

During epithelial–mesenchymal transition IP3Rs relocate from tight junctions to the leading edge of migrating pancreatic cancer cells and regulate dynamics of focal adhesions. STIM1-competent ER–PM junctions position closely behind IP3Rs and, together with IP3Rs, regulate cell migration.
Keywords: epithelial–mesenchymal transition, ER–PM junctions, focal adhesions, IP3 receptors, pancreatic ductal adenocarcinoma, PANC-1 cells
Abstract
Disconnection of a cell from its epithelial neighbours and the formation of a mesenchymal phenotype are associated with profound changes in the distribution of cellular components and the formation of new cellular polarity. We observed a dramatic redistribution of inositol trisphosphate receptors (IP3Rs) and stromal interaction molecule 1 (STIM1)-competent endoplasmic reticulum–plasma membrane junctions (ER–PM junctions) when pancreatic ductal adenocarcinoma (PDAC) cells disconnect from their neighbours and undergo individual migration. In cellular monolayers IP3Rs are juxtaposed with tight junctions. When individual cells migrate away from their neighbours IP3Rs preferentially accumulate at the leading edge where they surround focal adhesions. Uncaging of inositol trisphosphate (IP3) resulted in prominent accumulation of paxillin in focal adhesions, highlighting important functional implications of the observed novel structural relationships. ER–PM junctions and STIM1 proteins also migrate to the leading edge and position closely behind the IP3Rs, creating a stratified distribution of Ca2+ signalling complexes in this region. Importantly, migration of PDAC cells was strongly suppressed by selective inhibition of IP3Rs and store-operated Ca2+ entry (SOCE), indicating that these mechanisms are functionally required for migration.
INTRODUCTION
Pancreatic ductal adenocarcinoma (PDAC) is a leading cause of cancer-related death [1]. Epithelial–mesenchymal transition (EMT), migration and invasion are cellular processes that are crucially important for the formation of lethal metastases in this and other types of cancer (reviewed in [2–4]). Currently, understanding the fundamental contributions that Ca2+ signalling makes to cell migration is an important research avenue of potential clinical relevance [5–7]. It is particularly relevant for identifying putative therapeutic targets that could delay or prevent the formation of metastases (reviewed in [8]). Two prominent components of the Ca2+ signalling cascade are inositol trisphosphate receptors (IP3Rs) and the store-operated Ca2+ entry (SOCE) mechanism. IP3Rs are intracellular Ca2+-releasing channels [9] which mediate responses to numerous hormones and neurotransmitters (reviewed in [10,11]). SOCE restores the Ca2+ concentration in the ER ([Ca2+]ER) following its depletion due to the Ca2+-releasing activity of IP3Rs or other intracellular channels (reviewed in [12,13]). The endoplasmic reticulum–plasma membrane junctions (ER–PM junctions) are regions of close contact between the two organelles [14–16] which serve as hubs for cAMP [17,18], phospholipid [19] and Ca2+ [16,20,21] signalling. SOCE is a Ca2+ influx mechanism triggered by Ca2+ store depletion, which involves the oligomerization of stromal interaction molecule 1 (STIM1, an EF-hand-containing protein that serves as the ER Ca2+ sensor), translocation of STIM1 oligomers to ER–PM junctions and opening of PM Ca2+ channels [16,22–25]. STIM1-competent ER–PM junctions can thus be classed as platforms for SOCE. Orai proteins are pore-forming components of SOCE channels that are opened by STIM1 [22,24,26], although some types of transient receptor potential (TRP) channels are probably also involved [27]. Following [Ca2+]ER depletion, Orai proteins translocate to the PM component of the ER–PM junctions where they interact with STIM and form Ca2+-selective channels. Ca2+ is heavily buffered in the cytosol of most cell types (e.g. [28,29]) and therefore proximity of Ca2+ channels to their downstream targets is frequently crucial for the efficiency and specificity of signalling. In the case of migrating cells and the regulation of migration, the relative positioning of Ca2+ signalling complexes, proteins that define the leading edge and focal adhesions (structures responsible for interaction between the cell and extracellular matrix), is of particular interest.
In the present study we have characterized the drastic redistribution of Ca2+ signalling complexes and ER–PM junctions occurring when PDAC cells disconnect from their neighbours and develop a migratory phenotype. Furthermore, we have characterized novel stratified localization of the Ca2+ signalling complexes in the leading edge and their structural relationships with the components of migratory apparatus. Finally, we have revealed the functional importance of IP3Rs and of SOCE for cell migration.
MATERIALS AND METHODS
Reagents
Xestospongin-B was provided by Dr J. Molgó (Institut de Biologie et Technologies de Saclay, Saclay, France) and GSK-7975A was a gift from Dr Malcolm Begg (GlaxoSmithKline, Stevenage, U.K.). Cyclopiazonic acid (CPA) was from Tocris, thapsigargin (TG) and rapamycin were purchased from Calbiochem, caged Ins(1,4,5)P3/PM (cag-iso-2-145) was purchased from Sichem. RIPA lysis and extraction buffer, Halt protease inhibitor cocktail and EDTA supplements were purchased from Pierce-Thermo Scientific. Fura-2-acetoxymethyl ester (AM), Fluo-4-AM, 1,2-bis-(o-aminophenoxy)ethane-N,N,N′,N′-tetra-acetic acid (BAPTA)-AM, Hoechst 33342, Sytox Orange, propidium iodide, human IP3R siRNA oligomers, anti-GFP (rabbit and chicken), fluorophore-conjugated antibodies and Alexa Fluor 647 phalloidin were all purchased from Life Technologies.
Primary antibodies
Anti-IP3R1 [rabbit polyclonal raised against C-terminal amino acids 2735–2749 [30] was a gift from Professor J. Parys (Catholic University of Leuven, Leuven, Belgium)]. Anti-IP3R1 [D53A5 (rabbit)] was from Cell Signaling Technology. Anti-IP3R2 (rabbit polyclonal raised against C-terminal amino acids 2686–2702) was a gift from Professor D. Yule (University of Rochester, Rochester, NY, U.S.A.); anti-IP3R3 (mouse) was from BD Transduction Laboratories. Anti-pan-IP3R rabbit polyclonal raised against C-terminal epitopes was from Millipore. Anti-occludin (rabbit and mouse) was from Zymed Laboratories. Anti-vinculin (mouse), anti-β-actin (mouse) and horseradish peroxidase (HRP)-conjugated secondary antibodies (anti-rabbit and anti-mouse species) were from Sigma–Aldrich. Anti-E-cadherin (mouse) was from Santa Cruz Biotechnology.
Cell culture, constructs and transfection
PDAC cell line PANC-1 was obtained from the ATCC (ATCC number CRL-1469) and cultured in DMEM (Dulbecco's modified Eagle's medium) supplemented with 10% FBS (fetal bovine serum), 100 units/ml penicillin, 100 μg/ml streptomycin and 292 μg/ml glutamine. Cultured cells were maintained in a humidified incubator (Wolf Laboratories) at 37°C and 5% CO2.
DNA constructs coding for LL–FKBP–mRFP (where LL denotes a long linker, FKBP is FK506-binding protein and mRFP is monomeric RFP), CFP–FRB–LL (FRB is FKBP12–rapamycin-binding) [21] and for YFP–STIM1 with a TK (thymidine kinase) promoter [21] were gifts from Dr T. Balla (National Institute of Child Health and Human Development, Bethesda, MD, U.S.A.).
