy A. E.; Güssregen S.; Matter H.; Hennemann M.; Clark T. J. Chem. Inf. Model. 2013, 53, 1486–1502. 10.1021/ci400181b. [DOI] [PubMed] [Google Scholar]
- Lepšík M.; Řezáč J.; Kolář M.; Pecina A.; Hobza P.; Fanfrlík J. ChemPlusChem 2013, 78, 921–931. (see also references therein) 10.1002/cplu.201300199. [DOI] [PubMed] [Google Scholar]
- Vorlová B.; Nachtigallová D.; Jirásková-Vaníčková J.; Ajani H.; Jansa P.; Řezáč J.; Fanfrlík J.; Otyepka M.; Hobza P.; Konvalinka J.; Lepšík M. Eur. J. Med. Chem. 2015, 89, 189–197. 10.1016/j.ejmech.2014.10.043. [DOI] [PubMed] [Google Scholar]
- Fanfrlík J.; Bronowska A. K.; Řezáč J.; Přenosil O.; Konvalinka J.; Hobza P. J. Phys. Chem. B 2010, 114, 12666–12678. 10.1021/jp1032965. [DOI] [PubMed] [Google Scholar]
- Brahmkshatriya P. S.; Dobeš P.; Fanfrlík J.; Řezáč J.; Paruch K.; Bronowska A.; Lepšík M.; Hobza P. Curr. Comput.-Aided Drug Des. 2013, 9, 118–129. 10.2174/1573409911309010011. [DOI] [PubMed] [Google Scholar]
- Dral P. O.; Clark T. J. Phys. Chem. A 2011, 115, 11303–11312. 10.1021/jp204939x. [DOI] [PubMed] [Google Scholar]
- Ciammaichella A.; Dral P. O.; Clark T.; Tagliatesta P.; Sekita M.; Guldi D. M. Chem. - Eur. J. 2012, 18, 14008–14016. 10.1002/chem.201202245. [DOI] [PubMed] [Google Scholar]
- Jäger C. M.; Schmaltz T.; Novak M.; Khassanov A.; Vorobiev A.; Hennemann M.; Krause A.; Dietrich H.; Zahn D.; Hirsch A.; Halik M.; Clark T. J. Am. Chem. Soc. 2013, 135, 4893–4900. 10.1021/ja401320n. [DOI] [PubMed] [Google Scholar]
- Dral P. O. J. Mol. Model. 2014, 20, 2134. 10.1007/s00894-014-2134-7. [DOI] [PubMed] [Google Scholar]
- Grimme S. Angew. Chem., Int. Ed. 2013, 52, 6306–6312. 10.1002/anie.201300158. [DOI] [PubMed] [Google Scholar]
- Bauer C. A.; Grimme S. J. Phys. Chem. A 2014, 118, 11479–11484. 10.1021/jp5096618. [DOI] [PubMed] [Google Scholar]
- Bauer C. A.; Grimme S. Org. Biomol. Chem. 2014, 12, 8737–8744. 10.1039/C4OB01668H. [DOI] [PubMed] [Google Scholar]
- Grimme S.; Bauer C. A. Eur. Mass Spectrom. 2015, 21, 125–140. 10.1255/ejms.1313. [DOI] [PubMed] [Google Scholar]
- Polyak I.; Reetz M. T.; Thiel W. J. Am. Chem. Soc. 2012, 134, 2732–2741. 10.1021/ja2103839. [DOI] [PubMed] [Google Scholar]
- Acevedo O.; Jorgensen W. L. Acc. Chem. Res. 2010, 43, 142–151. (see also references therein) 10.1021/ar900171c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunaydin H.; Acevedo O.; Jorgensen W. L.; Houk K. N. J. Chem. Theory Comput. 2007, 3, 1028–1035. 10.1021/ct050318n. [DOI] [PubMed] [Google Scholar]
