

Abstract
The mediator complex subunit 12 gene (MED12) is responsible for an X-linked recessive intellectual disability syndrome that is characterized by dysmorphic features such as a long, narrow face and blepharophimosis, which is now recognized as an MED12-related syndrome. We identified a novel non-synonymous single-nucleotide variant, p.Ile1023Val, in a male patient with non-specific X-linked intellectual disability (XLID). Our results, together with the existence of similar reports, suggest a relationship between MED12 variants and XLID.
Obtaining a final diagnosis for a non-specific intellectual disability is challenging. However, the use of massive parallel sequencing with next-generation sequencers has made this task easier. This strategy has also revealed broader genotype–phenotype correlations. It is now possible to determine whether genes that are specific for a distinctive disorder might also be responsible for other clinical characteristics. We report herein a case of a male patient with non-specific intellectual disability who showed a novel mutation in the mediator complex subunit 12 gene (MED12),1 which is known to be responsible for FG syndrome (FGS; Opitz–Kaveggia syndrome, MIM#305450), Lujan–Fryns syndrome (intellectual disability with marfanoid habitus, MIM#309520), and X-linked Ohdo syndrome (Maat-Kievit–Brunner syndrome (MKBS); MIM#300895).
A 12-year-old boy was born to healthy parents at term with a birth weight of 3,700 g. He had two healthy older sisters, and there is no remarkable family clinical history. He has no history of seizures. Because he showed severe developmental delay since infancy, he was admitted to a special education school.
Presently, he is relatively small for his age, with a height of 142 cm (10th to 25th percentile), weight of 30.5 kg (3rd to 10th percentile) and occipitofrontal circumference of 52.5 cm (10th percentile). He has distinctive facial findings, with a long narrow face, high forehead, brushy arched eyebrows, large incisors and large ears; however, there is no blepharophimosis or hypertelorism. Finger pads are noted on all fingers. Although he could follow simple verbal commands from his family members, he is aphasic and requires support in his daily life activities. Although it is possible to make eye contact with him, communication is impossible with others. He is often constipated, which is likely because he shows stubbornness in his eating habits. Depending on his mood, he often refuses bathing. Although he does not exhibit aggressive behavior, he often shows panic in circumstances in which he does not get his way. From these findings, we concluded that he has a severe intellectual disability with autistic features.
To make a genetic diagnosis, blood samples were obtained from the patient and his parents after receiving informed consent. This study was performed in accordance with the Declaration of Helsinki Principles, and the ethics committee of Tokyo Women’s Medical University gave approval for this study. Genomic DNA was extracted from blood samples and used for further examination. Genomic copy number aberrations were examined using an Agilent SurePrint G3 Human CGH Microarray Kit 8×60 K (Agilent Technologies, Santa Clara, CA, USA) as described previously,2 which demonstrated no aberrations. Subsequently, targeted resequencing was performed using TruSight One v1.0 sequencing panel (Illumina, San Diego, CA, USA), which includes 125,396 probes aimed to capture 11,946,514-bp targeted exon regions consisting of 4,813 genes that are associated with known clinical phenotypes. After constructing the sequence library using 50 ng of genomic DNA, the MiSeq next-generation sequencer (Illumina) was used to sequence 151-bp paired-end reads according to the manufacturer’s instructions. The extracted data were mapped to a reference genome (GRCh37/hg19) using BWA Enrichment v1.0 cloud software (Illumina). On an average, 1.093 Gb of targeted aligned sequences and a mean coverage depth of 92 were obtained (the target coverage at 20× was 95.0%). The extracted variants were annotated and filtered using the Variant Studio software (Illumina; Figure 1a).
Figure 1.
