

Abstract
We report a highly enantioselective intermolecular Heck reaction of alkenyl triflates and acyclic primary or racemic secondary alkenols. The mild reaction conditions permit installation of a wide range of alkenyl groups at positions β, γ or δ to a carbonyl group in high enantioselectivity. The success of this reaction is attributed to the use of electron-withdrawing alkenyl triflates, which offer selective β-hydride elimination followed by migration of the catalyst through the alkyl chain to give the alkenylated carbonyl products. The synthetic utility of the process is demonstrated by a two-step modification of a reaction product to yield a tricyclic core structure, present in various natural products.
TOC image
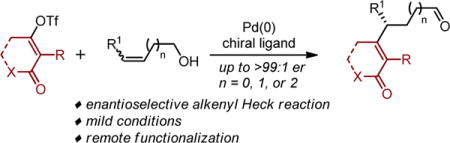
Installation of an alkenyl motif represents a widely used strategy for introducing molecular complexity found in natural products and pharamceuticals.1 Therefore, enantioselective alkenylation has been a cornerstone of synthetic methods development. However, most methods that enable the introduction of alkenyl/vinyl moieties enantioselectively are restricted to the use of biased α,β unsaturated carbonyls,2 allylic moeities,3 or other functional groups in close proximity to the reaction site.4 An alternative powerful strategy for the stereoselective introduction of alkenyl groups has been the enantioselective intramolecular Heck reaction of vinyl electrophiles. In spite of its extensive use in natural product synthesis,5 the effective installation of alkenyl units in an intermolecular fashion has been limited to the use of classical cyclic enol ether substrates.6 Herein, we report a method that overcomes this considerable limitation wherein a broad array of alkenyl electrophiles can be combined with acyclic alkenols of various chain lengths to yield alkenylated Heck type products in high enantioselectivity.
We have reported redox-relay Heck reactions of acyclic alkenols, wherein aryl groups derived from either aryldiazaonium salts7a or aryl boronic acids7b can be added enantioselectively to an alkenol followed by Pd-migration through the alkyl chain to form a carbonyl product (Figure 1a). This process provides a platform for setting absolute stereochemistry remotely to other functionality in a molecule, which was recently highlighted by the use of trisubstituted alkenes to generate quaternary centers at positions β-, γ-, δ-, ɛ- and ζ- to a carbonyl group.8 Even though the redox relay strategy leads to remote enantioselective functionalization, the limitation remains that only aryl groups can be effectively introduced using this methodology. In this regard, we sought to develop a general intermolecular enantioselective alkenylative relay Heck reaction, which would install a new alkene motif at distinct positions remote from a carbonyl (Figure 1b).
Figure 1.
