

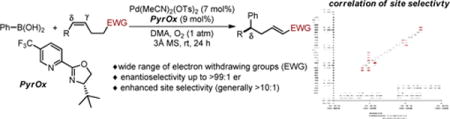
Abstract
A highly enantio- and site selective Pd-catalyzed arylation of alkenes linked to carbonyl derivatives to yield α,β-unsaturated systems is reported. The high site selectivity is attributed to both a solvent effect and the polarized nature of the carbonyl group, both of which have been analyzed through multidimensional analysis tools. The reaction can be performed in an iterative fashion allowing for a diastereoselective installation of two aryl groups along an alkyl chain.
Graphical abstract
Developing methods for site selective migratory insertion into relatively unbiased multi-substituted alkenes would be an enabling approach to synthesis.1 Of specific interest is precisely functionalizing internal alkenes with enantio- and site control for remotely installing groups in molecules.2–6 However, this prospect is difficult to achieve as alkenes of this sort are distantly located from a discriminating functional group, which removes obvious innate factors for governing site selectivity.1 In this regard, our discovery that alkenes positioned at various distances from an alcohol undergo site selective addition of a arly-Pd intermediate is unique (Figure 1a).7 The origin of this site selectivity has been investigated through empirical exploration of scope in combination with computational,8 labeling,8c and correlative studies.8d While the detailed origin of selectivity for the addition of the organometallic at the carbon distal to the alcohol is still under investigation, the vast majority of the studies point to a remote electronic effect that is modestly influenced by alkene substituent size. The electronic effect is thought to arise from reducing the dipole during the formation of the C–C bond through the orientation of the alcohol and the arene (compare TS A and B). Therefore, changing the substituent on the arene and the distance between the alcohol and the alkene influences site selectivity.
Figure 1.
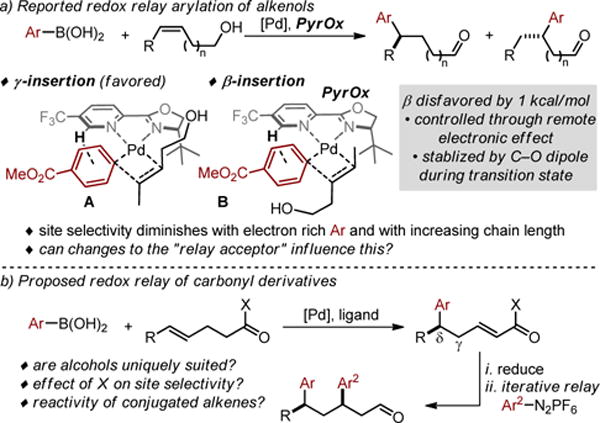
a) Previously reported enantioselective relay Heck reactions of alkenols including mechanistic analysis. b) Proposed new enantioselective relay Heck reaction of alkenes linked to carbonyl derivatives and resultant iterative Heck reaction.
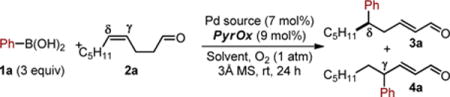
This hypothesis provides the impetus for further exploration of substrate scope in terms of replacing the alcohol with an alternative functional group. We were attracted to carbonyls due to three factors. First, the products of the enantioselective relay Heck reaction are remotely functionalized α,β-unsaturated carbonyls, which are envisioned to be useful for further elaboration and would be difficult to access using conventional strategies (Figure 1b). Second, the diversity of carbonyls would presumably allow for a wider range of observed site selectivity as a function of such groups, providing the basis for additional mechanistic understanding and enhancing selectivity.8 Finally, reduction of the resultant α,β-unsaturated carbonyl provides an allylic alcohol, which would be suitable for an iterative relay Heck reaction (Figure 1b).7a However, we also envisioned many potential issues including: further reaction of the α,β-unsaturated carbonyl products under the reaction conditions, isomerization of the alkenes into conjugation since Pd-hydrides are formed in these reactions, and the potential deleterious effect of these new functional groups on enantioselectivity and yield. Herein, we present the successful development of an enantioselective relay Heck reaction of alkenes linked to carbonyl derivatives, which allows access to an array of α,β-unsaturated carbonyl products with remotely installed aryl groups. Of particular interest, these arylations have enhanced site selectivity compared to the previous variants using alkenols and provide an opportunity for further interrogation of site selective migratory insertion of unbiased alkenes. Based on our previously reports,7 our study commenced with the reaction of phenylboronic acid (1a) and (Z)-dec-4-enal (2a) catalyzed by Pd ligated with PyrOx under oxidative Heck-type conditions (Table 1, entry 1). To our delight, the reaction performed well yielding the product in 73% and a 98:2 er without the need of a copper cocatalyst, thus allowing a reduction in the overall ligand loading. Under these conditions, the site selectivity was modest favoring the δ product in a 5.8:1 ratio. No reaction of the α,β-unsaturated aldehyde product is observed suggesting that the catalyst prefers reaction with electron rich alkenes.9 Control experiments confirmed the requirement of Pd (Table 1, entry 2), molecular sieves (entry 3), and PyrOx (entry 4). Slightly reduced yields are observed using air instead of pure O2 as the terminal oxidant albeit in excellent enantioselectivity (entry 5). As a significant goal of this project was to increase site selectivity, we further explored the choice of solvent. Interestingly, when DMA was used as solvent, the site selectivity significantly increased favoring the δ product in a 15:1 ratio with modestly enhanced yield and er (entry 6). Other solvents were also competent but did not improve the results further (entries 7–8).