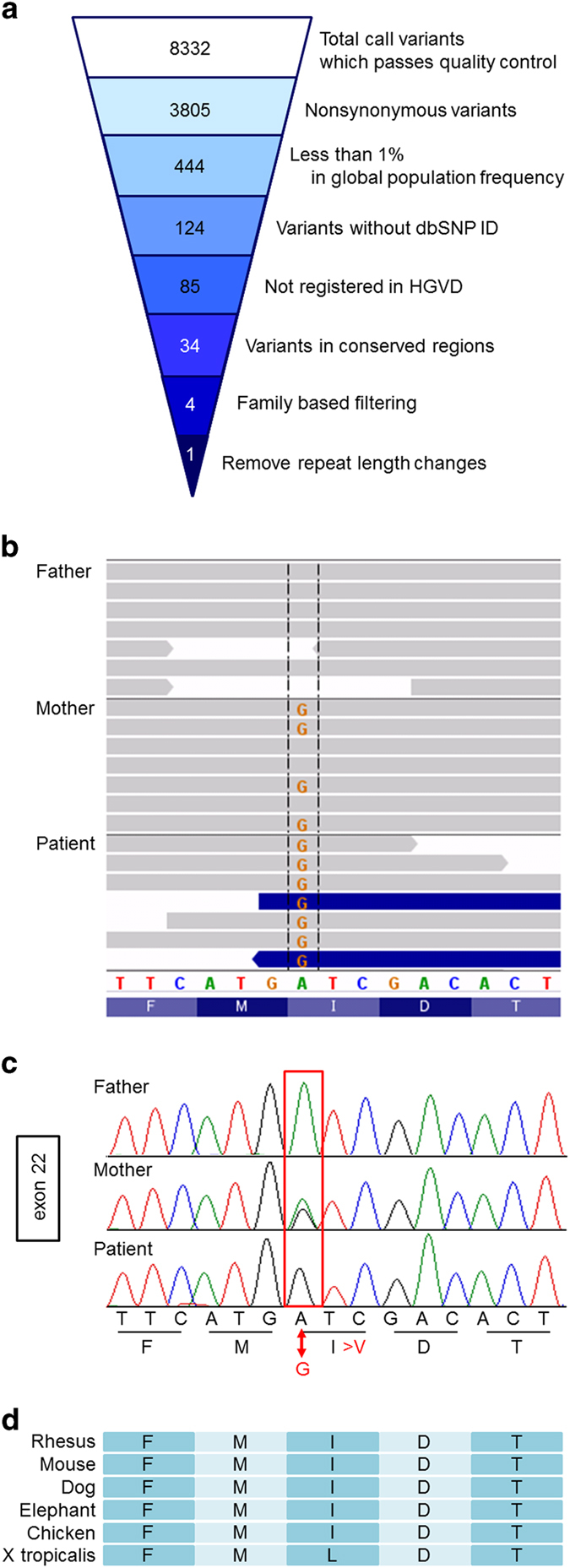
Sequencing results. (a) Filtering the candidate mutations. Numbers show the results: the top number indicates number of total variants that passed quality control. The second number indicates number of non-synonymous variants. The third number indicates number of the variants with <1% frequency in global population. The fourth number indicates the number of the variants without a dbSNP ID. The fifth number indicates the number of variants that are not registered in HGVD. The sixth number indicates the number of the variants in the conserved regions. The seventh number indicates the number of the variants after family-based filtering. The variant, which does not appear to have a de novo pattern, an autosomal recessive pattern or an X-linked recessive pattern, was filtered out. The bottom number indicates the number of variants after removal of the simple repeat length changes. (b) IGV presentation of the sequence results from family members. The present patient shows G at the indicated position. The mother is heterozygous for this variant. (c) Sanger sequencing confirmed all of the above results. (d) The affected amino acid is conserved among mammals and chicken. dbSNP, single-nucleotide polymorphism database; HGVD, human genetic variation database; IGV, integrative genomics viewer.
A single-nucleotide variant (SNV) in exon 22 of MED12, NM_005120.2(MED12_v001):c.3067A>G; NM_005120.2(MED12_i001):p.(Ile1023Val), was identified. The affected amino acid is conserved among mammals (Figure 1d). This variant is not reported in the dsSNP137 single-nucleotide polymorphism database, the exome variant server provided by the National Heart, Lung, and Blood Institute Exome Sequencing Project (NHLBI-ESP) 6500 (http://evs.gs.washington.edu/EVS/), or the human genetic variation database (HGVD; http://www.genome.med.kyoto-u.ac.jp/SnpDB). The HGVD is the database provided by Kyoto University,3 and it currently contains genetic variations that have been determined by exome sequencing of 1,208 individuals, and genotyping data of common variations obtained from a cohort of 3,248 individuals, thus indicating a novel variant in the general population. SIFT (http://sift.jcvi.org/www/SIFT_help.html) and PolyPhen2 predication (http://genetics.bwh.harvard.edu/pph2/) scores were tolerated (0.73) and benign (0.001); however, the CADD RawScore was 1.386821, and the CADD PHRED score was 15.4 (http://cadd.gs.washington.edu/info). Further, MutationTaster (http://www.mutationtaster.org/) defined this variant as disease causing (prob: 0.919680553678408). The filtered SNV in MED12 was checked visually using integrative genomics viewer (http://www.broadinstitute.org/igv/; Figure 1b) and confirmed by the standard PCR-direct sequencing method using the Big Dye Terminator (Life Technologies, Carlsbad, CA, USA) with specific primers (sense 5′-CAGGCCTTCTTCAACACTAC-3′ and antisense 5′-TAGTACCTCCCGGGTACCTC-3′; Figure 1c), as described previously.4 The patient was hemizygous for this SNV, which was confirmed to be inherited from his carrier mother.