In our study we utilized mCherry-labelled paxillin (Pax-mCh) and paxillin labelled with the Ca2+ sensor GCaMP5 (Pax-GCaMP5). The coding sequence for GCaMP5 was obtained from Addgene (plasmid 31788, originally generated by Douglas Kim and Loren Looger [31]). The GCaMP5 coding sequence was PCR-amplified using the following primer pair containing restriction endonuclease sites (underlined) to permit sub-cloning into the pcDNA3.1(+) backbone (Life Technologies) generating a new ‘pcDNA-GCaMP5-CT’ C-terminally tagging expression vector: sense (NotI) 5′-ATATGCGGCCGCATG-ACTGGTGGACAGCAAATG-3′; antisense (ApaI) 5′-ATA-TGGGCCCTCACTTCGCTGTCATCATTTGTAC-3′. Pax-GCaMP5 was created by PCR amplification of the chicken paxillin sequence obtained from Addgene (plasmid 15233, originally deposited by Rick Horwitz [32]) using the following primer pair containing restriction endonuclease sites (underlined) to permit subcloning into pcDNA-GCaMP5-CT: sense (EcoRI) 5′-ATATGAATTCACCATGGACGACCTC-GATGCC-3′; antisense (NotI) 5′-ATATGCGGCCGCTAC-AGAAGAGTTTGAGAAAGC-3′. Pax-mCh was created by PCR amplification of the chicken paxillin sequence using the following primer pair containing restriction endonuclease sites (underlined) for subcloning into the mCherry-N1 vector (Clontech): sense (XhoI) 5′-ATATCTCGAGACCATGGACG-ACCTCGATGCC-3′; antisense (EcoRI) 5′-ATATGAATTC-GACAGAAGAGTTTGAGAAAGCA-3′. All constructs were verified by automated sequencing (The Sequencing Service, University of Dundee, Dundee, U.K.).
To express the proteins of interest, cells were transfected at approximately 60–70% confluence with 1–2 μg of DNA per plasmid construct for 24 h using PromoFectin reagent (PromoKine) according to the manufacturer's instructions. For the knockdown of cellular proteins of interest, siRNA oligomers directed against human IP3R1, IP3R2 and IP3R3 isoforms were used. Cells were transfected at approximately 30–40% confluence with 50 nM per siRNA oligomer for 72 h using Lipofectamine 2000 (Life Technologies) according to the manufacturer's instructions.
Immunofluorescence and visualising ER–PM junctions
Cells were seeded on to 35-mm-diameter glass-bottom dishes from Mattek or ibidi.
For visualising ER–PM junctions, cells were transfected with both PM-targeted LL–FKBP–mRFP and ER-targeted CFP–FRB–LL constructs, and 24 h later, were treated with 100 nM rapamycin for 4–5 min at 37°C/5% CO2. For further details, see [21,33].
For visualising STIM1 puncta, cells were transfected with YFP–TK–STIM1, and 24 h later, were treated with 30 μM CPA for 1 h at 37°C/5% CO2 and imaged using a confocal microscope.
For immunostaining, cells were fixed using 4% (v/v) paraformaldehyde (PFA) in PBS for 10–15 min at room temperature (approximately 18–22°C) followed by three PBS washes, and subsequently permeabilized using 0.2% Triton X-100 in PBS for 5 min at room temperature, before an additional three PBS washes. Non-specific antibody binding was blocked for 1 h at room temperature in PBS containing 10% (v/v) goat serum and 1% (w/v) BSA prior to incubation with primary antibodies in PBS containing 5% (v/v) goat serum and 0.1% acetylated BSA for 1 h at room temperature or overnight at 4°C. Primary antibodies were used at the following dilutions: anti-IP3R1, 1:200; anti-IP3R2, 1:100; anti-pan-IP3R, 1:20; anti-vinculin, 1:200; anti-GFP, 1:200; anti-occludin, 1:100; anti-E-cadherin, 1:50. After the incubation with primary antibodies, cells were washed with PBS three times followed by the addition of appropriate species-specific fluorophore-conjugated secondary antibodies for 30 min at room temperature at dilutions of 1:500–1:1000 in PBS. Additional three PBS washes were carried out prior to imaging in PBS. In the specified experiments, Alexa Fluor 647 phalloidin was used (at 1:50 dilution).
Two different confocal microscopes were used to visualize the distribution of specific proteins and ER–PM junctions in fixed cells: Leica TCS SP2 (AOBS) confocal microscope with ×63 oil-immersion objective [NA (numerical aperture) − 1.4] and Zeiss LSM 710 confocal microscope with ×63 oil-immersion objective (NA − 1.4). The pinhole was set between 1 and 2 airy units.
Live-cell Ca2+ imaging and uncaging
To investigate inositol trisphosphate (IP3)-induced Ca2+ responses, cells were loaded with caged IP3 and with Ca2+ indicator Fluo-4 by incubation in the solution containing 1 μM caged IP3/PM and 5 μM Fluo-4-AM. A Zeiss LSM 510 confocal microscope with a ×63 water-immersion objective (NA 1.2) was utilized in these experiments; the 488 nm laser line was used to excite Fluo-4 (emission recorded at LP 505 nm), 351 nm and 364 nm laser lines were used for uncaging.
To investigate SOCE, PANC-1 cells were loaded with Fura-2 by incubation for 1 h in Fura-2-AM-containing solution. Cells were washed for 30 min to allow de-esterification of the probe. A Till Photonics Imaging system was used in these experiments. Fluorescence of cells loaded with Fura-2 was excited at 340 and 380 nm, and emission collected using a 510 nm bandpass filter. Data recorded in these experiments are expressed as the ratio of fluorescence excited by 340 nm (F340) and 380 nm (F380), after corresponding background subtraction. Changes of extracellular solution were made using gravity-fed perfusion system.
Migration assay
Boyden chambers were purchased from Corning. The pore size was 8 μm. Migration was measured in conditions of symmetrical FBS (1% FBS in both the upper and lower chambers) and asymmetrical FBS (0% FBS in the upper chamber and 5% FBS in the lower chamber). After seeding, PANC-1 cells were allowed to migrate for 6 h in a humidified environment at 37°C/5% CO2 in the presence or absence of the inhibitors of various components of the Ca2+ signalling cascade; Boyden chamber inserts were then fixed using 100% methanol and non-migrated cells were removed from the top-side of the inserts using cotton buds. The inserts were then rinsed two times in PBS, prior to the staining of migrated cells on the underside of the chamber inserts using 100 μg/ml propidium iodide. The inserts containing fixed and stained cells were imaged on a Leica AOBS TCS SP2 confocal microscope using a ×10 air objective with an NA of 0.3 as described in [34]. Five representative regions of interest were imaged per insert. Fluorescently stained migrated cells were counted using CellProfiler software cell counting algorithm.
Super-resolution imaging
For super-resolution imaging, PANC-1 cells were seeded into Lab-Tek chambered coverglass eight-well #1.0 with low thickness variation (Thermo Scientific). To visualize IP3R1, PANC-1 cells were fixed with 4% PFA for 10–15 min at room temperature and subsequently immunostained with anti-IP3R1 antibody, followed by the use of an appropriate species-specific Alexa Fluor 647-conjugated secondary antibody. To visualize ER–PM junctions at super-resolution level, PANC-1 cells co-transfected with PM-targeted LL–FKBP–mRFP and ER-targeted CFP–FRB–LL constructs for 24 h were fixed using 4% PFA for 10–15 min at room temperature after treatment with 100 nM rapamycin for 4–5 min at 37°C/5% CO2 to highlight the pre-existing ER–PM junctions without ER Ca2+ store depletion. Briefly, the heterodimerization of both ER- and PM-targeted constructs revealed the ER–PM junctions as punctate structures in both CFP and RFP fluorescence channels. To highlight the ER-targeted FRB–LL–CFP counterpart of the ER–PM junctions' puncta, cells were immunostained using anti-GFP antibody (which also recognizes CFP), and followed by appropriate species-specific Alexa Fluor 647-conjugated secondary antibody.
After immunostaining of IP3R1 or ER–PM junctions, samples were immersed and imaged in a dSTORM buffer containing 100 units/ml glucose oxidase, 2000 units/ml catalase, 50 mM mercaptoethylamine/HCl and 50 mg/ml glucose in PBS [35]. Wells were filled and sealed with a coverslip to exclude oxygen. Super-resolution microscopy was performed using a custom-built instrument, as described previously [35,36]. An Olympus IX71 microscope formed the basis of an inverted objective total internal reflection fluorescence (TIRF) instrument with a UAPON ×100 TIRF, NA − 1.49 objective. Laser illumination was provided by a 640 nm 150 mW diode laser (Toptica Photonic AG) and a 561 nm 200 mW optically pumped semiconductor laser (Coherent Europe). Additionally, a 405 nm laser diode (Mitsubishi Electronics) was available for re-activation of fluorophores if required. Laser power on the diode lasers was controlled directly, whereas a rotating quarter-wave plate was used to alter the power of the 561 nm laser. Low powers (1–10%) were used for field of view selection, context and conventional fluorescence images. Higher powers (50–100%) were used for dSTORM imaging. Fluorescence and excitation light were spectrally separated by the dichroic mirror and emission filter from a multi-edge filter set (LF405/488/561/635-A-000, Semrock), and an additional bandpass filter was used to remove cross-talk in each channel (FF01-676/37 with 640 nm excitation and FF01-600/37 with 561 nm excitation, both from Semrock).
