

Abstract
Several studies have explored the origin and development mechanism of oral lichen planus (OLP) with limited attention to the role of bacteria in the progression of this common oral disease. Here we utilized MiSeq sequencing of 16S rRNA gene amplicons to identify complex oral microbiota associated with OLP from saliva samples of two subtypes (reticular and erosive) of OLP patients and healthy controls. Our analyses indicated that the overall structure of the salivary microbiome was not significantly affected by disease status. However, we did observe evident variations in abundance for several taxonomic groups in OLP. Porphyromonas and Solobacterium showed significantly higher relative abundances, whereas Haemophilus, Corynebacterium, Cellulosimicrobium and Campylobacter showed lower abundances in OLP patients, as compared with healthy controls. In addition, we explored specific microbial co-occurrence patterns in OLP, and revealed significantly fewer linkers of Streptococcus comprising species in erosive OLP. Furthermore, the disease severity and immune dysregulation were also genus-associated, including with Porphyromonas that correlated to disease scores and salivary levels of interleukin (IL)-17 and IL-23. Overall, this study provides a general description of oral microbiome in OLP, and it will be useful for further investigation of their potential roles in the initiation and immune modulation of OLP.
Lichen planus is a common chronic mucocutaneous inflammatory disease affecting skin, oral, genital mucosa, scalp and nails. The prevalence of oral lichen planus (OLP) ranges from 0.1–4% in the general population, and females between 30 and 60 are more vulnerable to this disease1. Lesion manifestations can often persist for years alternating between periods of quiescence and exacerbation2. Intraorally, OLP presents as white striations, papules, plaques, mucosal atrophy, erosions or blisters, mainly affecting the buccal mucosa, tongue, gingiva and lips3. Among those clinical phenotypes, the reticular form is the most prevalent lesion characterized by the presence of Wickham striae and usually asymptomatic. Whereas the erosive type, although less frequent, always causes different degrees of discomfort and soreness, it may need ongoing concern due to its malignant potential4.
Despite of numerous literatures appraising the OLP origin and development mechanism, its aetiology remains uncertain, and the pathogenesis of this disease still needs more investigations. Several internal and external factors have been implicated, such as immunodeficiencies and microbial infection5. Several lines of evidences have suggested that immune dysregulation and complex cytokine network played important roles in the exacerbation and perpetuation of OLP6,7. For instance, a high proportion of the novel CD4+ T helper cells subset-Th17 cells were detected in OLP lesions8. And interleukin (IL)-17 and IL-23, the newly discovered Th17-associated cytokines, were found to be important proinflammatory factors involved in OLP9,10. In terms of infection with oral microorganisms, several viruses have been linked with OLP11,12,13,14,15. Besides that, Lundstrom et al. demonstrated Candida infection in 37–50% of OLP patients16. An association between Helicobacter pylori and OLP has been shown but remains controversial17,18,19. While not directly involved in the etiology of OLP, all of these factors could influence the composition of oral microbiota, which might in turn modulate immunity and thereby affect the progression of this disease. An early report noticed that intensive oral hygiene could provide improvement of some patients with atrophic and ulcerative gingival lichen planus20. Backman and Jontell demonstrated that the treatment of chlorhexidine could considerably relieve the symptoms of lichenoid lesions21. A recent study found significantly higher counts of Staphylococcus haemolyticus and Streptococcus agalactiae in OLP lesions22. These findings propelled the further exploration of the links between oral bacteria and OLP. Previously, we evaluated the differences in genetic profiling of salivary microbial community between OLP and matched healthy controls by means of polymerase chain reaction-based denaturing gradient gel electrophoresis10. To investigate the microbiota associated with OLP more extensively, in this study, we further applied with Illumina MiSeq sequencing to identify the microbiome in saliva samples from two subtypes (reticular and erosive) of OLP patients and healthy controls.
In the present study, we aimed to (1) characterize the overall structure of oral microbiota in saliva of OLP patients; (2) seek for OLP-associated microorganisms; (3) elucidate the changes in microbial interactions of OLP patients; and (4) identify key microbial populations associated with disease and immune status. Our results indicated that even though the microbial community composition has not dramatically altered, some abundant genera still have changed obviously. The interactions of microbial-microbial and microbial-immune might play a key role in OLP pathogenesis directly or indirectly.
Results
The phylogenetic structure of OLP microbial communities showed a greater variety and less specificity
The demographic and clinical characteristics of all subjects are shown in Table 1. Statistical analysis failed to reveal significant group differences in age and gender (P = 0.127 and P = 0.815 respectively).