a) Enantioselective arylative relay Heck reaction b) Proposed enantioselective alkenylative relay Heck reaction c) Mechanistic hypothesis d) Potential challenge e) Preliminary result
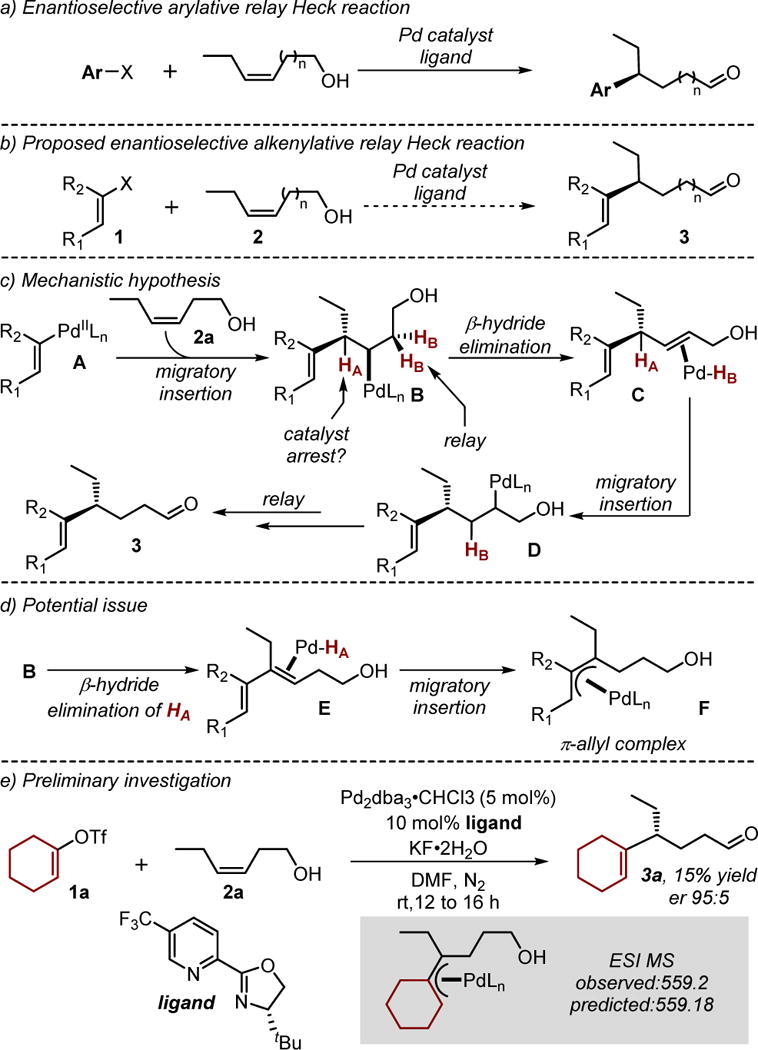
The dearth of examples in the area of intermolecular Heck reactions of alkenyl electrophiles is evidence that significant challenges exist to develop viable methods.9 These likely arise from issues in site selection with the first of which related to the initial migratory insertion of a relatively unbiased alkene into the Pd-alkenyl species akin to A proceeding to B in Figure 1c.10, 11 In our previous arylative redox relay Heck reactions,7 we observed that substrate control via remote electronic bias is responsible for effective site selection at the alkene carbon distal to the alcohol. This result is consistent with computational analysis wherein the polarization of the alkene in the transition state is stabilized by remote C–O dipole interactions.12 Therefore, we anticipated that site selective migratory insertion could be adequately addressed using this substrate class. Another problem we suspected to be of greater significance is that of site selective β-hydride elimination from an intermediate like B.11 If β-hydride elimination occurs at HB, the relay reaction is expected to proceed, revealing the carbonyl product through iterative β-hydride elimination and migratory insertion events (B→C→D→3). However, if HA undergoes β-hydride elimination yielding E (Figure 1d), migratory insertion of the alkene into the Pd-hydride would result in a π-allyl complex, which could arrest catalysis due to its relative stability. The inherent difference between installing an alkenyl versus an aryl group is the formation of both a more stabilized diene intermediate E compared to a styrene and a π-allyl compared to a π-benzyl intermediate. In fact, Pd-π-allyl intermediates have been exploited in our group for difunctionalization reactions of olefins using this precise strategy.13
To initiate the investigation, alkenyl triflates were considered to be likely suitable coupling partners, as their oxidative addition is facile and the resultant cationic PdII-alkenyl intermediate would be highly electrophilic, a requisite for the effective relay Heck reactions due to enhance alkene binding properties.7,10 Our initial efforts using a simple cyclohexenyl triflate 1a with (Z)-alkenol 2a, in the presence of a chiral pyrox ligand, resulted in the formation of the desired product 3a in 15% yield albeit a good enantiomeric ratio of 95:5 (Figure 1e).14 Attempts to improve the yield of this reaction were met with similar outcomes. Analyzing the reaction mixture with ESI-MS provided evidence of a species with a composition to that of the proposed π-allyl intermediate shown in Figure 1e.15 Such a π-allyl species was anticipated as a result of non-selective β-hydride elimination from an intermediate resembling B in figure 1c, as discussed above.
