

Abstract
Joubert syndrome (JS) is a rare autosomal recessive (AR), neurological condition characterized by dysgenesis of the cerebellar vermis with the radiological hallmark of molar tooth sign, oculomotor apraxia, recurrent hyperventilation and intellectual disability. Most cases display a broad spectrum of additional features, including polydactyly, retinal dystrophy and renal abnormalities, which define different subtypes of JS-related disorders (JSRDs). To date, 23 genes have been shown to cause JSRDs, and although most of the identified genes encode proteins involved in cilia function or assembly, the molecular mechanisms associated with ciliary signaling remain enigmatic. Arab populations are ethnically diverse with high levels of consanguinity (20–60%) and a high prevalence of AR disorders. In addition, isolated communities with very-high levels of inbreeding and founder mutations are common. In this article, we review the 70 families reported thus far with JS and JSRDs that have been studied at the molecular level from all the Arabic countries and compile the mutations found. We show that JS and the related JSRDs are genetically heterogeneous in Arabs, with 53 mutations in 15 genes. Thirteen of these mutations are potentially founder mutations for the region.
Demography of Arabs and consanguinity
The Arab countries comprise the 22 states and territories of the Arab League stretching from the Atlantic Ocean of North Africa in the west to the Arabian Sea of the Arabian Peninsula in the east. The incidence of congenital disorders among the 350 million inhabitants of these Arabic countries is influenced by their demographic and cultural characteristics. Most Arab populations are characterized by marriage at a young age, large extended family structure and advanced maternal and paternal ages. In addition, consanguineous marriages are favored, and intra-familial unions currently account for 20–60% of all marriages. Furthermore, first-cousin unions are popular and constitute almost one-quarter of all marriages.1 Consequently, these marriage traditions have resulted in the unequal distribution of founder mutations in most Arab populations.2 Moreover, isolated subpopulations with high levels of inbreeding have made the epidemiology of genetic disorders complicated. Consequently, some genetic conditions are confined to specific villages, families or tribal groups, and these communities face an increased burden of genetic disorders, particularly the rare autosomal recessive (AR) disorders.3 Numerous AR genes and loci have been identified in Arab families, with the majority associated with consanguinity.1,4,5
Joubert syndrome (JS) and Joubert syndrome-related disorders (JSRDs) are a large group of pleotropic conditions that affect different organs of the body. These conditions are characterized by dysgenesis of the cerebellar vermis and the appearance of the molar tooth sign (MTS)6 via neuroimaging. The most common clinical features of JS and JSRDs include hypotonia, ataxia, intellectual disability (ID), developmental delay, eye movement impairment and neonatal breathing difficulties. In addition, these disorders might involve multiorgan abnormalities such as liver fibrosis, retinopathy, nephronophthisis, polydactyly and cystic dysplastic kidneys. The prognosis for individuals with JS and JSRDs vary widely depending on the extent and severity of organ involvement. The prevalence of JSRDs in the USA has been estimated to be 1:100,000.7 However, this prevalence is likely to be an underestimate given the wide spectrum of clinical variability, particularly in individuals with milder symptoms.8 The prevalence of JSRDs has not been well defined in most Arab countries. However, an incidence of 1:5,000 births has been reported for the UAE,1 which is considerably higher than in the USA. JS and JSRDs have been found to be caused by defects in 23 genes (INPP5E, TMEM216, AHI1, NPHP1, CEP290 (NPHP6), TMEM67 (MKS3), RPGRIP1L, ARL13B, CC2D2A, OFD1, TTC21B, KIF7, TCTN1, TCTN2, TMEM237, CEP41, TMEM138, C5orf42, TCTN3, ZNF423, TMEM231, CSPP1 and PDE6D). All the products of these genes have some ciliary role; therefore, JS and JRSDs are classified as ciliopathies. Primary cilia are found in most cell types, including the cells found in the brain, kidneys and liver, and appear to have important roles in cellular chemosensation, mechanosensation and signaling.
Historical and diagnostic criteria
JS was named after Dr Marie Joubert, who was the first to describe siblings from a large French-Canadian family with ID, ataxia, abnormal eye movement and agenesis of the cerebellar vermis presenting with episodic tachypnea.9 Several years later, a pathognomonic midbrain–hindbrain abnormality, termed ‘molar tooth sign (MTS)’, was described via cranial magnetic resonance imaging of individuals with JS and JSRDs.7,10–12 A comprehensive review of previously reported and novel patients with JS established the first diagnostic criteria for this condition in 1992.13 Most recently, the term ‘Joubert Syndrome and related disorders’ has been adopted for a group of pleiotropic conditions that share the MTS characteristic, but may also have other distinctive features (see Figure 1).11 MTS is defined as a complex brainstem malformation that reflects aplasia or marked hypoplasia of the cerebellar vermis, thickened and elongated superior cerebellar peduncles and a deepened interpeduncular fossa that is apparent on axial magnetic resonance imaging at the midbrain–hindbrain junction.14 Based on a review by Saraiva and Baraitser in 1999, the primary diagnostic criteria of pure/classic JS include the following clinical features: (1) MTS on axial views from cranial magnetic resonance imaging images comprising cerebellar vermis hypoplasia, deepened interpeduncular fossa and thick, elongated superior peduncles; (2) ataxia and a variable degree of ID and developmental delay; (3) hypotonia in infancy; and (4) often one or both of the following clinical characteristics: irregular breathing in infancy (episodic neonatal apnea and/or tachypnea) and abnormal eye movements (nystagmus and/or oculomotor apraxia).8–10,13,15–17 In addition, autism represents a relatively common component of JS.18,19
Figure 1.

Clinical subtypes of JSRD adopted from Brancati et al. 17 The clinical classification scheme should not be considered as final given the extreme clinical variation and the variable onset of different features. Bold: major gene; CHS: congenital hepatic fibrosis; COACH: cerebellar vermis hypoplasia, oligophrenia, ataxia, coloboma and hepatic fibrosis; CORS: cerebello-oculo-renal syndromes; JS: Joubert syndrome; LCA: Leber congenital Amaurosis; MTS: molar tooth sign; NPHP: nephronophthisis.
Recently, a broad spectrum of JSRDs has been defined that encompasses pure JS associated with variable involvement of systematic abnormalities, including other central nervous anomalies, ocular coloboma, polydactyly of the hands and/or feet, liver fibrosis, cystic dysplastic kidneys, retinopathy and/or nephronophthisis.11,20,21 In 2008, a novel classification of JSRDs based on additional secondary criteria, which involves mainly three organs (eye, kidney and liver), was suggested, resulting in the designation of six subgroups16 as follows: (1) pure JS; (2) JS associated with retinopathy; (3) JS with renal involvement (either NPH or cystic dysplastic kidneys); (4) CORS: comprising JS with both retinal and renal involvement (JS + SLS); (5) COACH (MIM216360): including JS with both ocular coloboma and liver abnormalities; and (6) oral–facial–digital (OFD) VI (MIM277170): including JS with both orofacial and digital signs16 (Figure 1).
The multi-systemic nature of the clinical features of JS and JSRDs
In addition to the distinctive MTS as the hallmark characteristic, JS and JSRDs are associated with a broad range of additional features affecting multiple systems, which are described briefly below.