Images were acquired using an EMCCD camera (Andor iXon 897) using software written in LabVIEW (National Instruments). Typically dSTORM image ‘stacks’ were composed of 10000 frames, with 10 ms exposure time per frame. Super-resolution images were reconstructed from these image stacks using the open-source rainSTORM package [35,37,38].
IP3Rs knockdown and cell migration
Migration of PANC-1 cells after 72 h of siRNA knockdown of IP3R1 or IP3R2 or IP3R3 isoforms was measured using a Boyden chamber assay. siRNA sequences targeting each of the IP3R isoforms are: IP3R1 silencer select siRNA sense 5′-GCACGACAGUGAAAACGCAtt-3′, antisense 5′-UGCGUUUUCACUGUCGUGCct-3′; IP3R2 silencer select siRNA sense 5′-GGUGUCUAAUCAAGACGUAtt-3′, antisense 5′-UACGUCUUGAUUAGACACCag-3′; and IP3R3 silencer select siRNA sense 5′-GCAUGGAGCAGAUCGUGUUtt-3′, antisense 5′-AACACGAUCUGCUCCAUGCtg-3′. Following 72 h of treatment with the specified siRNA constructs, PANC-1 cells were allowed to migrate for 6 h in a humidified environment at 37°C/5% CO2. Boyden chamber inserts were then fixed using 100% methanol. Non-migrated cells were removed from the top-side of the chamber inserts using cotton buds and rinsed two times in PBS, prior to the staining and counting of migrated cells on the underside of the chamber inserts (see the Migration Assay section above for the description of staining and counting procedures).
Immunoblotting
Cells were treated with trypsin, removed from flasks and then collected by centrifugation. Cells were then lysed using RIPA lysis extraction buffer supplemented with Halt protease inhibitor cocktail and EDTA (Pierce-Thermo Scientific). Lysed samples were separated on a 4–12% NuPAGE Bis–Tris gradient gels and protein transferred on to nitrocellulose membranes by transverse electrophoresis. Nitrocellulose membranes were blocked in 3% (w/v) non-fat dried skimmed milk powder dissolved in PBS for 1 h at room temperature, and probed with primary antibodies including anti-IP3R1 (1:500 dilution), anti-IP3R2 (1:1000 dilution), anti-IP3R3 (1:500 dilution) and anti-β-actin (1:1000 dilution) at 4°C overnight. After overnight incubation, nitrocellulose membranes were incubated with appropriate species-specific HRP-conjugated secondary antibodies (1:400 dilution) for 1 h at room temperature. Bands were visualized using enhanced chemiluminescence (ECL) Western blotting substrate and a Bio-Rad Quantity One imaging system. Band intensities were quantified and analysed using ImageJ software (NIH). Blotting for β-actin was used as a loading control for siRNA knockdown experiments.
Image, data and statistical analyses
Image acquisition and preliminary analysis of confocal images was performed using either Leica LAS or Zeiss LSM 510 or Zeiss Zen software. Further analysis was performed using ImageJ software. Linear adjustments of contrast and brightness were applied if necessary using ImageJ. The ‘mask’ images used for illustrating the co-localization of the rapamycin-inducible linker components (images labelled ER–PM linkers) were created using the Co-localize RGB ImageJ plugin as described in [33].
In data presentation (for all components of the study) the error margins represent the S.E.M. The results were analysed using a Student's t test; P<0.05 was considered statistically significant and is indicated by the symbol * in the Figures.
RESULTS AND DISCUSSION
IP3Rs translocate from cell–cell contacts in cellular clusters to the leading edge of individual migrating cancer cells
In monolayers of PANC-1 cells, IP3R1 was observed primarily in the areas of cell–cell contacts (see Figure 1A and Supplementary Figure S1). Similar distribution was observed in smaller cellular clusters (Figure 1B). Fluorescence profiles measured along the lines selected to cross junctional regions demonstrate approximately 4–5-fold increased density of IP3R1 in these regions in comparison with the neighbouring regions of the cytoplasm (Figures 1A and 1B, and Supplementary Figures S1A and S1C, upper panel). In cell–cell contact regions IP3R1 was closely co-positioned with occludin, which we used as a marker of tight junctions (Figure 1C). The white colour in the ‘Merge’ panel of this figure indicates that in a part of this region the IP3R1 and occludin are in such close proximity that the distance between these proteins is below the resolution of a confocal microscope (i.e. less than 300 nm). Notably, in the monolayers of PANC-1 cells, E-cadherin (a component of adherence junctions and an important marker of the epithelial phenotype) was also preferentially found in the cell-cell contact regions (Supplementary Figure S2), confirming cellular connectivity.
Figure 1. The polarized distribution of IP3R1s in connected PANC-1 cells and in isolated migrating PANC-1 cells.

(A) IP3R1s are localized at the cell–cell contact sites of PANC-1 cells forming a confluent monolayer. (B) IP3R1s are also localized at the cell–cell contact sites of small clusters of PANC-1 cells. (C) IP3R1s co-localize with occludin (tight junction marker) at the cell–cell contact sites of PANC-1 cells. Fluorescence profiles (right panel) that illustrate juxtaposition of the proteins were measured along the line spanning the cell-cell contact region. (D) IP3R1 predominantly decorates the leading edge of a migrating PANC-1 cell (immunostained using anti-IP3R1 antibody). (E) IP3Rs predominantly decorate the leading edge of PANC-1 cell (immunostained using pan-IP3R antibody). (F) IP3R1 co-positions with actin-rich lamellipodia at the leading edge of migrating pancreatic adenocarcinoma cells. Other examples of specific localization of IP3Rs are shown in Supplementary Figures S1–S3. Scale bars represent 10 μm.
The observed proximity of IP3Rs to tight junctions was described before in non-transformed Madin–Darby canine kidney (MDCK) cells, where it was associated with the developed epithelial phenotype [30,39]. Interestingly, in our experiments this distribution was observed in the monolayers of cancer cells (PANC-1 cells) and even in relatively small clusters of these cells. Importantly, cells disconnected from the clusters or monolayers also clearly display polarized distribution of IP3R1, as these cells preferentially positioned their IP3R1s at the leading edge (Figure 1D, and Supplementary Figures S1A, right panels, and S1C, central panel). Notably, similar increased density of IP3R1 at the leading edge was observed in migrating PANC-1 cells after treatment with transforming growth factor (TGF)-β1, an agonist known to induce a migratory mesenchymal phenotype in this cell type [40,41] (see Supplementary Figure S3). Preferential positioning at the leading edge was also observed for IP3R2s (Supplementary Figure S4). Pan-IP3R antibodies, which were raised against a conserved C-terminal region common to all IP3R subtypes, also revealed a similarly polarized distribution of IP3Rs with preferential localization at the leading edge of individual migrating cells (Figure 1E and Supplementary Figure S1C, lower panel). The leading edge of migrating cells is characterized by the region of polymerized actin (reviewed in [4]). We therefore examined the relative positioning of the F-actin-enriched region and IP3R1; Figure 1(F) illustrates close apposition of the two proteins at the leading edge of isolated migrating PANC-1 cells. In addition to actin polymerization, cellular migration requires the formation of focal adhesions at the front of migrating cells; we therefore extended our analyses to examine the relative positioning of IP3Rs and focal adhesions.