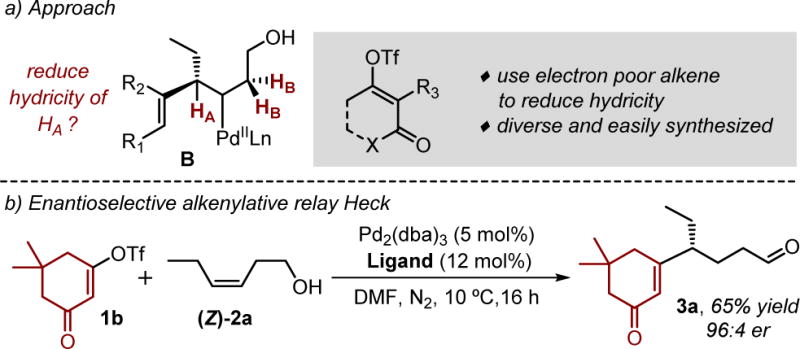
To tackle this issue, we reexamined putative intermediate B and considered approaches to circumvent catalyst arrest derived from formation of a π-allyl intermediate through β-hydride elimination of HA (Figure 2a). In the most simplistic terms, to promote the relay Heck process, the hydricity of HA needs to be reduced.16 As a general approach to accomplish this, the use of electron deficient alkenyl triflates derived from β-dicarbonyl compounds were considered, since the electron withdrawing nature of the resulting intermediate should avoid formation of the π-allyl intermediate. Finally, an attractive aspect of alkenyl triflates derived from β-dicarbonyl compounds is their modular, facile synthesis.17 To test this hypothesis, alkenyl triflate 1b and (Z)-2a were treated with a Pd(0) source in DMF under the conditions in Figure 1e. In this initial event, a modest, albeit significantly improved yield (53%) of the desired aldehyde 3a was observed in 92:8 er. Further improvement of the reaction conditions as depicted in Figure 2b allowed for a 65% yield and 96:4 er of 3a to be obtained (See SI for detailed optimization of the reaction).18 As an important note, the resulting electrophilic alkene in the product remains untouched in this reaction suggesting a high specificity for electron-rich alkenes using this catalyst.
Figure 2.
a) Approach to overcome the formation of π-allyl intermediates b) Successful enantioselective alkenylative relay Heck reaction
The scope of the enantioselective alkenylation reaction utilizing the redox-relay Heck strategy was evaluated using both (Z)- and (E)-alkenols as substrates with a wide range of alkenyl triflates (Table 1). Enantiomeric products are observed using the alkenol stereoisomers as demonstrated by the use of alkenyl triflate 2a. The (Z)-alkene yields the (R)-enantiomer of 4a in 96:4 er whereas 4:96 er is observed using the corresponding (E)-alkene. A simple cyclohexenone derived alkenyl triflate performs similarly providing the desired product 4b in good yield and enantioselectivity. A rather hindered tetrasubstituted alkenyl triflate led to the formation of 4c in good yield and excellent enantioselectivity using either stereoisomer of the alkenol substrate (er 1: >99 with (E)-alkene). Of particular interest, the reaction proceeds well, even in the presence of an alkenyl bromide resulting in a highly enantioselective alkenylative relay Heck reaction to form 4d.
Table 1.
Scope of the vinyl triflate coupling partner in the enantioselective alkenylative relay Heck reaction using homoallylic alcohols.

|
Yields are reported as an average of two parallel experiments. Ehantioselectivity determined by SFC equipped with a chiral column. Reaction performed on 0.5 mmol scale. Absolute configuration determined to be (R) for a synthetic derivative of 4b arising from the (Z)-alkene. All others were assigned by analogy.
Reaction performed at room temperature.
In order to probe the compatibility of an electron-withdrawing functional group other than ketone, several ester derived alkenyl triflates were evaluated (4e–4i). The first contained a Boc-protected amine in a ring and was effective albeit in modest yield to provide 4e with a 3:97 er using the (E)-alkene. A vinylogous cyclic ester derived alkenyl triflate was found to be an excellent substrate to afford 4f in good yields and high er (3:97) using the (E)-alkene. Next, (Z) and (E) acyclic enol triflates were stereoselectively synthesized using modular acetoacetate derivatives and applied to the relay Heck reaction.19 Although these alkenyl triflates were found to be less reactive, raising the temperature to room temperature provided the products in modest yields and good enantioselectivity (4g, 4h, 4i). The geometry of the alkene was preserved during the course of the reaction, showcasing the potential utility of this process. Finally, heteroaromatic derived alkenyl triflates are also effective coupling partners generating products 4j–4l in modest to good yield and high enantioselectivity. These building blocks are especially noteworthy as they incorporate privileged structures including the coumarin and napthoquinone moieties, which are often observed in biologically active compounds.20 Notably, the use of other electron withdrawing groups such as halogens and styrenyl units proved to be ineffective compared to a conjugated carbonyl system (See SI for details).