In 1974, Opitz and Kaveggia first reported an X-linked recessive intellectual disability syndrome characterized by dysmorphic features, including relative macrocephaly, hypertelorism, down-slanted palpebral fissures, a prominent forehead with frontal hair upsweep and broad thumbs and halluces.5 In 1984, Lujan et al.6 described four male patients with intellectual disability in a large family with marfanoid habitus and similar craniofacial changes, including a long narrow face, small mandible, high-arched palate and hypernasal voice. In 1993, X-linked Ohdo syndrome was first reported by Maat-Kievit et al.7 in familial patients with a low body weight; blepharophimosis; ptosis; wide, depressed nasal bridge; long, flat philtrum; thin vermilion; microstomia; micrognathia; cryptorchidism; scrotum hypoplasia; joint hyperextensibility; clinodactyly; overriding third toes; cafe-au-lait spots; developmental delay; deafness; and feeding problems.
In 2007, Risheg et al.1 reported that both the original family from which Opitz and Kaveggia derived the designation of FG and five other families had a recurrent MED12 mutation (p.Arg961Trp). This mutation was recurrently identified in a patient who was clinically diagnosed as FGS.8 Rump et al.9 also identified a different mutation (p.Gly958Glu) in a patient with the same clinical phenotype. Schwartz et al.10 identified a MED12 mutation (p.Asn1007Ser) in the original family with Lujan–Fryns syndrome. Vulto-van Silfhout et al.11 identified a mutation (p.Arg1148His) in the original family that was affected by MKBS as well as two additional mutations (p.Ser1165Pro and p.His1729Asn). Subsequently, Isidor et al.12 identified the same mutation (p.Arg1148His) in two male siblings associated with unique multiple anomalies that were similar to those of MKBS. These previously reported mutations are depicted on the primary structure of MED12 for a better understanding (Figure 2). It is now recognized that these three different clinical entities represent an allelic disorder derived from MED12 mutations (MED12-related syndrome).13
Figure 2.
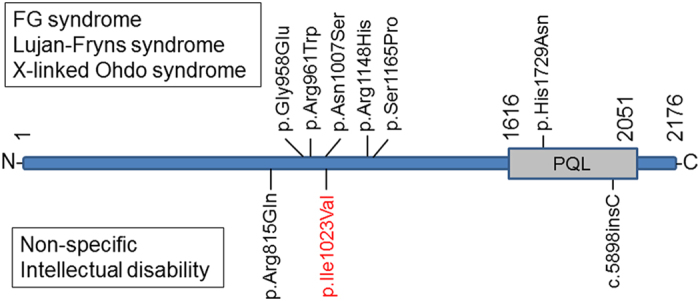
A schematic representation of the primary structure of MED12 and locations of identified mutations. The previously reported mutation identified in the patient with either FG syndrome, Lujan–Fryns syndrome or X-linked Ohdo syndrome is depicted in upper side. The mutation reported in the patient with XLID is in the bottom side. Most of the mutations are located on the narrow region. PQL indicates the domain with a high content of Pro, Gln and Leu residues, as reported by Kim et al.16 MED12, mediator complex subunit 12; XLID, X-linked intellectual disability.
Compared with these previous reports, the patient presented here displays non-specific intellectual disability, and only the long narrow face is common with MED12-related disorder. In 2013, Lesca et al.14 identified a single-nucleotide insertion in MED12 (c.5898insC) in a patient with non-specific intellectual disability. Subsequently, Tzschach et al.15 identified a MED12 variant (p.Arg815Gln) in a familial patient with moderate intellectual disability, short stature, and microcephaly (detailed information is unavailable) in a large cohort study that investigated X-linked intellectual disability (XLID). Most of the reported mutations are located in the narrow region between codon 815 and 1,165 (Figure 2). Although the functional importance of this narrow region is unknown, the localization of p.Ile1023Val, as identified in the present study, in this narrow region would suggest the pathogenesis of this variant. Based on these findings, we deduced that MED12 mutations are related not only to distinctive syndromes but also to non-specific XLID. Determination of the detailed genotype–phenotype correlation of MED12 is required to clarify this relationship.
Acknowledgments
We would like to express our gratitude to the patient and his family for their cooperation. This work was supported by a Grant-in-Aid for Scientific Research from Health Labor Sciences Research Grants from the Ministry of Health, Labor, and Welfare, Japan.