Focal adhesions are closely surrounded and regulated by IP3Rs at the leading edge of migrating cancer cells
Dual immunostaining of focal adhesions (using antibodies against vinculin) and IP3R1s revealed remarkable relative localization of the adhesions and the receptors. As expected, focal adhesions were preferentially localized close to the leading edge of the migrating cells; the leading edge was also enriched with IP3R1s (Figure 2A). Importantly, focal adhesions were not co-localized but were instead closely surrounded by the receptors forming ‘potholes’ on the background of IP3R1 immunostaining (see the fragment shown in the bottom panels in Figure 2(A) and the associated fluorescence profile). The preferential positioning of focal adhesions and IP3Rs as well as ‘potholes’ were observed in the smooth-shaped (lamellipodia-like) regions of the leading edge (Figure 2A and the central row of images in Figure 2B with the associated fluorescence profile) as well as in spiky, filopodia-like protrusions at the leading edge (Figure 2B and specifically the lower row of panels and the associated fluorescence profile). The intimate spatial relationship between focal adhesions and IP3Rs concentrated in this region should make these structures particularly sensitive to Ca2+ signals generated by IP3Rs at the leading edge of migrating cells and could explain the prominent effect of the inhibition of IP3Rs on cell migration (see the last subsection of the Results section). Using intracellular photorelease of IP3 from its caged precursor (uncaging) we directly probed the influence of this second messenger on the dynamics of focal adhesions. In these experiments we used cells expressing simultaneously Pax-mCh and Pax-GCaMP5. Paxillin was used in these experiments because it is an important regulatory component of focal adhesions [42].
Figure 2. IP3Rs encompass focal adhesions in migrating PANC-1 cells.

Non-transfected PANC-1 cells were fixed and stained with anti-IP3R1 and anti-vinculin antibodies. Scale bars represent 10 μm. (A) IP3R1s encompass focal adhesions in migrating pancreatic adenocarcinoma cells with a smooth leading edge. An expanded fragment of the cell is shown in the lower panels and the arrowheads indicate where the focal adhesions are encompassed by IP3R1s. Fluorescence profile (right panel) was measured along the line (shown in the top left and top central images) spanning the cell membrane of the leading edge and crossing a focal adhesion (specifically the lowest of three focal adhesions indicated by the yellow arrowhead in the expanded fragment). (B) The relationships between IP3R1s and focal adhesions in a cell with a complex leading edge composed of smooth (lamellipodia-like) regions and some spiky protrusions. The central panels correspond to the upper box in the cell image. These panels and the associated fluorescence profile represent an expanded fragment of a smooth region (similar to A), whereas the bottom panels show a representative spiky protrusion. In both cases the focal adhesions (indicated by arrowheads) are surrounded by IP3R1s. The associated fluorescence profiles were measured across the lines shown in the upper panels; the focal adhesions crossed by these lines are indicated by yellow arrowheads on central and lower panels.
Uncaging of IP3-induced rapid accumulation of paxillin in focal adhesions and the loss of paxillin from the cytosol (Figures 3A and 3B). These findings highlight the importance of IP3Rs for focal adhesion remodelling–the process immediately related to migration ([43], reviewed in [42]). Uncaging of IP3 also induced Ca2+ rises resolvable both in the cytosol and localized at focal adhesions (Figure 3A). Interestingly, inhibition of IP3Rs with xestospongin-B reduced the paxillin content of focal adhesions in unstimulated PANC-1 cells (Figure 3C), suggesting that IP3Rs are involved in the regulation of focal adhesion even when cells are not stimulated with IP3-producing agonists. These results suggest that focal adhesions are modulated by low ‘background’ activity of IP3Rs.
Figure 3. IP3Rs and remodelling of focal adhesions in PANC-1 cells.
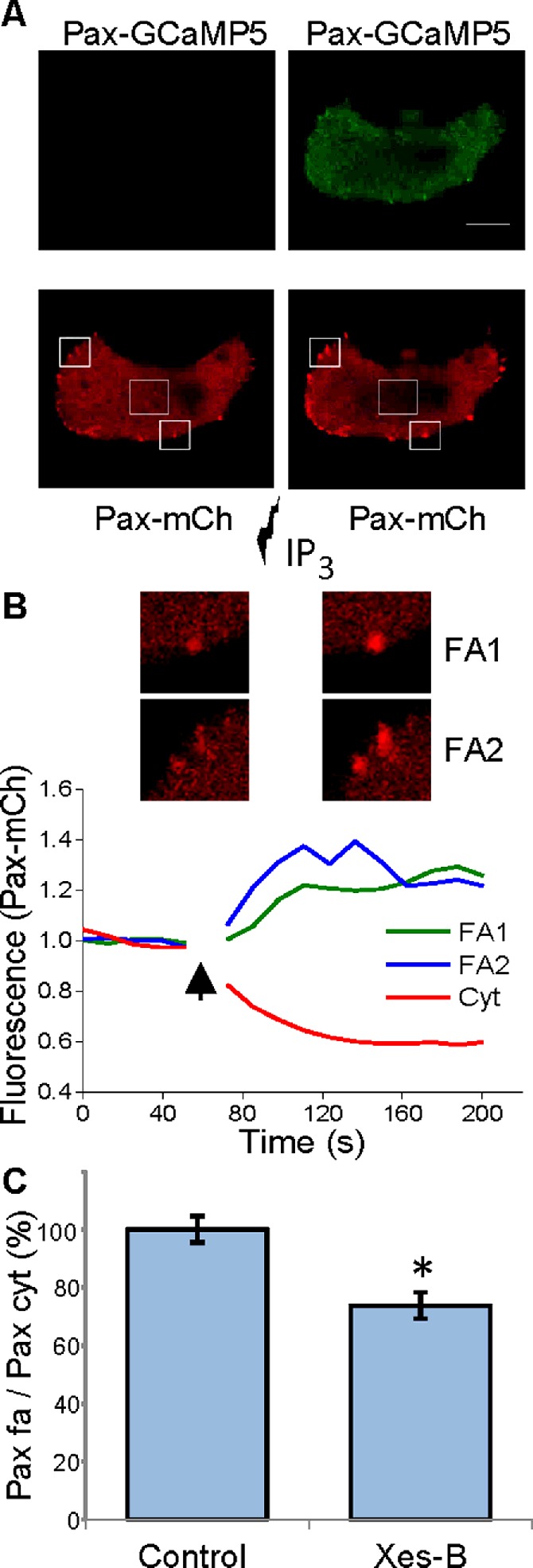
(A) IP3 uncaging induces paxillin accumulation in focal adhesions. Upper panels show the fluorescence of Pax-GCaMP5 before (left) and after (right) uncaging. The increase in GCaMP5 fluorescence indicates a Ca2+ rise; note the prominent response in focal adhesions. The lower panels show the distribution of Pax-mCh fluorescence before (left) and after (right) IP3 uncaging. Note the prominent increase in fluorescence in focal adhesions induced by the uncaging. (B) The graph shows the increase in fluorescence (i.e. Pax-mCh accumulation), recorded from the regions of interest containing focal adhesions (shown above the traces and highlighted by solid border boxes in A), and the decrease in Pax-mCh fluorescence in the cytosol (the region of interest for this analysis included the area of cytosol highlighted by dashed border box in A). The arrow indicates the period of uncaging (during this period the fluorescence recording was interrupted). The images are fragments of the cell shown in (A). (C) Xestospongin-B (Xes-B) produced a statistically significant decrease in paxillin content in the focal adhesions of unstimulated cells (i.e. cell are not treated with IP3-generating agonists). In these experiments cells were stained with anti-paxillin antibodies. The fluorescence of focal adhesions was normalized by the fluorescence of the cytosol. The normalized fluorescence of paxillin in focal adhesions was measured in untreated cells (control, n=39) and in cells incubated with xestospongin-B for 1 h (n=32). The results for both groups are shown as mean values ± S.E.M.
Non-excitable cells rely on SOCE as the main [Ca2+]ER-reloading mechanism for efficient calcium signalling mediated by IP3Rs. To activate SOCE, STIM1 has to translocate to the ER–PM junctions [44]. Indeed the presence of STIM1-competent ER–PM junctions in the proximity of the leading edge has recently been reported [7,33]. We therefore next investigated the relative positioning of IP3R1, STIM1 and ER–PM junctions.
