

Abstract
Background
Analysis of myocardium has revealed mechanistic insights into arrhythmogenic cardiomyopathy but cardiac samples are difficult to obtain from probands and especially from family members. To identify a potential surrogate tissue, we characterized buccal mucosa cells.
Methods and Results
Buccal cells from patients, mutation carriers and controls were immunostained and analyzed in a blinded fashion. In additional studies, buccal cells were grown in vitro and incubated with SB216763. Immunoreactive signals for the desmosomal protein plakoglobin and the major cardiac gap junction protein Cx43 were markedly diminished in buccal mucosa cells from arrhythmogenic cardiomyopathy patients with known desmosomal mutations when compared with controls. Plakoglobin and Cx43 signals were also reduced in most family members who carried disease alleles but showed no evidence of heart disease. Signal for the desmosomal protein plakophilin-1 was reduced in buccal mucosa cells in patients with PKP2 mutations but not in those with mutations in other desmosomal genes. Signal for the desmosomal protein desmoplakin was reduced in buccal mucosa cells from patients with mutations in DSP, DSG2 or DSC2 but not in PKP2 or JUP. Abnormal protein distributions were reversed in cultured cells incubated with SB216763, a small molecule that rescues the disease phenotype in cardiac myocytes.
Conclusions
Buccal mucosa cells from arrhythmogenic cardiomyopathy patients exhibit changes in the distribution of cell junction proteins similar to those seen in the heart. These cells may prove useful in future studies of disease mechanisms and drug screens for effective therapies in arrhythmogenic cardiomyopathy.
Keywords: arrhythmogenic right ventricular cardiomyopathy, connexin43, desmosome cardiomyopathy, diagnosis, buccal mucosa cells, plakoglobin
Introduction
Arrhythmogenic cardiomyopathy is a familial myocardial disease characterized by a high incidence of ventricular arrhythmias and increased risk of sudden death, especially in young individuals and athletes.1 It was originally described as a right ventricular disease and referred to as arrhythmogenic right ventricular cardiomyopathy/dysplasia (ARVC/D), but recognition of left dominant and biventricular forms has given rise to the more general name of arrhythmogenic cardiomyopathy (ACM).2 Mutations in one or more desmosomal genes (PKP2, DSP, JUP, DSG2, DSC2) can be found in ∼60% of patients who fulfill criteria for the clinical diagnosis of ACM.3 However, genetic penetrance and disease expression may be highly variable, and identifying affected family members at risk of adverse events remains challenging.
Previous studies of myocardial tissue samples from patients with ACM have revealed changes in the distribution of various cell-cell junction proteins.4-6 The most consistent changes occur in the desmosomal protein plakoglobin (γ-catenin)4,5 and the major ventricular gap junction protein Cx43.4-6 Normally, both of these proteins are highly concentrated at intercalated discs in cardiac myocytes but their junctional immunoreactive signals are markedly reduced in the great majority of patients with ACM. Redistribution of plakoglobin from junctional to intracellular and/or intranuclear sites has been linked to abnormal Wnt/β-catenin signaling in ACM,7 and loss of junctional Cx43 may affect electrical conduction and, thereby, play a role in the highly arrhythmogenic phenotype.4,8 A potential pathogenic role for aberrant Wnt/β-catenin signaling in ACM is supported by recent studies showing that key features of the disease phenotype can be prevented or reversed by SB216763, a small molecule annotated as an inhibitor of glycogen synthase kinase-3β (GSK3β).9
Most myocardial samples available from probands with ACM are formalin-fixed, paraffin-embedded tissues obtained at autopsy or by endomyocardial biopsy, which precludes studies in fresh tissue or living cells. While studies of myocardium from family members of probands might prove useful in diagnosis and risk stratification, such samples are understandably difficult to obtain. A few studies have used cardiac myocytes differentiated from induced pluripotent stems cells obtained from ACM probands and family members,9-11 but this approach is expensive, technically demanding, and time-consuming. To find a surrogate tissue for the heart that can be studied in ACM probands and family members, we have investigated buccal mucosa cells which can be easily and safely obtained from large numbers of subjects at minimal cost. Here, we report that buccal mucosa cells from ACM patients exhibit many features previously documented in cardiac myocytes. Similar features are seen in buccal mucosa cells in a subset of family members who carry disease alleles but show no clinical evidence of heart disease. These results suggest that buccal mucosa cells may be a simple, inexpensive source of patient material for subsequent studies of disease mechanisms in ACM.
Methods
Patients, family members, controls
We obtained buccal mucosa samples from a total of 101 subjects categorized into 4 groups. Group 1 consisted of 39 patients identified in the Johns Hopkins ARVC Registry, and in the Nikos Protonotarios Medical Center and the 1st Department of Cardiology of the University of Athens Medical School in Greece. All of these patients fulfilled international task force criteria for the diagnosis of ACM12 and had a documented mutation in a desmosomal gene. The specific mutations are included in Supplemental Table 1. Group 2 included 40 healthy individuals with no history of heart disease who served as controls. Group 3 consisted of 15 family members (some related to patients in Group 1) who were known to carry a disease allele (see Supplemental Table 1) but showed no evidence of heart disease. Group 4 consisted of 7 patients with other types of heart diseases including hypertrophic cardiomyopathy (HCM, n=4), dilated cardiomyopathy (DCM; n=2) and ischemic heart disease (n=1). All subjects provided written informed consent, and sample collection protocols were approved by appropriate institutional review boards. Standard HIPAA protocols were followed, and database information was stored in password-encrypted files. All buccal mucosa samples were coded and analyzed in a blinded fashion.