Table 1.
Optimization of the enantioselective relay Heck reaction of 2a.
| |||||
|---|---|---|---|---|---|
| Entry | Pd source | Solvent | yield of 3a (%)a | er (3a)b | δ:γc |
| 1 | Pd(MeCN)2(OTs)2 | DMF | 73 | 98:2 | 5.8 |
| 2 | None | DMF | – | – | – |
| 3 | Pd(MeCN)2(OTs)2d | DMF | trace | – | – |
| 4 | Pd(MeCN)2(OTs)2e | DMF | trace | – | – |
| 5 | Pd(MeCN)2(OTs)2f | DMF | 65 | 98:2 | 5.2 |
| 6 | Pd(MeCN)2(OTs)2 | DMA | 78 | 99:1 | 15 |
| 7 | Pd(MeCN)2(OTs)2 | 1,4-dioxane | 73 | 98:2 | 7.8 |
| 8 | Pd(MeCN)2(OTs)2 | THF | 38 | 98:2 | 9.3 |
Conditions: 1a (0.75 mmol, 3.0 eq), 2a (0.25 mmol, 1.0 eq), [Pd] (0.01875 mmol, 7.5 mol%), ligand (0.0225 mmol, 9 mol%), solvent (3 mL), O2 (1 atm), rt, 24 h.
Reported yields are of isolated 3a as a >20:1 ratio of the E-alkene isomer.
Determined by SFC equipped with a chiral stationary phase.
Determined by NMR.
In the absence of 3Å MS.
In the absence of ligand.
Using air (1 atm). DMF = N,N-dimethylformamide; DMA = N,N-dimethylacetamide, THF = tetrahydrofuran.
This observed improvement in site selectivity is notable and inspired us to investigate the potential origin of the solvent effect.10 Therefore, an array of various substituted amide solvents were evaluated in the reaction (Figure 2). To correlate these results, we performed multidimensional analysis relating the measured ΔΔG‡ for site selectivity to various parameters describing the different amide solvents (see Supporting Information for details). An excellent correlation was achieved relating the IR intensity of the carbonyl stretch (IC=O) and Sterimol B1 value of the substituent R to the measured site selectivity.11 This relationship was validated using Leave-One-Out analysis with a Q2=0.96. As illustrated by the coefficients in the regression model, decreasing the IC=O and increasing the Sterimol B1 of the R group of the solvent results in higher site selectivity. This suggests that the polarization model for site selectivity described in the introduction is impacted by both the dipole and orientation of the solvent. It should be noted that improved site selectivity does not necessary lead to higher yields and DMA was found to balance both of these requirements.
Figure 2.
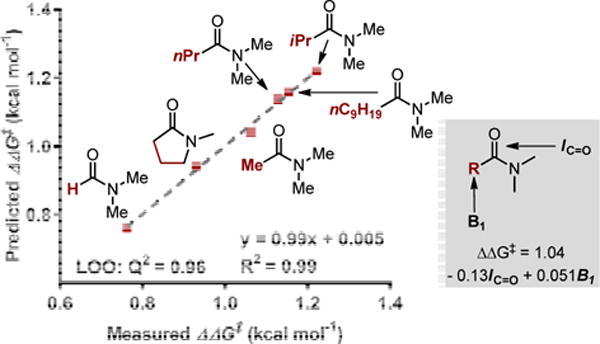
Multidimensional correlation of solvent parameters with site selectivity.
