

Abstract
Aims
Recent studies have demonstrated RAMP ®, a complete starter feed, to have beneficial effects for animal performance. However, how RAMP may elicit such responses is unknown. To understand if RAMP adaptation results in changes in the rumen bacterial community that can potentially affect animal performance, we investigated the dynamics of rumen bacterial community composition in corn‐adapted and RAMP‐adapted cattle.
Methods and Results
During gradual acclimation of the rumen bacterial communities, we compared the bacterial community dynamics in corn and RAMP‐adapted using 16S rRNA gene amplicon sequencing. Significant shifts in bacterial populations across diets were identified. The shift in corn‐adapted animals occurred between adaptation step3 and step4, whereas in RAMP‐adapted cattle, the shift occurred between step2 and step3. As the adaptation program progressed, the abundance of OTUs associated with family Prevotellaceae and S24‐7 changed in corn‐adapted animals. In RAMP‐adapted animals, OTUs belonging to family Ruminococcaceae and Lachnospiraceae changed in abundance.
Conclusions
Rumen bacteria can be acclimated faster to high concentrate diets, such as RAMP, than traditional adaptation programs and the speed of bacterial community acclimation depends on substrate composition.
Significance and Impact of the Study
These findings may have implications for beef producers to reduce feedlot costs, as less time adapting animals would result in lower feed costs. However, animal feeding behavior patterns and other factors must be considered.
Keywords: 16S rRNA, bacteria, microbial community, RAMP, rumen microbiology
Introduction
Complex and diverse microbial communities mediate the cycling of nutrients within the ruminant gut. The composition of this complex microbial community is shaped by the physical, chemical, and predatory environment and potentially by genetic factors of the host (Hungate 1966; Church 1993; Benson et al. 2010; Spor et al. 2011; Carlisle et al. 2013; McKnite et al. 2012; Schluter and Foster 2012). In turn, the microbial community regulates the environment and nutrient availability, including the release of energy to the host in the form of volatile fatty acids (VFAs) (Church 1993). The diversity of the rumen microbial community is both ecologically and biochemically important for ruminant health and production. It is estimated that 80% of the animal's energy needs and 50% of the animal's protein needs are obtained through microbial digestion of the feed and microbial cell protein (Doi and Kosugi 2004). However, dysbiosis of this complex microbial ecosystem can have devastating consequences for the host. When the rumen microbial population is exposed to large amounts of highly fermentable substrates over a short period of time, the rate of VFA production will exceed the rate of VFA absorption leading to ruminal acidosis. Ruminal acidosis can cause damage to the rumen epithelium and lead to poor animal performance due to decreased VFA absorption (Owens et al. 1998; Bevans et al. 2005; Nagaraja and Lechtenberg 2007; Nagaraja and Titgemeyer 2007; Mao et al. 2013; Sato 2015). To overcome this metabolic disorder, feeding programs are implemented in the feedlot industry to gradually adapt cattle from a high forage diet to a high concentrate diet by progressively increasing the proportion of grain and decreasing the amount of roughage (Vasconcelos and Galyean 2007). As an alternative to the traditional adaptation, Sweet Bran® (Cargill Corn Milling, Blair, NE) has been effectively used to adapt cattle to high grain diets with improved average daily gain (ADG) and gain/feed (G/F) (Huls et al. 2009). Based on this premise, a complete starter feed called RAMP® (Cargill Corn Milling, Blair, NE), with a high level of Sweet Bran and a minimal amount of forage, has been developed to rapidly adapt feedlot cattle to high concentrate diets. RAMP is intended to serve as an alternative to a mixture of grain and forage in order to eliminate a significant portion of the forage needed in feedlots, in turn reducing feed costs for producers. Research using RAMP for grain adaptation has shown an increase in hot carcass weight (HCW ‐ weight immediately after slaughter) compared to cattle adapted with traditional grain adaptation methods (Buttrey et al. 2012; MacDonald and Luebbe 2012). Although these studies have shown beneficial effects for animal performance and HCW, there are no reports demonstrating how RAMP may elicit such responses. One hypothesis is RAMP‐adaptation may change the rumen microbial community by selecting certain populations of microbes, which may have a positive impact on performance. However, only a few studies have investigated microbial community changes during traditional adaptation programs (Tajima et al. 2001a,b; Nagaraja and Titgemeyer 2007; Fernando et al. 2010; Sun et al. 2010), and no study has evaluated the impact of feeding RAMP on microbial community composition. Therefore, the influence of gradual adaptation and inclusion of RAMP on microbial community composition and in turn animal performance are poorly understood.
Previous investigations of traditional adaptation strategies have detected significant changes in ruminal fermentation (pH and VFA profiles) and microbial community composition, but fail to identify core bacterial communities that emerge during adaptation due to sequencing depth and community analysis methodology (Tajima et al. 2001a,b; Bevans et al. 2005; Nagaraja and Titgemeyer 2007; Sun et al. 2010). In addition, although diet is a major factor shaping the rumen microbial community (Fernando et al. 2010), how fast the community acclimates or can be acclimated to a high concentrate diet is unknown. Understanding strategies to adapt cattle faster would result in significant savings to the producer. Here, we utilize 16S rRNA gene amplicon sequencing as a first step towards understanding rumen bacterial community dynamics during grain adaptation and the effect of feeding RAMP on rumen bacterial community composition.
Materials and methods
Experimental design and dietary formulations
All animal care and management procedures were approved by the University of Nebraska‐Lincoln Institutional Animal Care and Use Committee. Six ruminally fistulated steer calves (BW = 255 ± 30 kg, fistulated at the age of 10 months and were 18 months old at the time of study) on a 35 day metabolism trial were utilized to evaluate the effects of different grain adaptation strategies on rumen bacterial community composition. Adaptation programs included a corn‐based control and RAMP (Tables S1‐1 and S1‐2). Diets for each period were mixed in a stationary ribbon mixer (Model S‐5 Mixer; H.C. Davis Inc, Bonner Springs, KS). Mixed feed and wet feed ingredients were stored in a walk‐in 4°C cooler to maintain freshness until feeding. Steers were individually housed in box stalls and fed once daily at 08:00 h with ad libitum access to feed and water. Feed refusals were collected daily, weighed, and a 10% representative sample was retained and dried in a forced‐air oven at 60°C for 48 h to obtain dry matter intake (DMI).