Repositioning of IP3Rs from cell–cell contacts to the leading edge of migrating cells is accompanied by the accumulation of ER–PM junctions and STIM1 puncta in the adjacent cytoplasmic region
In migrating PANC-1 cells, the region with the increased density of IP3R1s at the leading edge was closely followed by the region with a high density of STIM1 puncta (Figure 4A). Similar relative positioning was seen for IP3R1s and co-localized ER and PM linkers (white dots highlighting the ER–PM junctions in Figure 4B), suggesting that the ER–PM junctions migrating just behind the IP3Rs are STIM/SOCE-competent. The similarity of distributions of STIM1 puncta and ER–PM junctions (identified by the linkers) is consistent with other studies that observed the co-localization of STIM1 with ER–PM junctions [21,33]. The preferential localization of IP3R1s and ER–PM junctions at the front of migrating cells was also confirmed using super-resolution microscopy. In these experiments, employing the dSTORM technique, we observed that the leading edge of migrating cells indeed had increased density of IP3R1s (Figure 4C) and higher concentration of ER–PM junctions (Figure 4D). Note the increased resolution of dSTORM images (in the x–y plane) in comparison with diffraction-limited (in the x–y plane) TIRF images taken from the same cellular regions (insets in Figures 4C and 4D). The actual size of both the ER–PM junctions and clusters of IP3Rs is significantly smaller than the limit of resolution of diffraction-limited microscopy but the preferential localization at the leading edge was observed using all types of microscopy. dSTORM imaging, which has considerably improved axial and lateral resolution in comparison with conventional microscopy, confirmed that both IP3R1s and ER–PM junctions can be observed close to the leading edge and in the immediate proximity to the ventral membrane of the migrating cells (i.e. portion of the membrane that is involved in forming contacts with the substratum and that is sliding along the substratum). A number of recent studies reported the importance of Ca2+ signalling for cell migration and invasion [5–7,45–47]. The Ca2+ responses have been shown to both potentiate [7,46] and suppress [7] migration, depending on cell type and extracellular environment. Considering the observed prominent stratified localization of IP3Rs and STIM1/ER–PM junctions near the leading edge of migrating PANC-1 cells and the proximity of these structures to the components of migratory apparatus (e.g. focal adhesions and actin fibres) we next decided to test the importance of IP3Rs and SOCE for the migration of this cell type.
Figure 4. Relative positioning of IP3R1 and STIM1/ER–PM junctions in migrating PANC-1 cells.

(A) In migrating PANC-1 cells, IP3R1s decorate the leading edge, whereas STIM1 puncta concentrate in the adjacent region (just behind the leading edge). PANC-1 cells were transfected with TK–YFP–STIM1 and then treated with 30 μM CPA to reveal STIM1 puncta. Cells were then fixed and immunostained using antibodies against IP3R1. All images in (A) and (B) show confocal sections taken from ventral parts of the cells located in the immediate proximity to the coverslip. Scale bars represent 10 μm. (B) In migrating PANC-1 cells IP3R1s decorate the leading edge, whereas ER–PM junctions concentrate in the adjacent region (behind the leading edge). PANC-1 cells were simultaneously transfected with linker constructs ER-targeted CFP–FRB–LL and PM-targeted LL–FKBP–mRFP. Cells expressing both linkers were treated with 100 nM rapamycin to reveal ER–PM junctions. Cells were then fixed and immunostained using antibodies against IP3R1. It is informative to compare the observed relative positioning of ER–PM junctions and IP3R1 in migrating cells with that in cellular clusters. We found that on confocal sections closest to the coverslip ER–PM junctions were preferentially localized at the cell periphery. Interestingly, some ER–PM junctions were found just behind the IP3R1s that decorated cell–cell contacts (Supplementary Fig-ure S5). (C) Super-resolution microscopy of IP3R1s at the leading edge of a PANC-1 cell. Left panel: the leading edge of a cell immunostained using antibodies against IP3R1s and imaged using a TIRF microscope (here and in D ‘diffraction-limited’ refers to its lateral resolution). Scale bar represents 1 μm. The fragment, highlighted as a square in the left panel, was then imaged using dSTORM and the result is shown in the central panel. Scale bar represents 1 μm. Right panel (Merge): co-positioning of the two images. Expanded fragments in the right part of (C) (small panels) are taken from the peripheral regions indicated by arrowheads in the ‘Merge’ (image left arrowhead corresponds to the upper set of images). (D) Super-resolution microscopy of ER–PM junctions near the leading edge of PANC-1 cells. PANC-1 cells simultaneously transfected with both linker constructs (PM-targeted FBKP–LL–mRFP and ER-targeted FRB–LL–CFP) were fixed after treatment with 100 nM rapamycin to highlight the pre-existing ER–PM junctions without ER Ca2+ store depletion. PANC-1 cells were then stained using anti-GFP antibody (which also recognizes CFP) to reveal ER-targeted FRB–LL–CFP accumulated in ER–PM junctions. Left panel: the localization of ER–PM junctions visualized using TIRF mode. Scale bar represents 1 μm. The PM border outline was generated using the Threshold and Wand (tracing) tool function of ImageJ (see Supplementary Figure S6). The fragment, highlighted as a square in the left panel, was then imaged using dSTORM and the result is shown in the central panel. Scale bar represents 1 μm. Right panel (Merge): the co-positioning of the two images. Expanded fragments in the right part of (D) (small panels) are taken from the peripheral regions highlighted in the Merge image by arrowheads (upper arrowhead corresponds to the upper set of images). Note the improvement of resolution in comparison with diffraction-limited images (C and D, lateral dSTORM resolution in these experiments was approximately 60–70 nm).
Inhibition of IP3Rs and STIM–Orai channels suppresses migration of PANC-1 cells
The selective inhibitor of IP3Rs–xestospongin-B [48]–effectively suppressed cytosolic Ca2+ responses induced by IP3 uncaging in PANC-1 cells (Figure 5A). SOCE in this cell type was significantly (by 61±1%, n=151) inhibited by 30 μM GSK-7975A (Figure 5B), a selective inhibitor of SOCE mediated by STIM–Orai interaction [49]. Note that 10 μM GSK-7975A produced only a slightly weaker inhibition than 30 μM (inhibited by 53±1%, n=162; results not shown) and 100 μM was not more effective than 30 μM (n=145; results not shown). Migration in our experiments was tested using Boyden chambers. In the absence of FBS, PANC-1 cells migrate very inefficiently (leftmost bars in Figures 5C and 5D). We therefore investigated the effect of the inhibitors on migration of these cells in the presence of FBS using symmetrical FBS distribution (1% FBS in both the upper and lower chambers, Figure 5C) and asymmetrical FBS distribution (0% FBS in the upper chamber and 5% FBS in the lower chamber; this configuration can be considered as a model of chemotactic migration, see Figure 5D). Xestospongin-B significantly inhibi-ted migration of PANC-1 cells in conditions of symmetrical FBS (Figure 5C). The effects of this IP3R inhibitor on migration were even stronger for cells migrating along the gradient of FBS (Figure 5D); in this condition xestospongin-B inhibited migration by 74±9%. These findings are consistent with the results of IP3R-knockdown experiments that suggested the involvement of IP3Rs (particularly IP3R1 and possibly IP3R2) in migration (Figure 6). GSK-7975A supressed migration of PANC-1 cells (Figures 5C and 5D) and, as for xestospongin-B, the effect was particularly prominent in the experiments with asymmetrical FBS (Figure 5D, in these experiments GSK-7975A inhibited migration by 84±3%). Both xestospongin-B and GSK-7975A also inhibited cell migration as measured by wound-healing assay (Supplementary Figure S7). Neither xestospongin-B nor GSK-7975A induced substantial cellular toxicity (Supplementary Figure S8). Strong inhibition of migration by xestospongin-B and GSK-7975A suggest that the striking accumulation of IP3Rs and STIM/SOCE-competent ER–PM junctions in the leading edge of PANC-1 cells has a clear function, which is to provide signals important for migration of this type of cancer cells. There was a difference between the effect of xestospongin-B and GSK-7975A on the paxillin content of focal adhesions. Incubation for 1 h with xestospongin-B produced a statistically significant decrease in paxillin content in the focal adhesions of unstimulated PANC-1 cells (see Figure 3C); we did not, however, observe changes in paxillin content in focal adhesions following 1 h of application of 30 μM GSK-7975A (results not shown; n=44 for GSK-7975A treated and n=39 for the control group). It is therefore conceivable that the two inhibitors utilize different mechanisms for supressing the cell migration and that Ca2+ release and influx regulate different processes contributing to the cell migration.