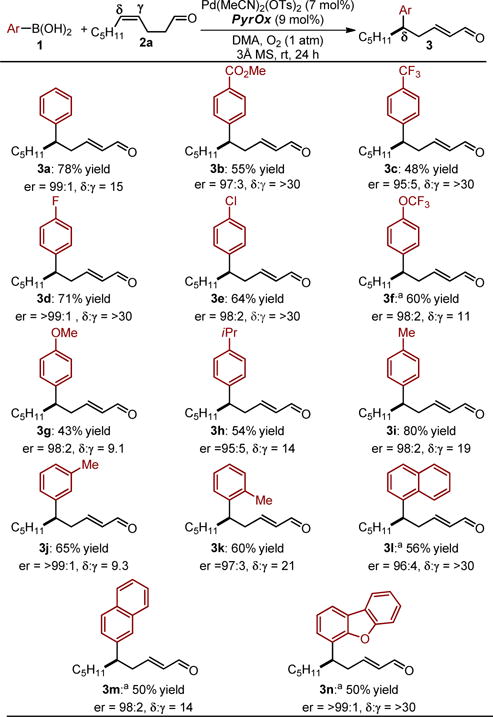
Using DMA as solvent under the optimal conditions, the scope of aryl substituted boronic acid (1) was probed in the formation of various δ-aryl substituted α,β-unsaturated aldehydes (Table 2). In general, modest to good yields using either electron rich or deficient aryl substituted boronic acids is observed as a single geometrical alkene isomer. In all cases, the enantioselectivity are excellent. Previously, we found a significant reduction of site selectivity when electron rich aryl boronic acids were employed. In fact, the best site selectivity previously observed for the formation of a δ-aryl product was 7:1 with an electron poor boronic acid. In this system, site selectivity is >9:1 favoring the δ-product for both electron rich and poor partners with electron poor coupling partners providing even higher levels of control (often >30:1). Of note, some yields were improved by the addition of Cu(OTf)2 (see table).12,13
Table 2.
Aryl boronic acid scope.
|
Standard reaction conditions: 1 (0.75 mmol, 3.0 eq), 2a (0.25 mmol, 1.0 eq), Pd(MeCN)2(OTs)2 (0.019 mmol, 7.5 mol%), ligand (0.023 mmol, 9 mol%), DMA (3 mL), 3Å Ms (50 mg), O2 (1 atm), rt, 24 h. Yields are of isolated 3, >20:1 E-alkene product. Enantioselectivty determined by SFC equiped with a chiral stationary phase.
Cu(OTf)2 (0.013 mmol, 5 mol%), ligand (0.035 mmol, 14 mol%) were used.
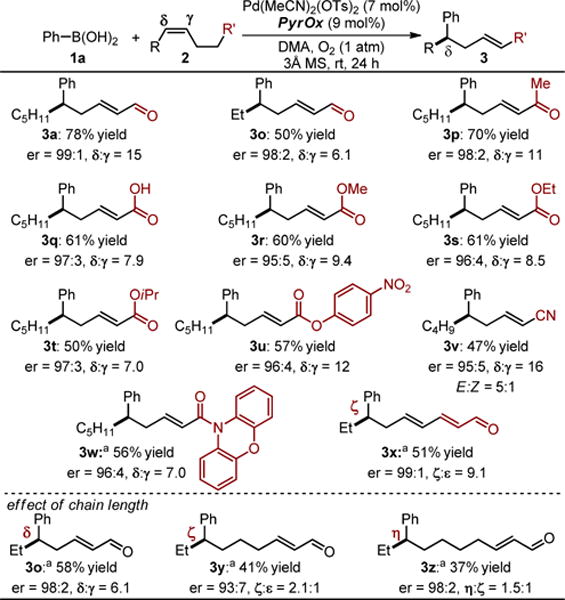
The second stage of scope evaluation was focused on the nature of the carbonyl linked to the alkene (Table 3). As discussed above, we believed that this would be an opportunity to further probe the origin of site selectivity. In general, a wide range of functional groups are compatible with the reaction including a ketone (3p) and a carboxylic acid (3q). Various esters are excellent substrates for the reaction (3r-t) including the highly versatile and sensitive p-nitrophenol derived substrate (3u). Both a nitrile (3v) and amide (3w) perform well in the reaction although a 5:1 mixture of alkene geometrical isomers is observed using the smaller nitrile. Finally, a substrate containing an α,β-unsaturated aldehyde yields the conjugated product 3x with a ζ-aryl substitution in excellent enantioselectivity and site selectivity. Unfortunately, site selectivity and yield diminish significantly as the chain length is extended between the alkene and the aldehyde (3y–z) similar to our previous report.7b
Table 3.