Table 1. Demographics and clinical parameters of all subjects.
| Characteristics | Healthy subjects (n = 18) | Reticular OLP patients (n = 19) | Erosive OLP patients (n = 18) |
|---|---|---|---|
| Age (Mean ± SD) | 39.72 ± 11.02 | 43.42 ± 9.56 | 46.72 ± 9.80 |
| Male/Female | 5/13 | 7/12 | 5/13 |
| Disease severity score | 0 | 1~3 | 4~5 |
| Body mass index (Mean ± SD) | 20.22 ± 1.37 | 21.14 ± 1.12 | 20.56 ± 1.78 |
| Diabetes | 0 | 0 | 0 |
| Cigarette smoking | 0 | 0 | 0 |
| Alcohol drinking | 0 | 0 | 0 |
| No. of teeth (Mean ± SD) | 29.78 ± 1.77 | 28.43 ± 2.25 | 28.78 ± 2.58 |
| Plaque index (Mean ± SD) | 1.32 ± 0.43 | 1.22 ± 0.51 | 1.40 ± 0.47 |
| Gingival index (Mean ± SD) | 1.08 ± 0.54 | 1.29 ± 0.45 | 1.23 ± 0.52 |
| Bleeding on probing (+/−) | 1/18 | 2/18 | 1/17 |
| Probing depth/mm (Mean ± SD) | 2.01 ± 0.71 | 2.21 ± 0.40 | 2.17 ± 0.56 |
| Clinical attachment loss/mm (Mean ± SD) | 0.67 ± 0.50 | 0.73 ± 0.54 | 0.75 ± 0.43 |
To investigate the changes in structure and composition of salivary microbial communities of OLP, 55 samples were sequenced with Illumina MiSeq technology. After preprocessing, 2,706,496 high quality sequences with average length of 253 bp were obtained in this study, with an average of 49209 sequences per sample. From these sequences, 13 known phyla and 418 genera were identified, and a total of 8717 OTUs were detected, of which 4006 were singletons (detected in only one sample) and were excluded from further statistical analysis. Good’s estimator of coverage was 99.85%, indicating that the 16S rRNA sequences identified in this study likely represent the majority of bacterial sequences present in saliva samples. The sequencing data for details are available in Supplementary Table S1.
Principal coordinates analysis (PCoA) plot based on Bray-Curtis distances indicated no obvious separation among healthy subjects, reticular OLP and erosive OLP (Supplementary Figure S1). The dissimilarity tests among groups did not show marked differences as well (P > 0.05, Table 2). Oral microbial diversity was similar between OLP patients and healthy controls (P > 0.05, Fig. 1A–C, Supplementary Table S1). There was a slightly higher richness detected in erosive OLP than that in other two groups, but no statistical significance was found. The rarefaction analysis showed a total of 1989, 2054, and 2823 OTUs (at 239,000 sequencing depth) in healthy controls, reticular OLP and erosive OLP respectively, indicating that more phylotypes would likely be detected in erosive OLP than other groups after exploring larger number of sequences (Fig. 1D).
Table 2. Comparison of the overall microbial community structure using three nonparametric statistical methods.
| Groups | MRPP | P-value | Adonis | P-value | ANOSIM | P-value |
|---|---|---|---|---|---|---|
| H vs R | 0.511 | 0.252 | 0.036 | 0.209 | 0.011 | 0.294 |
| H vs E | 0.513 | 0.158 | 0.040 | 0.155 | 0.021 | 0.210 |
| R vs E | 0.520 | 0.302 | 0.032 | 0.263 | 0.008 | 0.326 |
(H, healthy control; R, reticular OLP; E, erosive OLP).
Figure 1. Alpha Diversity analysis for healthy controls (H), reticular OLP (R) and erosive OLP (E).

No significant differences between healthy and diseased samples were observed for various alpha diversity measures (P > 0.05). The scatter plots were shown with black spots and horizontal lines representing the mean and standard deviation, respectively. (A) Shannon Index. (B) Number of Operational taxonomic units (OTUs) generated with a minimum pairwise identity of 97%. (C) Chao1 richness. (D) Rarefaction curves of the number of OTUs in each sample.
To investigate the existence of a disease-specific microbiome in saliva of OLP patients, we evaluated the genera and OTUs which were found in at least 50% of subjects with a mean relative abundance of >1% and >0.3% respectively. As a result, the core genera are shown in Fig. 2A. In this range, Porphyromonas was only found in erosive OLP patients, while Leptotrichia was detected in reticular and erosive OLP patients. Although Actinomyces was detected in both reticular OLP and healthy controls, this genus was indeed more abundant in reticular OLP. Additionally, using the BLAST method against the HOMD to assign the sequences to bacterial species, we found that Prevotella melaninogenica (OTU5115) was unique in reticular OLP and Veillonella parvula (OTU8105) dominated in reticular and erosive OLP when compared with healthy controls (Fig. 2B).
Figure 2. The core microbiota of saliva samples in three experimental groups.

The model included the genera (above 1% of relative abundance) and OTUs (above 0.3% of relative abundance) in healthy controls (green), reticular OLP (blue) and erosive OLP (red), which were found in at least 50% of subjects. Colored labels indicate genera and OTUs having a greater relative abundance in healthy controls (green), reticular OLP (blue), and erosive OLP (red). Species corresponding to statistically different OTUs are listed in the square box. Statistical differences were calculated by Student’s t test. a: H vs R; b: H vs E; c: R vs E. Superscript letters in red indicate P < 0.05, and those in black indicate P < 0.1.