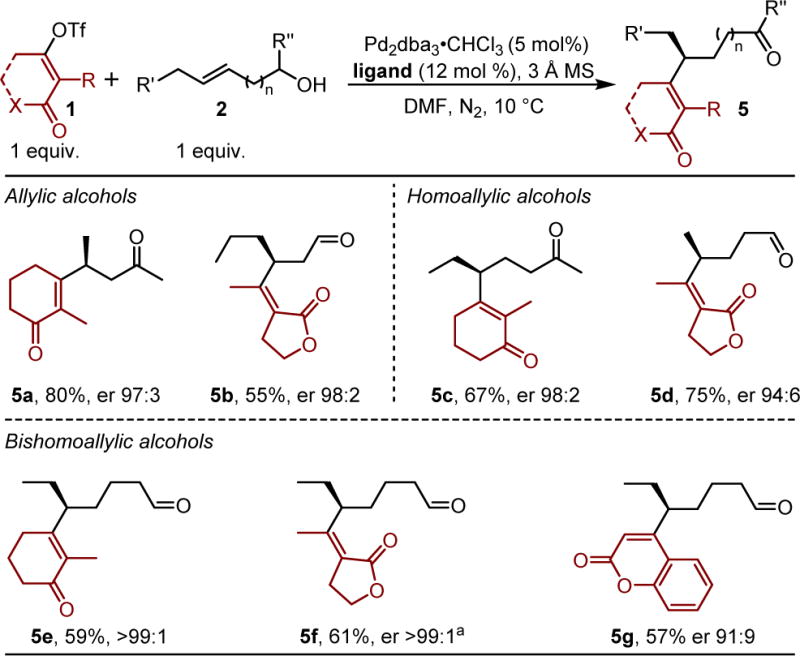
The scope of this method was further evaluated by varying the alkenyl alcohol (Table 2). A racemic secondary trans-allylic alcohol was converted to 5a in high yield and er of 97:3. The use of a similar allylic alcohol and vinylogous ester derived alkenyl triflate provided the β-substituted aldehyde 5b in high enantioselectivity. A racemic secondary homoallylic alcohol substrate was subjected to the alkenylation reaction, to give the γ-substituted ketone 5c in good yield and 98:2 er. A methyl substituted homoallylic alcohol was converted to 5d in high yield and good er. With an aim of setting chiral centers more remotely from the carbonyl functional group, a trans-bishomoallylic alcohol was subjected to the standard conditions using three different alkenyl triflates. Indeed, the alkenyl groups were installed enantioselectively, giving products 5e–5g in moderate yields. Exceptional enantioselectivity was observed for the formation of 5e and 5f but a reduction in enantioselectivity was observed using the coumarin derived alkenyl triflate. Additionally, a lower enantioselectivity was observed for product 5f, when the cis-bishomoallylic alcohol was used as substrate. Unfortunately, trisubstituted alkenols are unreactive under the current reaction conditions.
Table 2.
Evaluation of alkenol substrates in the enantioselective alkenylative relay Heck reaction
|
Yields are reported as an average of two parallel experiments. Enantiomeric ratio determined by SFC equipped with a chiral column. Reaction performed on 0.5 mmol scale.
Using the Z-alkene, a 57% yield and er of 10:90 was observed.
In general, the alkenyl Heck relay reaction with (E)-alkenes provides higher enantioselectivity compared to the use of (Z)-alkene substrates, especially when sterically hindered tetrasubstituted alkenyl triflates were employed. This is in contrast to what has been observed in the enantioselective arylation of acyclic alkenols, wherein similar levels of enantioselectivity was observed for both (Z) and (E)-alkenes.7 While a precise explanation for these differences is not clear at present, the absolute configuration of product 4b arising from the (Z)-alkene was determined to be (R), which is the same face from which the arene adds in our previous reports. This outcome indicates that the transition state during the migratory insertion step for the alkenylative and arylative Heck reactions are likely to be similar, as the ligand employed is the same in both reactions.