Evaluation of functional group tolerance of the alkene substituent.
|
Standard reaction conditions: 1a (0.75 mmol, 3.0 eq), 2 (0.25 mmol, 1.0 eq), Pd(MeCN)2(OTs)2 (0.019 mmol, 7.5 mol%), ligand (0.023 mmol, 9 mol%), DMA (3 mL), 3Å Ms (50 mg), O2 (1 atm), rt, 24 h. Yields of isolated 3, >20:1 E-alkene product. Enantioselectivty determined by SFC equiped with a chiral stationary phase.
Yields are a combination of 3 and 4, >20:1 E-alkene product.
On the basis of the range of selectivity observed as a function of the carbonyl in the substrate, we explored how the carbonyl may be impacting site selectivity. To accomplish this, the substrates containing only the C5H11 group with different carbonyls were submitted for geometry minimizations and IR frequency calculations.8d Various parameters were compiled including point charges on a number of atoms and IR frequencies and intensities of the functional groups. A good correlation was found between the NBO charge of the carbonyl oxygen (Figure 3)14 and the measured site selectivity. The aldehyde substrate corresponding to product 3a with the least negative NBO charge on the carbonyl oxygen resulted in the highest site selectivity, while the ester substrate corresponding to product 3t, which has the greatest negative NBO charge on the carbonyl oxygen, resulted in the lowest site selectivity. Despite the distance of the functional group from the alkene, the optimized geometries of the substrates illustrate bending of the substrate to allow alignment of the carbonyl with the alkene (see Supporting Information). Additionally, the greatest NBO charge difference between the alkene carbons (Cδ=Cγ) was observed for the substrate that provided the highest site selectivity (2a), illustrating the remote effect of alkene polarization on the migratory insertion step.
Figure 3.
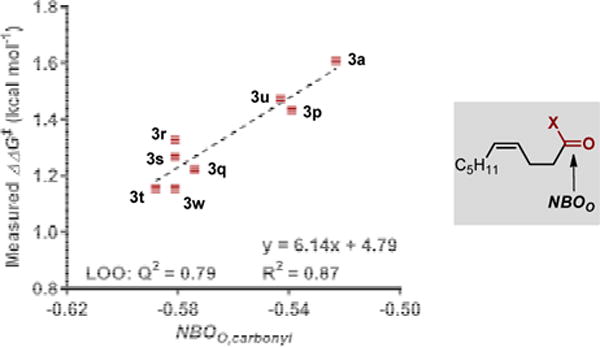
Correlation of observed site selectivity to the NBO charge of the carbonyl oxygen.
As this method provides direct access to highly enantiomerically-enriched α,β-unsaturated aldehydes, we envisioned exploiting these products in iterative relay Heck reactions. This three step approach would facilitate the 1,3 difunctionalization of a simple alkyl chain in a modular fashion. To test this possibility, the carbonyl of product 3a was reduced to yield an allylic alcohol (5), which upon treatment with an aryl diazonium salt under conditions from our previously reported relay Heck reaction7a forms product 6 in high diastereoselectivity (eq. 1). Using the enantiomer of ligand provides access to the other diastereomer in good albeit modestly reduced diastereoselectivity (eq. 2), suggesting mainly catalyst controlled addition.
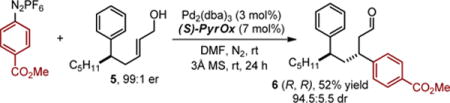 |
(1) |
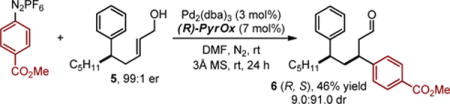 |
(2) |
In conclusion, we have reported a unique method to produce α,β-unsaturated carbonyls containing an aryl group δ to the carbonyl in high enantioselectivity. The scope of the reaction is broad in both the aryl coupling partners and the carbonyl derivatives. Of particular interest is the evolving understanding of substrate substituent effects on site selective migratory insertion. The subtle influence of the remote carbonyl has been studied through correlative tools providing further evidence that polarization of the substrate during migratory insertion is essential for effective site control. This important finding should allow us to design more versatile catalysts and systems to achieve greater site selection at even more remote sites. Exploring this goal in the context of further expansion of the enantioselective relay Heck reaction is underway.