Changes of oral microbiota at the phylum, genus and OTU levels in OLP patients
The salivary microbial community composition of patients with OLP and healthy individuals were analyzed at different taxonomic levels. The overall microbial community composition of three groups at the phylum level was shown in Fig. 3A. As the histogram displayed, the most predominant phylum of all groups was Firmicutes, followed by Proteobacteria, Actinobacteria, Bacteroidetes and Fusobacteria. The phylum Spirochaetes was more abundant in erosive OLP over reticular OLP (P = 0.02), and yet Actinobacteria showed more abundance in reticular OLP over erosive OLP (P = 0.01). Meanwhile, the abundance of some genera differed significantly. Only the genera with average relative abundance >0.1% were demonstrated in Fig. 3B,C. As compared with healthy controls, the relative abundance of genus Porphyromonas was increased in erosive OLP, and Solobacterium was more abundant in reticular OLP. In contrast, Haemophilus, Corynebacterium, Cellulosimicrobium and Campylobacter showed significantly overabundance in healthy group than diseased groups. Nonetheless, few species had significantly different abundance levels among three groups. Haemophilus parainfluenzae (OTU1493), Rothia aeria (OTU104) and Streptococcus parasanguinis (OTU8510) were less abundant in erosive OLP, while Prevotella melaninogenica (OTU5115) had significantly higher levels in reticular OLP (Fig. 2B).
Figure 3. Relative abundance of bacterial phyla and predominant genera (>0.1%) among healthy controls (H), reticular OLP (R) and erosive OLP (E).

(A) Phylum level. Compared with reticular OLP, Spirochaetes was more abundant, and yet Actinobacteria was less abundant in erosive OLP. (B) Comparison among top 8 abundant genera. (C) Comparison among top 9 to 30 abundant genera. a: H vs R; b: H vs E; c: R vs E; Superscript letters in red indicate P < 0.05, and those in black indicate P < 0.1.
Co-occurrence patterns of salivary microbiota in OLP
The goal of network inference is to identify combinations of microorganisms demonstrating significant co-occurrence or mutual exclusion patterns across samples and to predict ecological relationships. By analyzing and then visualizing the spatial Pearson’s correlations between the bacterial species detected from saliva samples, decreased network complexity, as measured by average connectivity, could be observed for microbial co-occurrence networks in reticular and erosive OLP groups. For example, we captured 1094 associations among 282 nodes in healthy group, 1185 associations among 335 nodes in reticular OLP group, and 985 associations among 324 nodes in erosive OLP group at the OTU level (Supplementary Figure S2). In the network, pairwise relationships were represented by edges connecting two nodes. The nodes with the most numerous linkers, which were defined as hub nodes, were different among three groups. The hub species were Atopobium sp. (OTU7853 co-occurring with 29 OTUs) in healthy controls, Prevotella melaninogenica (OTU661 co-occurring with 31 OTUs) in reticular OLP, and Solobacterium moorei (OTU771 co-occurring with 24 OTUs) in erosive OLP, respectively. In addition, to further explore the biological significance of co-occurrence relationships of oral microbiota, we identified highly connected microbial clusters of the genus Streptococcus in the network (Fig. 4). Interestingly, clear reduction of Streptococcus co-occurrence network patterns could be observed from healthy individuals to patients with reticular and erosive OLP. In healthy controls, the genus Streptococcus comprising species were clustered into two modules (module A and B) associated with OTU7975 and OTU7832 respectively. In reticular OLP, there was only one module (module C) of Streptococcus associated with OTU8419, but corresponding nodes did not form any modules in erosive OLP. Also, we found that the OTUs representing the genus Streptococcus had significantly fewer linkers in erosive OLP than those in reticular OLP and healthy controls (P < 0.05 and P < 0.001, respectively). This indicated that Streptococcus might have played an important role in the development of OLP.
Figure 4. Co-occurrence networks of connected microbial clusters (modules) of Streptococcus comprising species.

Each node represents an OTU colored by the genus-level phylotypes, and each edge represents a significant co-occurrence relationship colored by the sign of the association (red negative, black positive). Two highly connected microbial clusters (Module A,B) in healthy controls (H), and one module (Module C) in reticular OLP (R) were observed in the networks.
We further employed the co-occurrence and co-exclusion analysis of the top 30 abundant bacterial genera in detail. Two subtypes of OLP patients were combined into the OLP group for this analysis. Out of 26 abundant genera which were shared between the diseased and healthy groups, we found significant changes in the ecologically competitive interactions of these bacteria (Fig. 5). In healthy controls, Treponema and Filifactor were the most positively correlated (ρ = 0.945), whereas Capnocytophage and Streptococcus were the most negatively correlated (ρ = −0.601). But in OLP, the most positively correlated genera were Solobacterium and Peptostreptococcus (ρ = 0.839), while Streptococcus and Prevotella were the most negatively correlated (ρ = −0.540). Besides, Opponent co-occurrence patterns were observed for several microbial genera in diseased and healthy status. For example, Treponema and Actinobacillus were correlated negatively (ρ = −0.249) in healthy group, but their correlation converted to be positive (ρ = 0.564) in OLP group. The same pattern was also observed for the genera Actinomyces and Granulicatella.
Figure 5. Co-occurrence and co-exclusion analysis of the bacterial genera.

The analysis was calculated by Pearson correlations among the top 30 abundant bacterial genera. The co-occurrence of the abundant genera in OLP and healthy samples was shown on the left and right, respectively. The correlation values ranged from −1.00 (green) to 1.00 (red).