- Alexandrova A. N.; Rothlisberger D.; Baker D.; Jorgensen W. L. J. Am. Chem. Soc. 2008, 130, 15907–15915. 10.1021/ja804040s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexandrova A. N.; Jorgensen W. L. J. Phys. Chem. B 2009, 113, 497–504. 10.1021/jp8076084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dewar M. J. S.; Thiel W. J. Am. Chem. Soc. 1977, 99, 4899–4907. 10.1021/ja00457a004. [DOI] [Google Scholar]
- Dewar M. J. S.; Thiel W. J. Am. Chem. Soc. 1977, 99, 4907–4917. 10.1021/ja00457a005. [DOI] [Google Scholar]
- Dewar M. J. S.; Zoebisch E. G.; Healy E. F.; Stewart J. J. P. J. Am. Chem. Soc. 1985, 107, 3902–3909. 10.1021/ja00299a024. [DOI] [Google Scholar]
- Stewart J. J. P. J. Comput. Chem. 1989, 10, 209–220. 10.1002/jcc.540100208. [DOI] [Google Scholar]
- Stewart J. J. P. J. Comput. Chem. 1989, 10, 221–264. 10.1002/jcc.540100209. [DOI] [Google Scholar]
- Thiel W.; Voityuk A. A. Theor. Chim. Acta 1992, 81, 391–404. 10.1007/BF01134863. [DOI] [Google Scholar]
- Thiel W.; Voityuk A. A. Theor. Chim. Acta 1996, 93, 315. 10.1007/BF01127509. [DOI] [Google Scholar]
- Thiel W.; Voityuk A. A. J. Phys. Chem. 1996, 100, 616–626. 10.1021/jp952148o. [DOI] [Google Scholar]
- Winget P.; Horn A. H. C.; Selçuki C.; Martin B.; Clark T. J. Mol. Model. 2003, 9, 408–414. 10.1007/s00894-003-0156-7. [DOI] [PubMed] [Google Scholar]
- Rocha G. B.; Freire R. O.; Simas A. M.; Stewart J. J. P. J. Comput. Chem. 2006, 27, 1101–1111. 10.1002/jcc.20425. [DOI] [PubMed] [Google Scholar]
- Repasky M. P.; Chandrasekhar J.; Jorgensen W. L. J. Comput. Chem. 2002, 23, 1601–1622. (see also references therein and in the Supporting Information) 10.1002/jcc.10162. [DOI] [PubMed] [Google Scholar]
- Tubert-Brohman I.; Guimarães C. R. W.; Repasky M. P.; Jorgensen W. L. J. Comput. Chem. 2004, 25, 138–150. 10.1002/jcc.10356. [DOI] [PubMed] [Google Scholar]
- Stewart J. J. P. J. Mol. Model. 2007, 13, 1173–1213. 10.1007/s00894-007-0233-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart J. J. P. J. Mol. Model. 2013, 19, 1–32. 10.1007/s00894-012-1667-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiel W. Adv. Chem. Phys. 1996, 93, 703–757. 10.1002/9780470141526.ch10. [DOI] [Google Scholar]
- Sattelmeyer K. W.; Tubert-Brohman I.; Jorgensen W. L. J. Chem. Theory Comput. 2006, 2, 413–419. 10.1021/ct050174c. [DOI] [PubMed] [Google Scholar]
- Kolb M.Ein neues semiempirisches Verfahren auf Grundlage der NDDO-Näherung: Entwicklung der Methode, Parametrisierung und Anwendungen. Ph.D. thesis, Bergische Universität-Gesamthochschule Wuppertal, Elberfeld, Germany, 1991. [Google Scholar]
- Kolb M.; Thiel W. J. Comput. Chem. 1993, 14, 775–789. 10.1002/jcc.540140704. [DOI] [Google Scholar]