Buccal mucosa smears were prepared from each subject. In most cases, enough smears were prepared to allow immunostaining for multiple proteins, but in a few cases only a limited number of smears were available. In selected cases, additional samples were obtained for preparation of buccal mucosal cell cultures.
Buccal mucosa sampling and preparation of smears
A clean cotton-tipped swab (Q-tip) was used for collection of the specimen. Material was collected from the inside of the cheek by using slight rolling and scraping motions for about 30 seconds on each side. Immediately after collection, the buccal mucosa material was smeared on microscope slides, which were immediately dipped in 70% ethanol for ∼1 minute to fix the sample. The slides were allowed to air dry and stored at room temperature before being immunostained.
Buccal mucosa culture protocol
The oral cavity was first rinsed thoroughly with antiseptic mouthwash. Buccal mucosa cells were collected with a sterile toothbrush which was lightly scraped on the inside of both cheeks for ∼2 minutes. The oral cavity was washed with 15 ml of sterile saline which was collected in a centrifuge tube. The toothbrush itself was washed in an additional 10ml of saline. The same process was repeated one or more times. The resultant cell suspension was centrifuged at 3000g for 5 minutes. The pellet was washed once in sterile PBS, re-centrifuged and subsequently re-suspended in 20ml of PBS supplemented with 5% penicillin/streptomycin and 7mM fungizone for 30 minutes. The cells were then washed twice in PBS, reconstituted in KGM™-CD chemically defined keratinocyte growth medium (LONZA) and seeded on matrigel-coated chamber slides, where they were maintained at 37°C in 5% CO2 for up to 7 days. Selected cultures were incubated with SB216763 (5mM) for 24 hours before being immunostained.
Immunohistochemistry
Buccal mucosa smears were immunostained with one of the following primary antibodies using established protocols: mouse monoclonal anti-plakoglobin, mouse monoclonal anti-plakophilin-1, mouse monoclonal anti-desmoplakin or rabbit polyclonal anti-Cx43 4,9 Slides were then incubated with Cy3-conjugated secondary antibodies (Jackson ImmnunoResearch) for 2 hours at room temperature and counterstained with DAPI to label nuclei.
Buccal mucosa cultures were washed in PBS, fixed in 4% paraformaldehyde and, after being washed 3 additional times in PBS, were immunostained as described above with mouse monoclonal anti-plakoglobin (Sigma) or mouse monoclonal anti-Cx43 (Millipore) primary antibodies. All immunostained preparations were imaged at 40× using a ZEISS inverted confocal microscope.
Statistical analysis
Differences between groups in the distribution of immunoreactive signals were assessed in a pairwise fashion with the Fisher's exact test. Bonferroni correction was used to correct p-values due to multiple comparisons, as appropriate. p<0.05 was considered significant. SPSS 21.0 software was used for all analyses.
Results
Immunohistochemical analysis of expression of cell-cell junction proteins in normal buccal mucosa cells
Before characterizing expression of cell-cell junction proteins in buccal mucosa cells in ACM patients, we optimized immunostaining conditions in cells from normal subjects. For each primary antibody, we determined the minimal concentration (i.e., maximum antibody dilution) that still led to bright immunofluorescent signal in normal cells. This essential step was designed to optimally demonstrate any potential reduction in signal intensity in ACM patient cells. Once suitable primary antibody dilutions had been determined in a pilot study of normal cells, we used these conditions throughout the study. Detailed information about primary antibody sources and antibody optimization strategies are included in Supplemental Table 2 and Supplemental Figure 1.
As shown in representative images in Figure 1, buccal smears consisted of flattened squamous cells with small central nuclei and clearly identifiable edges. The immunostaining protocol resulted in abundant fluorescent signal in normal cells for the desmosomal proteins plakoglobin, desmoplakin and plakophilin-1, and the gap junction protein Cx43. Signal for each of these proteins was present at the cell surface and, depending on the protein being studied, was also seen within the cell. No signal was detected in buccal mucosa cells stained for plakophilin-2, the plakophilin expressed in cardiac myocytes. To determine whether the anti-plakophilin-1 antibody used in this study cross-reacted with plakophilin-2, we immunostained sections of human skin (which expresses only plakophilin-1) and sections of human and mouse myocardium (which express only plakophilin-2) with antibodies against each protein (see Supplemental Table 2 for details about antibody sources and dilutions). As shown in Supplemental Figure 2, each antibody produced bright signal only in the expected tissue and showed no evidence of cross-reactivity.
Figure 1.