Genera associated with the clinical parameters
The concentrations of IL-17 and IL-23 were measured by means of enzyme-linked immunosorbent assay (ELISA) and the results were shown in Supplementary Table S2. To further investigate the association between oral microorganisms and the clinical data, Pearson correlation coefficients were calculated (Supplementary Table S3). A significant correlation was observed between some detected genera and clinical parameters, including the clinical scores and salivary levels of IL-17 and IL-23 (Fig. 6). First, the genera including Treponema, Porphyromonas and Abiotrophia were positively correlated with clinical scores, whereas Oribacterium was negatively correlated with clinical scores. Second, it was observed that many genera were correlated with immunologic factors involved in the inflammation response in OLP. For example, Treponema, Porphyromonas, Gemella and Parvimonas showed significantly positive correlations with the salivary levels of IL-17, whereas Abiotrophia, Porphyromonas, Paraprevotella and Fusobacterium were found to be positively correlated with the salivary levels of IL-23. Finally, some of these genera associated with clinical data had significant correlations with more than one parameter, such as Porphyromonas, which was correlated positively with the clinical scores and salivary levels of IL-17 and IL-23 at the meantime.
Figure 6. The bacterial genera associated with disease scores and proinflammatory cytokines.
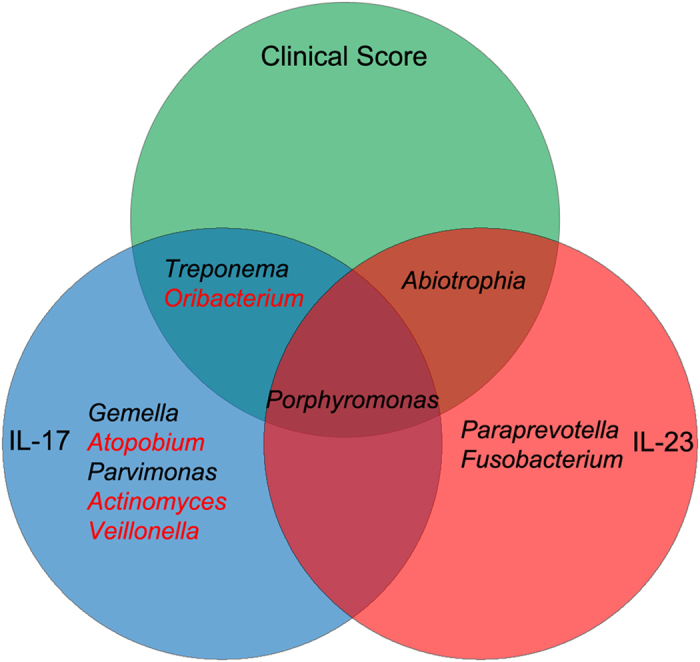
The model displayed all genera associated with each of the three clinical parameters of OLP (P < 0.05). The genera name in red were correlated negatively with clinical data, and those in black were correlated positively with clinical data.
Discussion
The role of the viral infection in OLP has already come into notice. However, there is a paucity of knowledge regarding the role of bacteria in the progression of this common oral disease. In the present study, we described the complexity of microbial communities in OLP, and our results uncovered crucial yet striking salivary microbiomic characteristics associated with disease. The microbial community structure displayed a greater variety and less specificity in OLP patients. And the samples from erosive OLP displayed even higher phylogenetic diversity than healthy controls, as its rarefaction curves did not yet reach a plateau with current sequencing. These results are not in agreement with findings of a recent study10. This discrepancy may be due to differences in sequencing methods, bioinformatic analyzing methods and the number of subjects included in different studies. Our results implicated the association between changes in the microbial enrichment and OLP.
Among three groups, five major phyla constituted the predominant salivary microbiome (that is, Firmicutes, Proteobacteria, Actinobacteria, Bacteroidetes and Fusobacteria), consistent with previous surveys23,24. Significant differences of oral microbiome at the phylum and genus levels were identified between OLP patients and healthy individuals. The relative abundances of phylum Actinobacteria, Spirochaetes and its genus Treponema differed between two subtypes of OLP patients, and these microbial taxa may be associated with disease classification of OLP. Porphyromonas was discovered to be dominant in erosive OLP, which was also considered to be the core genus in periodontitis25,26. As the implementation of our sample collection had excluded those subjects with periodontal inflammation or other oral diseases, we could assume that the abundance shift of Porphyromonas had something to do with OLP state. Our findings supported the results of a previous study, in which higher periodontopathogens’ percentages were detected in OLP groups than those in the non-OLP groups27. This periodontopathogenic genus with high percentage may also play an important role in the etiology of OLP.