- Weber W.Ein neues semiempirisches NDDO-Verfahren mit Orthogonalisierungskorrekturen: Entwicklung des Modells, Parametrisie-rung und Anwendungen. Ph.D. thesis, Universität Zürich, Zürich, Switzerland, 1996. [Google Scholar]
- Weber W.; Thiel W. Theor. Chem. Acc. 2000, 103, 495–506. 10.1007/s002149900083. [DOI] [Google Scholar]
- Scholten M.Semiempirische Verfahren mit Orthogonalisierungs-korrekturen: Die OM3 Methode. Ph.D. thesis, Universität Düsseldorf, Düsseldorf, Germany, 2003. [Google Scholar]
- Dral P. O.; Wu X.; Spörkel L.; Koslowski A.; Weber W.; Steiger R.; Scholten M.; Thiel W.. J. Chem. Theory Comput. 2016, preceding paper in this issue; DOI: 10.1021/acs.jctc.5b01046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin B.; Clark T. Int. J. Quantum Chem. 2006, 106, 1208–1216. 10.1002/qua.20856. [DOI] [Google Scholar]
- Grimme S. J. Comput. Chem. 2006, 27, 1787–1799. 10.1002/jcc.20495. [DOI] [PubMed] [Google Scholar]
- Grimme S.; Antony J.; Ehrlich S.; Krieg H. J. Chem. Phys. 2010, 132, 154104. 10.1063/1.3382344. [DOI] [PubMed] [Google Scholar]
- Grimme S.; Ehrlich S.; Goerigk L. J. Comput. Chem. 2011, 32, 1456–1465. 10.1002/jcc.21759. [DOI] [PubMed] [Google Scholar]
- Tuttle T.; Thiel W. Phys. Chem. Chem. Phys. 2008, 10, 2159–2166. 10.1039/b718795e. [DOI] [PubMed] [Google Scholar]
- Risthaus T.; Grimme S. J. Chem. Theory Comput. 2013, 9, 1580–1591. 10.1021/ct301081n. [DOI] [PubMed] [Google Scholar]
- Sure R.; Grimme S. J. Chem. Theory Comput. 2015, 11, 3785–3801. 10.1021/acs.jctc.5b00296. [DOI] [PubMed] [Google Scholar]; Correction see:Sure R.; Grimme S. J. Chem. Theory Comput. 2015, 11, 5990. 10.1021/acs.jctc.5b01016. [DOI] [PubMed] [Google Scholar]
- Otte N.; Scholten M.; Thiel W. J. Phys. Chem. A 2007, 111, 5751–5755. 10.1021/jp0700130. [DOI] [PubMed] [Google Scholar]
- Silva-Junior M. R.; Thiel W. J. Chem. Theory Comput. 2010, 6, 1546–1564. 10.1021/ct100030j. [DOI] [PubMed] [Google Scholar]
- Korth M.; Thiel W. J. Chem. Theory Comput. 2011, 7, 2929–2936. 10.1021/ct200434a. [DOI] [PubMed] [Google Scholar]
- Tirado-Rives J.; Jorgensen W. L. J. Chem. Theory Comput. 2008, 4, 297–306. 10.1021/ct700248k. [DOI] [PubMed] [Google Scholar]
- Thiel W.MNDO2005, version 7.0; Max-Planck-Institut für Kohlenforschung: Mülheim an der Ruhr, Germany, 2005.
- Stewart J. J. P.MOPAC2012, versions 13.326L and 15.027L; http://OpenMOPAC.net; Stewart Computational Chemistry, Colorado Springs, CO, USA.
- Maia J. D. C.; Carvalho G. A. U.; Mangueira C. P. Jr.; Santana S. R.; Cabral L. A. F.; Rocha G. B. J. Chem. Theory Comput. 2012, 8, 3072–3081. 10.1021/ct3004645. [DOI] [PubMed] [Google Scholar]
- Andrienko G. A.ChemCraft, http://www.chemcraftprog.com, 2014.