Neurological features
The cardinal neurological features of JSRDs are altered respiratory patterns in the neonatal period, abnormal ocular movements and hypotonia evolving into ataxia and developmental delay, which is also often associated with ID. Early hypotonia is observed in almost all JSRD patients during either the neonatal period or infancy. The association of hypotonia, along with other peculiar features such as an irregular breathing pattern and altered eye movements, should suggest the diagnosis of JSRD and prompt clinicians to request a brain magnetic resonance imaging. The typical respiratory abnormalities are represented either by short alternate episodes of apnea and hyperpnea or by episodic hyperpnea alone.19 The presence of abnormal eye movements such as oculomotor apraxia (the most characteristic and frequent abnormalities and manifests as an inability to follow objects visually with compensatory head movements), decreased smooth pursuit and cancellation of the vestibulo-ocular reflex. Primary position nystagmus is also common, associated occasionally with strabismus and ptosis.19 Developmental abilitiesare delayed in all JSRD patients to variable degrees of severity. Mild-to-severe ID is common; however, it is not a mandatory feature of JSRDs, and in exceptional cases, patients may have borderline or even normal intellect.19 Speech dyspraxia is typical and likely due to the cerebellar malformation.12 Behavioral disturbances, when present, include impulsivity,temper tantrums and autismin in a few reported children.12 A wide range of central nervous system malformations have been reported associated with a higher incidence of epilepsy as rare feature of JSRDs.19
Ophthalmological features
The retina is frequently affected in JSRDs, mostly in the form of retinal dystrophy. The clinical spectrum can range from congenital retinal blindness (Leber congenital amaurosis (LCA)) to retinal dystrophy characterized by a progressive course and variably conserved vision.19 Colobomas mostly affect the posterior segment of the eye, but iris colobomas have also been reported.19 Nystagmus is often present at birth and may improve with age. Strabismus, amblyopia and ptosis may require medical or surgical intervention. In addition, third nerve palsy has also been observed.12
Renal features
Renal disease has been reported in patients with JSRDs, (25–30%)12,19 and presents as nephronophthisis. Cystic dysplasia, multiple cysts of various sizes in immature kidneys with fetal lobulation, may be present at birth.12 Juvenile nephronophthisis may remain asymptomatic for several years or present with subtle and often unrecognized signs, such as polyuria, polydipsia, anemia and growth failure. Acute or chronic renal insufficiency manifests either late in the first decade of life or early in the second. End-stage renal failure is usually reached by the end of the second decade, requiring dialysis or kidney transplantation.19 An infantile variant of NPH manifests within the first years of life and takes a more rapid and severe course.19
Hepatic features
Approximately 6% of JSRD patients present with liver disease12 manifesting as congenital hepatic fibrosis.12,19 Liver disease may present with elevated serum liver enzymes and early onset of hepatosplenomegaly or with more severe manifestations such as portal hypertension, esophageal varices and liver cirrhosis.19 The association of JS with congenital hepatic fibrosis was previously referred to using the acronym COACH.19
Skeletal features
Skeletal defects may include polydactyly, which occurs in 8–16% of patients.19 Postaxial polydactyly that variably affects the hands and feet is the most common type, but preaxial polydactyly of the toes is observed in some cases.12,19 In addition, mesoaxial polydactyly has been described in rare cases, in which other signs of OFD-type VI syndrome (Varadi–Papp syndrome; OMIM 277170) are present.12,19 Mild-to-severe scoliosis may manifest in JSRDs because of the degree of hypotonia in early infancy; structural anomalies of the vertebrae are uncommon.19
Miscellaneous features
Although JSRDs are not typically dysmorphic syndromes, a ‘typical’ dysmorphic facial appearance, including a broad forehead, arched eyebrows, ocular hypertelorism and open, tent-shaped mouth, has been observed. A recent study has outlined the presence of peculiar age-related cranio-facial features and distinct anthropometric facial patterns.12,19 Oral frenula and tongue hamartomas have been described in the OFD VI group of disorders.12 Endocrine abnormalities such as micropenis, isolated growth hormone deficiency or panhypopituitarism have been reported.12 Congenital heart defects are not typically associated with JSRDs but have been reported occasionally.12 Gastrointestinal system manifestations, including Hirschsprung's disease, have been described in a small number of JSRD patients.19
Molecular aspects of JS and JSRDs in Arab families
We focus on reviewing the mutation spectrum that causes JS and JSRDs in Arabs. Mutations in 15 out of 23 genes have been found in Arab patients with JSRDs (Tables 1 and 2). Table 2 summarizes the genes with pathogenic mutations in 72 Arab families with JSRDs identified so far. Details of these genes and the families are briefly described in the following section.
Table 1. List of genes responsible for JS and JSRDs among the Arabs.
| JS Type | Gene symbol | Protein name | Locus | Inheritance | Alternative names | Phenotype MIM nb. | ||
|---|---|---|---|---|---|---|---|---|
| JBTS1 | INPP5E | Inositol polyphosphate 5-phosphatase | 9q34.3 | AR | Joubert–Boltshauser syndrome Cerebelloparenchymal disorder iv; CPD4 Cerebellooculorenal syndrome 1; CORS1 | 213300 | ||
| JBTS3 | AHI1 | Jouberin | 6q23.3 | AR | 608629 | |||
| JBTS4 | NPHP1 | Nephrocystin-1 | 2q13 | AR | 609583 | |||
| JBTS5 | CEP290 | Centrosomal protein of 290 kDa | 12q21.32 | AR | 610188 | |||
| JBTS6 | TMEM67 | Meckelin | 8q22.1 | AR | 610688 | |||
| JBTS7 | RPGRIP1L | Protein fantom | 16q12.2 | AR | 611560 | |||
| JBTS9 | CC2D2A | Coiled-coil and C2 domain containing protein 2A | 4p15.32 | Joubert syndrome 9/15, digenic, included | 612285 | |||
| JBTS12 | KIF7 | Kinesin-like protein KIF7 | 15q26.1 | AR | Hallux duplication, postaxial polydactyly and absence of corpus callosum Schinzel Acrocallosal syndrome Joubert syndrome 12/15, digenic, included | 200990 | ||
| JBTS13 | TCTN1 | Tectonic-1 | 12q24.11 | AR | 614173 | |||
| JBTS14 | TMEM237 | Transmembrane protein 237 | 2q33.1 | AR | 614424 | |||
| JBTS15 | CEP41 | Centrosomal protein of 41 kDa | 7q32.2 | AR | Joubert syndrome 12/15, digenic, included | 614464 | ||
| JBTS16 | TMEM138 | Transmembrane protein 138 | 11q12.2 | AR | 614465 | |||
| JBTS17 | C5orf42 | Uncharacterized protein | 5p13.2 | AR | 614615 | |||
| JBTS18 | TCTN3 | Tectonic 3 | 10q24.1 | AR | 614815 | |||
| TBTS21 | CSPP1 | centrosome/spindle pole-associated protein | 8q13.1-q13.2 | AR | 615636 |
Abbreviations: AR, autosomal recessive; nb, number; JBTS, Joubert syndrome.
Table 2. List of pathogenic mutations responsible for JS and JSRDs among Arabs.