Before the start of the study, steers were maintained on a common diet for 2 weeks to help establish a similar rumen microbial community composition within all animals. Following the common diet, steers were fed each step‐up diet for 7 days before transitioning to the next step of the adaptation program. Following adaptation, all animals were fed a common finishing diet for 7 days, resulting in 5 periods over 35 days. On day 7 of each period, total rumen contents were collected 6 h after feeding for bacterial community analysis. During the trial, an animal on the corn‐adaptation program had abnormally low intake (~50% of other treatment animals) during periods 1–3 and was removed from the study. Reduced feed intake was determined to be due to pre‐existing health problems. For this reason, data collected from this animal were not included in the analysis. Therefore, the data presented are from five animals (two on corn‐based adaptation and three on RAMP‐adaptation).
Wireless, submersible pH probes (Dascor Inc, Escondido, CA) were placed into the rumen of each steer to monitor ruminal pH. Each ruminal pH probe was weighted to ensure the electrode remained in the ventral sac of the rumen. Before trial initiation, each pH probe was calibrated by submersing probes in pH 4 and 7 standard solutions. Ruminal pH was recorded every min for each period. Prior to feeding the next adaptation diet, the pH probes were removed from the rumen for approximately 2 h to download pH data and recalibrate the probes. Ruminal pH measurements from each period were adjusted using beginning and ending calibration values to ensure accurate pH measurements. Ruminal pH variance, time below pH of 5·6, and area below pH of 5·6 were calculated as described previously (Cooper et al. 1999; Erickson et al. 2003; Vander Pol et al. 2009).
Rumen sampling and DNA isolation
On day 7 of each period, total rumen contents (partially digested feed particles (solid) and rumen fluid) were collected from each animal through the rumen fistula 6 h after feeding. Briefly, total rumen contents were removed, mixed, and a homogeneous sample was collected for bacterial community analysis after 7 days of acclimation to each diet. Collected samples were snap frozen in liquid nitrogen and stored at −80°C until DNA extraction. DNA was extracted from 0·5 g of rumen sample (representing both the solid and liquid fraction) using the QIAamp DNA stool kit (Qiagen, Valencia, CA) according to the manufacture's protocol with a few modifications as described previously (Fernando et al. 2010). The isolated DNA was stored at −80°C until used for 16S rRNA gene amplification.
16S rRNA library preparation and sequencing of the V1–V3 region
The V1–V3 region of the 16S rRNA gene specific for eubacterial communities was amplified from extracted total rumen DNA using universal primers 27F and 518R (Rosenzweig and Ragsdale 2011). The designed primers contained Roche 454 A and B sequencing adaptors at the 5′ end followed by a five nucleotide key and the universal 16S rRNA gene primer sequence at the 3′ end (Martinez et al. 2009).
The V1–V3 region of the 16S rRNA gene was amplified in a 25 μl volume. A PCR reaction contained 0·5 Units of Phusion DNA polymerase (Finnzymes Inc., Woburn, MA), 1× reaction buffer, 200 μmol l−1 dNTPs (Invitrogen, Carlsbad, CA), 200 nmol l−1 of each primer, and 20–50 ng of nucleic acid template or no‐template control. The following cycling conditions were used for PCR amplification of the 16S rRNA gene: an initial denaturation of 98°C for 3 min; 30 cycles of 98°C for 30 s, 52°C for 30 s, and 72°C for 1 min; and a final extension of 72°C for 2 min. Following amplification, PCR products were analysed on a 1·8% agarose gel to confirm correct product size and estimate quantity for multiplexing. The amount of PCR product amplified was measured by evaluating the band density using genetools automated image analysis software (Syngene, Frederrick, MD). The pooled 16S rRNA gene library was gel purified (QIAquick Gel Extraction Kit; Qiagen) and quality and concentration were evaluated using the Bioanalyzer (Agilent, Palo Alto, CA). Following amplification, pooling, and quality control of the V1–V3 16S rRNA libraries, sequencing was performed on a 454 pyrosequencer in the 5′‐V3‐V1‐3′ direction (454 Life Sciences, Branford, CT). Emulsion PCR, bead recovery, bead deposition, and sequencing were done as described by the manufacturer.
Data processing and microbial community analysis
Raw reads from 454 pyrosequencing were subjected to initial quality filtering. Briefly, reads not matching the following criteria were removed: (i) having a complete forward primer and barcode; (ii) no ambiguous bases (‘N’); (iii) sequence length between 200 nt and 500 nt; (iv) average quality score ≥20. The resulting reads and an R markdown file to reproduce the following analysis is available at https://github.com/FernandoLab/. Additional quality filter steps to remove sequences with two or more errors in the barcode, more than one error in the forward primer, and more than two errors in the reverse primer were applied during demultiplexing with qiime (ver. 1.9.0) (Caporaso et al. 2010). Subsequently, mothur (v.1.35.1) (Schloss et al. 2009) and the fastx‐toolkit (http://hannonlab.cshl.edu/fastx_toolkit/) were used to remove sequences <400 bp and trim remaining sequences to a fixed length of 400 bp, respectively, to improve operational taxonomic unit (OTU) picking (Edgar 2013). OTUs were identified by clustering reverse complemented sequences based on 97% similarity according to the UPARSE pipeline with both de novo and reference based chimera removal of nonsingleton reads (Edgar 2013). OTU clustering and all subsequent analyses were performed on the combined data set, which included samples from both RAMP‐adapted and corn‐adapted animals.
Representative sequences for each OTU were assigned taxonomy with the ‘mothur’ assignment method trained on the greengenes database (ver. 13_8) (McDonald et al. 2012) using qiime. Due to the large unknown bacterial diversity in the rumen and the fragment length (400 bp), only a portion of the OTUs could be classified at the genus level (44·3% of OTUs, 62·7% of the reads). However, a majority of the reads were classified at family level (66·5% of OTUs, 83·6% of total reads). Therefore, results are discussed at family level except in the case of Prevotellaceae where nearly all Prevotellaceae OTUs were classified to the genus Prevotella. OTUs with phylum‐level classification to Cyanobacteria were removed as no photosynthesis occurs in the rumen and these assignments are likely due to the high homology of plant chloroplast rRNA to the Cyanobacteria 16S rRNA gene. The Chao1 index and the number of observed OTUs were used to calculate rarefaction curves. Representative sequences of each OTU were aligned using the silva database (release 119) within mothur. The resulting alignment was used to generate a phylogenetic tree with clearcut (Sheneman et al. 2006). Shifts in the rumen bacterial communities were visualized using principle coordinate analysis based on unweighted UniFrac distances calculated from subsampled OTU tables (Hamady et al. 2010; Lozupone et al. 2011).