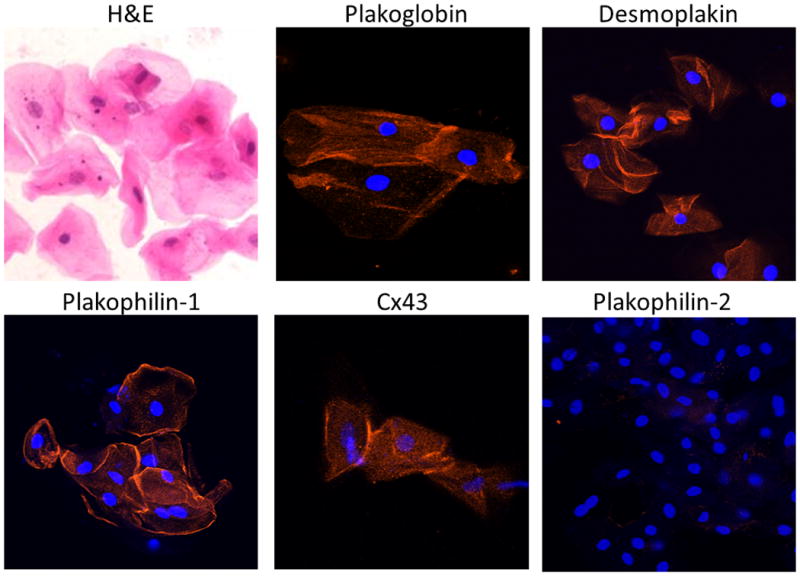
Representative images of normal buccal mucosa smears. Cells stained with hematoxylin and eosin (H&E) show typical squamous morphology with central nuclei and clearly delineated cell borders. Buccal mucosa cells immunostained with antibodies against desmosomal and gap junction proteins show strong immunoreactive signals for plakoglobin, desmoplakin, plakophilin-1 and Cx43 concentrated at the edges of the cells. No apparent signal is seen for plakophilin-2 which is expressed in the heart but not in epithelial tissues. Cell nuclei (blue) are stained with DAPI.
Expression of cell-cell junction proteins in buccal mucosa cells from ACM patients with desmosomal mutations
To determine whether changes in cell-cell junction proteins occur in buccal mucosa cells in ACM, we tested samples from patients with clinically documented ACM and a known mutation in a desmosomal gene (Group 1). This included patients with autosomal dominant mutations in PKP2 (n=25), DSG2 (n=4), DSC2 (n=2), and DSP (n=2); and 6 patients with the homozygous mutation in JUP that causes the cardiocutaneous syndrome Naxos disease13 (total n=39). We immunostained these samples for plakoglobin and Cx43 which usually show reduced junctional signal in the hearts of patients with ACM.4 As shown in Table 1 and in representative images in Figure 2, signal for plakoglobin was diminished or absent in 20 of 25 patients with PKP2 mutations and in all 14 patients with other desmosomal mutations. Signal for Cx43 was reduced in 30 of 31 patient samples (additional smears for Cx43 staining were not available in 8 patients) (Table 1 and Figure 2). The only case with apparently normal Cx43 staining was a Naxos disease patient in whom plakoglobin signal was depressed. Group 1 ACM patient samples were compared to 40 control samples (Group 2) including 11 from the island of Naxos where patients with the recessive mutation in JUP originated. Strong signals were seen for plakoglobin in all 40 controls and for Cx43 in 39 controls (additional smears were not available in 1 control subject) (Table 1, Figures 1 and 2).
Figure 2.
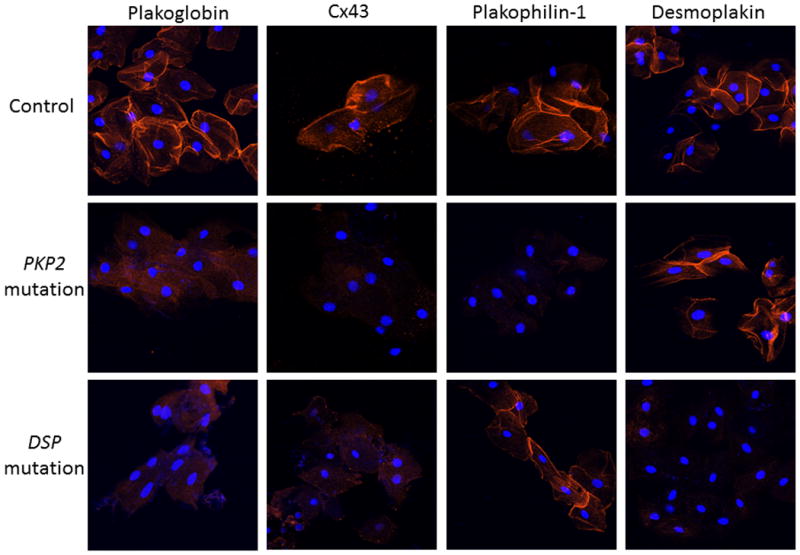
Representative images of buccal mucosa smears from a control, and ACM patients with mutations in PKP2 or DSP. Both patients show loss of junctional signal for plakoglobin and Cx43 compared to controls. Cells from the patient with a PKP2 mutation show loss of signal for plakophilin-1 but not desmoplakin, whereas cells from the patient with a DSP mutation show loss of signal for desmoplakin but not plakophilin-1. Cell nuclei (blue) are stained with DAPI.