| JS type | Genes | Fam. ID | No. Aff. | Allele | DNA change | Pt. change | Cons. | Note | Ethnicity | Reference |
|---|---|---|---|---|---|---|---|---|---|---|
| JBTS1 | INPP5E | MTI-007 MTI-134 | 5 (1 died) 1 | CH | c.1537C>T c.1543G>A | p.R512W p.R515W | Yes | Emirati (Omani) | 1 | |
| MTI-498 | 1 | H | c.1543G>A | p.R515W | Yes | Emirati (Omani) | This study | |||
| JS_D | 1 | H | c.1535G>A | p.R512Q | Yes | Yamani | This study | |||
| JBS-011 | 2 | CH | c.1600T>G c.1862G>A | p.Y534D p.R621Q | No | Algerian | 2 | |||
| MTI-008 | 4 | H | c.1688G>A | p.R563H | Yes | Emirati | 1 | |||
| MTI-627 | 3 (1 died) | H | c.1738A>G | p.K580E | Yes | Egyptian | 1 | |||
| MTI-888 | 2 | H | c.1921T>C | p.C641R | Yes | Egyptian | 2 | |||
| MTI-1521 | 2 | H | c.1921T>C | p.C641R | Yes | Egyptian | 2 | |||
| JBTS3 | AHI1 | Pedigree 1 | 3 | H | c.1051C>T | p.R351* | Yes | Saudi | 3 | |
| JS_A | 1 | H | c.1051C>T | p.R351* | Yes | Emirati | This study | |||
| Pedigree 2 | 2 | c.1303C>T | p.R435* | Yes | Saudi | 3 | ||||
| JS_F2 | 1 | H | c.1328T>A | p.V443D | Yes | Saudi | 4 | |||
| JS_F10 | 2 | H | c.1328T>A | p.V443D | Yes | Saudi | 4 | |||
| Pedigree 3 | 1 | H | c.1328T>A | p.V443D | Yes | Saudi | 3 | |||
| 115 | 2 | H | c.1328T>A | p.V443D | Yes | Kuwaiti | 5 | |||
| MTI-1501 | 1 | H | c.1922T>A | p.I641N | Yes | novel | Emirati | This study | ||
| JS_B | 2 | Ht | c.1922T>A | p.I641N | No | digenic | F (Emirati) M (Syrian) | This study | ||
| K8103 | 2 | H | c.2156A>G | p.D719G | Yes | Saudi | 6 | |||
| MTI-115 | 2 | H | c.1190_1191delTG | Fs*408 | Yes | Kuwaiti | 5 | |||
| ND | ND | ND | c.3263_3264delGG | ND | Yes | Egyptian | 7 | |||
| MTI-10 | 1 | H | c.787dupC | Fs*270 | Yes | Palestinian | 5 | |||
| JBTS4 | NPHP1 | A2229 | 1 | H | c.143G>A | p.R48K | Yes | Arab | ||
| JBTS5 | CEP290 | A1332 | 1 | Ht | c.164_167delCTCA | p.T55fsX57 | Yes | Syrian | 8 | |
| JS_3 | 1 | H | c.4714G>T | p.E1572* | Yes | Saudi | 4 | |||
| JS_F11 | 1 | H | c.5668G>T | p.G1890* | Yes | Emirati | 9 | |||
| JS_F12 | 1 | H | c.5668G>T | p.G1890* | Yes | Saudi | 10 | |||
| JS_C | H | c.5668G>T | p.G1890* | Yes | Omani | This study | ||||
| MTI-587 | 1 | H | c.5668G>T | p.G1890* | Yes | Emirati | This study | |||
| MTI-012 | 1 | H | c.5668G>T | p.G1890* | Yes | Emirati | This study | |||
| MTI-1001 | 3 | CH | c.5668G>T c.5932G>A | p.G1890* p.R1978* | No | novel | Emirati | This study | ||
| MTI-133 | 2 (2 died) | H | c.5824C>T | p.Q1942* | Yes | Palestinian | 11 | |||
| JBTS6 | TMEM67 | A1371 | 2 | H | c.1888T>C | p.S630P | Yes | Moroccan | 12 | |
| A1421-21 | ND | Ht | c.2461G>A | p.G821S | No | digenic | Egyptian | 13 | ||
| JS-05 | 1 | H | c.2439+5G>C | p.I775_A813del | ND | Algerian | 14 | |||
| JBTS7 | RPGRIP1L | JS_F1 | 3 | H | c.1649A>G | p.Q550R | Yes | Saudi | 4 | |
| JBTS9 | CC2D2A | JBS-006 | 1 | H | c.2161C>T | p.P721S | Yes | Algerian | 15 | |
| A1421-21 | 1 | Ht | c.3055C>T | p.R1019* | No | digenic | Egyptian | 13 | ||
| ND | ND | H | c.3056G>A | p.R1019* | Yes | Egyptian | ||||
| UW36 | 1 | c.3364C>T | p.P1122S | Yes | Saudi | 16 | ||||
| UW48 | 3 (2 died) | c.3364C>T | p.P1122S | Yes | Saudi | 16 | ||||
| F871 | 1 | H | c.4652T>C | p.L1551P | Yes | Saudi | 16 | |||
| UW50 | 2 | c.4582C>T | p.R1528C | Yes | Levarten Arab | 16 | ||||
| MTI-127 | 1 | CH | c.4258G>A c.1412delG | p.R1528H p.K472Rfs* | Yes | Novel | Emirati | This study | ||
| JBTS12 | KIF7 | E | 2 | H | c.217delG | p.A73Pfs*109 | Yes | Egyptian | 17 | |
| Fam9 | 1 | H | c.233_234del | p.L78Pfs*2 | Yes | Egyptian | 18 | |||
| Fam1 | 3 | H | c.2896_2897del | p.A966Pfs*81 | Yes | Algerian | 18 | |||
| Fam8 | 1 | H | c.2896_2897del | p.A966Pfs*81 | Yes | Algerian | 18 | |||
| JBTS13 | TCTN1 | E1 & E2 | 2 | c.217delG | ND | Yes | Egyptian | 17 | ||
| JS_F8 | 2 | c.342-2A>G | p.G115Kfs*8 | Yes | Saudi | 4 | ||||
| JS_F9 | 1 | c.342-2A>G | p.G115Kfs*8 | Yes | Saudi | 4 | ||||
| JBTS14 | TMEM237 | Family L | 1 | c.1066dupC | p.Q356Pfs*23 | Yes | Jordanian | 19 | ||
| JS_F4 | 2 | c.869+1G>A | ND | Yes | Saudi | 4 | ||||
| MTI-131 | 2 | H | c.953_954AGdel | p.Q318Pfs*5 | Yes | novel | Emirati | This study | ||
| JBTS15 | CEP41 | MTI-429 | 5 (1 died) | H | c.33+2T>G | ND | Yes | Egyptian | 20 | |
| MTI-1491 | 2 | H | c.97+3_97+5delGAG | ND | Yes | Egyptian | 20 | |||
| JBTS16 | TMEM138 | MTI-656 | 1 | c.376G>A | p.A126T | Yes | Egyptian | 21 | ||
| MTI-998 | 1 | c.376G>A | p.A126T | Yes | Egyptian | 21 | ||||
| MTI-129 | 3 | c.380C>T | p.A127V | Yes | Emirati | 21 | ||||
| MTI-499 | 7 (6 died) | c.389A>G | p.Y130C | Yes | Oman | 21 | ||||
| JS_D | 2 | c.389A>G | p.Y130C | Yes | Emirati | This study | ||||
| MTI-1479 | 1 | c.389A>G | p.Y130C | Yes | Emirati | This study | ||||
| MTI-006 | 2 | c.128+5G>A | ND | Yes | Emirati | 21 | ||||
| MTI-381 | 3 (1 died) | c.128+5G>A | ND | ND | OFD VI | Emirati | 21 | |||
| JBTS17 | C5orf42 | JS_F5 | 1 | c.7978C>T | p.R2660* | Yes | Saudi | 4 | ||
| JS_F6 | 2 | c.7988_7989delGA | p.G2663Afs*40 | Yes | Saudi | 4 | ||||
| JS_F7 | 2 | c.7988_7989delGA | p.G2663Afs*40 | Yes | Saudi | 4 | ||||
| JBTS18 | TCTN3 | JS_B | 2 | c.1437G>C | p.R479S | No | digenic | F (Emirati) M (Syrian) | This study | |
| JBTS21 | CSPP1 | ND | 1 | H | c.2527_2528delAT | p.M843Efs*25 | Yes | Lebanese | 22 | |
| ND | 3 (1 died) | H | c.2244_2247delAAGA | p.E750Lfs*7 | Yes | Saudi | 23 | |||
| ND | 1 | H | c.2773C>T | p.R925* | Yes | Libyan | 24 |
Underline, families with digenic inheritance; bold, suspected founder mutations.
Abbreviations: Aff., Affected; CH, compound heterozygous; Cons, consanguinity; F, Female; Fam, family; H, Homozygous; Ht, Heterozygous; JBTS, JS, Joubert Syndrome; M, Male; ND, not determined; No, Number; Pt, Protein; fs, frameshift; * Stop codon.
INPP5E: This gene encodes an inositol polyphosphate-5-phosphatase (EC 3.1.3.36) enzyme that has a role in regulating synaptic vesicle recycling, insulin signaling and embryonic development.22 The INPP5E protein localizes to the primary cilia and is involved in signal transduction. The JBTS1 locus harboring INPP5E was first mapped in two Emirati families showing retinopathy and a proven MTS.23,24 Knockdown of Inppe5 in mice results in decreased primary cilia stability, leading to disorders in multiple organs, including the absence of eyes, polydactyly, exencephaly and renal cysts.25 Homozygous mutations in 20 affected individuals from nine Arab families (three Egyptian, four Emirati, one Yemeni and one Algerian) have been reported26 (Table 2). The major phenotypic features associated with INPP5E mutations include retinal disease associated with renal cystic disease and hepatic fibrosis.