Statistical analyses
Data for ruminal pH and DMI were analysed separately within each period using the glimmix procedure of SAS. All data were analysed using a repeated measures analysis with ‘day’ repeated. An autoregressive‐1 (AR‐1) covariance structure was selected for the model. The model included day and treatment as fixed effects and animal nested within treatment was considered a random effect. A Kenward‐Rogers denominator degrees of freedom adjustment was utilized.
To estimate sample coverage, we calculated the Good's coverage for each sample by subsampling to an equal sequencing depth (subsampling was done to the sample with the lowest number of reads (2160 reads)). The mean Good's coverage for 10 different subsampling events is reported. Global changes in bacterial community composition within each adaptation strategy were evaluated using unweighted UniFrac distance matrices as input for a two‐way permanova where animal and diet were used as main effects (adonis function of the vegan package (Dixon 2003) in r (Team 2007)). The unweighted UniFrac distance matrices were computed from subsampled OTU tables at a depth of 2160 reads. Throughout the study, P‐values <0·05 were used to determine significance. Mann–Whitney U tests (R function wilcox.test) were utilized as a post hoc test to identify community shifts between two subsequent steps within an adaptation strategy. This was performed by comparing all pairwise distances relative to step1 (e.g. step1‐step2 distances compared to step1‐step3 distances to test for a shift in bacterial community composition from step2 to step3). Box and whisker plots for all pairwise distances were generated to visualize bacterial community shifts (Wickham 2009). Changes in OTU relative abundance that occurred during significant shifts in bacterial community composition were identified using LEfSe, which implements a Kruskal–Wallis sum‐rank test followed by linear discriminate analysis to identify significantly different features with biological relevance (Segata et al. 2011). For this analysis, all bacterial communities on either side of the community shift were compared to identify OTUs with a significant change in abundance. For instance, in corn‐adapted animals, we identified a single shift in community composition between step3 and step4 and tested for OTU‐level differences between step1‐step2‐step3 and step4‐Finisher.
Heatmap generation and shared OTU calculations
All heatmaps were generated in r using the heatmap.2 function (Ploner 2014; Warnes et al. 2015) with OTU relative abundance as input. Bray‐Curtis dissimilarity was used to compute the distance between rows or columns and hierarchical clustering (average linkage) to generate the dendrograms. All Venn diagrams were constructed in r using the venn function in the gplots package of r. The percentage of shared OTUs was calculated using the lowest total OTU count among the compared samples (as this would represent the maximum number of OTUs that could be shared).
Results
DMI and pH
A numerical increase in DMI was observed in RAMP‐adapted animals compared to corn‐adapted animals (Fig. S1), but no significant increase in DMI was detected over the 35 day study (P > 0·05). In addition, no differences were observed within any adaptation step for average ruminal pH, minimum pH, or maximum pH (P > 0·05). Ruminal pH change and variance were similar between treatments for all periods. No differences were observed among treatments during any period for time below pH 5·6 and area below pH 5·6 (P > 0·05) (Fig. S1).
Rumen bacterial community composition and species richness
As the rumen microbial community is poorly understood and with relatively few reference rumen microbial genomes available, we employed a database independent, OTU‐based approach to analyse the rumen bacterial communities. 272 630 sequences passing quality control from 25 samples were clustered into 1084 OTUs. Rarefaction curves were generated for each step within RAMP and corn‐based adaptation programs (Fig. S2). The rarefaction curves did not plateau, but display a diminishing rate of identifying new OTUs with the addition of new sequence reads. To ensure the sampling effort used was adequate to investigate global community and species‐level changes across the two adaptation strategies, we performed a Good's coverage test which revealed the lowest estimate to be 0·951 (suggesting that the sequencing depth used in this study would enable us to characterize ~95% of the rumen bacterial population). Corn‐based adaptation displayed high bacterial diversity in step1, step2, and step3, whereas, there was a decrease in diversity in step4 and finisher (Fig. S2a). However, in RAMP‐adapted animals, bacterial diversity remained consistent throughout the adaptation program. The bacterial diversity in RAMP‐adapted animals was similar to the bacterial diversity observed in step4 and finisher of corn‐adapted animals (Fig. S2a). To better understand the variation in microbial community diversity at each step of adaptation, the Chao1 index and the number of OTUs observed at an even sampling depth were plotted for each adaptation step (Fig. S2b). This clearly demonstrates the large shift in bacterial diversity in corn‐adapted animals after adaptation step3 is due to a change in dietary substrate and not the result of an outlying animal.
Taxonomic analysis of representative OTU sequences detected 15 (9 with a maximum relative abundance >1%) phyla within the rumen from RAMP‐adapted and corn‐adapted animals. Bacteroidetes and Firmicutes were the most abundant phyla regardless of adaptation program or stage of adaptation.
Global shifts in the rumen bacterial community during adaptation
Changes in bacterial community composition during adaptation to high concentrate diets were evaluated using unweighted UniFrac distances (see methods for details). In both cases, permanova analysis displayed a high F‐statistic (P < 0·05) for the dietary factor, suggesting a significant change in bacterial community composition during adaptation to high concentrate diets (Fig. 1a). To identify significant changes in the bacterial community between adaptation steps, we compared unweighted UniFrac distances between subsequent adaptation diets relative to adaptation step1 (see methods for details). In the traditional corn‐based adaption, a single shift in the bacterial community was detected between adaptation step3 and step4 (P < 0·05). Whereas, adaptation step1, step2, and step3 displayed similar bacterial community composition (P > 0·05). No differences between the bacterial communities of step4 and finisher of corn‐based adaptation were identified (P > 0·05) (Fig. 1a,b). This was consistent with the decrease in alpha diversity observed during corn‐adaptation between step3 and step4 (Fig. S2). Similar comparisons of RAMP‐adapted animals revealed two significant shifts in bacterial community composition. Community shifts were detected between adaptation step1 and step2 (P < 0·05) and between step2 and step3 (P < 0·05). The microbial communities in RAMP‐adapted animals were similar for adaptation step3, step4, and finisher (P > 0·05) (Fig. 1a,c).
Figure 1.