Moreover, analysis at the OTU level confirmed the disease-associated species, which increased in relative abundance and prevalence, including Veillonella parvula and Prevotella melaninogenica. Also, higher counts of Veillonella parvula was examined at sites with evidence of OLP lesions using DNA-DNA hybridization in a previous study22. Prevotella species, the Gram-negative rod-shaped anaerobes, were also implicated involved in many other oral disease, such as endodontic disease28, periodontal infection29 and dental caries30. Prevotella melaninogenica, as the hub species with significantly higher abundance, could be regarded as the core bacteria in reticular OLP. Findings reported here underlined the potential role of this reticular OLP-associated species played in the disease. While in this study, Haemophilus parainfluenzae, Rothia aeria and Streptococcus parasanguinis were less abundant in erosive OLP compared with healthy controls. Indeed, in a recent study, Said et al.31 reported that the salivary microbiome of patients with inflammatory bowel disease comprised higher proportions of Veillonella and Prevotella and lower proportions of Haemophilus and Streptococcus than those in healthy controls, and they indicated that the dysbiotic genera Veillonella and Prevotella could elicit an inflammatory response in mucosa. Although the abundance of the genera Veillonella and Prevotella were not significantly different between OLP patients and healthy individuals, they might be important for the pathogenesis of OLP at the species level. However, further investigation is required to clarify the role of these dysbiotic oral microbiota in the progression of this disease.
Oral microorganisms rarely survive in isolation, but instead coexist in complex ecology with various symbiotic relationships32. Previous studies identified that individual microbial interactions were not only essential for community stability but also involved in dysbiosis and overgrowth of pathogens linked to disease24,33. In our study, the co-occurrence analysis revealed significant changes in the interactions of salivary microbiota in OLP. For example, in diseased group, the suspected bacteria Porphyromonas with higher abundance turned to be correlated significantly with Eubacterium, which was also found to be involved in periodontitis25 and peri-implantitis34. These shifts of the symbiotic and antagonistic relationships suggested the existence of microbial dysbiosis in OLP. Groups of densely connected molecules usually have an important biological significance in the interaction networks35. Our network analyses showed significantly decreased interactions of Streptococcus comprising species, also supporting the imbalance of oral microbiota in OLP. Equally, the decrease of Streptococcaceae comprising species associated with oral health was observed in ulcerated sites of recurrent aphthous stomatitis (RAS) patients36. Potential effect of Streptococci-based products in controlling oral inflammatory conditions has been suggested recently37. Our findings may not exclude a role for these products in preventing the exacerbation of OLP.
There is an increasing body of evidence suggesting that perturbations to the structure of complex commensal communities can modulate innate and adaptive immune response and lead to the development of immune mediated diseases, with inflammation arising upon the reduction of the proportion of symbiont microbiota or the increase in the number of pathogenic microbiota36,38. With the identification of Th17 cell, the importance for its defense against extracellular and intracellular bacterial infection has been demonstrated in a mass of researches39,40. Previously, we demonstrated the potential association between oral microbial diversity and levels of IL-17 in saliva of OLP patients10. In this study, we further explored 7 genera that correlated positively to the Th17-associated cytokines (IL-17 and IL-23), and these bacteria might participate in dysimmunity and inflammation maintaining in OLP. Most notably, out of these genera, the anaerobic bacteria Porphyromonas positively correlated to three clinical parameters deserves more attractions. As one of the most frequently encountered species in human, Porphyromonas gingivalis has recently been demonstrated on its ability to subvert host immunity and promote dysbiosis of oral microbiota41. Besides, Porphyromonas gingivalis was also indicated to enhance the Th17 cell responses for the development of atherosclerosis42. Together with detection of significantly higher abundance of Porphyromonas in erosive OLP, our findings highlight the urgency of the further research on the role of this bacteria played in OLP. Additionally, we postulated that some microbiota colonized in saliva of OLP patients might be associated with the severity of disease. The genera Treponema, Porphyromonas and Abiotrophia showed significantly positive correlations with the clinical scores, suggesting these bacteria might be involved in the exacerbation of OLP and their proportion may signify the degree of severity of this disease. Treponema, Porphyromonas, Parvimonas and Fusobacterium have recently been reported to be the core microbiota in periodontitis25. The potential roles these periodontopathogenic microorganisms play in the progression of OLP was also reported recently27, and our results agreed with those of the previous study. In addition, higher levels of Treponema and Porphyromonas in erosive OLP compared to those in reticular OLP and healthy individuals also supported their association with disease severity. While the genus Abiotrophia, which is often associated with culture-negative infective endocarditis43, was found to be correlated with disease severity and IL-23 levels in our study. We assume that these relevant microorganisms, together or separately, might play a direct role in the initiation of OLP or act as triggers of immune response directly or indirectly following the formation of OLP lesions.
In conclusion, we preliminarily identified the microbial taxa associated with OLP and delineated different features of salivary microbiome between OLP and healthy individuals, providing a comprehensive cross-sectional description of oral microbiota in OLP. Our study afforded deep insight into the dysbiosis in the microbial community of OLP, suggesting a potential microbial role in progression of this disease. Also, Our findings could provide the basis for further exploring the bacteria suspected of being associated with OLP in the future studies.
Methods
Subject recruitment and sample collection
Subjects with reticular OLP (n = 19) and erosive OLP (n = 18) aged between 22–62 years were selected during follow-up clinical examinations from the West China Hospital of Stomatology, Sichuan University, and the OLP patients were diagnosed in accordance with the clinical classification and definition of OLP by the World Health Organization44. The degree of severity of the disease was based on the clinical scores evaluated by a semiquantitative scoring system45 in line with the site, area and presence of lesions. Additionally, 18 healthy volunteers who met the requirements of having matched gender and age with the OLP patients were recruited as controls. All subjects were willing to consent to the clinical examination and saliva sampling. They had neither detectable periodontal inflammation and visible caries lesions, nor any severe systemic disorders and smoking history. Moreover, they had not received treatment for OLP or taken prescription drugs at least two months before sampling. This study was approved by the local ethics committee of West China Hospital of Stomatology, Sichuan University. Informed consent was obtained from all study participants. The methods were carried out in accordance with the approved guidelines.