Many of the products accessed during this study are clearly unique chiral building blocks. To highlight the anticipated synthetic utility of this method, product 4b was further elaborated through two synthetic steps to access the tricyclic core of various natural products, including elisabethin A, elisapterosin A and colombiasin A.21 As depicted in Scheme 1, Horner-Wadsworth-Emmons olefination of the aldehyde 4b delivers diene 6 in high (E)-selectivity,22 which serves as a precursor for an intramolecular Diels-Alder reaction. Treatment of 6 under the conditions reported by Danishefsky and coworkers yields the tricylic product 7 as a single diastereomer.23 In all, four steps are required from the β-dicarbonyl precursor including a highly enantioselective alkenylative relay Heck and a diastereoselective intramolecular Diels-Alder reaction to access these interesting structures.
Scheme 1.
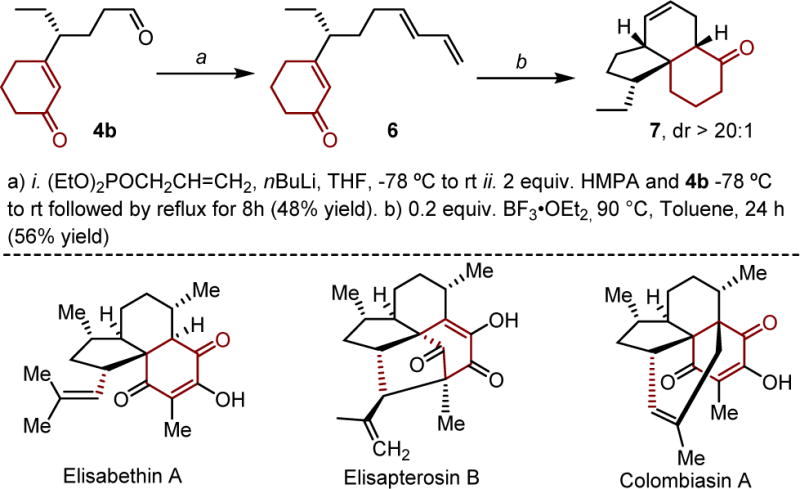
Processing of a relay Heck product to resemble a natural product core structure.
In summary, we have successfully developed a method to install electron deficient alkenyl groups in high enantioselectivity at various distances from a resultant carbonyl by using a redox-relay strategy. The success of this reaction is attributed to the use of electron-withdrawing alkenyl triflates, which leads to improved selectivity during the β-hydride elimination step to reinforce migration of the palladium catalyst towards the alcohol. This prevents trapping of the catalyst as a proposed π-allyl species. The relatively mild conditions and ability to remotely install alkenyl groups to the carbonyl set the stage for further synthetic applications of this method. Future work is aimed at this goal and further detailing the mechanistic underpinnings of this process.
Supplementary Material
Acknowledgments
The work was supported by National Institute of Health (NIGMS R01GM063540).
Footnotes
Notes
The authors declare no competing financial interests.