Table 1. Summary of immunohistochemical findings in buccal mucosa smears in 4 groups of subjects.
| Depressed plakoglobin signal | Depressed Cx43 signal | Depressed plakophilin-1 signal | Depressed desmoplakin signal | ||
|---|---|---|---|---|---|
|
| |||||
| GROUP 1 | ACM patients (n=39) | ||||
| PKP2 (n=25) | 20/25 | 19/19 | 19/19 | 0/19 | |
| DSG2 (n=4) | 4/4 | 2/2 | 0/2 | 2/2 | |
| DSC2 (n=2) | 2/2 | 2/2 | 0/2 | 2/2 | |
| DSP (n=2) | 2/2 | 2/2 | 0/2 | 2/2 | |
| JUP (n=6) | 6/6 | 5/6 | 0/6 | 0/6 | |
| Total: 34/39*# | Total: 30/31* | Total: 19/31* | Total: 6/31 | ||
|
| |||||
| GROUP 2 | Controls (n=40) | Total: 0/40 | Total: 0/39 | Total: 0/39 | Total: 0/39 |
|
| |||||
| GROUP 3 | Unaffected carriers (n=15) | ||||
| 2157del2 JUP (n=12) | 10/12 | 11/12 | 0/11 | 0/11 | |
| DSC2 (n=1) | 1/1 | 1/1 | 0/1 | 1/1 | |
| DSP (n=2) | 1/2 | 2/2 | 0/2 | 2/2 | |
| Total: 12/15*† | Total: 14/15* | Total: 0/14 | Total: 3/14 | ||
|
| |||||
| GROUP 4 | Patients with other CMs (n=7) | ||||
| HCM (n=4) | 0/4 | 4/4 | |||
| DCM (n=2) | 0/2 | 2/2 | |||
| Ischemia (n=1) | 0/1 | 1/1 | |||
| Total: 0/7 | Total: 7/7* | ||||
p<0.001 vs. controls
p<0.001 vs. patients with other heart diseases
We have previously reported that intercalated disc signals for other desmosomal proteins including plakophilin-2 and desmoplakin are reduced in the hearts of some but not all patients with ACM.4 To determine whether signals for plakophilin and desmoplakin are altered in buccal mucosa cells from ACM patients, we immunostained samples with antibodies against plakophilin-1 and desmoplakin. As shown in Table 1 and in representative images in Figure 2, signal for plakophilin-1 was depressed in all ACM patients with PKP2 mutations in whom smears were available for study (n=19), but in none of 12 ACM patients with mutations in other desmosomal genes including autosomal dominant mutations in DSG2 (n=2), DSC2 (n=2) or DSP (n=2), and the homozygous recessive mutation in JUP (n=6). Additional smears for plakophilin-1 staining were available in 4 of 5 patients with PKP2 mutations who showed normal plakoglobin signal. In all 4 cases, signal for plakophilin-1 was virtually absent (see Table 1).
Signal for desmoplakin was reduced in ACM patients with mutations in DSP (n=2), DSC2 (n=2) or DSG2 (n=2) but not in patients with mutations in PKP2 (n=19) or the homozygous mutation in JUP (n=6) (Table 1, Figure 2).
Expression of cell-cell junction proteins in buccal mucosa cells from ACM mutation carriers
Opportunities to characterize molecular pathology in myocardial samples from gene carriers with no clinical evidence of ACM are obviously limited. We were able to prepare buccal mucosa smears from 12 carriers of the recessive mutation in Naxos disease and from 3 carriers of other disease alleles including those with dominant mutations in DSP (n=2) and DSC2 (n=1) (Group 2; total n=15). None of these subjects exhibited clinical features of ACM although some of the Naxos disease carriers had curly hair. As shown in Table 1 and Supplemental Figure 3, signal for plakoglobin was depressed in 10 of the 12 Naxos disease gene carriers and in 2 of 3 carriers of other mutations. Signal for Cx43 was reduced in 11 of the 12 Naxos carriers and in all 3 carriers of the other mutations. Signal for plakophilin-1 was normal in all of these disease gene carriers (none of whom carried mutations in PKP2). Signal for desmoplakin was reduced in carriers of DSC2 (n=1) and DSP mutations (n=2) but not in the Naxos disease carriers (n=12).