AHI1: This gene encodes Jouberin, a component of a protein complex in the basal body that forms a barrier to restrict protein diffusion between the plasma and ciliary membranes in the transition zone at the base of cilia.27 Jouberin is strongly expressed in the embryonic hindbrain and forebrain, suggesting a role in both cerebral and cortical development.28 Ahi1-null mice displayed a hypoplastic cerebellum with an underdeveloped vermis and a mildly defective foliation pattern, similar to the clinical features of JBTS3 in humans.29 Clinical analysis indicated that AHI1 mutations were associated with retinal abnormalities ranging from retinitis pigmentosa (RP) to blindness; ~80% of patients presented with retinal dystrophy with no kidney or liver changes16 (Figure 1). The proportion of JS patients with mutations in AHI1 is estimated at 10%,30,31 including 12 Arab families from Saudi Arabia, UAE, Kuwait, Egypt and Palestine (Table 2). Three different homozygous mutations in the AHI1 gene were reported in three unrelated families living in the same geographical region of Saudi Arabia.32
CEP290: This gene encodes a centrosomal protein of 290 kDa (known as nephrocystin-6) that localizes to the centrosome and cilia. Nephrocystin-6 is involved in renal cyst formation by modulating the activity of the ATF4 transcription factor. CEP290 interacts with other ciliary proteins such as CC2D2A and meckelin.33–35 Knockdown of cep290 in zebrafish resulted in abnormal cerebellar, renal, and retinal development, whereas naturally occurring mutations in rd16 mice and Abyssinian cats caused progressive retinal degeneration in the absence of renal or cerebellar defects.36,37 To date, over 100 mutations have been identified in CEP290. Approximately 10% of JSRD patients have mutations in the CEP290 gene and display a strong association for both retinal and renal involvement.16 Eight different mutations have been identified in 13 Arab families. Mutations in this gene were the most common type found in the UAE, particularly the potential founder mutation p.G1890* (Table 2 and Figure 2).
Figure 2.

Distribution of all reported mutations in JSRD-associated genes in the Arab world. The Arab world map showing the different distribution of genes and mutations responsible for Joubert and related conditions in the Arab populations. Bold: presumed founder mutations; c´Compound heterozygous mutations; Đdigenic inheritance; del: deletion; Fs: frameshift; *stop codon.
NPHP1: This gene encodes nephrocystin-1, a protein that interacts with the AHI1 protein and other nephronophthisis disease-causing proteins, such as INVS, NPHP3 and NPHP4. Nephrocystin-1 localizes to the primary cilium, to cell–cell adherens junctions, and to the basal body, where it plays essential roles in the control of cell division and cell–cell and cell–matrix adhesion signaling.38 Mutations in NPHP1 result in JBTS4 characterized by JS and nephronophthisis. The MTS in these patients have a distinctive elongated appearance without thickened superior cerebellar peduncles.31 A common ~290 kb deletion involving NPHP1 has been associated with rare cases of JSRD.39 In addition, some individuals with more severe phenotypes than nephronophthisis or SLS carry the homozygous NPHP1 deletion as well as a heterozygous mutation in the AHI1 or CEP290 genes, suggesting that these genes contribute as modifiers.39 This deletion appears to be absent or very rare among Arabs, as only one missense mutation (p.R48K) has been reported (Table 2, Figure 2).
TMEM family of genes (TEME67, TMEM138, TMEM216, TMEM231 and TMEM237)
The tetraspanin super family (Tspan) of proteins contains over 30 genes40–42 that regulate signaling pathways involved in cell adhesion, migration and fusion.43,44 Most recently, five members of the TMEM family of proteins have been implicated in JS, JSRDs and other ciliopathies: TMEM67, TMEM138, TMEM216, TMEM231 and TMEM237.45
TMEM67/MKS3: This gene encodes the meckelin protein, which is expressed in the kidneys, liver, retina, hindbrain, developing sphenoid bone and the brain midline and is strongly expressed in the cartilage of developing limbs, particularly in the digits.46 It was first discovered in five families with Meckel–Gruber syndrome (MIM 607361).45 As reported by Romano et al.,6 Baala et al. 47 found mutations in TMEM67 in two siblings with JS and designated this distinct form as JBTS6 (MIM 610688). Moreover, mutations were identified in patients with the COACH syndrome (MIM 216360).48–50 The clinical features of these patients were consistent with JS associated with congenital hepatic fibrosis, defining the COACH syndrome as subtype of JS with liver involvement. Of note, TMEM67 is involved in other forms of ciliopathy, such as Nephronophthisis 11, and as modifier gene for Bardet–Biedl syndrome. Interestingly, molecular analysis of patients with nephronophthisis and hepatic fibrosis (NPHP11; #212840) and a cohort of nephronophthisis cases without liver involvement showed that liver fibrosis is a specific feature of TMEM67 mutations and is independent of neurological involvement49 (Figure 1). Therefore, MKS3, JBTS6, COACH and NPHP11 represent a broad spectrum of allelic disorders caused by biallelic mutations in the TMEM67 gene. To date, three different TMEM67 mutations have been reported in families from Morocco, Algeria and Egypt (Table 2, Figure 2).
TMEM237/ALS2CR4: This gene was originally described in the Hutterite population with Meckel syndrome.51 However, some patients with JSRDs were found to have mutations in this gene.51,52 JBTS14 is characterized by severe ID, abnormal breathing difficulties in infancy, MTS, renal cysts, abnormal eye movements and early death in many patients and is categorized as JS with oculorenal disease.52 In addition, a distinctive optic disc anomaly was reported in a large Austrian family.53 Encephalocele, hydrocephalus and cystic kidney disease are common in JBTS14 phenotype. Furthermore, tmem237 knockdown in zebrafish caused gastrulation defects consistent with ciliary dysfunction, similar to defects resulting from knockdown of transition zone proteins, including Mks3 (TMEM67; MIM 609884) and Tmem216 (MIM 613277).52 These findings suggest that TMEM237, TMEM216 and MKS3 function as a module to regulate ciliogenesis and WNT signaling.52 Mutations in TMEM237 have been reported in three Arab families from Jordan, Saudi Arabia and the UAE (Table 2, Figure 2).
TMEM138: The involvement of this gene was identified after the exclusion of TMEM216 linked to the JBTS2 locus in six families.54 Screening for mutations in positional genes identified homozygous mutations in TMEM138.54 The JBTS16 phenotype was indistinguishable from that of JBTS2 by MTS, oculomotor apraxia, variable coloboma and rare kidney involvement.54 Mutations in 20 individuals from 8 Arab families have been reported (Table 2, Figure 2).54 Both the TMEM138 and TMEM216 genes are aligned in a head-to-tail orientation in higher vertebrates, with a conserved intergenic region,54 which mediates the coordinated expression of TMEM138 and TMEM216 via the RFX4 protein.54 The knockdown of Tmem216 affects the vesicular trafficking of Tmem138 and Cep290, whereas the knockdown of Tmem138 showed little effect on the vesicular movement of Tmem216.54 Tmem138 localizes to the ciliary axoneme/basal body, while Tmem216 localizes to the basal body and the Golgi apparatus surrounding the base of cilium.54 Both proteins localize to different vesicle pools that move toward the primary cilia over time.54 Only Tmem138 vesicles showed co-localization with the endogenous Cep290 in IMCD3 cells.54
RPGRIP1L: This gene encodes protein fantom, which localizes to the primary cilia and centrosomes in ciliated Madin-Darby canine kidney II cells.55 Recent functional studies in C. elegans showed that Rpgrip1l form a complex with the Mks1, Mksr1, Mksr, Tmem67, Cc2d2a, Nphp1 and Nphp4 proteins. This complex establishes attachments between the basal body and the transition zone membrane and functions as a docking site to restrict vesicle fusion of vesicles containing ciliary proteins.56 RPGRIP1L is highly expressed in adult human testis and kidney and in fetal eye, brain, and kidney.57 JBTS7 is characterized by renal disease (NPHP) in addition to the classical neurological abnormalities of JS55 (Table 1, Figure 1). Moreover, a case of COACH syndrome displaying ID, MTS, nephronophthisis and congenital hepatic fibrosis also displayed mutations in the RPGRIP1 gene, which was defined as a COACH subtype of JS with liver involvement.50 Furthermore, truncated mutations in this gene cause Meckel syndrome, a more severe phenotype (generally lethal). Overall, all reported RPGRIP1 mutations are estimated to contribute to 2–4% of JSRDs,8,55,57 and only one mutation has been found, in a Saudi Arabian family (Table 2, Figure 2).