Footnotes
The authors declare no conflict of interest.
References
- Risheg H , Graham JM Jr. , Clark RD , Rogers RC , Opitz JM , Moeschler JB et al. A recurrent mutation in MED12 leading to R961W causes Opitz-Kaveggia syndrome. Nat Genet 2007; 39: 451–453. [DOI] [PubMed] [Google Scholar]
- Yamamoto T , Shimojima K , Shimada S , Yokochi K , Yoshitomi S , Yanagihara K et al. Clinical impacts of genomic copy number gains at Xq28. Hum Genome Variat 2014; 1: 14001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narahara M , Higasa K , Nakamura S , Tabara Y , Kawaguchi T , Ishii M et al. Large-scale East-Asian eQTL mapping reveals novel candidate genes for LD mapping and the genomic landscape of transcriptional effects of sequence variants. PLoS ONE 2014; 9: e100924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto T , Shimojima K , Nishizawa T , Matsuo M , Ito M , Imai K . Clinical manifestations of the deletion of Down syndrome critical region including DYRK1A and KCNJ6. Am J Med Genet A 2011; 155A: 113–119. [DOI] [PubMed] [Google Scholar]
- Opitz JM , Kaveggia EG . Studies of malformation syndromes of man 33: the FG syndrome. An X-linked recessive syndrome of multiple congenital anomalies and mental retardation. Z Kinderheilkd 1974; 117: 1–18. [DOI] [PubMed] [Google Scholar]
- Lujan JE , Carlin ME , Lubs HA . A form of X-linked mental retardation with marfanoid habitus. Am J Med Genet 1984; 17: 311–322. [DOI] [PubMed] [Google Scholar]
- Maat-Kievit A , Brunner HG , Maaswinkel-Mooij P . Two additional cases of the Ohdo blepharophimosis syndrome. Am J Med Genet 1993; 47: 901–906. [DOI] [PubMed] [Google Scholar]
- Lyons MJ , Graham JM Jr. , Neri G , Hunter AG , Clark RD , Rogers RC et al. Clinical experience in the evaluation of 30 patients with a prior diagnosis of FG syndrome. J Med Genet 2009; 46: 9–13. [DOI] [PubMed] [Google Scholar]
- Rump P , Niessen RC , Verbruggen KT , Brouwer OF , de Raad M , Hordijk R . A novel mutation in MED12 causes FG syndrome (Opitz-Kaveggia syndrome). Clin Genet 2011; 79: 183–188. [DOI] [PubMed] [Google Scholar]
- Schwartz CE , Tarpey PS , Lubs HA , Verloes A , May MM , Risheg H et al. The original Lujan syndrome family has a novel missense mutation (p.N1007S) in the MED12 gene. J Med Genet 2007; 44: 472–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vulto-van Silfhout AT , de Vries BB , van Bon BW , Hoischen A , Ruiterkamp-Versteeg M , Gilissen C et al. Mutations in MED12 cause X-linked Ohdo syndrome. Am J Hum Genet 2013; 92: 401–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isidor B , Lefebvre T , Le Vaillant C , Caillaud G , Faivre L , Jossic F et al. Blepharophimosis, short humeri, developmental delay and hirschsprung disease: expanding the phenotypic spectrum of MED12 mutations. Am J Med Genet A 2014; 164A: 1821–1825. [DOI] [PubMed] [Google Scholar]
- Graham JM Jr. , Schwartz CE . MED12 related disorders. Am J Med Genet A 2013; 161A: 2734–2740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesca G , Moizard MP , Bussy G , Boggio D , Hu H , Haas SA et al. Clinical and neurocognitive characterization of a family with a novel MED12 gene frameshift mutation. Am J Med Genet A 2013; 161A: 3063–3071. [DOI] [PubMed] [Google Scholar]
- Tzschach A , Grasshoff U , Beck-Woedl S , Dufke C , Bauer C , Kehrer M et al. Next-generation sequencing in X-linked intellectual disability. Eur J Hum Genet (e-pub ahead of print 4 February 2015; doi:10.1038/ejhg.2015.5). [DOI] [PMC free article] [PubMed]
- Kim S , Xu X , Hecht A , Boyer TG . Mediator is a transducer of Wnt/beta-catenin signaling. J Biol Chem 2006; 281: 14066–14075. [DOI] [PubMed] [Google Scholar]
Data Citations
- Yamamoto Toshiyuki.HGV Database. 2015. 10.6084/m9.figshare.hgv.586. [DOI]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Citations
- Yamamoto Toshiyuki.HGV Database. 2015. 10.6084/m9.figshare.hgv.586. [DOI]