Supporting Information
Experimental procedures and characterization data for new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Nicolaou KC, Sorensen EJ. Classics in Total Synthesis. VCH; Weinheim: 1996. [Google Scholar]; (b) Nicolaou KC, Snyder SA. Classics in Total Synthesis II. More Targets, Strategies, Methods. Wiley-VCH; Weinheim: 2003. [Google Scholar]; (c) Saklani A, Kutty SK. Drug Discovery Today. 2008;13:161. doi: 10.1016/j.drudis.2007.10.010. [DOI] [PubMed] [Google Scholar]; (d) Liu KK-C, Sakya LS. Mini-Rev Med Chem. 2004;4:1105. doi: 10.2174/1389557043402900. [DOI] [PubMed] [Google Scholar]
- 2.For reports on asymmetric conjugate additions, see:; (a) Nakao Y, Chen IH, Hiyama T, Ichikawa I, Duan W, Shintani R, Hayashi T. J Am Chem Soc. 2007;129:9137. doi: 10.1021/ja071969r. [DOI] [PubMed] [Google Scholar]; (b) Oi S, Sato T, Inoue Y. Tetrahedron Lett. 2004;45:5051. [Google Scholar]; (c) Nicolaou KC, Tang W, Dagneau P, Faraoni R. Angew Chem Int Ed. 2005;44:3874. doi: 10.1002/anie.200500789. [DOI] [PubMed] [Google Scholar]; (d) Hayashi T, Yamasaki K. Chem Rev. 2003;103:2829. doi: 10.1021/cr020022z. [DOI] [PubMed] [Google Scholar]
- 3.For reports on asymmetric allylic displacement, see:; (a) Hamilton J, Sarlah D, Carreira EM. J Am Chem Soc. 2013;135:994. doi: 10.1021/ja311422z. [DOI] [PubMed] [Google Scholar]; (b) Akiyama K, Gao F, Hoveyda AH. Angew Chem Int Ed. 2010;49:419. doi: 10.1002/anie.200905223. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Gao F, McGrath KP, Lee Y, Hoveyda AH. J Am Chem Soc. 2010;132:14315. doi: 10.1021/ja106829k. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Gao F, Carr JL, Hoveyda AH. Angew Chem, Int Ed. 2012;51:6613. doi: 10.1002/anie.201202856. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Shintani R, Takatsu K, Takeda M, Hayashi T. Angew Chem, Int Ed. 2011;50:8656. doi: 10.1002/anie.201103581. [DOI] [PubMed] [Google Scholar]; (f) Jung B, Hoveyda AH. J Am Chem Soc. 2012;134:1490. doi: 10.1021/ja211269w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Other methods for the enantioselective introduction of alkenyl groups, see; (a) Biradar DB, Gau H-M. Org Lett. 2009;11:499. doi: 10.1021/ol801999u. [DOI] [PubMed] [Google Scholar]; (b) Pu L, Yu H-B. Chem Rev. 2001;101:757. doi: 10.1021/cr000411y. and references within. [DOI] [PubMed] [Google Scholar]; (c) Page JP, RajanBabu TV. J Am Chem Soc. 2012;134:6556. doi: 10.1021/ja301640e. and references within. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Stevens JM, MacMillan DWC. J Am Chem Soc. 2013;135:11756. doi: 10.1021/ja406356c. and references within. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Cherney AH, Reisman SE. J Am Chem Soc. 2014;136:14365. doi: 10.1021/ja508067c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dounay AB, Overman LE. Chem Rev. 2003;103:2945. doi: 10.1021/cr020039h. and references therein. [DOI] [PubMed] [Google Scholar]
- 6.(a) Ozawa F, Kobatake Y, Hayashi T. Tetrahedron Lett. 1993;34:2505. [Google Scholar]; (b) Kilroy TC, Cozzi PG, Nicole E, Patrick J. Synthesis. 2004;11:1879. [Google Scholar]; (c) Loiseleur O, Hayashi M, Schmees N, Pflatz A. Synthesis. 1997;11:1338. [Google Scholar]; (d) Loiseleur O, Meier P, Pfaltz A. Angew Chem Int Ed Engl. 1996;35:200. [Google Scholar]; (e) Gilbertson SR, Fu Z. Org Lett. 2001;3:161. doi: 10.1021/ol006747b. [DOI] [PubMed] [Google Scholar]; (f) Gilbertson SR, Denov DG, Rheingold AL. Org Lett. 2000;2:2885. doi: 10.1021/ol006323h. [DOI] [PubMed] [Google Scholar]; (g) Kaukoranta P, Kallstrom K, Andersson PG. Adv Syn Catal. 2007;349:2595. [Google Scholar]; (h) Tietze LF, Thede K, Sannicolo F. Chem Comm. 1999:1811. [Google Scholar]; (i) Hennessy AJ, Malone YM, Guiry PJ. Tetrahedron Lett. 2000;41:2261. [Google Scholar]