CC2D2A: This gene encodes a component of a protein complex that comprises Mks1, Tmem216, Tmem67, Cep290, Rpgrip1l, Tctn1 and Tctn2, and it is located in the basal body of the cilia.27,58 CC2D2A is highly expressed in prostate, pancreas, kidney, lung and liver tissues.59 The phenotypic spectrum is highly variable, ranging from pure JS to JS associated with RP and COACH.33,59 Mutations in this gene are responsible for 9% of all JSRDs.33,50 Genotype–phenotype studies showed that null mutations result in the Meckel phenotype, whereas missense and/or hypomorphic mutations are responsible for JSRDs.34,60 Furthermore, mutations in this gene were associated with ventriculomegaly and seizures in some cases.35 A possible digenic inheritance has been reported in a Swiss patient with heterozygous mutations in the CC2D2A and CEP41 genes, consistent with JBTS9 and JBTS15, respectively.61 Similarly, a potential digenic inheritance in one Egyptian family was reported. The patient had heterozygous mutations in the CC2D2A and TMEM67 genes, consistent with JBTS9 and JBTS6, respectively62 (Table 2, Figure 2). Mutations in CC2D2A have been reported in Arabs from Saudi Arabia, Egypt, Algeria and Syria (Table 2, Figure 2).
KIF7: This gene encodes a cilia-associated protein belonging to the kinesin family that has a major role in the hedgehog signaling pathway and the regulation of microtubule acetylation and stabilization.63,64 Individuals with KIF7 mutations often have oro-facio-digital manifestations associated with or without agenesis/hypoplasia of the corpus callosum, hydrocephalus and macrocephaly. The digital anomalies consist of postaxial polydactyly of the hands and preaxial (and/or postaxial) polydactyly of the feet. Overlapping genetically related disorders include hydrolethalus syndrome (HLS; MIM 614120) and acrocallosal syndrome (ACLS; MIM 200990), with several patients exhibiting MTS along with features of HLS and ACLS, leading to diagnostic ambiguity.65 ACLS is a rare disorder characterized by anencephaly as the primary manifestation together with a variety of developmental abnormalities, ID and preaxial polydactyly.63,65 Patients displaying the major features of ACLS present with MTS, eliciting a subtype of JSRD and suggesting that ACLS and JBTS12 are overlapping ciliopathies.65 HLS is a lethal disorder caused by mutation of the HYLS1 gene, particularly in the Finnish population. A homozygous mutation was identified in KIF7 in four affected fetuses of consanguineous Algerian pedigrees who showed the typical features of HLS in addition to MTS.65 Additionally, digenic inheritance of heterozygous mutations in the CEP290 and KIF7 genes has been reported in a German patient with JS.54 In another German patient, a heterozygous 12 bp deletion in KIF7 and two missense mutations in TMEM67 gene were detected.63 Two Arab families from Egypt and Algeria were found to have mutations in KIF7 (Table 2, Figure 2). In addition, a missense mutation in KIF7 has been reported to cause an AR syndrome presenting with macrocephaly, multiple epiphyseal dysplasia and distinctive facial appearance in two Omani families.66
TCTN1: This gene encodes Tectonic-1 that localizes to the membrane-spanning transition zone complex, a region between the basal body and the ciliary axoneme that regulates ciliogenesis.58,67 Knockdown of Tctn1 in mice resulted in lethal holoprosencephaly and disruption of the nodal flow, laterality defects and neural tube dorsalization. This finding suggests that Tctn1 plays a dual role in the repression and activation of the hedgehog signaling pathway.67 Moreover, null mice for Tcnt2 and Cc2d2a displayed similar phenotypes, suggesting a common role for these three genes in tissue-specific ciliogenesis.58 Furthermore, Tctn1 has been shown to be a member of a JSRD and Meckel-associated protein complex, similar to Mks1, Tmem216, Tmem67, Cep290 Tctn2 and Cc2d2a. To date, only three pedigrees from Bangladesh and Saudi Arabia harboring TCTN1 mutations have been reported (Table 2, Figure 2). JBTS13 patients display cerebellar vermis hypoplasia and MTS without renal or ophthalmological abnormalities.58,68 Of note, only one case has been reported to exhibit bilateral fronto-temporal pachygyria and coarsening of the cerebral gyri.58
TCTN3: This gene encodes tectonic-3, which is part of the transition zone complex at the cilium/plasma membrane border.69 TCTN3 was first linked to a severe prenatal form of OFD IV (Mohr–Majewski) syndrome characterized by long bone bowing, tibial hypoplasia, polydactyly, cystic kidneys, orofacial anomalies and encephalocele. Moreover, a less severe phenotype manifesting vermis agenesis and MTS as well as variable digital and axial skeletal anomalies, including kyphoscoliosis and horseshoe kidney,69 was reported in a Turkish family with JSRD.69 We report here a digenic inheritance in a non-consanguineous Emirati family (family JS_B); the father is Emirati, while the mother is Syrian. Two heterozygous missense mutations, p.R479S and p.I641N, were detected in TCTN3 and AHI1, respectively, by exome sequencing (Table 2, Figure 2). Both mutations were novel, and computational analysis suggested that they are potentially pathogenic. Although TCTN3 is not critical for cilia biogenesis in the kidneys, this protein plays a major role in GLI3 processing and function in the sonic hedgehog pathway.69
CEP41: This gene encodes a protein with two coiled-coil domains and a rhodanese-like domain that localizes to the centrioles and cilia.61 Knockdown in zebrafish showed peripheral heart edema and tail defects, which is consistent with a ciliopathy.61 In this organism, Cep41 is expressed in various ciliary organs such as brain, ear, heart and kidney.61 However, null mice demonstrated a wide phenotypic spectrum, ranging from wild type to exencephaly, dilated pericardial sac and lethality.61 JBTS15 is characterized by ataxia, hypotonia, delayed psychomotor development, and ID with variable features, including breathing difficulties, polydactyly and oculomotor apraxia.61 Moreover, some patients carrying CEP41 mutations display retinal and kidney disease. All mutations identified in the CEP41 gene are splice site mutations.61 However, heterozygous mutations in CEP41 associated with heterozygous mutations in other ciliary genes (such as KIF7 and CC2D2A) have been detected in patients with JS, BBS and Meckel syndromes.61 These findings suggest possible digenic inheritance and a role for CEP41 as a modifier gene in other ciliopathies.61 A single mutation in this gene was reported in two Arab families with JSRDs from Egypt (Table 2, Figure 2).
C5orf42: This gene encodes a protein of unknown function that has features of a transmembrane protein and a putative coiled-coil domain.70 This protein is expressed in a broad variety of tissues and might play a role in neurodevelopment. Several compound heterozygous mutations have been detected in unrelated families of French-Canadian descent with JBTS17.70 The clinical manifestations showed a distinct phenotype that most closely resembles a pure JS with an absence of retinal or renal involvement.70,71 Only one case has been reported to exhibit preaxial and postaxial polydactyly.70 This gene is the cause of JS in the original family reported by Joubert et al.9 in 1969. Three families from Saudi Arabia carrying one possible founder mutation (p.G2663Afs*40) have been reported (Table 2, Figure 2).