Clinical correlations with staining patterns in ACM probands and family members
All 5 of the ACM patients in Group 1 whose buccal mucosa cells showed apparently normal plakoglobin signal had mutations in PKP2. Three of these patients came to medical attention because the disease had been diagnosed in a family member. Two had mild disease with ventricular ectopy and ECG abnormalities, but no sustained arrhythmias or evidence of structural remodeling. They fulfilled criteria for the diagnosis of ACM mainly on the basis of family history/mutation status. The third patient also came to medical attention because ACM had been diagnosed in her brother. This subject showed ectopy and had an abnormal 12-lead ECG and signal-averaged ECG but otherwise showed no structural abnormalities and was largely asymptomatic until experiencing an episode of VT storm, 12 years after the diagnosis of ACM was made. The other two ACM patients with PKP2 mutations and normal plakoglobin signal in buccal mucosa cells were Greek individuals both of whom showed marked disease with sustained arrhythmias and right ventricular remodeling.
Twelve of 15 family members (Group 3) showed reduced plakoglobin signal. Two family members with apparently normal signal levels were heterozygous for the recessive mutation in JUP that causes Naxos disease. One of these carriers came from a family in which affected individuals exhibited a very mild form of the cardiomyopathy with minimal arrhythmias and no heart failure phenotype even in older individuals. The other carrier of the Naxos disease mutation whose buccal mucosa cells appeared normal had straight hair and no evidence of heart disease whereas 3 other carriers in the same family (all of whom showed reduced plakoglobin signal) had curly hair but no evidence of heart disease. The third family member with normal plakoglobin and Cx43 signal in buccal mucosa cells was a 14-year old carrier of a disease allele in DSP who showed no evidence of disease.
Expression of cell-cell junction proteins in buccal mucosa cells from patients with other heart diseases
We have previously reported that plakoglobin signal at intercalated discs is normal in patients with ischemic heart disease or dilated or hypertrophic cardiomyopathies.4 This indicates that among this disease spectrum, redistribution of plakoglobin appears to be specific for ACM. By contrast, reduced signal for Cx43 at intercalated discs occurs in various forms of heart disease including both ischemic and non-ischemic cardiomyopathies. Here, we stained buccal mucosa smears from patients with HCM (n=4), DCM (n=2) and ischemic heart disease (n=1) (Group 4). As shown in Table 1, signal for plakoglobin was normal in all of these patients, whereas Cx43 signal was depressed in all cases.
Studies in cultured buccal mucosa cells
We obtained sufficient material from 6 ACM patients and 2 gene carriers to establish buccal mucosa cell cultures. Cultured cells from all patients and unaffected gene carriers showed loss of junctional signal for plakoglobin and Cx43 while plakoglobin signal accumulated in cell nuclei. In both patients and gene carriers, incubation of cultured cells with SB216763 for 24 hours normalized plakoglobin and Cx43 signal distributions. Representative images are shown in Figure 3.
Figure 3.
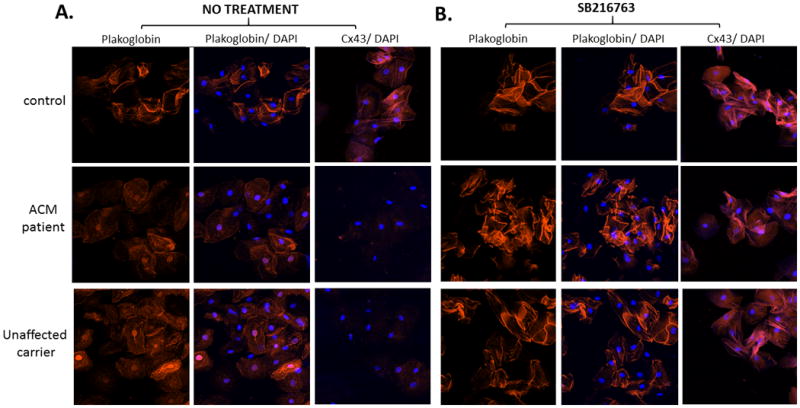
Representative images of cultured buccal mucosa cells obtained from a control subject, an ACM patient and a clinically unaffected carrier of a PKP2 mutation, before (A) and after (B) exposure to SB216763. Cultured cells from both the patient and carrier showed loss of junctional signal for plakoglobin, accumulation of nuclear plakoglobin and loss of junctional Cx43. Incubation with SB216763 for 24 hours normalized plakoglobin and Cx43 signal distributions. Cell nuclei (blue) are stained with DAPI.
Discussion
Exfoliated buccal mucosa cells have been used as a source of material for various genetic tests and in studies of oral neoplasia,14 but their use in studies of cardiovascular disease has been limited. Previous applications have included analysis of telomere length in buccal cells from patients with ischemic heart failure,15 and measurements of intracellular magnesium levels in patients undergoing radiofrequency catheter ablation for atrial fibrillation.16 To the best of our knowledge, the present study is the first analysis of buccal mucosa cells in patients with cardiomyopathy. Similarly, while gap junctions and desmosomes have been documented in buccal mucosa cells,14,17 most previous studies have centered largely on expression of cell-cell junction proteins in oral cancers or in response to carcinogenic agents such as cigarette smoke.14,17,18
The results of the present study suggest a close relationship between patterns of altered distribution of intercalated disc proteins in cardiac myocytes and cell-cell junction proteins in buccal mucosa cells in patients with ACM. Previous studies have reported that a majority of patients with ACM, but not other cardiomyopathies, show reduced plakoglobin signal at cardiac myocyte intercalated discs.4-6,8 Here, we saw a similar reduction in junctional signal for plakoglobin in buccal mucosa cells in 34 of 39 ACM patients but in none of 40 controls or 7 patients with other forms of heart disease. Five ACM patients with mutations in PKP2 showed normal plakoglobin signal, of which 3 had relatively mild disease.