Supplementary Material
Acknowledgments
This research reported in this publication was supported by the national Institutes of Health under award number RO1GM063540.
Footnotes
Supporting Information. Experimental procedures, analytical data for products, NMR spectra of products. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.For selected reviews, see:; (a) Sigman MS, Werner EW. Acc Chem Res. 2012;45:874. doi: 10.1021/ar200236v. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Cartney DMc, Guiry PJ. Chem Soc Rev. 2011;40:5122. doi: 10.1039/c1cs15101k. [DOI] [PubMed] [Google Scholar]; (c) Shibasaki M, Vogl EM, Ohshima T. Adv Synth Catal. 2004;346:1533. [Google Scholar]; (d) Dounay AB, Overman LE. Chem Rev. 2003;103:2945. doi: 10.1021/cr020039h. [DOI] [PubMed] [Google Scholar]; (e) Beletskaya IP, Cheprakov AV. Chem Rev. 2000;100:3009. doi: 10.1021/cr9903048. [DOI] [PubMed] [Google Scholar]
- 2.For selected reviews, see:; (a) Sun Y-W, Zhu P-L, Xu Q, Shi M. RSC Adv. 2013;3:3153. [Google Scholar]; (b) Hawner C, Alexakis A. Chem Commun. 2010;46:7295. doi: 10.1039/c0cc02309d. [DOI] [PubMed] [Google Scholar]; (c) Harutyunyan SR, den Hartog T, Geurts K, Minnaard AJ, Feringa BL. Chem Rev. 2008;108:2824. doi: 10.1021/cr068424k. [DOI] [PubMed] [Google Scholar]; (d) Hayashi T, Yamasaki K. Chem Rev. 2003;103:2829. doi: 10.1021/cr020022z. [DOI] [PubMed] [Google Scholar]
- 3.For other strategies to construct remote chiral center, see:; (a) Zultanski SL, Fu GC. J Am Chem Soc. 2011;133:15362. doi: 10.1021/ja2079515. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Smith SW, Fu GC. J Am Chem Soc. 2009;131:14231. doi: 10.1021/ja9061823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.For selected examples of using a redox relay strategy, see:; (a) Renata H, Zhou Q, Baran PS. Science. 2013;339:59. doi: 10.1126/science.1230631. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Aspin S, Goutierre A-S, Larini P, Jazzar R, Baudoin O. Angew Chem, Int Ed. 2012;51:10808. doi: 10.1002/anie.201206237. [DOI] [PubMed] [Google Scholar]; (c) Stokes BJ, Opra SM, Sigman MS. J Am Chem Soc. 2012;134:11408. doi: 10.1021/ja305403s. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Weinstein AB, Stahl SS. Angew Chem, Int Ed. 2012;51:11505. doi: 10.1002/anie.201206702. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) McDonald RI, White PB, Weinstein AB, Tam CP, Stahl SS. Org Lett. 2011;13:2830. doi: 10.1021/ol200784y. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) McDonald RI, Stahl SS. Angew Chem, Int Ed. 2010;49:5529. doi: 10.1002/anie.200906342. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Melpolder JB, Heck RF. J Org Chem. 1976;41:265. [Google Scholar]; (h) Heck RF. J Am Chem Soc. 1968;90:5526. [Google Scholar]
- 5.For recent examples of isomerization/migration in Heck-type reactions, see:; (a) Kochi T, Hamasaki T, Aoyama Y, Kawasaki J, Kakiuchi F. J Am Chem Soc. 2012;134:16544. doi: 10.1021/ja308377u. [DOI] [PubMed] [Google Scholar]; (b) Crawley ML, Phipps KM, Goljer I, Mehlmann JF, Lundquist JT, Ullrich JW, Yang C, Mahaney PE. Org Lett. 2009;11:1183. doi: 10.1021/ol900036y. [DOI] [PubMed] [Google Scholar]