CSPP1: This gene encodes a protein that localizes to microtubules that accumulate around the centrosome.72 CSPP1 is highly expressed in both the adult and fetal human brain, particularly in the cerebellum.72 Knockdown of cspp1a in zebrafish results in a curved body shape, dilated ventricles and pronephric cysts, which is consistent with a ciliopathy.73 JBTS21 is characterized by MTS, hypotonia, developmental delay, eye movement abnormalities and abnormal breathing.73–75 However, some cases display features of asphyxiating thoracic dystrophy (Jeune syndrome).73 Immunostaining of patients’ fibroblasts revealed defects in ciliogenesis and evidence of decreased trafficking of the ciliary proteins ARL13B and ADCY3 to the axoneme compared with controls.73 Three Arab families from Saudi Arabia, Lebanon and Libya carrying mutations in CSPP1 have been reported (Table 2, Figure 2). The patients from Saudi Arabia showed more severe phenotypes that were characterized by hydranencephaly, occipital encephalocele, wide cranial sutures, anophthalmia, single nostril and hyperechogenic kidneys.74
JS and JSRDS are genotypically heterogeneous in Arab populations
We have presented a comprehensive review of the genes and mutations causing JS and JSRDs in Arab families. Despite the high level of consanguinity and the presence of isolated communities within most Arab populations, there is considerable genetic and allelic heterogeneity within these populations with only a few potential founder mutations (Table 2, Figure 2).
To date, 23 genes have been linked to the broad phenotypic spectrum of JS, with potentially more genes remaining unknown. Mutations in 15 of these genes have been reported in Arab families (Table 2). Interestingly, the most common mutated genes in Arabs are INPP5E, AHI1, CEP290, CC2D2A and TMEM138 (Table 2, Figure 2), each with suspected founder mutations. Moreover, some genes have a high incidence in a particular region or country compared with the other ciliopathy genes. For example, mutations in KIF7 and TMEM67 are found primarily in North African Arab countries (Figures 2 and 3). On the other hand, mutations in C5orf42 and RPGRIP1L have only been detected in Saudi Arabia. Mutations in AHI1 and CEP290 predominantly occur in the Arabian Gulf region (Figure 2). It is worth noting that mutations in TCTN1 have been found exclusively in Arab families thus far (Table 2, Figure 2, and Figure 3). Overall, the incidence of these mutations in Arabs is not very high; therefore, each of these genes should be considered relevant when dealing with suspected cases (Table 2, Figure 2, and Figure 3).
Figure 3.
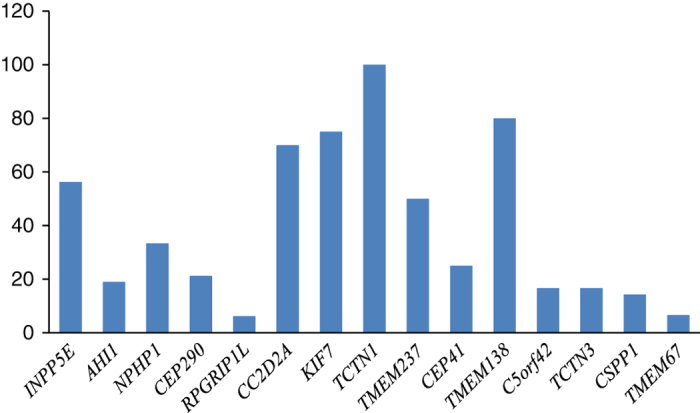
Percentage of reported mutations in Arab populations per total mutations in each gene. The bar chart represents the percentage of mutations reported in this study and extracted from the HGMD database (http://www.biobase-international.com/product/hgmd) in Arab populations per total reported mutations in each gene individually.
Conclusions
This review has focused on the genes and mutations involved in JSRDs among Arabs. We found a wide genetic and genotypic heterogeneity along the JS spectrum and JSRDs in Arabs. Most of the underlying genes encode proteins expressed in the primary cilium and/or basal body and centrosome. Physical interactions between several ciliary proteins have been shown; however, the exact signaling mechanisms and disrupted developmental pathways remain unclear. Moreover, the link between ciliogenesis and brain development is still not well defined. Therefore, identifying the molecular pathogenesis associated with JSRDs could alter prognosis and medical management and provide preventive guidance for these ciliopathies. Many of the ciliary genes described above are not available for clinical testing but can instead be studied in research laboratories. Owing to the diversity of genes and mutations underlying JSRDs, whole exome and/or whole genome sequencing may be an appropriate starting point for molecular diagnosis. We anticipate that many families with JSRDs in Arab countries remain unstudied; therefore, the spectrum and distribution of the causative mutations might change as we learn more about this condition. This review should be of significant importance to clinicians and scientists working on genetic conditions in Arab populations.
Acknowledgments
The authors acknowledge financial support from the National Research Foundation (NRF), the United Arab Emirates University and the Sheikh Hamdan Bin Rashid Al Maktoum Award for Medical Research.
The authors declare no conflict of interest.
References
- Al-Gazali L, Hanan H. Consanguinity and dysmorphology in Arabs. Human Heredity 2014; 77: 93–107. [DOI] [PubMed] [Google Scholar]
- Tadmouri GO, Nair P, Obeid T, Al Ali MT, Al Khaja N, Hamamy HA. Consanguinity and reproductive health among Arabs. Reprod Health 2009; 6: 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Gazali L, Hamamy H, Al-Arrayad S. Genetic disorders in the Arab world. BMJ 2006; 333: 831–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bener A, Eihakeem AA, Abdulhadi K. Is there any association between consanguinity and hearing loss. Int J Pediatr Otorhinolaryngol 2005; 69: 327–333. [DOI] [PubMed] [Google Scholar]
- Al-Gazali L, Ali BR. Mutations of a country: a mutation review of single gene disorders in the United Arab Emirates (UAE). Hum Mutat 2010; 31: 505–520. [DOI] [PubMed] [Google Scholar]
- Romano S, Boddaert N, Desguerre I, Hubert L, Salomon R, Seidenwurm D et al. Molar tooth sign and superior vermian dysplasia: a radiological, clinical, and genetic study. Neuropediatrics 2006; 37: 42–45. [DOI] [PubMed] [Google Scholar]
- Parisi MA, Doherty D, Chance PF, Glass IA. Joubert syndrome (and related disorders) (OMIM 213300). Eur J Hum Genet 2007; 15: 511–521. [DOI] [PubMed] [Google Scholar]
- Parisi MA. Clinical and molecular features of Joubert syndrome and related disorders. Am J Med Genet C Semin Med Genet 2009; 151C: 326–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joubert M, Eisenring JJ, Andermann F. Familial dysgenesis of the vermis: a syndrome of hyperventilation, abnormal eye movements and retardation. Neurology 1968; 18: 302–303. [PubMed] [Google Scholar]
- Maria BL, Hoang KB, Tusa RJ, Mancuso AA, Hamed LM, Quisling RG et al. ‘Joubert syndrome’ revisited: key ocular motor signs with magnetic resonance imaging correlation. J Child Neurol 1997; 12: 423–430. [DOI] [PubMed] [Google Scholar]
- Gleeson JG, Keeler LC, Parisi MA, Marsh SE, Chance PF, Glass IA et al. Molar tooth sign of the midbrain-hindbrain junction: occurrence in multiple distinct syndromes. Am J Med Genet A 2004; 125A: 125–134. [DOI] [PubMed] [Google Scholar]
- Parisi MA, Pinter JD, Glass IA, Field K, Maria BL, Chance PF et al. Cerebral and cerebellar motor activation abnormalities in a subject with Joubert syndrome: functional magnetic resonance imaging (MRI) study. J Child Neurol 2004; 19: 214–218. [PubMed] [Google Scholar]
- Saraiva JM, Baraitser M. Joubert syndrome: a review. Am J Med Genet 1992; 43: 726–731. [DOI] [PubMed] [Google Scholar]