Four of the ACM patients whose buccal mucosa cells were analyzed in the present study had previous endomyocardial biopsies, which had been analyzed by immunostaining and reported in a study in 2009.4 Three showed depressed plakoglobin signal in both their heart tissue and buccal mucosa. The remaining patient showed apparently normal signal in the heart in 2009 but depressed signal in buccal mucosa cells in the present study. This individual has a mutation in PKP2. He was diagnosed after the sudden death of his teenage daughter and has exhibited clear evidence of ACM since before 2009.
An unexpected aspect of the present study is the apparently strong link between reduced cell surface signal for specific desmosomal proteins in buccal mucosa cells and the mutant gene of interest. For example, reduced intercalated disc signal for plakophilin-2 occurs in the hearts of only some ACM patients,4 and there has been no previous association between loss of plakophilin-2 signal and the presence of a disease-causing mutation in PKP2. By contrast, every patient in the present study with a PKP2 mutation showed loss of plakophilin-1 signal in their buccal mucosa cells (including the 5 patients in whom plakoglobin signal was normal) whereas none of the ACM patients with mutations in other genes showed a similar loss. This association is even more remarkable when one considers that plakophilin-1 and plakophilin-2 are encoded by separate genes on separate chromosomes.19,20 Plakophilin-2 is expressed in the heart and in basal cells of the epidermis but not in keratinocytes or buccal mucosa cells.19,20 Whether progenitors of buccal mucosa cells express plakophilin-2 is not known, but our results implicate common regulatory mechanisms. We also observed loss of buccal cell desmoplakin signal only in patients with mutations in DSP, DSC2 or DSG2 but not with the more common mutations in PKP2 or in patients with the highly penetrant cardiomyopathy caused by the recessive JUP mutation in Naxos disease. A similar association has not been reported previously in studies of myocardium in ACM patients. Desmoplakin interacts extensively with the cytoplasmic domains of the desmosomal cadherins (desmocollins and desmogleins),21 and our results suggest that mutations in any of their genes have the potential to disrupt normal localization of desmoplakin in buccal mucosa cells. Future studies will be required to define mechanisms responsible for these observations, and to determine whether immunohistochemical analysis of buccal mucosa cells will be useful as a diagnostic test for ACM and whether it can predict mutational status.
Because of the relative ease of preparing buccal smears, we were able to analyze changes in distribution of cell-cell junction proteins in family members who carried ACM disease alleles but showed no clinical evidence of heart disease (albeit in buccal mucosa cells rather than in the heart). In most cases, signals for plakoglobin and Cx43 were reduced. The few carriers who showed normal signals apparently belonged to families with milder disease expression. The implications of these changes are unclear, but they suggest that redistribution of junctional proteins does not necessarily correlate with clinical expression of disease. Whether such changes in family members might predict future development of disease or whether the absence of such changes in gene carriers predicts less virulent disease in affected probands remains unknown. We could only study a relatively small number of family members and did not undertake a detailed analysis of multiple members of well characterized pedigrees. However, in view of the ease of obtaining buccal mucosa samples, future studies may better define the potential diagnostic and risk-stratification value of analyzing buccal smears in affected families.
The fact that buccal mucosa cells can be maintained in culture provides an opportunity to conduct studies in living cells. Here, we showed that the GSK3β inhibitor SB216763 can restore apparently normal cell surface signal for plakoglobin and Cx43 in cells from ACM, as previously shown in cultured neonatal rat ventricular myocytes transfected to express ACM mutations.8 This suggests that similar disease mechanisms occur in cardiac myocytes and buccal mucosa cells, at least with respect to the distribution of junctional proteins. Interestingly, loss of junctional plakoglobin signal in cultured buccal mucosa cells from ACM patients and gene carriers was accompanied by nuclear accumulation of plakoglobin. Previous studies in mouse models of ACM have reported nuclear accumulation of plakoglobin which has been implicated in aberrant Wnt/β-catenin signaling.7 Nuclear plakoglobin in ACM patient myocardium has not been reported although this may be related to technical (e.g., tissue fixation) rather than biological factors. Cultured buccal mucosa cells from probands and gene carriers may be useful in future studies to characterize alterations in signaling mechanisms in ACM, and in drug screens to evaluate potential therapies in patient-specific cells.
Of course, it must be emphasized that buccal mucosa cells are not cardiac myocytes and, at best, can only be considered surrogates. Whether they may be useful clinically for diagnosis or risk-stratification will require much more extensive, prospective studies. And, the extent to which they may reveal insights into disease mechanisms in the heart remains to be further defined. Nevertheless, we show here that buccal mucosa cells from patients with ACM have a molecular phenotype that is remarkably similar to that seen in the heart. Similar features of this phenotype are seen in family members. These observations, therefore, raise the possibility that buccal mucosa cells may be a useful source of patient material to gain insights into disease mechanisms and better understand potential genetic, epigenetic and environmental factors that determine genotype-phenotype relationships. These and other questions will be topics of future research.