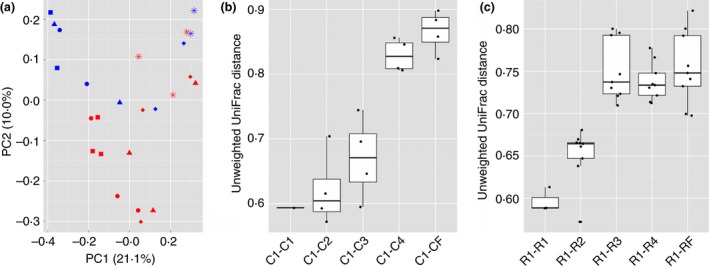
Principle coordinate analysis and unweighted UniFrac distances relative to step1 diet. Principle coordinate analysis of unweighted UniFrac distances between four step‐up diets and the finisher diet during corn‐adaptation and RAMP‐adaptation programs (a). Unweighted UniFrac distances relative to step1 were used to identify shifts in bacterial community composition between subsequent steps (compared using Mann–Whitney U tests) in the adaptation programs. A single shift in the community composition was identified between step3 and step4 during corn‐based adaptation (b) while two earlier shifts, step1 to step2 and from step2 to step3, were detected during RAMP‐adaptation (c). C1–C4 represents adaptation steps during corn‐based adaptation; R1–R4 represents adaptation steps during RAMP‐based adaptation. CF and RF represent corn‐ and RAMP‐adapted animals on the same finisher diet, respectively. ( ), C1; (
), C1; ( ), C2; (
), C2; ( ), C3; (
), C3; ( ), C4; (
), C4; ( ), CF; (
), CF; ( ), R1; (
), R1; ( ), R2; (
), R2; ( ), R3; (
), R3; ( ), R4; (
), R4; ( ), RF.
), RF.
To visualize the shifts in bacterial community composition identified above, all OTUs with a maximum relative abundance >1% were subjected to hierarchical clustering (Fig. 2). Hierarchical clustering of OTUs produced two main clusters. One of the clusters contained bacterial communities from step1, step2, and step3 of corn‐adapted animals, as well as, step1 and step2 from RAMP‐adapted animals. The other cluster contained bacterial communities from step4 and finisher diets of corn‐adapted animals. Additionally, the second cluster consisted of bacterial communities from step3, step4, and finisher diets from RAMP‐adapted animals. Bacterial communities collected from the finisher diets of the two adaptation programs clustered together, as expected, due to the identical composition of the diets.
Figure 2.
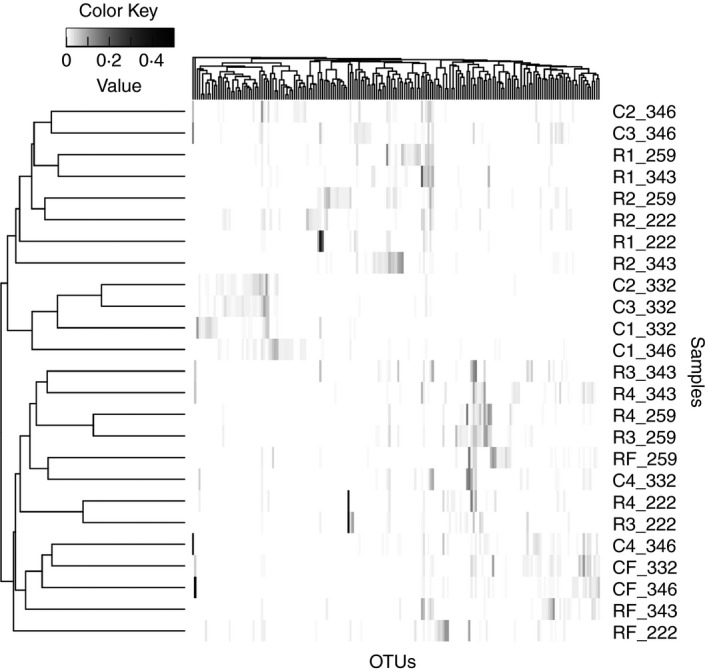
Hierarchical clustering revealed two groupings of samples from corn‐adapted and RAMP‐adapted step‐up diets. One cluster contains samples prior to the shift in bacterial community composition during corn‐adaptation (step1, step2, and step3) along with step1 and step2 of RAMP‐adaptation. The second cluster contains ‘acclimated’ communities from step4 and finisher of corn‐adapted animals and step3, step4, and finisher of RAMP‐adapted animals. Only operational taxonomic units (OTUs) with a maximum relative abundance >1% are shown. Samples are labeled with their step‐up diets followed by an underscore and the animal identifier. C1–C4 represents adaptation steps during corn‐based adaptation; R1–R4 represents adaptation steps during RAMP‐based adaptation. CF and RF represent samples from the same finisher diet.
OTU‐level changes in the rumen bacterial community during adaptation
We utilized LEfSe to identify changes in OTU abundance before and after significant shifts in bacterial community composition. In corn‐adapted animals, 74 OTUs were identified as significantly higher in bacterial communities from adaptation step1‐step2‐step3, while an additional 46 OTUs were significantly higher in adaptation step4‐finisher communities (Fig. 3a and Table S2‐1). A majority of the OTUs with a high abundance in step1‐step2‐step3 belonged to family S24‐7 or were unclassified, while OTUs in high abundance in step4‐finisher belonged to family Prevotellaceae.
Figure 3.

Heatmaps for operational taxonomic units (OTUs) identified as having a significantly different abundance prior to or after an identified bacterial community shift during corn‐based (a) and RAMP‐based adaptation (b, c) to high concentrate diets. OTUs are ordered by decreasing linear discriminant analysis score (as calculated by LEfSe). Only OTUs with a maximum relative abundance >1% are shown. Samples are labelled with their step‐up diets followed by an underscore and the animal identifier. C1–C4 represents adaptation steps during corn‐based adaptation; R1–R4 represents adaptation steps during RAMP‐based adaptation. CF and RF represent samples from the same finisher diet.
In RAMP‐adapted animals, two significant shifts in the bacterial community were identified. Relative to step2, 24 OTUs were detected in increased abundances adaptation step1. Family‐level classification of these OTUs revealed the predominance of Ruminococcaceae and Lachnospiraceae populations (Fig. 3b and Table S2‐2). Similarly, relative to step1, 13 OTUs were significantly higher in abundance in adaptation step2 and many of these OTUs belonged to family Prevotellaceae. Change in bacterial populations from adaptation step2 to step3‐step 4‐finisher in RAMP‐adapted animals was associated with 107 OTUs that were in higher abundance in step 2, many of which belonged to families Lachnospiraceae, and Prevotellaceae (Fig. 3c and Table S2‐3). Only 11 OTUs were detected in increased abundances in communities from adaptation step3‐step4‐finisher compared to step2. A majority of these OTUs belonged to family Prevotellaceae (Fig. 3c and Table S2‐3).