- Maria BL, Quisling RG, Rosainz LC, Yachnis AT, Gitten J, Dede D et al. Molar tooth sign in Joubert syndrome: clinical, radiologic, and pathologic significance. J Child Neurol 1999; 14: 368–376. [DOI] [PubMed] [Google Scholar]
- Maria BL, Boltshauser E, Palmer SC, Tran TX. Clinical features and revised diagnostic criteria in Joubert syndrome. J Child Neurol 1999; 14: 583–590. [DOI] [PubMed] [Google Scholar]
- Valente EM, Brancati F, Dallapiccola B. Genotypes and phenotypes of Joubert syndrome and related disorders. Eur J Med Genet 2008; 51: 1–23. [DOI] [PubMed] [Google Scholar]
- Brancati F, Dallapiccola B, Valente EM. Joubert Syndrome and related disorders. Orphanet J Rare Dis 2010; 5: 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holroyd S, Reiss AL, Bryan RN. Autistic features in Joubert syndrome: a genetic disorder with agenesis of the cerebellar vermis. Biol Psychiatry 1991; 29: 287–294. [DOI] [PubMed] [Google Scholar]
- Ozonoff S, Williams BJ, Gale S, Miller JN. Autism and autistic behavior in Joubert syndrome. J Child Neurol 1999; 14: 636–641. [DOI] [PubMed] [Google Scholar]
- Satran D, Pierpont ME, Dobyns WB. Cerebello-oculo-renal syndromes including Arima, Senior-Löken and COACH syndromes: more than just variants of Joubert syndrome. Am J Med Genet 1999; 86: 459–469. [PubMed] [Google Scholar]
- Kher AS, Chattopadhyay A, Divekar A, Khambekar K, Bharucha BA. Joubert syndrome with polydactyly and optic coloboma in two sibs. Indian J Pediatr 1994; 61: 729–732. [DOI] [PubMed] [Google Scholar]
- Dyson JM, Fedele CG, Davies EM, Becanovic J, Mitchell CA. Phosphoinositide phosphatases: just as important as the kinases. Subcell Biochem 2012; 58: 215–279. [DOI] [PubMed] [Google Scholar]
- Saar K, Al-Gazali L, Sztriha L, Rueschendorf F, Nur-E-Kamal M, Reis A et al. Homozygosity mapping in families with Joubert syndrome identifies a locus on chromosome 9q34.3 and evidence for genetic heterogeneity. Am J Hum Genet 1999; 65: 1666–1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valente EM, Marsh SE, Castori M, Dixon-Salazar T, Bertini E, Al-Gazali L et al. Distinguishing the four genetic causes of Jouberts syndrome-related disorders. Ann Neurol 2005; 57: 513–519. [DOI] [PubMed] [Google Scholar]
- Jacoby M, Cox JJ, Gayral S, Hampshire DJ, Ayub M, Blockmans M et al. INPP5E mutations cause primary cilium signaling defects, ciliary instability and ciliopathies in human and mouse. Nat Genet 2009; 41: 1027–1031. [DOI] [PubMed] [Google Scholar]
- Bielas SL, Silhavy JL, Brancati F, Kisseleva MV, Al-Gazali L, Sztriha L et al. Mutations in INPP5E, encoding inositol polyphosphate-5-phosphatase E, link phosphatidyl inositol signaling to the ciliopathies. Nat Genet 2009; 41: 1032–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chih B, Liu P, Chinn Y, Chalouni C, Komuves LG, Hass PE et al. A ciliopathy complex at the transition zone protects the cilia as a privileged membrane domain. Nat Cell Biol 2012; 14: 61–72. [DOI] [PubMed] [Google Scholar]
- Dixon-Salazar T, Silhavy JL, Marsh SE, Louie CM, Scott LC, Gururaj A et al. Mutations in the AHI1 gene, encoding jouberin, cause Joubert syndrome with cortical polymicrogyria. Am J Hum Genet 2004; 75: 979–987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lancaster MA, Gopal DJ, Kim J, Saleem SN, Silhavy JL, Louie CM et al. Defective Wnt-dependent cerebellar midline fusion in a mouse model of Joubert syndrome. Nat Med 2011; 17: 726–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valente EM, Brancati F, Silhavy JL, Castori M, Marsh SE, Barrano G et al. AHI1 gene mutations cause specific forms of Joubert syndrome-related disorders. Ann Neurol 2006; 59: 527–534. [DOI] [PubMed] [Google Scholar]
- Parisi MA, Doherty D, Eckert ML, Shaw DW, Ozyurek H, Aysun S et al. AHI1 mutations cause both retinal dystrophy and renal cystic disease in Joubert syndrome. J Med Genet 2006; 43: 334–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferland RJ, Eyaid W, Collura RV, Tully LD, Hill RS, Al-Nouri D et al. Abnormal cerebellar development and axonal decussation due to mutations in AHI1 in Joubert syndrome. Nat Genet 2004; 36: 1008–1013. [DOI] [PubMed] [Google Scholar]
- Gorden NT, Arts HH, Parisi MA, Coene KL, Letteboer SJ, van Beersum SE et al. CC2D2A is mutated in Joubert syndrome and interacts with the ciliopathy-associated basal body protein CEP290. Am J Hum Genet 2008; 83: 559–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tallila J, Jakkula E, Peltonen L, Salonen R, Kestilä M. Identification of CC2D2A as a Meckel syndrome gene adds an important piece to the ciliopathy puzzle. Am J Hum Genet 2008; 82: 1361–1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachmann-Gagescu R, Ishak GE, Dempsey JC, Adkins J, O'Day D, Phelps IG et al. Genotype-phenotype correlation in CC2D2A-related Joubert syndrome reveals an association with ventriculomegaly and seizures. J Med Genet 2012; 49: 126–137. [DOI] [PubMed] [Google Scholar]
- Coppieters F, Lefever S, Leroy BP, De Baere E. CEP290, a gene with many faces: mutation overview and presentation of CEP290base. Hum Mutat 2010; 31: 1097–1108. [DOI] [PubMed] [Google Scholar]
- Sayer JA, Otto EA, O'Toole JF, Nurnberg G, Kennedy MA, Becker C et al. The centrosomal protein nephrocystin-6 is mutated in Joubert syndrome and activates transcription factor ATF4. Nat Genet 2006; 38: 674–681. [DOI] [PubMed] [Google Scholar]
- Hildebrandt F, Attanasio M, Otto E. Nephronophthisis: disease mechanisms of a ciliopathy. J Am Soc Nephrol 2009; 20: 23–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tory K, Lacoste T, Burglen L, Morinière V, Boddaert N, Macher MA et al. High NPHP1 and NPHP6 mutation rate in patients with Joubert syndrome and nephronophthisis: potential epistatic effect of NPHP6 and AHI1 mutations in patients with NPHP1 mutations. J Am Soc Nephrol 2007; 18: 1566–1575. [DOI] [PubMed] [Google Scholar]
- Maecker HT, Todd SC, Levy S. The tetraspanin superfamily: molecular facilitators. FASEB J 1997; 11: 428–442. [PubMed] [Google Scholar]
- Todd SC, Doctor VS, Levy S. Sequences and expression of six new members of the tetraspanin/TM4SF family. Biochim Biophys Acta 1998; 1399: 101–104. [DOI] [PubMed] [Google Scholar]
- Serru V, Dessen P, Boucheix C, Rubinstein E. Sequence and expression of seven new tetraspans. Biochim Biophys Acta 2000; 1478: 159–163. [DOI] [PubMed] [Google Scholar]
- Junge HJ, Yang S, Burton JB, Paes K, Shu X, French DM et al. TSPAN12 regulates retinal vascular development by promoting Norrin- but not Wnt-induced FZD4/beta-catenin signaling. Cell 2009; 139: 299–311. [DOI] [PubMed] [Google Scholar]
- Hübner K, Windoffer R, Hutter H, Leube RE. Tetraspan vesicle membrane proteins: synthesis, subcellular localization, and functional properties. Int Rev Cytol 2002; 214: 103–159. [DOI] [PubMed] [Google Scholar]
- Smith UM, Consugar M, Tee LJ, McKee BM, Maina EN, Whelan S et al. The transmembrane protein meckelin (MKS3) is mutated in Meckel–Gruber syndrome and the wpk rat. Nat Genet 2006; 38: 191–196. [DOI] [PubMed] [Google Scholar]
- Dawe HR, Smith UM, Cullinane AR, Gerrelli D, Cox P, Badano JL et al. The Meckel–Gruber syndrome proteins MKS1 and meckelin interact and are required for primary cilium formation. Hum Mol Genet 2007; 16: 173–186. [DOI] [PubMed] [Google Scholar]