Supplementary Material
What is Known.
Analysis of myocardium has revealed mechanistic insights into arrhythmogenic cardiomyopathy but cardiac samples are difficult to obtain from probands and especially from family members.
To identify a potential surrogate tissue, we used immunohistochemistry to characterize buccal mucosa cells from arrhythmogenic cardiomyopathy patients.
What the Study Adds.
Buccal mucosa cells from arrhythmogenic cardiomyopathy patients showed changes in the distribution of cell junction proteins similar to those seen in the heart. These changes were normalized in cultured buccal mucosa cells by a small molecule shown previously to rescue the disease phenotype in experimental models.
These results suggest that patients with arrhythmogenic cardiomyopathy also exhibit an epithelial cell phenotype.
Buccal mucosa cells may prove useful in future studies of disease mechanisms and drug screens to identify new therapies in arrhythmogenic cardiomyopathy.
Acknowledgments
We are grateful to the patients, family members and normal volunteers who participated in this study.
Funding Source: This work was supported by NIH grant HL116906. The Johns Hopkins ARVD/C program is supported by the Leyla Erkan Family Fund for ARVD Research; the Dr. Satish, Rupal, and Robin Shah ARVD Fund at Johns Hopkins; the Dr. Francis P. Chiaramonte Private Foundation; the Bogle Foundation; the Healing Hearts Foundation; the Campanella family; the Patrick J. Harrison Family; the Peter French Memorial Foundation; the St. Jude Medical Foundation; and the Wilmerding Endowments. Dr. Te Riele was supported by the Mark Josephson and Hein Wellens Research Fellowship from the Heart Rhythm Society.
Footnotes
Conflict of Interest Disclosures: A patent application has been filed for the use of buccal mucosa cells in the diagnosis and management of ACM and related heart diseases.
References
- 1.Basso C, Corrado D, Marcus FI, Nava A, Thiene G. Arrhythmogenic right ventricular cardiomyopathy. Lancet. 2009;373:1289–1300. doi: 10.1016/S0140-6736(09)60256-7. [DOI] [PubMed] [Google Scholar]
- 2.Sen-Chowdhry S, Morgan RD, Chambers JC, McKenna WJ. Arrhythmogenic cardiomyopathy: etiology, diagnosis and treatment. Annu Rev Med. 2010;61:233–253. doi: 10.1146/annurev.med.052208.130419. [DOI] [PubMed] [Google Scholar]
- 3.Marcus FI, Edson S, Towbin JA. Genetics of arrhythmogenic right ventricular cardiomyopathy: a practical guide for physicians. J Am Coll Cardiol. 2013;61:1945–1948. doi: 10.1016/j.jacc.2013.01.073. [DOI] [PubMed] [Google Scholar]
- 4.Asimaki A, Tandri H, Huang H, Halushka MK, Gautam S, Basso C, Thiene G, Tsatsopoulou A, Protonotarios N, McKenna WJ, Calkins H, Saffitz JE. A new diagnostic test for arrhythmogenic right ventricular cardiomyopathy. N Eng J Med. 2009;360:1075–1084. doi: 10.1056/NEJMoa0808138. [DOI] [PubMed] [Google Scholar]
- 5.Noorman M, Hakim S, Kessler E, Groneweg JA, Cox MG, Asimaki A, van Rijen HV, van Stuijvenberg L, Chkourko H, van der Heyden MA, Vos MA, de Jonge N, van der Smagt JJ, Dooijes D, Vink A, de Weger RA, Varro A, de Bakker JM, Saffitz JE, Hund TJ, Mohler PJ, Delmar M, Hauer RN, van Veen TA. Remodeling of the cardiac sodium channel, connexin43 and plakoglobin at the intercalated disk in patients with arrhythmogenic cardiomyopathy. Heart Rhythm. 2013;10:412–419. doi: 10.1016/j.hrthm.2012.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fidler LM, Wilson GJ, Liu F, Cui X, Scherer SW, Taylor GP, Hamilton RM. Abnormal connexin43 in arrhythmogenic right ventricular cardiomyopathy caused by plakophilin-2 mutations. J Cell Mol Med. 2009;13:4219–4228. doi: 10.1111/j.1582-4934.2008.00438.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Garcia-Gras E, Lombardi R, Giocondo MJ, Willerson JT, Schneider MD, Khoury DS, Marian AJ. Suppression of canonical Wnt/beta-catenin signaling by nuclear plakoglobin recapitulates phenotype of arrhythmogenic right ventricular cardiomyopathy. J Clin Invest. 2006;116:2012–2021. doi: 10.1172/JCI27751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kaplan SR, Gard JJ, Protonotarios N, Tsatsopoulou A, Spilliopoulou C, Anastasakis A, Squarcioni CP, McKenna WJ, Thiene G, Basso C, Brousse N, Fontaine G, Saffitz JE. Remodeling of myocyte gap junctions in arrhythmogenic right ventricular cardiomyopathy due to a deletion in plakoglobin (Naxos disease) Heart Rhythm. 2004;1:3–11. doi: 10.1016/j.hrthm.2004.01.001. [DOI] [PubMed] [Google Scholar]