Figure 5. Inhibition of Ca2+ signalling complexes suppresses PANC-1 cell migration.

(A) Xestospongin-B (Xes-B) blocks IP3-induced Ca2+ release in pancreatic adenocarcinoma cells. Ca2+ releases from internal stores were measured in cells loaded with caged IP3 in the absence (control; red trace, 28 cells) or presence of 50 μM xestospongin B (blue trace, 29 cells). The traces were composed of normalized Fluo-4 fluorescence measurements (mean values ± S.E.M. for individual time points). Pulses of UV light induced the IP3 uncaging (i.e. release of IP3 from its caged precursor) and subsequent releases of Ca2+ from internal stores into the cytosol. The intensities of the black arrows indicate the duration of uncaging (3, 8 and 20 s). In these experiments nominally Ca2+-free extracellular solution was used to reveal cytosolic Ca2+ responses occurring specifically due to Ca2+ release from the intracellular stores. (B) GSK-7975A inhibits SOCE in PANC-1 cells. Cells were pre-treated with TG for approximately 20 min before the beginning of the experiments in order to permanently deplete the ER Ca2+ stores, and maintained in nominally Ca2+-free solution. Increase in extracellular [Ca2+] to 1.8 mM resulted in Ca2+ influx via the SOCE pathway. Red trace shows control experiment (192 cells). Green trace (151 cells) shows significant inhibition of SOCE by 30 μM GSK-7975A. The green arrow indicates the moment of GSK-7975A addition. The traces were composed from normalized mean values ± S.E.M. for individual time points of Fura-2 fluorescence ratio (F340/F380) measurements. (C and D) Xestospongin-B and GSK-7975A suppress migration of PANC-1 cells. PANC-1 cells were subjected to both symmetric (1% FBS in both chambers) (C) and asymmetric (0% FBS in the upper chamber and 5% FBS in the lower chamber) (D) Boyden chamber migration assay for 6 h at 37°C in the absence (Control) or presence of the inhibitors. 0% FBS conditions (in both chambers, leftmost bars on both panels) were employed for comparison (this effectively inhibits cell migration). The number of migrated cells in each inhibitor-treated group was normalized to that in the control group for every individual experiment. Means ± S.E.M. were obtained from at least three independent experiments. In all cases the number of cells migrated in the presence of an inhibitor was statistically different from that in the control conditions.
Figure 6. Cellular depletion of IP3Rs suppresses pancreatic adenocarcinoma cell migration.

siRNA knockdown of IP3R1 (A), IP3R2 (B) but not IP3R3 (C) inhibits migration of PANC-1 cells. The cells were treated for 72 h post-transfection with IP3R siRNAs (R1 siRNA, R2 siRNA and R3 siRNA) or non-targeting siRNA (NC siRNA) oligomer sequences. After this treatment cell migration was measured using Boyden chamber assay with asymmetric FBS content (0% FBS in the upper chamber and 5% FBS in the lower chamber). In these experiments cells migrated for 6 h at 37°C/5% CO2. The number of migrated cells in each siRNA group was normalized to that in the corresponding non-targeting control group. At least three independent experiments were performed for each condition. Western blot profiles showing successful knockdown of IP3Rs by siRNA (lower panels). IP3Rs and actin protein levels were quantified using the ImageJ densitometry tool. Actin blot was used as a loading control. The amount of IP3R proteins was obtained as a function of normalization to the amount of actin protein. At least three independent experiments were performed for each condition. Molecular masses are indicated in kDa. IB, immunoblot.
It is interesting to note that in primary normal pancreatic acinar cells IP3Rs are preferentially positioned in close proximity to the tight junctions in the cell–cell contact regions near the apical part of the cells [14,50–53]. Indeed, we verified and confirmed this previously reported distribution using the same antibodies as employed for immunostaining of IP3Rs in PANC-1 cells (see Supplementary Figure S9). PDAC cells probably originate from pancreatic acinar cells (evidence for this has recently been reviewed in [54]). Therefore the changes in the distribution of IP3Rs from cell–cell contacts to the leading edge, that were characterized using a cellular model in our study, are likely to reflect the pathophysiological process associated with EMT in vivo. In other words the present study suggests a novel switch of polarity and function of Ca2+ signalling complexes during EMT associated with cancerogenesis. Ca2+ signalling complexes move from the intercellular contact regions (proximal to the apical part of the cell) to the leading edge and change function from regulating vital physiological processes (exocytosis and fluid secretion) to regulating migration of cancer cells. It is important to note that the leading edge attracts not only IP3Rs but also ER–PM junctions and STIM1 and that both Ca2+ release and Ca2+ influx are important for migration.
The present study describes prominent changes in the distribution of Ca2+ signalling complexes that develop when cancer cells disconnect from their neighbours and form a migratory ‘mesenchymal’ phenotype. The observed preferential distribution of IP3Rs and STIM1/ER–PM junctions signifies the formation of novel structural and functional (signalling) polarity in migrating PDAC cells. Disrupting this polarized distribution could present a mechanism for inhibiting migration and invasion of PDAC cells and ultimately suppressing the formation of metastasis of this type of cancer.
Acknowledgments
Help from Dr Alex Burdyga, Dr Judy Coulson, Dr Tobias Zech, Dr Michael Chvanov and Dr Dayani Rajamanoharan is gratefully acknowledged. We are also grateful for help from Dr Jean Sathish and Carolyn Rainer (Flow Cytometry & Cell Sorting Technology Directorate at the University of Liverpool).
Abbreviations
- AM
acetoxymethyl ester
- BAPTA
1,2-bis-(o-aminophenoxy)ethane-N,N,N′,N′-tetra-acetic acid
- [Ca2+]C
Ca2+ concentration in the cytosol
- [Ca2+]ER
Ca2+ concentration in the ER
- CPA
cyclopiazonic acid
- EMT
epithelial–mesenchymal transition
- ER
endoplasmic reticulum
- FKBP
FK506-binding protein
- FRP
FKBP12rapamycin-binding
- HRP
horseradish peroxidase
- IP3
inositol trisphosphate
- IP3R
inositol trisphosphate receptor
- LL
long linker
- mRFP
monomeric RFP
- NA
numerical aperture
- Pax-GCaMP5
paxillin labelled with Ca2+ sensor GCaMP5
- Pax-mCh
mCherry-labelled paxillin
- PDAC
pancreatic ductal adenocarcinoma
- PFA
paraformaldehyde
- PM
plasma membrane
- SOCE
store-operated Ca2+ entry
- STIM
stromal interaction molecule
- TG
thapsigargin
- TIRF
total internal reflection fluorescence
- TK
thymidine kinase
AUTHOR CONTRIBUTION
Emmanuel Okeke made major contributions to the experimental part of the project and data analyses; Tony Parker, Hayley Dingsdale, Matthew Concannon and Svetlana Voronina made contributions to the experimental part of the project; Muhammad Awais and Emmanuel Okeke developed protocols for IP3 uncaging and imaging focal adhesion dynamics; Emmanuel Okeke, Daniel Metcalf and Alex Knight designed and conducted experiments involving dSTORM; Lee Haynes designed constructs and conducted preliminary experiments; Jordi Molgó advised on IP3R inhibitors and provided xestospongin-B; Malcolm Begg provided GSK-7975A, advised on the properties and application of this compound and contributed to critical discussion of the data; Emmanuel Okeke, Alex E. Knight, Robert Sutton, Lee Haynes and Alexei V. Tepikin designed the project. Emmanuel Okeke, Lee Haynes and Alexei Tepikin wrote the paper.