Distribution of shared OTUs and sequences during adaptation
To understand how microbial communities acclimate to a high concentrate diet, we evaluated the number of OTUs shared between each stage of the adaptation programs (Fig. 4 and Table S4). Thirty‐three (14·3%) OTUs were shared between all bacterial communities from corn‐adapted animals. Step1 of corn‐based adaptation shared 299 (62·8%), 304 (63·9%), 113 (35·8%), and 50 (21·7%) OTUs with step2, step3, step4, and finisher, respectively. Adaptation step2 shared 376 (76·9%), 173 (54·7%), and 103 (44·8%) OTUs with step3, step4, and finisher, respectively. Adaptation step3 shared 202 (63·9%) and 124 (53·9%) OTUs with step4 and finisher, respectively. When looking across identified bacterial community shifts during corn‐based adaptation, step1, step2, and step3 shared 249 (52·3%) OTUs, while step4 and finisher shared 161 (70·0%) OTUs. To emphasize the shift in bacterial community composition between step3 and step4 during corn‐based adaptation, we compared sequences shared between adaptation step1 and step3 to sequences shared between step1 and step4 (Table S3). Between step1 and step3, 70·1% of the sequences were shared, whereas only 30·7% of the sequences were shared between adaptation step1 and step4, clearly demonstrating that a change in the bacterial community occurred between step3 and step4 during corn‐adaptation.
Figure 4.
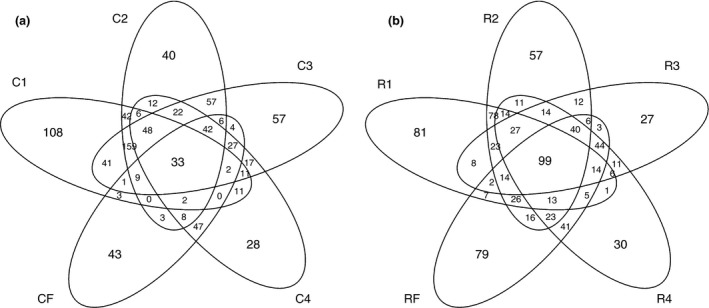
Venn diagrams displaying the distribution of operational taxonomic units (OTUs) among step‐up diets during corn‐adaptation (a) and RAMP‐adaptation (b) to high concentrate diets. C1–C4 represents adaptation steps during corn‐based adaptation; R1–R4 represents adaptation steps during RAMP‐based adaptation. CF and RF represent samples from the same finisher diet.
In RAMP‐adapted animals, 99 (28·3%) OTUs were shared between all four step‐up diets and the finisher diet. Step1 of RAMP‐adaptation shared 294 (70·3%), 193 (55·1%), 179 (45·5%), and 180 (43·1%) OTUs with adaptation step2, step3, step4, and finisher, respectively. Adaptation step2 shared 235 (67·1%), 241 (61·3%), and 237 (54·9%) OTUs with step3, step4, and finisher, respectively. Step3 shared 255 (72·9%) and 222 (63·4%) OTUs with step4 and finisher, respectively. Step4 and finisher shared 279 (71·0%) OTUs. Adaptation step3, step4, and finisher shared 197 OTUs (56·3%). In addition, adaptation step1, step2 and step3 shared 163 (46·6%) OTUs. Shared sequences between each stage in both corn‐adapted and RAMP‐adapted animals show a similar pattern to the proportion of OTUs shared (Table S3).
To investigate how bacterial populations from two different adaptation methods differ when progressing towards a common finisher diet, we looked at shared OTUs and sequences between the same stage of the step‐up regime in each adaptation program. More OTUs and sequences were shared as sampling progressed further along in the adaptation program (Table S4). At step1 of adaptation, RAMP‐adapted and corn‐adapted animals shared 221 (52·9%) OTUs which represented 63·4% of the sequences. In step2, 282 OTUs were shared (59·6%) which represented 72·3% of the sequences. In step3, step4, and finisher, 210 (60·0%), 215 (68·0%), and 207 (90·0%) OTUs were shared which represented 68·8%, 87·8%, and 81·2% of the sequences, respectively.
Discussion
Adapting cattle to high concentrate diets is a common practice in the beef cattle industry to increase ADG while minimizing the risk of rumen acidosis (Bevans et al. 2005; Nagaraja and Titgemeyer 2007; Sun et al. 2010). During this adaptation phase, the breakdown of readily fermentable substrates by the rumen microbial community leads to many changes in the rumen, including changes in microbial community composition, decreased pH, and increased VFA concentrations (Fernando et al. 2010; Sun et al. 2010). Therefore, adaptation programs are critical to decrease the risk of ruminal acidosis while maintaining or increasing animal performance. The changes in bacterial species composition during adaptation help maintain pH while increasing feed efficiency. Studies have evaluated bacterial community composition changes during adaptation to high‐concentrate diets, however, these investigations fail to identify core bacterial communities that emerge during adaptation (Tajima et al. 2001a,b; Nagaraja and Titgemeyer 2007; Sun et al. 2010). In addition, how fast the community adapts or can be adapted to a high concentrate diet is unknown. Therefore, the objective of this study was to understand and compare the shifts in the rumen bacterial community composition during adaptation within and between a traditional corn‐based adaptation and using a complete starter ration (RAMP).
There was no significant difference in DMI in RAMP‐adapted and corn‐adapted animals, suggesting intake was not affected and did not impact microbial community composition. Therefore, changes in the bacterial community detected could be due to the differences in diet composition. This was also supported by the pH data, where no significant differences were identified between the two adaptation programs (Fig. S1).
Alpha diversity estimates show that with increasing level of concentrate in the diet, the diversity of bacterial communities decreased (Fig. S2). This suggests, with increasing amounts of fermentable substrates, the bacterial community diversity decreases when compared to higher forage diets (initial steps in corn‐based adaptation). RAMP is a complete starter diet, which is a high‐energy feed containing a high level of Sweet Bran and low level of forage. Therefore, the drop in bacterial diversity during RAMP‐adaptation compared to the first three steps of corn‐adaptation may be due to the effect of Sweet Bran in the RAMP diet. In comparisons between high forage diets and grain fed animals, Fernando et al. (2010) observed a similar pattern of decreasing diversity with increasing amounts of starch (Fernando et al. 2010).