- Baala L, Romano S, Khaddour R, Saunier S, Smith UM, Audollent S et al. The Meckel–Gruber syndrome gene, MKS3, is mutated in Joubert syndrome. Am J Hum Genet 2007; 80: 186–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brancati F, Iannicelli M, Travaglini L, Mazzotta A, Bertini E, Boltshauser E et al. MKS3/TMEM67 mutations are a major cause of COACH Syndrome, a Joubert syndrome related disorder with liver involvement. Hum Mutat 2009; 30: E432–E442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otto EA, Tory K, Attanasio M, Zhou W, Chaki M, Paruchuri Y et al. Hypomorphic mutations in meckelin (MKS3/TMEM67) cause nephronophthisis with liver fibrosis (NPHP11). J Med Genet 2009; 46: 663–670. [DOI] [PubMed] [Google Scholar]
- Doherty D, Parisi MA, Finn LS, Gunay-Aygun M, Al-Mateen M, Bates D et al. Mutations in 3 genes (MKS3, CC2D2A and RPGRIP1L) cause COACH syndrome (Joubert syndrome with congenital hepatic fibrosis). J Med Genet 2010; 47: 8–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boycott KM, Parboosingh JS, Scott JN, McLeod DR, Greenberg CR, Fujiwara TM et al. Meckel syndrome in the Hutterite population is actually a Joubert-related cerebello-oculo-renal syndrome. Am J Med Genet A 2007; 143A: 1715–1725. [DOI] [PubMed] [Google Scholar]
- Huang L, Szymanska K, Jensen VL, Janecke AR, Innes AM, Davis EE et al. TMEM237 is mutated in individuals with a Joubert syndrome related disorder and expands the role of the TMEM family at the ciliary transition zone. Am J Hum Genet 2011; 89: 713–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janecke AR, Müller T, Gassner I, Kreczy A, Schmid E, Kronenberg F et al. Joubert-like syndrome unlinked to known candidate loci. J Pediatr 2004; 144: 264–269. [DOI] [PubMed] [Google Scholar]
- Lee JH, Silhavy JL, Lee JE, Al-Gazali L, Thomas S, Davis EE et al. Evolutionarily assembled cis-regulatory module at a human ciliopathy locus. Science 2012; 335: 966–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delous M, Baala L, Salomon R, Laclef C, Vierkotten J, Tory K et al. The ciliary gene RPGRIP1L is mutated in cerebello-oculo-renal syndrome (Joubert syndrome type B) and Meckel syndrome. Nat Genet 2007; 39: 875–881. [DOI] [PubMed] [Google Scholar]
- Williams CL, Li C, Kida K, Inglis PN, Mohan S, Semenec L et al. MKS and NPHP modules cooperate to establish basal body/transition zone membrane associations and ciliary gate function during ciliogenesis. J Cell Biol 2011; 192: 1023–1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arts HH, Doherty D, van Beersum SE, Parisi MA, Letteboer SJ, Gorden NT et al. Mutations in the gene encoding the basal body protein RPGRIP1L, a nephrocystin-4 interactor, cause Joubert syndrome. Nat Genet 2007; 39: 882–888. [DOI] [PubMed] [Google Scholar]
- Garcia-Gonzalo FR, Corbit KC, Sirerol-Piquer MS, Ramaswami G, Otto EA, Noriega TR et al. A transition zone complex regulates mammalian ciliogenesis and ciliary membrane composition. Nat Genet 2011; 43: 776–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noor A, Windpassinger C, Patel M, Stachowiak B, Mikhailov A, Azam M et al. CC2D2A, encoding a coiled-coil and C2 domain protein, causes autosomal-recessive mental retardation with retinitis pigmentosa. Am J Hum Genet 2008; 82: 1011–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mougou-Zerelli S, Thomas S, Szenker E, Audollent S, Elkhartoufi N, Babarit C et al. CC2D2A mutations in Meckel and Joubert syndromes indicate a genotype-phenotype correlation. Hum Mutat 2009; 30: 1574–1582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JE, Silhavy JL, Zaki MS, Schroth J, Bielas SL, Marsh SE et al. CEP41 is mutated in Joubert syndrome and is required for tubulin glutamylation at the cilium. Nat Genet 2012; 44: 193–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otto EA, Ramaswami G, Janssen S, Chaki M, Allen SJ, Zhou W et al. Mutation analysis of 18 nephronophthisis associated ciliopathy disease genes using a DNA pooling and next generation sequencing strategy. J Med Genet 2011; 48: 105–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dafinger C, Liebau MC, Elsayed SM, Hellenbroich Y, Boltshauser E, Korenke GC et al. Mutations in KIF7 link Joubert syndrome with Sonic Hedgehog signaling and microtubule dynamics. J Clin Invest 2011; 121: 2662–2667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liem KF, He M, Ocbina PJ, Anderson KV. Mouse Kif7/Costal2 is a cilia-associated protein that regulates Sonic Hedgehog signaling. Proc Natl Acad Sci USA . 2009; 106: 13377–13382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putoux A, Thomas S, Coene KL, Davis EE, Alanay Y, Ogur G et al. KIF7 mutations cause fetal hydrolethalus and acrocallosal syndromes. Nat Genet 2011; 43: 601–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali BR, Silhavy JL, Akawi NA, Gleeson JG, Al-Gazali L. A mutation in KIF7 is responsible for the autosomal recessive syndrome of macrocephaly, multiple epiphyseal dysplasia and distinctive facial appearance. Orphanet J Rare Dis 2012; 7: 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiter JF, Skarnes WC. Tectonic, a novel regulator of the Hedgehog pathway required for both activation and inhibition. Genes Dev 2006; 20: 22–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alazami AM, Alshammari MJ, Salih MA, Alzahrani F, Hijazi H, Seidahmed MZ et al. Molecular characterization of Joubert syndrome in Saudi Arabia. Hum Mutat 2012; 33: 1423–1428. [DOI] [PubMed] [Google Scholar]
- Thomas S, Legendre M, Saunier S, Bessières B, Alby C, Bonnière M et al. TCTN3 mutations cause Mohr-Majewski syndrome. Am J Hum Genet 2012; 91: 372–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srour M, Schwartzentruber J, Hamdan FF, Ospina LH, Patry L, Labuda D et al. Mutations in C5ORF42 cause Joubert syndrome in the French Canadian population. Am J Hum Genet 2012; 90: 693–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srour M, Hamdan FF, Schwartzentruber JA, Patry L, Ospina LH, Shevell MI et al. Mutations in TMEM231 cause Joubert syndrome in French Canadians. J Med Genet 2012; 49: 636–641. [DOI] [PubMed] [Google Scholar]
- Patzke S, Stokke T, Aasheim HC. CSPP and CSPP-L associate with centrosomes and microtubules and differently affect microtubule organization. J Cell Physiol 2006; 209: 199–210. [DOI] [PubMed] [Google Scholar]
- Tuz K, Bachmann-Gagescu R, O'Day DR, Hua K, Isabella CR, Phelps IG et al. Mutations in CSPP1 cause primary cilia abnormalities and Joubert syndrome with or without Jeune asphyxiating thoracic dystrophy. Am J Hum Genet 2014; 94: 62–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaheen R, Shamseldin HE, Loucks CM, Seidahmed MZ, Ansari S, Ibrahim Khalil M et al. Mutations in CSPP1, encoding a core centrosomal protein, cause a range of ciliopathy phenotypes in humans. Am J Hum Genet 2014; 94: 73–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akizu N, Silhavy JL, Rosti RO, Scott E, Fenstermaker AG, Schroth J et al. Mutations in CSPP1 lead to classical Joubert syndrome. Am J Hum Genet 2014; 94: 80–86. [DOI] [PMC free article] [PubMed] [Google Scholar]