- Dewar M. J. S.; Hashmall J. A.; Venier C. G. J. Am. Chem. Soc. 1968, 90, 1953–1957. 10.1021/ja01010a005. [DOI] [Google Scholar]
- Dewar M. J. S.; Dieter K. M. J. Am. Chem. Soc. 1986, 108, 8075–8086. 10.1021/ja00285a033. [DOI] [Google Scholar]
- Chase M. W. Jr.; Curnutt J. L.; Downey J. R. Jr.; McDonald R. A.; Syverud A. N.; Valenzuela E. A. J. Phys. Chem. Ref. Data 1982, 11, 695. 10.1063/1.555666. [DOI] [Google Scholar]
- Thiel W.; Green D. G.. The MNDO94 Code: Parallelization of a Semiempirical Quantum-Chemical Program. In Methods and Techniques in Computational Chemistry: METECC-95; Clementi E., Corongiu G., Eds.; NIC Series; STEF: Cagliari, Italy, 1995; Vol. 3, pp 141–168. [Google Scholar]
- Thiel W.Semiempirical methods. In Modern methods and algorithms of quantum chemistry; Grotendorst J., Ed.; NIC Series; John von Neumann Institute for Computing: Jülich, Germany, 2000; Vol. 3, pp 261–283. [Google Scholar]
- Wu X.; Koslowski A.; Thiel W. J. Chem. Theory Comput. 2012, 8, 2272–2281. 10.1021/ct3001798. [DOI] [PubMed] [Google Scholar]
- Wu X.Semiempirical Quantum Chemistry on High-Performance Heterogeneous Computers. Ph.D. thesis, Universität Düsseldorf, Düsseldorf, Germany, 2013. [Google Scholar]
- Grimme S.; Antony J.; Ehrlich S.; Krieg H.. DFT-D3, versions 3.0 and 3.1; http://www.thch.uni-bonn.de/tc/index.php?section/downloads&subsection/getd3. Universität Bonn, Bonn, Germany, 2014.
- Higgins D.; Thomson C.; Thiel W. J. Comput. Chem. 1988, 9, 702–707. 10.1002/jcc.540090616. [DOI] [Google Scholar]
- Scano P.; Thomson C. J. Comput. Chem. 1991, 12, 172–174. 10.1002/jcc.540120205. [DOI] [Google Scholar]
- Haworth N. L.; Smith M. H.; Bacskay G. B.; Mackie J. C. J. Phys. Chem. A 2000, 104, 7600–7611. 10.1021/jp000475c. [DOI] [Google Scholar]
- Curtiss L. A.; Raghavachari K.; Redfern P. C.; Pople J. A. J. Chem. Phys. 1997, 106, 1063–1079. 10.1063/1.473182. [DOI] [Google Scholar]
- Curtiss L. A.; Raghavachari K.; Redfern P. C.; Pople J. A. J. Chem. Phys. 2000, 112, 7374–7383. 10.1063/1.481336. [DOI] [Google Scholar]
- Redfern P. C.; Zapol P.; Curtiss L. A.; Raghavachari K. J. Phys. Chem. A 2000, 104, 5850–5854. 10.1021/jp994429s. [DOI] [Google Scholar]
- Karton A.; Daon S.; Martin J. M. Chem. Phys. Lett. 2011, 510, 165–178. 10.1016/j.cplett.2011.05.007. [DOI] [Google Scholar]
- Goerigk L.; Grimme S. J. Chem. Theory Comput. 2011, 7, 291–309. (see also references therein) 10.1021/ct100466k. [DOI] [PubMed] [Google Scholar]
- Korth M.; Grimme S. J. Chem. Theory Comput. 2009, 5, 993–1003. 10.1021/ct800511q. [DOI] [PubMed] [Google Scholar]
- Karton A.; Tarnopolsky A.; Lamére J.-F.; Schatz G. C.; Martin J. M. L. J. Phys. Chem. A 2008, 112, 12868–12886. 10.1021/jp801805p. [DOI] [PubMed] [Google Scholar]
- Curtiss L. A.; Raghavachari K.; Trucks G. W.; Pople J. A. J. Chem. Phys. 1991, 94, 7221. 10.1063/1.460205. [DOI] [Google Scholar]