- 9.Asimaki A, Kapoor S, Plovie E, Karin Arndt A, Adams E, Liu Z, James CA, Judge DP, Calkins H, Churko J, Wu JC, MacRae CA, Kleber AG, Saffitz JE. Identification of a new modulator of the intercalated disc in a zebrafish model of arrhythmogenic cardiomyopathy. Sci Transl Med. 2014;6:240ra74. doi: 10.1126/scitranslmed.3008008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kim C, Wong J, Wen J, Wang S, Wang C, Spiering S, Kan NG, Forcales S, Puri PL, Leone TC, Marine JE, Calkins H, Kelly DP, Judge DP, Chen HS. Studying arrhythmogenic right ventricular dysplasia with patient-specific iPSCs. Nature. 2013;494:105–110. doi: 10.1038/nature11799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Caspi O, Huber I, Gepstein A, Arbel G, Maizels K. Boulos M, Gepstein L. Modeling of arrhythmogenic right ventricular cardiomyopathy with human induced pluripotent stem cells. Circ Cardiovasc Genet. 2013;6:557–568. doi: 10.1161/CIRCGENETICS.113.000188. [DOI] [PubMed] [Google Scholar]
- 12.Marcus FI, McKenna WJ, Sherrill D, Basso C, Bauce D, Bluemke DA, Calkins H, Corrado D, Cox MG, Daubert JP, Fontaine G, Gear K, Hauer R, Nava A, Picard MH, Protonotarios N, Saffitz JE, Sanborn DM, Steinberg JS, Tandri H, Thiene G, Towbin JA, Tsatsopoulou A, Wichter T, Zareba W. Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia: proposed modification of the task force criteria. Circulation. 2010;121:1533–1541. doi: 10.1161/CIRCULATIONAHA.108.840827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McKoy G, Protonotarios N, Crosby A, Tsatsopoulou A, Anastasakis A, Coonar A, Norman M, Baboonian C, Jeffery S, McKenna WJ. Identification of a deletion in plakoglobin in arrhythmogenic right ventricular cardiomyopathy with palmoplantar keratoderma and woolly hair (Naxos disease) Lancet. 2000;355:2119–2124. doi: 10.1016/S0140-6736(00)02379-5. [DOI] [PubMed] [Google Scholar]
- 14.Presland EB, Dale BA. Epithelial structural proteins of the skin and oral cavity: function in health and disease. Crit Rev Oral Biol Med. 2000;11:383–408. doi: 10.1177/10454411000110040101. [DOI] [PubMed] [Google Scholar]
- 15.Wong LS, Huzen J, de Boer RA, van Gilst WH, van Veldhuisen DJ, van der Harst P. Telomere length of circulating leukocyte subpopulations and buccal cells in patients with ischemic heart failure and their offspring. PLoS One. 2011;6:e23118. doi: 10.1371/journal.pone.0023118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shah SA, Clyne CA, Henyan N, Migeed M, Yarlaqadda R, Silver BB, Kluger J, White CM. Impact of magnesium sulfate on serum magnesium concentrations and intracellular electrolyte concentrations among patients undergoing radio frequency catheter ablation. Conn Med. 2008;72:261–265. [PubMed] [Google Scholar]
- 17.Frank DK, Szymkowiak B, Hughes CA. Connexin expression and gap junctional intercellular communication in human squamous cell carcinoma of the head and neck. Otolaryngol Head Neck Surg. 2006;135:736–743. doi: 10.1016/j.otohns.2006.06.1242. [DOI] [PubMed] [Google Scholar]
- 18.Brockmeyer P, Jung K, Perske C, Schliephake H, Hemmerlein B. Membrane connexin 43 acts as an independent prognostic marker in oral squamous cell carcinoma. Int J Oncol. 2014;45:273–281. doi: 10.3892/ijo.2014.2394. [DOI] [PubMed] [Google Scholar]
- 19.Hartzfeld M. Plakophilins: multifunctional proteins or just regulators of desmosomal adhesion? Biochim Biophys Acta. 2007;1773:69–77. doi: 10.1016/j.bbamcr.2006.04.009. [DOI] [PubMed] [Google Scholar]
- 20.Neuber S, Muhmer M, Wratten D, Koch PJ, Moll R, Schmidt A. The desmosomal plaque proteins of the plakophilin family. Dermatol Res Pract. 2010;101425 doi: 10.1155/2010/101452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Al-Amoudy A, Castano-Diez D, Devos DP, Russell RB, Johnson GT, Frangakis AS. The three-dimensional molecular structure of the desmosome. Proc Natl Acad Sci USA. 2011;108:6480–6485. doi: 10.1073/pnas.1019469108. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.