According to the standard techniques described by Navazesh46, about 5 ml spontaneous, whole unstimulated saliva (WUS) was collected in a sterile DNA-free conical tube from each subject between 8:00~11:00 am. Subjects were refrained from drinking and eating for at least 2 h before sampling. All samples were transported to laboratory on ice within 2 h, and stored at −80 °C before further processing.
Cytokines assay
The levels of IL-17 and IL-23 in saliva were detected as described previously10.
DNA extraction
Genomic DNA was extracted from saliva samples using the QIAamp DNA micro Kit (Qiagen, Valencia, CA, USA) according to the manufacturer’s instructions with minor modifications by adding a lysozyme (3 mg ml−1, 1.5 h) treatment step. Quality of the extracts was evaluated by the absorbance ratios at A260/A280 and A260/A230 using spectrophotometry (NanoDrop 1000, Thermo Scientific Inc, Wilmington, DE, USA). Only DNA samples with A260/A280 >1.7 and A260/A230 >1.8 were used in downstream experiments. The final concentrations of DNA were quantified using the Pico-Green kit (Invitrogen, Carlsbad, CA, USA). DNA samples were frozen at −20 °C until further analysis.
Illumina sequencing and bioinformatics analysis of 16S rRNA gene amplicons
Bacterial 16S rRNA genes from each sample were amplified using F515 (primer seq) and R806 (primer seq) primers targeting V4 hypervariable regions and then sequenced with the Illumina MiSeq technology at the Institute for Environmental Genomics, University of Oklahoma (Norman, OK, USA). The MiSeq 500 cycles 250 × 2 V2 kit was used for sequencing 16S rRNA V4 on MiSeq machine (Illumina, San Diego, CA). Data preprocessing and OTU clustering was performed as described previously25. Taxonomic assignment was carried out using the RDP classifier with confidence cutoff value of 50%47.
The sequence data were further analyzed with the following methods: (A) Principal coordinate analysis (PCoA) was used to determine the degree of dissimilarity between pairs of bacterial communities using Bray-Curtis distance method. Three different nonparametric analyses for multivariate data implemented by the “vegan” package in R were also used for community functional structure comparison, including multiresponse permutation procedure (MRPP), nonparametric multivariate analysis of variance (adonis) using distance matrices and analysis of similarities (ANOSIM). (B) Alpha-diversity indices were estimated by the Shannon diversity index, the number of observed OTUs, and Chao1 species richness estimates. Rarefaction curves were applied to calculate the species richness based on the bacterial OTUs and were then compared among three groups by the mothur package48. (C) Microbial-microbial co-occurrence networks were constructed by the MENA pipeline49. A Pearson correlation cutoff of 0.74 was determined by random matrix theory approach by observing a transition point of nearest-neighbor spacing distribution of eigenvalues from Gaussian to Poisson distribution, which are two universal extreme distributions. In such networks, OTUs were represented by network nodes while correlations were transformed into links between them. The co-occurrence networks were then visualized using Cytoscape 3.2.0 with a force-directed algorithm50, and network topological parameters were computed using NetworkAnalyzer51. (D) Co-occurrence patterns of the 30 most abundant taxonomic groups across samples were explored by calculating the Pearson correlation coefficients. Results were clustered and visualized by the MeV package52.
Student’s t-test was used to compare microbial diversity and relative abundances of each bacterial taxon. Pearson correlations were used for the analyses of co-occurrence and bacterial associations with the clinical data.
Additional Information
Accession codes: The data are available at the NCBI Sequence Read Archive (SRA) under accession no. SRP067603 (http://www.ncbi.nlm.nih.gov/sra). http://www.nature.com/srep
How to cite this article: Wang, K. et al. Preliminary analysis of salivary microbiome and their potential roles in oral lichen planus. Sci. Rep. 6, 22943; doi: 10.1038/srep22943 (2016).
Supplementary Material
Acknowledgments
This research was funded by the National Natural Science Foundation of China (81430011, 81172579), National Key Technologies R&D Programme of the Twelves Five-Year Plan, the Ministry of Science and Technology of China (2012BAI07B03) and Sichuan Provincial Department of science and technology project (2013SZ0039).
Footnotes
The authors declare no competing financial interests.
Author Contributions Conception and design of the experiments: Y.L., L.X., X.Z. and K.W. Conduction of the experiments: K.W., W.L. and J.V.N. Data processing and analysis: Q.T., K.W. and Y.G. Volunteers recruitment and samples collection: J.H., Y.Z., Y.G. and Y.Q. Paper writing: K.W. and W.L. Revision of the manuscript: Y.L., Q.T., L.X., J.L. J.V.N., J.Z. and X.Z.