- Parthiban S.; Martin J. M. L. J. Chem. Phys. 2001, 114, 6014–6029. 10.1063/1.1356014. [DOI] [Google Scholar]
- Zhao Y.; Truhlar D. G. J. Phys. Chem. A 2006, 110, 10478–10486. 10.1021/jp0630626. [DOI] [PubMed] [Google Scholar]
- Goerigk L.; Grimme S. J. Chem. Theory Comput. 2010, 6, 107–126. 10.1021/ct900489g. [DOI] [PubMed] [Google Scholar]
- Guner V.; Khuong K. S.; Leach A. G.; Lee P. S.; Bartberger M. D.; Houk K. N. J. Phys. Chem. A 2003, 107, 11445–11459. 10.1021/jp035501w. [DOI] [Google Scholar]
- Ess D. H.; Houk K. N. J. Phys. Chem. A 2005, 109, 9542–9553. 10.1021/jp052504v. [DOI] [PubMed] [Google Scholar]
- Grimme S.; Mück-Lichtenfeld C.; Würthwein E.-U.; Ehlers A. W.; Goumans T. P. M.; Lammertsma K. J. Phys. Chem. A 2006, 110, 2583–2586. 10.1021/jp057329x. [DOI] [PubMed] [Google Scholar]
- Dinadayalane T. C.; Vijaya R.; Smitha A.; Sastry G. N. J. Phys. Chem. A 2002, 106, 1627–1633. 10.1021/jp013910r. [DOI] [Google Scholar]
- Zhao Y.; Lynch B. J.; Truhlar D. G. J. Phys. Chem. A 2004, 108, 2715–2719. 10.1021/jp049908s. [DOI] [Google Scholar]
- Zhao Y.; González-García N.; Truhlar D. G. J. Phys. Chem. A 2005, 109, 2012–2018. 10.1021/jp045141s. [DOI] [PubMed] [Google Scholar]
- Neese F.; Schwabe T.; Kossmann S.; Schirmer B.; Grimme S. J. Chem. Theory Comput. 2009, 5, 3060–3073. 10.1021/ct9003299. [DOI] [PubMed] [Google Scholar]
- Zhao Y.; Tishchenko O.; Gour J. R.; Li W.; Lutz J. J.; Piecuch P.; Truhlar D. G. J. Phys. Chem. A 2009, 113, 5786–5799. 10.1021/jp811054n. [DOI] [PubMed] [Google Scholar]
- Grimme S.; Steinmetz M.; Korth M. J. Org. Chem. 2007, 72, 2118–2126. 10.1021/jo062446p. [DOI] [PubMed] [Google Scholar]
- Huenerbein R.; Schirmer B.; Moellmann J.; Grimme S. Phys. Chem. Chem. Phys. 2010, 12, 6940–6948. 10.1039/c003951a. [DOI] [PubMed] [Google Scholar]
- Piacenza M.; Grimme S. J. Comput. Chem. 2004, 25, 83–99. 10.1002/jcc.10365. [DOI] [PubMed] [Google Scholar]
- Woodcock H. L.; Schaefer H. F.; Schreiner P. R. J. Phys. Chem. A 2002, 106, 11923–11931. 10.1021/jp0212895. [DOI] [Google Scholar]
- Schreiner P. R.; Fokin A. A.; Pascal R. A. Jr.; de Meijere A. Org. Lett. 2006, 8, 3635–3638. 10.1021/ol0610486. [DOI] [PubMed] [Google Scholar]
- Lepetit C.; Chermette H.; Gicquel M.; Heully J.-L.; Chauvin R. J. Phys. Chem. A 2007, 111, 136–149. 10.1021/jp064066d. [DOI] [PubMed] [Google Scholar]
- Lee J. S. J. Phys. Chem. A 2005, 109, 11927–11932. 10.1021/jp040705d. [DOI] [PubMed] [Google Scholar]
- Grimme S. J. Chem. Phys. 2006, 124, 034108. 10.1063/1.2148954. [DOI] [PubMed] [Google Scholar]
- Johnson E. R.; Mori-Sánchez P.; Cohen A. J.; Yang W. J. Chem. Phys. 2008, 129, 204112. 10.1063/1.3021474. [DOI] [PubMed] [Google Scholar]
- Krieg H.; Grimme S. Mol. Phys. 2010, 108, 2655–2666. 10.1080/00268976.2010.519729. [DOI] [Google Scholar]