Despite progressing towards a common finishing diet, the two adaptation strategies displayed unequal shifts in bacterial community composition over time (Fig. 2). Hierarchical clustering further demonstrated step1, step2, and step3 of corn‐adaptation, as well as, step1 and step2 of RAMP‐adaptation have a similar bacterial community composition. This suggests comparable changes in the bacterial communities were occurring during these stages of adaptation. Likewise, step4 and finisher of corn‐adaptation displayed a similar bacterial community composition to step3, step4, and finisher during RAMP‐adaptation. Results from hierarchical clustering, pairwise Mann–Whitney U tests, and the number of significant OTUs identified around microbial shifts point to one major breakpoint in the rumen bacterial community composition during each adaptation program. In corn‐adapted animals, this shift occurs from step3 to step4, whereas in RAMP‐adapted animals, this shift occurs earlier, from step2 to step3. Moreover, we compared RAMP‐adapted and corn‐adapted rumen bacterial communities by investigating the proportion of shared OTUs and sequences within each adaptation program (Fig. 4 and Table S3). The number of OTUs and sequences shared among samples on either side of a bacterial community shift in RAMP‐adapted and corn‐adapted animals were higher than across the shift, confirming the shifts identified. Interestingly, when comparing the same step‐up stage across adaptation methods (i.e. step1 of corn‐based adaptation compared to step1 of RAMP‐adaptation), the number of OTUs and sequences shared increase as the adaptation program progressed closer to the finisher diet. With community shifts in corn‐adapted and RAMP‐adapted animals occurring at different steps, it indicates substrate concentration and type likely dictate how fast the bacterial community acclimates to a high concentrate diet.
In addition, our data demonstrate the size of bacterial community shift during RAMP‐adaption is smaller compared to the shifts observed during corn‐adaptation. This is supported by the smaller unweighted UniFrac distance from step1 to finisher in RAMP‐adapted animals compared to corn‐adapted animals (Fig. 1). Therefore, the bacterial community in RAMP‐adapted animals may be adapted faster than in corn‐based adapted animals. Furthermore, a greater number of OTUs are shared among all stages of RAMP‐adaption compared to corn‐based adaptation. Thus, it is possible the stability of the bacterial community during RAMP‐adaption may help maintain rumen ecosystem function, resulting in increased ADG previously observed in RAMP fed animals (Huls et al. 2009).
With increasing levels of concentrate in corn‐based adaptation, a larger number of OTUs belonging to family Prevotellaceae were detected, where, all 30 differentially abundant OTUs identified as Prevotellaceae could be assigned to the genus Prevotella. Previous studies evaluating Prevotella populations (Tajima et al. 2001a,b; Fernando et al. 2010) have demonstrated a similar increase in Prevotella during adaptation. We also noted a decrease in family S24‐7 as the adaptation steps progressed. Family S24‐7 has been detected in dairy and beef cattle (McCann et al. 2014; Lima et al. 2015), however, the role of S24‐7 in the rumen is poorly understood. Bacteria belonging to family S24‐7 have been identified in mice fed high fat diets and gluco‐oligosaccharides (Serino et al. 2012). Therefore, it is possible family S24‐7 is capable of starch utilization (Serino et al. 2012).
In RAMP‐adapted animals, two shifts in the bacterial community were detected (Fig. 3b,c). OTUs high in abundance in RAMP step1, but decreased in abundance by step2 were predominantly Ruminococcaceae and Lachnospiraceae. These two families are resident microbiota within the rumen bacterial community (Church 1993). Their detection at initial stages of adaptation and the absence of these families in corn‐adapted animals suggests these microbes are better able to utilize Sweet Bran as a substrate. From step2 to step3, family Prevotellaceae and Lachnospiraceaea increased in RAMP‐adapted animals. This increase in Prevotellaceae has been shown previously and is similar to the shifts in bacterial populations from corn‐adapted animals (Tajima et al. 2001a,b; Fernando et al. 2010). The increase in Lachnospiraceaea during early stages of RAMP‐adaptation may be due to the effect of Sweet Bran in the diet, suggesting RAMP increases Lachnospiraceae abundance at a greater rate than observed in corn‐adapted animals.
The differentially abundant OTUs detected at points of bacterial community shifts in both corn‐adapted and RAMP‐adapted animals show a trend where the bacterial community changes towards increasing Prevotellacea. Prevotella is a well‐studied rumen bacterial population often found in high‐energy diets (Tajima et al. 2001a,b; Fernando et al. 2010). Prevotella are known to be efficient starch, protein, and amino acid digesters (Flint et al. 2012), so their high abundance during later stages of adaptation is not surprising.
Despite differences in nutrient composition, feedlots currently use a standard number of days to adapt cattle to high grain diets. However, the difference in composition of the finishing diet compared to the starting diet may influence bacterial acclimation. Although this study clearly demonstrates rumen bacterial communities can be acclimated faster to high concentrate diets than current adaptation programs, there are many other factors to consider before adapting animals faster. One critical consideration is the need to adapt the animal's feeding pattern from bulk fill, where the tension receptors indicate when to stop feeding on a high forage diet, to a high concentrate diet where chemo‐receptors signal when to stop eating (Church 1993). In summary, this preliminary study demonstrates rumen microbes can be adapted faster to high concentrate diets than current practices, and the rate of microbial community acclimation will depend on substrate composition. These findings may have implications for beef producers to reduce feedlot costs, as less time adapting animals would result in lower feed costs, but a larger investigation with other phenotypic characteristic including animal feeding behaviour patterns and the absorption rate of VFAs in the ruminal epithelium is needed to evaluate consequences of faster adaptation programs.
Conflict of Interest
The authors have declared that no conflict of interest.
Supporting information
Figure S1. Daily dry matter intake (DMI) and average ruminal pH of steers adapted using traditional corn‐adaptation with alfalfa hay or an adaptation system using a complete starter feed (RAMP; Cargill Corn Milling, Blair, NE).
Figure S2. Rarefaction curves based on observed number of OTUs (a) and Chao1 index (b) and alpha diversity indices calculated based on even sampling depth (c and d) display a drop in bacterial diversity after step3 in corn‐adaptation, while fairly constant diversity is maintained during RAMP‐adaptation.
Table S1‐1. Traditional (corn‐based) adaptation fed to steers. Table S1‐2. RAMP‐based adaptation diets fed in a 1‐ration system to steers.
Table S2‐1. Significant OTUs associated with corn‐based adaptation step‐up diets prior to and after the shift in microbial community composition identified between step3 and step4. Table S2‐2. Significant OTUs associated with RAMP‐adaptation step‐up diets prior to and after the shift in microbial community composition identified between step1 and step2. Table S2‐3. Significant OTUs associated with RAMP‐adaptation step‐up diets prior to and after the shift in microbial community composition identified between step2 and step3.