- 6.For recent examples of highly regioselective or asymmetric Heck reactions see:; (a) Holder JC, Zou L, Marziale AN, Liu P, Lan Y, Gatti M, Kikushima K, Houk KN, Stoltz BM. J Am Chem Soc. 2013;135:14996. doi: 10.1021/ja401713g. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Qin L, Ren X, Lu Y, Li Y, Zhou J. Angew Chem, Int Ed. 2012;51:5915. doi: 10.1002/anie.201201806. [DOI] [PubMed] [Google Scholar]; (c) Werner EW, Sigman MS. J Am Chem Soc. 2011;133:9692. doi: 10.1021/ja203164p. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Zhu C, Falck JR. Angew Chem, Int Ed. 2011;50:6626. doi: 10.1002/anie.201101857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (e).Yoo KS, O’Neil J, Sakaguchi S, Giles R, Lee JH, Jung KW. J Org Chem. 2010;75:95. doi: 10.1021/jo901977n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.(a) Werner EW, Mei T-S, Burckle AJ, Sigman MS. Science. 2012;338:1455. doi: 10.1126/science.1229208. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Mei T-S, Werner EW, Burckle AJ, Sigman MS. J Am Chem Soc. 2013;135:6830. doi: 10.1021/ja402916z. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Mei T-S, Patel HH, Sigman MS. Nature. 2014;508:340. doi: 10.1038/nature13231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.(a) Xu L, Hilton MJ, Zhang X, Norrby P-O, Wu Y-D, Sigman MS, Wiest O. J Am Chem Soc. 2014;136:1960. doi: 10.1021/ja4109616. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Dang Y, Qu S, Wang Z-X, Wang X. J Am Chem Soc. 2014;136:986. doi: 10.1021/ja410118m. [DOI] [PubMed] [Google Scholar]; (c) Hilton MJ, Xu L-P, Norrby P-O, Wu Y-D, Wiest O, Sigman MS. J Org Chem. 2014;79:11841. doi: 10.1021/jo501813d. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Milo A, Bess EN, Sigman MS. Nature. 2014;507:210. doi: 10.1038/nature13019. [DOI] [PubMed] [Google Scholar]
- 9.Patel HH, Sigman MS. J Am Chem Soc. 2015;137:3462. doi: 10.1021/ja5130836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.For examples demonstrating the relationship between selectivity and solvent, see:; (a) Reichardt C. Angew Chem, Int Ed. 1979;18:98. [Google Scholar]; (b) MacManus DA, Vulfson EN. Enzyme Microb Technol. 1997;20:225. [Google Scholar]
- 11.Verloop A. Drug Design. Academic Press; New York: 1976. [Google Scholar]
- 12.For reviews on Pd-catalyzed aerobic oxidative reactions, see:; (a) Campbell AN, Stahl SS. Acc Chem Res. 2012;45:851. doi: 10.1021/ar2002045. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Wu W, Jiang H. Acc Chem Res. 2012;45:1736. doi: 10.1021/ar3000508. [DOI] [PubMed] [Google Scholar]; (c) Shi Z, Zhang C, Tang C, Jiao N. Chem Soc Rev. 2012;41:3381. doi: 10.1039/c2cs15224j. [DOI] [PubMed] [Google Scholar]; (d) Sigman MS, Jensen DR. Acc Chem Res. 2006;39:221. doi: 10.1021/ar040243m. [DOI] [PubMed] [Google Scholar]; (e) Gligorich KM, Sigman MS. Angew Chem Int Ed. 2006;45:6612. doi: 10.1002/anie.200602138. [DOI] [PubMed] [Google Scholar]; (f) Punniyamurthy T, Velusamy S, Iqbal J. Chem Rev. 2005;105:2329. doi: 10.1021/cr050523v. [DOI] [PubMed] [Google Scholar]; (g) Stahl SS. Angew Chem, Int Ed. 2004;43:3400. doi: 10.1002/anie.200300630. [DOI] [PubMed] [Google Scholar]
- 13.For selected reviews on Cu-catalyzed aerobic oxidative reactions, see:; Zhang C, Tang C, Jiao N. Chem Soc Rev. 2012;41:3464. doi: 10.1039/c2cs15323h. [DOI] [PubMed] [Google Scholar]
- (b).Wendlandt AE, Suess AM, Stahl SS. Angew Chem, Int Ed. 2011;50:11062. doi: 10.1002/anie.201103945. [DOI] [PubMed] [Google Scholar]
- 14.(a) Foster JP, Weinhold F. J Am Chem Soc. 1980;102:7211. [Google Scholar]; (b) Reed AE, Weinstock RB, Weinhold F. The Journal of chemical physics. 1985;83:735. [Google Scholar]; (c) Reed AE, Curtiss LA, Weinhold F. Chem Rev. 1988;88:899. [Google Scholar]; (d) Glendening ED, Landis CR, Weinhold F. Wiley Interdisciplinary Reviews: Computational Molecular Science. 2012;2:1. doi: 10.1002/wcms.74. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Weinhold F. J Comput Chem. 2012;33:2363. doi: 10.1002/jcc.23060. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.