- Schwabe T.; Grimme S. Phys. Chem. Chem. Phys. 2007, 9, 3397–3406. 10.1039/b704725h. [DOI] [PubMed] [Google Scholar]
- Grimme S. Angew. Chem., Int. Ed. 2006, 45, 4460–4464. 10.1002/anie.200600448. [DOI] [PubMed] [Google Scholar]
- Bryantsev V. S.; Diallo M. S.; van Duin A. C. T.; Goddard W. A. J. Chem. Theory Comput. 2009, 5, 1016–1026. 10.1021/ct800549f. [DOI] [PubMed] [Google Scholar]
- Jurečka P.; Šponer J.; Černý J.; Hobza P. Phys. Chem. Chem. Phys. 2006, 8, 1985–1993. 10.1039/b600027d. [DOI] [PubMed] [Google Scholar]
- Takatani T.; Hohenstein E. G.; Malagoli M.; Marshall M. S.; Sherrill C. D. J. Chem. Phys. 2010, 132, 144104. 10.1063/1.3378024. [DOI] [PubMed] [Google Scholar]
- Tsuzuki S.; Honda K.; Uchimaru T.; Mikami M. J. Chem. Phys. 2006, 124, 114304. 10.1063/1.2178795. [DOI] [PubMed] [Google Scholar]
- Řeha D.; Valdés H.; Vondrášek J.; Hobza P.; Abu-Riziq A.; Crews B.; de Vries M. S. Chem. - Eur. J. 2005, 11, 6803–6817. 10.1002/chem.200500465. [DOI] [PubMed] [Google Scholar]
- Gruzman D.; Karton A.; Martin J. M. L. J. Phys. Chem. A 2009, 113, 11974–11983. 10.1021/jp903640h. [DOI] [PubMed] [Google Scholar]
- Csonka G. I.; French A. D.; Johnson G. P.; Stortz C. A. J. Chem. Theory Comput. 2009, 5, 679–692. 10.1021/ct8004479. [DOI] [PubMed] [Google Scholar]
- The Minnesota databases, http://comp.chem.umn.edu/db/. Accessed on Jul. 24, 2014.
- Peverati R.; Truhlar D. G. Philos. Trans. R. Soc., A 2014, 372, 20120476. 10.1098/rsta.2012.0476. [DOI] [PubMed] [Google Scholar]
- Zhao Y.; Schultz N. E.; Truhlar D. G. J. Chem. Theory Comput. 2006, 2, 364–382. 10.1021/ct0502763. [DOI] [PubMed] [Google Scholar]
- Peverati R.; Truhlar D. G. J. Chem. Phys. 2011, 135, 191102. 10.1063/1.3663871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo S.; Zhao Y.; Truhlar D. G. Phys. Chem. Chem. Phys. 2011, 13, 13683–13689. 10.1039/c1cp20834a. [DOI] [PubMed] [Google Scholar]
- Zhao Y.; Schultz N. E.; Truhlar D. G. J. Chem. Phys. 2005, 123, 161103. 10.1063/1.2126975. [DOI] [PubMed] [Google Scholar]
- Zhao Y.; Truhlar D. G. J. Phys. Chem. A 2005, 109, 5656–5667. 10.1021/jp050536c. [DOI] [PubMed] [Google Scholar]
- Lynch B. J.; Zhao Y.; Truhlar D. G. J. Phys. Chem. A 2003, 107, 1384–1388. 10.1021/jp021590l. [DOI] [Google Scholar]
- Peverati R.; Truhlar D. G. Phys. Chem. Chem. Phys. 2012, 14, 13171–13174. 10.1039/c2cp42025b. [DOI] [PubMed] [Google Scholar]
- Li R.; Peverati R.; Isegawa M.; Truhlar D. G. J. Phys. Chem. A 2013, 117, 169–173. 10.1021/jp3079106. [DOI] [PubMed] [Google Scholar]
- Zhao Y.; Truhlar D. G. J. Chem. Phys. 2006, 125, 194101. 10.1063/1.2370993. [DOI] [PubMed] [Google Scholar]
- Izgorodina E. I.; Coote M. L.; Radom L. J. Phys. Chem. A 2005, 109, 7558–7566. 10.1021/jp052021r. [DOI] [PubMed] [Google Scholar]
- Peverati R.; Zhao Y.; Truhlar D. G. J. Phys. Chem. Lett. 2011, 2, 1991–1997. 10.1021/jz200616w. [DOI] [Google Scholar]