FUNDING
This work was supported by the Wellcome Trust [grant numbers 092790/Z/10/Z and 092792/Z/10/Z]; the National Institute for Health Research (UK) grant to the NIHR Liverpool Pancreas Biomedical Research Unit; and the Chemical and Biological Metrology programme of the National Measurement System [grant number BA21 (to A.K. and D.M.)].
References
- 1.Siegel R., Naishadham D., Jemal A. Cancer statistics, 2013. CA Cancer J. Clin. 2013;63:11–30. doi: 10.3322/caac.21166. [DOI] [PubMed] [Google Scholar]
- 2.Hanahan D., Weinberg R.A. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 3.Lamouille S., Xu J., Derynck R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014;15:178–196. doi: 10.1038/nrm3758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ridley A.J. Life at the leading edge. Cell. 2011;145:1012–1022. doi: 10.1016/j.cell.2011.06.010. [DOI] [PubMed] [Google Scholar]
- 5.Evans J.H., Falke J.J. Ca2+ influx is an essential component of the positive-feedback loop that maintains leading-edge structure and activity in macrophages. Proc. Natl. Acad. Sci. U.S.A. 2007;104:16176–16181. doi: 10.1073/pnas.0707719104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tsai F.C., Meyer T. Ca2+ pulses control local cycles of lamellipodia retraction and adhesion along the front of migrating cells. Curr. Biol. 2012;22:837–842. doi: 10.1016/j.cub.2012.03.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tsai F.C., Seki A., Yang H.W., Hayer A., Carrasco S., Malmersjo S., Meyer T. A polarized Ca2+, diacylglycerol and STIM1 signalling system regulates directed cell migration. Nat. Cell Biol. 2014;16:133–144. doi: 10.1038/ncb2906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Prevarskaya N., Skryma R., Shuba Y. Calcium in tumour metastasis: new roles for known actors. Nat. Rev. Cancer. 2011;11:609–618. doi: 10.1038/nrc3105. [DOI] [PubMed] [Google Scholar]
- 9.Streb H., Irvine R.F., Berridge M.J., Schulz I. Release of Ca2+ from a nonmitochondrial intracellular store in pancreatic acinar cells by inositol-1,4,5-trisphosphate. Nature. 1983;306:67–69. doi: 10.1038/306067a0. [DOI] [PubMed] [Google Scholar]
- 10.Berridge M.J. Inositol trisphosphate and calcium signalling mechanisms. Biochim. Biophys. Acta. 2009;1793:933–940. doi: 10.1016/j.bbamcr.2008.10.005. [DOI] [PubMed] [Google Scholar]
- 11.Yule D.I. Subtype-specific regulation of inositol 1,4,5-trisphosphate receptors: controlling calcium signals in time and space. J. Gen. Physiol. 2001;117:431–434. doi: 10.1085/jgp.117.5.431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hogan P.G., Lewis R.S., Rao A. Molecular basis of calcium signaling in lymphocytes: STIM and ORAI. Annu. Rev. Immunol. 2010;28:491–533. doi: 10.1146/annurev.immunol.021908.132550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Parekh A.B., Putney J.W., Jr Store-operated calcium channels. Physiol. Rev. 2005;85:757–810. doi: 10.1152/physrev.00057.2003. [DOI] [PubMed] [Google Scholar]
- 14.Lur G., Haynes L.P., Prior I.A., Gerasimenko O.V., Feske S., Petersen O.H., Burgoyne R.D., Tepikin A.V. Ribosome-free terminals of rough ER allow formation of STIM1 puncta and segregation of STIM1 from IP(3) receptors. Curr. Biol. 2009;19:1648–1653. doi: 10.1016/j.cub.2009.07.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Orci L., Ravazzola M., Le Coadic M., Shen W.W., Demaurex N., Cosson P. From the Cover: STIM1-induced precortical and cortical subdomains of the endoplasmic reticulum. Proc. Natl. Acad. Sci. U.S.A. 2009;106:19358–19362. doi: 10.1073/pnas.0911280106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wu M.M., Buchanan J., Luik R.M., Lewis R.S. Ca2+ store depletion causes STIM1 to accumulate in ER regions closely associated with the plasma membrane. J. Cell Biol. 2006;174:803–813. doi: 10.1083/jcb.200604014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lefkimmiatis K., Srikanthan M., Maiellaro I., Moyer M.P., Curci S., Hofer A.M. Store-operated cyclic AMP signalling mediated by STIM1. Nat. Cell Biol. 2009;11:433–442. doi: 10.1038/ncb1850. [DOI] [PubMed] [Google Scholar]
- 18.Willoughby D., Everett K.L., Halls M.L., Pacheco J., Skroblin P., Vaca L., Klussmann E., Cooper D.M. Direct binding between Orai1 and AC8 mediates dynamic interplay between Ca2+ and cAMP signaling. Sci. Signal. 2012;5:ra29. doi: 10.1126/scisignal.2002299. [DOI] [PubMed] [Google Scholar]
- 19.Chang C.L., Hsieh T.S., Yang T.T., Rothberg K.G., Azizoglu D.B., Volk E., Liao J.C., Liou J. Feedback regulation of receptor-induced Ca2+ signaling mediated by E-Syt1 and Nir2 at endoplasmic reticulum-plasma membrane junctions. Cell Rep. 2013;5:813–825. doi: 10.1016/j.celrep.2013.09.038. [DOI] [PubMed] [Google Scholar]
- 20.Kar P., Samanta K., Kramer H., Morris O., Bakowski D., Parekh A.B. Dynamic assembly of a membrane signaling complex enables selective activation of NFAT by Orai1. Curr. Biol. 2014;24:1361–1368. doi: 10.1016/j.cub.2014.04.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Varnai P., Toth B., Toth D.J., Hunyady L., Balla T. Visualization and manipulation of plasma membrane-endoplasmic reticulum contact sites indicates the presence of additional molecular components within the STIM1-Orai1 complex. J. Biol. Chem. 2007;282:29678–29690. doi: 10.1074/jbc.M704339200. [DOI] [PubMed] [Google Scholar]
- 22.Feske S., Gwack Y., Prakriya M., Srikanth S., Puppel S.H., Tanasa B., Hogan P.G., Lewis R.S., Daly M., Rao A. A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function. Nature. 2006;441:179–185. doi: 10.1038/nature04702. [DOI] [PubMed] [Google Scholar]
- 23.Liou J., Kim M.L., Heo W.D., Jones J.T., Myers J.W., Ferrell J.E., Jr, Meyer T. STIM is a Ca2+ sensor essential for Ca2+-store-depletion-triggered Ca2+ influx. Curr. Biol. 2005;15:1235–1241. doi: 10.1016/j.cub.2005.05.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Park C.Y., Hoover P.J., Mullins F.M., Bachhawat P., Covington E.D., Raunser S., Walz T., Garcia K.C., Dolmetsch R.E., Lewis R.S. STIM1 clusters and activates CRAC channels via direct binding of a cytosolic domain to Orai1. Cell. 2009;136:876–890. doi: 10.1016/j.cell.2009.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Roos J., DiGregorio P.J., Yeromin A.V., Ohlsen K., Lioudyno M., Zhang S., Safrina O., Kozak J.A., Wagner S.L., Cahalan M.D., et al. STIM1, an essential and conserved component of store-operated Ca2+ channel function. J. Cell Biol. 2005;169:435–445. doi: 10.1083/jcb.200502019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yuan J.P., Zeng W., Dorwart M.R., Choi Y.J., Worley P.F., Muallem S. SOAR and the polybasic STIM1 domains gate and regulate Orai channels. Nat. Cell Biol. 2009;11:337–343. doi: 10.1038/ncb1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Worley P.F., Zeng W., Huang G.N., Yuan J.P., Kim J.Y., Lee M.G., Muallem S. TRPC channels as STIM1-regulated store-operated channels. Cell Calcium. 2007;42:205–211. doi: 10.1016/j.ceca.2007.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mogami H., Gardner J., Gerasimenko O.V., Camello P., Petersen O.H., Tepikin A.V. Calcium binding capacity of the cytosol and endoplasmic reticulum of mouse pancreatic acinar cells. J. Physiol. 1999;518:463–467. doi: 10.1111/j.1469-7793.1999.0463p.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhou Z., Neher E. Mobile and immobile calcium buffers in bovine adrenal chromaffin cells. J. Physiol. 1993;469:245–273. doi: 10.1113/jphysiol.1993.sp019813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dingli F., Parys J.B., Loew D., Saule S., Mery L. Vimentin and the K-Ras-induced actin-binding protein control inositol-(1,4,5)-trisphosphate receptor redistribution during MDCK cell differentiation. J. Cell Sci. 2012;125:5428–5440. doi: 10.1242/jcs.108738. [DOI] [PubMed] [Google Scholar]