Table S3. Proportion of shared sequences between corn‐based adaptation and RAMP‐adaption step‐up diets.
Table S4. Proportion of shared OTUs between corn‐based adaptation and RAMP‐adaption step‐up diets.
Acknowledgements
This manuscript is based upon the work supported by USDA Agriculture and Food Research Initiative (AFRI) under grant No: 2012‐68002‐19823) to SCF; Cargill Corn Wet Milling, Blair, NE to GEE; University of Nebraska Agricultural Research Division; and in part, by the funds provided through the Hatch Act to SCF and GEE. The funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript.
References
- Benson, A.K. , Kelly, S.A. , Legge, R. , Ma, F. , Low, S.J. , Kim, J. , Zhang, M. , Oh, P.L. et al (2010) Individuality in gut microbiota composition is a complex polygenic trait shaped by multiple environmental and host genetic factors. Proc Natl Acad Sci USA 107, 18933–18938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bevans, D.W. , Beauchemin, K.A. , Schwartzkopf‐Genswein, K.S. , McKinnon, J.J. and McAllister, T.A. (2005) Effect of rapid or gradual grain adaptation on subacute acidosis and feed intake by feedlot cattle. J Anim Sci 83, 1116–1132. [DOI] [PubMed] [Google Scholar]
- Buttrey, E.K. , Luebbe, M.K. , Bondurant, R.G. and MacDonald, J.C. (2012) Case Study: grain adaptation of yearling steers to steam‐flaked corn‐based diets using a complete starter feed. Prof Anim Sci 7, 482–488. [Google Scholar]
- Caporaso, J.G. , Kuczynski, J. , Stombaugh, J. , Bittinger, K. , Bushman, F.D. , Costello, E.K. , Fierer, N. , Pena, A.G. et al (2010) QIIME allows analysis of high‐throughput community sequencing data. Nat Methods 7, 335–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlisle, E.M. , Poroyko, V. , Caplan, M.S. , Alverdy, J. , Morowitz, M.J. and Liu, D. (2013) Murine gut microbiota and transcriptome are diet dependent. Ann Surg 257, 287–294. [DOI] [PubMed] [Google Scholar]
- Church, D.C. (1993) The Ruminant Animal Digestive Physiology and Nutrition. Prospect Heights, IL: Waveland Press, Inc. [Google Scholar]
- Cooper, R.J. , Klopfenstein, T.J. , Stock, R.A. , Milton, C.T. , Herold, D.W. and Parrott, J.C. (1999) Effects of imposed feed intake variation on acidosis and performance of finishing steers. J Anim Sci 77, 1093–1099. [DOI] [PubMed] [Google Scholar]
- Dixon, P. (2003) VEGAN, a package of R functions for community ecology. J Veg Sci 14, 927–930. [Google Scholar]
- Doi, R.H. and Kosugi, A. (2004) Cellulosomes: plant‐cell‐wall‐degrading enzyme complexes. Nat Rev Microbiol 2, 541–551. [DOI] [PubMed] [Google Scholar]
- Edgar, R.C. (2013) UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat Methods 10, 996–998. [DOI] [PubMed] [Google Scholar]
- Erickson, G.E. , Milton, C.T. , Fanning, K.C. , Cooper, R.J. , Swingle, R.S. , Parrott, J.C. , Vogel, G. and Klopfenstein, T.J. (2003) Interaction between bunk management and monensin concentration on finishing performance, feeding behavior, and ruminal metabolism during an acidosis challenge with feedlot cattle. J Anim Sci 81, 2869–2879. [DOI] [PubMed] [Google Scholar]
- Fernando, S.C. , Purvis, H.T. , Najar, F.Z. , Sukharnikov, L.O. , Krehbiel, C.R. , Nagaraja, T.G. , Roe, B.A. and DeSilva, U. (2010) Rumen microbial population dynamics during adaptation to a high‐grain diet. Appl Environ Microbiol 76, 7482–7490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flint, H.J. , Scott, K.P. , Duncan, S.H. , Louis, P. and Forano, E. (2012) Microbial degradation of complex carbohydrates in the gut. Gut Microbes 3, 289–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamady, M. , Lozupone, C. and Knight, R. (2010) Fast UniFrac: facilitating high‐throughput phylogenetic analyses of microbial communities including analysis of pyrosequencing and PhyloChip data. ISME J 4, 17–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huls, T.J. , Lubbe, M.K. , Griffin, W.A. , Erickson, G.E. , Klopfenstein, T.J. and Stock, R.A. (2009) Using Wet Corn Gluten Feed to Adapt Cattle to Finishing Diets. Lincoln: University of Nebraska. [Google Scholar]
- Hungate, R.E. (1966) The Rumen and Its Microbes. New York: Academic Press Inc. [Google Scholar]
- Lima, F.S. , Oikonomou, G. , Lima, S.F. , Bicalho, M.L. , Ganda, E.K. , Filho, J.C. , Lorenzo, G. , Trojacanec, P. et al (2015) Prepartum and postpartum rumen fluid microbiomes: characterization and correlation with production traits in dairy cows. Appl Environ Microbiol 81, 1327–1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lozupone, C. , Lladser, M.E. , Knights, D. , Stombaugh, J. and Knight, R. (2011) UniFrac: an effective distance metric for microbial community comparison. ISME J 5, 169–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDonald, J.C. and Luebbe, M.K. (2012) Grain adaptation for feedlot cattle using high concentrations of wet corn gluten feed in steam‐flaked corn‐based diets. Prof Anim Sci 28, 9. [Google Scholar]
- Mao, S.Y. , Zhang, R.Y. , Wang, D.S. and Zhu, W.Y. (2013) Impact of subacute ruminal acidosis (SARA) adaptation on rumen microbiota in dairy cattle using pyrosequencing. Anaerobe 24, 12–19. [DOI] [PubMed] [Google Scholar]
- Martinez, I. , Wallace, G. , Zhang, C. , Legge, R. , Benson, A.K. , Carr, T.P. , Moriyama, E.N. and Walter, J. (2009) Diet‐induced metabolic improvements in a hamster model of hypercholesterolemia are strongly linked to alterations of the gut microbiota. Appl Environ Microbiol 75, 4175–4184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCann, J.C. , Drewery, M.L. , Sawyer, J.E. , Pinchak, W.E. and Wickersham, T.A. (2014) Effect of postextraction algal residue supplementation on the ruminal microbiome of steers consuming low‐quality forage. J Anim Sci 92, 5063–5075. [DOI] [PubMed] [Google Scholar]
- McDonald, D. , Price, M.N. , Goodrich, J. , Nawrocki, E.P. , DeSantis, T.Z. , Probst, A. , Andersen, G.L. , Knight, R. et al (2012) An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J 6, 610–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKnite, A.M. , Perez‐Munoz, M.E. , Lu, L. , Williams, E.G. , Brewer, S. , Andreux, P.A. , Bastiaansen, J.W. , Wang, X. et al (2012) Murine gut microbiota is defined by host genetics and modulates variation of metabolic traits. PLoS One 7, e39191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagaraja, T.G. and Lechtenberg, K.F. (2007) Acidosis in feedlot cattle. Vet Clin North Am Food Anim Pract 23, 333–350, viii‐ix. [DOI] [PubMed] [Google Scholar]
- Nagaraja, T.G. and Titgemeyer, E.C. (2007) Ruminal acidosis in beef cattle: the current microbiological and nutritional outlook. J Dairy Sci 90(Suppl 1), E17–E38. [DOI] [PubMed] [Google Scholar]
- Owens, F.N. , Secrist, D.S. , Hill, W.J. and Gill, D.R. (1998) Acidosis in cattle: a review. J Anim Sci 76, 275–286. [DOI] [PubMed] [Google Scholar]
- Ploner, A. (2014) Heatplus: Heatmaps with row and/or column covariates and colored clusters. R package version 2.14.0.