- Zhao Y.; Lynch B. J.; Truhlar D. G. Phys. Chem. Chem. Phys. 2005, 7, 43–52. 10.1039/b416937a. [DOI] [PubMed] [Google Scholar]
- Zheng J.; Zhao Y.; Truhlar D. G. J. Chem. Theory Comput. 2009, 5, 808–821. 10.1021/ct800568m. [DOI] [PubMed] [Google Scholar]
- Zhao Y.; Truhlar D. G. J. Chem. Theory Comput. 2005, 1, 415–432. 10.1021/ct049851d. [DOI] [PubMed] [Google Scholar]
- Ramakrishnan R.; Dral P. O.; Rupp M.; von Lilienfeld O. A. Sci. Data 2014, 1, 140022. 10.1038/sdata.2014.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Řezáč J.; Hobza P. J. Chem. Theory Comput. 2013, 9, 2151–2155. 10.1021/ct400057w. [DOI] [PubMed] [Google Scholar]
- Řezáč J.; Riley K. E.; Hobza P. J. Chem. Theory Comput. 2011, 7, 2427–2438. 10.1021/ct2002946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Řezáč J.; Riley K. E.; Hobza P. J. Chem. Theory Comput. 2011, 7, 3466–3470. 10.1021/ct200523a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janowski T.; Pulay P. J. Am. Chem. Soc. 2012, 134, 17520–17525. 10.1021/ja303676q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrd J. N.; Bartlett R. J.; Montgomery J. A. Jr. J. Phys. Chem. A 2014, 118, 1706–1712. 10.1021/jp4121854. [DOI] [PubMed] [Google Scholar]
- Thiel W. Tetrahedron 1988, 44, 7393–7408. 10.1016/S0040-4020(01)86235-9. [DOI] [Google Scholar]
- Karton A.; Yu L.-J.; Kesharwani M. K.; Martin J. M. L. Theor. Chem. Acc. 2014, 133, 1483. 10.1007/s00214-014-1483-8. [DOI] [Google Scholar]
- GMTKN30—A database for general main group thermochemistry, kinetics, and non-covalent interactions, http://www.thch.uni-bonn.de/tc/index.php?section=downloads&subsection=GMTKN30. Accessed on Jul. 24,2014.
- Anacker T.; Friedrich J. J. Comput. Chem. 2014, 35, 634–643. 10.1002/jcc.23539. [DOI] [PubMed] [Google Scholar]
- Fanourgakis G. S.; Aprà E.; Xantheas S. S. J. Chem. Phys. 2004, 121, 2655. 10.1063/1.1767519. [DOI] [PubMed] [Google Scholar]
- Stewart J. J. P.Accuracy of PM7: Set of Individual Molecules, http://openmopac.net/PM7_accuracy/molecules.html. Accessed on Jul. 30, 2014.
- Curtiss L. A.; Redfern P. C.; Raghavachari K. J. Chem. Phys. 2007, 127, 124105. 10.1063/1.2770701. [DOI] [PubMed] [Google Scholar]
- Ruddigkeit L.; van Deursen R.; Blum L.; Reymond J.-L. J. Chem. Inf. Model. 2012, 52, 2864–2875. 10.1021/ci300415d. [DOI] [PubMed] [Google Scholar]
- BEGDB: Benchmark Energy and Geometry DataBase, http://www.begdb.com/. Accessed on Aug. 4, 2014.
- Grimme S. Chem. - Eur. J. 2012, 18, 9955–9964. 10.1002/chem.201200497. [DOI] [PubMed] [Google Scholar]
- Lüttschwager N. O. B.; Wassermann T. N.; Mata R. A.; Suhm M. A. Angew. Chem., Int. Ed. 2013, 52, 463–466. 10.1002/anie.201202894. [DOI] [PubMed] [Google Scholar]
- Wu X.; Thiel W.; Pezeshki S.; Lin H. J. Chem. Theory Comput. 2013, 9, 2672–2686. 10.1021/ct400224n. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.