References
- Lodi G. et al. Current controversies in oral lichen planus: report of an international consensus meeting. Part 1. Viral infections and etiopathogenesis. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 100, 40–51 (2005). [DOI] [PubMed] [Google Scholar]
- Ismail S. B., Kumar S. K. & Zain R. B. Oral lichen planus and lichenoid reactions: etiopathogenesis, diagnosis, management and malignant transformation. J Oral Sci. 49, 89–106 (2007). [DOI] [PubMed] [Google Scholar]
- Eisen D. The evaluation of cutaneous, genital, scalp, nail, esophageal, and ocular involvement in patients with oral lichen planus. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 88, 431–436 (1999). [DOI] [PubMed] [Google Scholar]
- Mollaoglu N. Oral lichen planus: a review. Br J Oral Maxillofac Surg. 38, 370–377 (2000). [DOI] [PubMed] [Google Scholar]
- Masaki M., Sato T., Sugawara Y., Sasano T. & Takahashi N. Detection and identification of non-Candida albicans species in human oral lichen planus. Microbiol Immunol. 55, 66–70 (2011). [DOI] [PubMed] [Google Scholar]
- Tao X. A. et al. FOXP3 T regulatory cells in lesions of oral lichen planus correlated with disease activity. Oral Dis. 16, 76–82 (2010). [DOI] [PubMed] [Google Scholar]
- Pekiner F. N., Demirel G. Y., Borahan M. O. & Ozbayrak S. Cytokine profiles in serum of patients with oral lichen planus. Cytokine. 60, 701–706 (2012). [DOI] [PubMed] [Google Scholar]
- Xie S., Ding L., Xiong Z. & Zhu S. Implications of Th1 and Th17 Cells in Pathogenesis of Oral Lichen Planus. J Huazhong Univ Sci Technolog Med Sci. 32, 451–457 (2012). [DOI] [PubMed] [Google Scholar]
- Wang H. et al. Interaction Between Oral Lichen Planus and Chronic Periodontitis with Th17-Associated Cytokines in Serum. Inflammation. 36, 696–704 (2013). [DOI] [PubMed] [Google Scholar]
- Wang K. et al. Analysis of oral microbial community and Th17-associated cytokines in saliva of patients with oral lichen planus. Microbiol Immunol. 59, 105–113 (2015). [DOI] [PubMed] [Google Scholar]
- Ficarra G. et al. White lichenoid lesions of the buccal mucosa in patients with HIV infection. Oral Surg Oral Med Oral Pathol. 76, 460–466 (1993). [DOI] [PubMed] [Google Scholar]
- Sun A. et al. Human cytomegalovirus as a potential etiologic agent in recurrent aphthous ulcers and Behcet’s disease. J Oral Pathol Med. 25, 212–218 (1996). [DOI] [PubMed] [Google Scholar]
- Sand L., Jalouli J., Larsson P. A. & Hirsch J. M. Human papilloma viruses in oral lesions. Anticancer Res. 20, 1183–1188 (2000). [PubMed] [Google Scholar]
- Sand L. P., Jalouli J., Larsson P. A. & Hirsch J. M. Prevalence of Epstein-Barr virus in oral squamous cell carcinoma, oral lichen planus, and normal oral mucosa. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 93, 586–592 (2002). [DOI] [PubMed] [Google Scholar]
- Konidena A. & Pavani B. V. Hepatitis C virus infection in patients with oral lichen planus. Niger J Clin Pract. 14, 228–231 (2011). [DOI] [PubMed] [Google Scholar]
- Lundström I. M., Anneroth G. B. & Holmberg K. Candida in patients with oral lichen planus. Int J Oral Surg. 13, 226–238 (1984). [DOI] [PubMed] [Google Scholar]
- Shimoyama T., Horie N., Kato T., Kaneko T. & Komiyama K. Helicobacter pylori in oral ulcerations. J Oral Sci. 42, 225–229 (2000). [DOI] [PubMed] [Google Scholar]
- Attia E. A., Abdel Fattah N. S. & Abdella H. M. Upper gastrointestinal findings and detection of Helicobacter pylori in patients with oral lichen planus. Clin Exp Dermatol. 35, 355–360 (2010). [DOI] [PubMed] [Google Scholar]
- Hulimavu S. R. et al. No evidence for Helicobacter pylori in oral lichen planus. J Oral Pathol Med. 43, 576–578 (2014). [DOI] [PubMed] [Google Scholar]
- Holmstrup P., Schiotz A. W. & Westergaard J. Effect of dental plaque control on gingival lichen planus. Oral Surg Oral Med Oral Pathol. 69, 585–590 (1990). [DOI] [PubMed] [Google Scholar]
- Backman K. & Jontell M. Microbial-associated oral lichenoid reactions. Oral Dis. 13, 402–406 (2007). [DOI] [PubMed] [Google Scholar]
- Bornstein M. M., Hakimi B. & Persson G. R. Microbiological findings in subjects with asymptomatic oral lichen planus: a cross-sectional comparative study. J Periodontol. 79, 2347–2355 (2008). [DOI] [PubMed] [Google Scholar]
- Keijser B. J. et al. Pyrosequencing analysis of the oral microflora of healthy adults. J Dent Res. 87, 1061–1020 (2008). [DOI] [PubMed] [Google Scholar]