- Rosenzweig, A.C. and Ragsdale, S.W. (2011) Methods in Methane Metabolism. Burlington, MA: Elsevier. [Google Scholar]
- Sato, S. (2015) Subacute ruminal acidosis (SARA) challenge, ruminal condition and cellular immunity in cattle. Jpn J Vet Res 63(Suppl 1), S25–S36. [PubMed] [Google Scholar]
- Schloss, P.D. , Westcott, S.L. , Ryabin, T. , Hall, J.R. , Hartmann, M. , Hollister, E.B. , Lesniewski, R.A. , Oakley, B.B. et al (2009) Introducing mothur: open‐source, platform‐independent, community‐supported software for describing and comparing microbial communities. Appl Environ Microbiol 75, 7537–7541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schluter, J. and Foster, K.R. (2012) The evolution of mutualism in gut microbiota via host epithelial selection. PLoS Biol 10, e1001424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segata, N. , Izard, J. , Waldron, L. , Gevers, D. , Miropolsky, L. , Garrett, W.S. and Huttenhower, C. (2011) Metagenomic biomarker discovery and explanation. Genome Biol 12, R60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serino, M. , Luche, E. , Gres, S. , Baylac, A. , Berge, M. , Cenac, C. , Waget, A. , Klopp, P. et al (2012) Metabolic adaptation to a high‐fat diet is associated with a change in the gut microbiota. Gut 61, 543–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheneman, L. , Evans, J. and Foster, J.A. (2006) Clearcut: a fast implementation of relaxed neighbor joining. Bioinformatics 22, 2823–2824. [DOI] [PubMed] [Google Scholar]
- Spor, A. , Koren, O. and Ley, R. (2011) Unravelling the effects of the environment and host genotype on the gut microbiome. Nat Rev Microbiol 9, 279–290. [DOI] [PubMed] [Google Scholar]
- Sun, Y.Z. , Mao, S.Y. and Zhu, W.Y. (2010) Rumen chemical and bacterial changes during stepwise adaptation to a high‐concentrate diet in goats. Animal 4, 210–217. [DOI] [PubMed] [Google Scholar]
- Tajima, K. , Aminov, R.I. , Nagamine, T. , Matsui, H. , Nakamura, M. and Benno, Y. (2001a) Diet‐dependent shifts in the bacterial population of the rumen revealed with real‐time PCR. Appl Environ Microbiol 67, 2766–2774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tajima, K. , Nagamine, T. , Matsui, H. , Nakamura, M. and Aminov, R.I. (2001b) Phylogenetic analysis of archaeal 16S rRNA libraries from the rumen suggests the existence of a novel group of archaea not associated with known methanogens. FEMS Microbiol Lett 200, 67–72. [DOI] [PubMed] [Google Scholar]
- Team, R.D.C. (2007) R: A Language and Environment for statistical Computing. Vienna, Austria: R Foundation for Statistical Computing. [Google Scholar]
- Vander Pol, K.J. , Luebbe, M.K. , Crawford, G.I. , Erickson, G.E. and Klopfenstein, T.J. (2009) Performance and digestibility characteristics of finishing diets containing distillers grains, composites of corn processing coproducts, or supplemental corn oil. J Anim Sci 87, 639–652. [DOI] [PubMed] [Google Scholar]
- Vasconcelos, J.T. and Galyean, M.L. (2007) Nutritional recommendations of feedlot consulting nutritionists: the 2007 texas tech university survey. J Anim Sci 85, 2772–2781. [DOI] [PubMed] [Google Scholar]
- Warnes, G.R. , Bolker, B. , Bonebakker, L. , Gentleman, R. , Huber, W. , Liaw, A. , Lumley, T. , Maechler, M. et al (2015) gplots: Various R Programming Tools for Plotting Data. R package. version 2.17.0.
- Wickham, H. (2009) ggplot2: Elegant Graphics for Data Analysis. NY: Springer. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Daily dry matter intake (DMI) and average ruminal pH of steers adapted using traditional corn‐adaptation with alfalfa hay or an adaptation system using a complete starter feed (RAMP; Cargill Corn Milling, Blair, NE).
Figure S2. Rarefaction curves based on observed number of OTUs (a) and Chao1 index (b) and alpha diversity indices calculated based on even sampling depth (c and d) display a drop in bacterial diversity after step3 in corn‐adaptation, while fairly constant diversity is maintained during RAMP‐adaptation.
Table S1‐1. Traditional (corn‐based) adaptation fed to steers. Table S1‐2. RAMP‐based adaptation diets fed in a 1‐ration system to steers.
Table S2‐1. Significant OTUs associated with corn‐based adaptation step‐up diets prior to and after the shift in microbial community composition identified between step3 and step4. Table S2‐2. Significant OTUs associated with RAMP‐adaptation step‐up diets prior to and after the shift in microbial community composition identified between step1 and step2. Table S2‐3. Significant OTUs associated with RAMP‐adaptation step‐up diets prior to and after the shift in microbial community composition identified between step2 and step3.
Table S3. Proportion of shared sequences between corn‐based adaptation and RAMP‐adaption step‐up diets.
Table S4. Proportion of shared OTUs between corn‐based adaptation and RAMP‐adaption step‐up diets.