- 31.Akerboom J., Chen T.W., Wardill T.J., Tian L., Marvin J.S., Mutlu S., Calderon N.C., Esposti F., Borghuis B.G., Sun X.R., et al. Optimization of a GCaMP calcium indicator for neural activity imaging. J. Neurosci. 2012;32:13819–13840. doi: 10.1523/JNEUROSCI.2601-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Laukaitis C.M., Webb D.J., Donais K., Horwitz A.F. Differential dynamics of alpha 5 integrin, paxillin, and alpha-actinin during formation and disassembly of adhesions in migrating cells. J. Cell Biol. 2001;153:1427–1440. doi: 10.1083/jcb.153.7.1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dingsdale H., Okeke E., Awais M., Haynes L., Criddle D.N., Sutton R., Tepikin A.V. Saltatory formation, sliding and dissolution of ER-PM junctions in migrating cancer cells. Biochem. J. 2013;451:25–32. doi: 10.1042/BJ20121864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Burdyga A., Conant A., Haynes L., Zhang J., Jalink K., Sutton R., Neoptolemos J., Costello E., Tepikin A. cAMP inhibits migration, ruffling and paxillin accumulation in focal adhesions of pancreatic ductal adenocarcinoma cells: effects of PKA and EPAC. Biochim. Biophys. Acta. 2013;1833:2664–2672. doi: 10.1016/j.bbamcr.2013.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Metcalf D.J., Edwards R., Kumarswami N., Knight A.E. Test samples for optimizing STORM super-resolution microscopy. J. Vis. Exp. 2013:e50579. doi: 10.3791/50579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ferraro F., Kriston-Vizi J., Metcalf D.J., Martin-Martin B., Freeman J., Burden J.J., Westmoreland D., Dyer C.E., Knight A.E., Ketteler R., Cutler D.F. A two-tier Golgi-based control of organelle size underpins the functional plasticity of endothelial cells. Dev. Cell. 2014;29:292–304. doi: 10.1016/j.devcel.2014.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rees E., Erdelyi M., Pinotsi D., Knight A.E., Metcalf D., Kaminski C.F. Blind assessment of localisation microscope image resolution. Optical Nanoscopy. 2012;1:12. doi: 10.1186/2192-2853-1-12. [DOI] [Google Scholar]
- 38.Rees E.J., Miklos Erdelyi M., Kaminski Schierle G.S., Knight A., Kaminski C.F. Elements of image processing in localization microscopy. J. Optics. 2013;15:094012. doi: 10.1088/2040-8978/15/9/094012. [DOI] [Google Scholar]
- 39.Colosetti P., Tunwell R.E., Cruttwell C., Arsanto J.P., Mauger J.P., Cassio D. The type 3 inositol 1,4,5-trisphosphate receptor is concentrated at the tight junction level in polarized MDCK cells. J. Cell Sci. 2003;116:2791–2803. doi: 10.1242/jcs.00482. [DOI] [PubMed] [Google Scholar]
- 40.Horiguchi K., Shirakihara T., Nakano A., Imamura T., Miyazono K., Saitoh M. Role of Ras signaling in the induction of snail by transforming growth factor-beta. J. Biol. Chem. 2009;284:245–253. doi: 10.1074/jbc.M804777200. [DOI] [PubMed] [Google Scholar]
- 41.Nakajima S., Doi R., Toyoda E., Tsuji S., Wada M., Koizumi M., Tulachan S.S., Ito D., Kami K., Mori T., et al. N-cadherin expression and epithelial-mesenchymal transition in pancreatic carcinoma. Clin. Cancer Res. 2004;10:4125–4133. doi: 10.1158/1078-0432.CCR-0578-03. [DOI] [PubMed] [Google Scholar]
- 42.Deakin N.O., Turner C.E. Paxillin comes of age. J. Cell Sci. 2008;121:2435–2444. doi: 10.1242/jcs.018044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kim D.H., Wirtz D. Focal adhesion size uniquely predicts cell migration. FASEB J. 2013;27:1351–1361. doi: 10.1096/fj.12-220160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Carrasco S., Meyer T. STIM proteins and the endoplasmic reticulum-plasma membrane junctions. Annu. Rev. Biochem. 2011;80:973–1000. doi: 10.1146/annurev-biochem-061609-165311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nicol X., Hong K.P., Spitzer N.C. Spatial and temporal second messenger codes for growth cone turning. Proc. Natl. Acad. Sci. U.S.A. 2011;108:13776–13781. doi: 10.1073/pnas.1100247108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wei C., Wang X., Chen M., Ouyang K., Song L.S., Cheng H. Calcium flickers steer cell migration. Nature. 2009;457:901–905. doi: 10.1038/nature07577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Motiani R.K., Hyzinski-Garcia M.C., Zhang X.X., Henkel M.M., Abdullaev I.F., Kuo Y.H., Matrougui K., Mongin A.A., Trebak M. STIM1 and Orai1 mediate CRAC channel activity and are essential for human glioblastoma invasion. Pflugers Arch. 2013;465:1249–1260. doi: 10.1007/s00424-013-1254-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jaimovich E., Mattei C., Liberona J.L., Cardenas C., Estrada M., Barbier J., Debitus C., Laurent D., Molgó J. Xestospongin B, a competitive inhibitor of IP3-mediated Ca2+ signalling in cultured rat myotubes, isolated myonuclei, and neuroblastoma (NG108–15) cells. FEBS Lett. 2005;579:2051–2057. doi: 10.1016/j.febslet.2005.02.053. [DOI] [PubMed] [Google Scholar]
- 49.Derler I., Schindl R., Fritsch R., Heftberger P., Riedl M.C., Begg M., House D., Romanin C. The action of selective CRAC channel blockers is affected by the Orai pore geometry. Cell Calcium. 2013;53:139–151. doi: 10.1016/j.ceca.2012.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lee M.G., Xu X., Zeng W.Z., Diaz J., Wojcikiewicz R.J. H., Kuo T.H., Wuytack F., Racymaekers L., Muallem S. Polarized expression of Ca2+ channels in pancreatic and salivary gland cells–correlation with initiation and propagation of [Ca2+](i) waves. J. Biol. Chem. 1997;272:15765–15770. doi: 10.1074/jbc.272.25.15765. [DOI] [PubMed] [Google Scholar]
- 51.Nathanson M.H., Fallon M.B., Padfield P.J., Maranto A.R. Localization of the type-3 inositol 1,4,5-trisphosphate receptor in the Ca2+ wave trigger zone of pancreatic acinar-cells. J. Biol. Chem. 1994;269:4693–4696. [PubMed] [Google Scholar]
- 52.Yule D.I., Ernst S.A., Ohnishi H., Wojcikiewicz R.J. H. Evidence that zymogen granules are not a physiologically relevant calcium pool–defining the distribution of inositol 1,4,5-trisphosphate receptors in pancreatic acinar cells. J. Biol. Chem. 1997;272:9093–9098. doi: 10.1074/jbc.272.14.9093. [DOI] [PubMed] [Google Scholar]
- 53.Lur G., Sherwood M.W., Ebisui E., Haynes L., Feske S., Sutton R., Burgoyne R.D., Mikoshiba K., Petersen O.H., Tepikin A.V. InsP(3) receptors and Orai channels in pancreatic acinar cells: co-localization and its consequences. Biochem. J. 2011;436:231–239. doi: 10.1042/BJ20110083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rooman I., Real F.X. Pancreatic ductal adenocarcinoma and acinar cells: a matter of differentiation and development? Gut. 2012;61:449–458. doi: 10.1136/gut.2010.235804. [DOI] [PubMed] [Google Scholar]