- Zaura E., Keijser B. J., Huse S. M. & Crielaard W. Defining the healthy “core microbiome” of oral microbial communities. BMC Microbiol. 9, 259 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y. et al. Phylogenetic and functional gene structure shifts of the oral microbiomes in periodontitis patients. ISME J. 8, 1879–1891 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abusleme L. et al. The subgingival microbiome in health and periodontitis and its relationship with community biomass and inflammation. ISME J. 7, 1016–1025 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ertugrul A. S., Arslan U., Dursun R. & Hakki S. S. Periodontopathogen profile of healthy and oral lichen planus patients with gingivitis or periodontitis. Int J Oral Sci. 5, 92–97 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocas I. N. & Siqueira J. F. Jr. Prevalence of new candidate pathogens Prevotella baroniae, Prevotella multisaccharivorax and as-yet-uncultivated Bacteroidetes clone X083 in primary endodontic infections. J Endod. 35, 1359–1362 (2009). [DOI] [PubMed] [Google Scholar]
- Serrano C. et al. Antibiotic resistance of periodontal pathogens obtained from frequent antibiotic users. Acta Odontol Latinoam. 22, 99–104 (2009). [PubMed] [Google Scholar]
- Yang F. et al. Saliva microbiomes distinguish caries-active from healthy human populations. ISME J. 6, 1–10 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Said H. S. et al. Dysbiosis of salivary microbiota in inflammatory bowel disease and its association with oral immunological biomarkers. DNA Res. 21, 15–25 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faust K. et al. Microbial Co-occurrence Relationships in the Human Microbiome. Plos Comput Biol. 8, e1002606 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank D. N., Zhu W., Sartor R. B. & Li E. Investigating the biological and clinical significance of human dysbioses. Trends Microbiol. 19, 427–434 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng H. et al. Subgingival microbiome in patients with healthy and ailing dental implants. Sci Rep. 5, 10948 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman M. E. Modularity and community structure in networks. Proc Natl Acad Sci USA 103, 8577–8582 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hijazi K. et al. Mucosal Microbiome in Patients with Recurrent Aphthous Stomatitis. J Dent Res. 94, 87S–94S (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pradeep K., Kuttappa M. A. & Prasana K. R. Probiotics and oral health: an update. SADJ. 69, 20–24 (2014). [PubMed] [Google Scholar]
- Petersen C. & Round J. L. Defining dysbiosis and its influence on host immunity and disease. Cell Microbiol. 16, 1024–1033 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubino S. J., Geddes K. & Girardin S. E. Innate IL-17 and IL-22 responses to enteric bacterial pathogens. Trends Immunol. 33, 112–118 (2012). [DOI] [PubMed] [Google Scholar]
- Tanabe S. The effect of probiotics and gut microbiota on Th17 cells. Int Rev Immunol. 32, 511–525 (2013). [DOI] [PubMed] [Google Scholar]
- Maekawa T. et al. Porphyromonas gingivalis manipulates complement and TLR signaling to uncouple bacterial clearance from inflammation and promote dysbiosis. Cell Host Microbe. 15, 768–778 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Y., Kobayashi R., Hashizume-Takizawa T. & Kurita-Ochiai T. Porphyromonas gingivalis infection enhances Th17 responses for development of atherosclerosis. Arch Oral Biol. 59, 1183–1191 (2014). [DOI] [PubMed] [Google Scholar]
- Fukui Y. et al. Metagemonic analysis for detecting pathogens in culture-negative infective endocarditis. J Infect Chemother. 21, 882–884 (2015). [DOI] [PubMed] [Google Scholar]
- van der Meij E. H. & van der Waal I. Lack of clinicopathologic correlation in the diagnosis of oral lichen planus based on the presently available diagnostic criteria and suggestions for modifications. J Oral Pathol Med. 32, 507–512 (2003). [DOI] [PubMed] [Google Scholar]
- Society of Oral Medicine, Chinese Stomatological Association. Standard for evaluation of effect for oral lichen planus (erosive, atrophic) (trial implementation). Zhonghua Kou Qiang Yi Xue Za Zhi. 40, 92–93 (2005). [PubMed] [Google Scholar]
- Navazesh M. Methods for collecting saliva. Ann N Y Acad Sci. 694, 72–77 (1993). [DOI] [PubMed] [Google Scholar]
- Wang Q., Garrity G. M., Tiedje J. M. & Cole J. R. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol. 73, 5261–5267 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schloss P. D. et al. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol. 75, 7537–7541 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng Y. et al. Molecular ecological network analyses. BMC Bioinformatics. 13, 113 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smoot M. E., Ono K., Ruscheinski J., Wang P. L. & Ideker T. Cytoscape 2.8: new features for data integration and network visualization. Bioinformatics. 27, 431–432 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Assenov Y., Ramírez F., Schelhorn S. E., Lengauer T. & Albrecht M. Computing topological parameters of biological networks. Bioinformatics. 24, 282–284 (2008). [DOI] [PubMed] [Google Scholar]
- Mar J. C., Matigian N. A., Quackenbush J. & Wells C. A. attract: A method for identifying core pathways that define cellular phenotypes. PLoS One. 6, e25445 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.