- 7.(a) Werner EW, Mei TS, Burkle AJ, Sigman MS. Science. 2012;338:1455. doi: 10.1126/science.1229208. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Mei TS, Werner EW, Burkle AJ, Sigman MS. J Am Chem Soc. 2013;135:6830. doi: 10.1021/ja402916z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mei TS, Patel HH, Sigman MS. Nature. 2014;508:340. doi: 10.1038/nature13231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cartney DMc, Guiry P. Chem Soc Rev. 2011;40:5122. doi: 10.1039/c1cs15101k. [DOI] [PubMed] [Google Scholar]
- 10.(a) Shibasaki M, Vogl EM. J Organomet Chem. 1999;576:1. [Google Scholar]; (b) Beletskaya IP, Cheprakov AV. Chem Rev. 2000;100:3009. doi: 10.1021/cr9903048. [DOI] [PubMed] [Google Scholar]
- 11.Shibasaki M, Boden CDJ, Kojima A. Tetrahedron. 1997;53:7371. [Google Scholar]
- 12.Xu L, Hilton MJ, Zhang X, Norrby P-O, Wu Y-D, Sigman MS, Wiest O. J Am Chem Soc. 2014;136:1960. doi: 10.1021/ja4109616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.(a) Liao L, Jana R, Urkalan KB, Sigman MS. J Am Chem Soc. 2011;133:5784. doi: 10.1021/ja201358b. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Saini V, Sigman MS. J Am Chem Soc. 2012;134:11372. doi: 10.1021/ja304344h. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) McCammant MS, Liao L, Sigman MS. J Am Chem Soc. 2013;135:4167. doi: 10.1021/ja3110544. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Stokes BJ, Bischoff AJ, Sigman MS. Chem Sci. 2014;5:2336. doi: 10.1039/C4SC00602J. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Stokes BJ, Opra SM, Sigman MS. J Am Chem Soc. 2012;134:11408. doi: 10.1021/ja305403s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.For examples of application of chiral pyrox ligands in Pd-catalyzed reactions, see:; (a) Kikushima K, Holder JC, Gatti M, Stoltz BM. J Am Chem Soc. 2011;133:6902. doi: 10.1021/ja200664x. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) McDonald RI, White PB, Weinstein AB, Tam CP, Stahl SS. Org Lett. 2011;13:2830. doi: 10.1021/ol200784y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rauf W, Brown JM. Chem Comm. 2013;49:8430. doi: 10.1039/c3cc44842h. [DOI] [PubMed] [Google Scholar]
- 16.Werner EW, Sigman MS. J Am Chem Soc. 2011;133:9692. doi: 10.1021/ja203164p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chassaing S, Specklin S, Weibel J-M, Pale P. Tetrahedron. 2012;68:7245. [Google Scholar]
- 18.The starting materials are generally consumed in the reactions. The remainder of the mass balance mainly consists of a mixture of alkene products that are derived from β-insertion into the alkenol.
- 19.Babinski D, Soltani O, Frantz DE. Org Lett. 2008;10:2902. doi: 10.1021/ol8010002. [DOI] [PubMed] [Google Scholar]
- 20.(a) Paya M, Hoult JRS. Gen Pharmac. 1996;4:713. doi: 10.1016/0306-3623(95)02112-4. [DOI] [PubMed] [Google Scholar]; (b) Sendl A, Chen JL, Jolad SD, Stoddart C, Rozhon E, Kernan M. J Nat Prod. 1996;59:808. doi: 10.1021/np9601871. [DOI] [PubMed] [Google Scholar]
- 21.Elisabethin:; (a) Rodríguez AD, González E, Huang SD. J Org Chem. 1998;63:7083. doi: 10.1021/jo981385v. [DOI] [PubMed] [Google Scholar]; Elisapterosin:; (b) Rodríguez AD, Ramírez C, Rodríguez II, Barnes CL. J Org Chem. 2000;65:1390. doi: 10.1021/jo9914869. [DOI] [PubMed] [Google Scholar]; Columbiasin:; (c) Rodríguez AD, Ramírez C. Org Lett. 2000;2:507. doi: 10.1021/ol991362i. [DOI] [PubMed] [Google Scholar]
- 22.(a) Wang Y, West FG. Synthesis. 2002;1:99. [Google Scholar]; (b) Kraus GA, Kim J. Org Lett. 2004;18:3116. doi: 10.1021/ol048847d. [DOI] [PubMed] [Google Scholar]
- 23.Ross AG, Li X, Danishefsky SJ. J Am Chem Soc. 2012;134:16080. doi: 10.1021/ja307708q. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.