

Abstract
While cancer cell invasion and metastasis is dependent on cancer cell-stroma, cancer cell-blood vessel, and cancer cell-lymphatic vessel interactions, our understanding of these interactions remain largely unknown. A need exists for physiologically-relevant models that more closely mimic the complexity of cancer cell dynamics in a real tissue environment. The objective of this study was to combine laser-based cell printing and tissue culture methods to create a novel ex vivo model in which cancer cell dynamics can be tracked during angiogenesis in an intact microvascular network. Laser direct-write (LDW) was utilized to reproducibly deposit breast cancer cells (MDA-MB-231 and MCF-7) and fibroblasts into spatially-defined patterns on cultured rat mesenteric tissues. In addition, heterogeneous patterns containing co-printed MDA-MB-231/fibroblasts or MDA-MB-231/MCF-7 cells were generated for fibroblast-directed and collective cell invasion models. Printed cells remained viable and the cells retained the ability to proliferate in serum-rich media conditions. Over a culture period of five days, time-lapse imaging confirmed fibroblast and MDA-MB-231 cell migration within the microvascular networks. Confocal microscopy indicated that printed MDA-MB-231 cells infiltrated the tissue thickness and were capable of interacting with endothelial cells. Angiogenic network growth in tissue areas containing printed cancer cells was characterized by significantly increased capillary sprouting compared to control tissue areas containing no printed cells. Our results establish an innovative ex vivo experimental platform that enables time-lapse evaluation of cancer cell dynamics during angiogenesis within a real microvascular network scenario.
Keywords: matrix assisted pulsed laser evaporation direct-write, MAPLE DW, mesentery, breast cancer, angiogenesis
Introduction
Investigation of the underlying mechanisms involved in cancer cell dynamics and the pre-clinical development of therapies requires experimental models that include cancer cells, stromal cells, blood vessels, and lymphatic vessels in a relevant 3-D microenvironment. The recognized need to incorporate multiscale complexity has motivated the development of biomimetic 3-D culture systems,1,2 ex vivo tissue explant models,3 and microfluidic devices.4,5 None of the current models, however, enable simultaneous investigation of cancer cell migration and angiogenesis in intact microvascular networks – a requirement that more closely reflects an in vivo scenario. And despite recent advances in imaging techniques to track cell movement in animal models, such as the use of optically-transparent transgenic zebrafish6 or installation of anatomic viewing windows for high-resolution intravital microscopy,7 the ability to localize distinct groups of cancer cells proximal to vessels and follow individual cell infiltration in 3-D space remains elusive. Thus, a gap exists between current in vivo and in vitro models.
In an attempt to bridge this gap, the objective of this study was to develop an innovative ex vivo experimental platform that enables time-lapse imaging of cancer cell dynamics during angiogenesis within a real microvascular network scenario by combining two novel approaches – laser direct-write (LDW) cell printing and the rat mesentery culture model. We have shown that the rat mesentery culture model is advantageous because it can be used for 1) real time imaging in the same tissue,8,9 2) quantification of endothelial cell sprouting at specific locations within a microvascular network during growth factor-induced angiogenesis,8,10 and 3) investigating functional effects of pericytes on endothelial cell sprouting.8 We have also shown that lymphatic vessels in our model maintain their lymphatic identity and can be induced to undergo lymphangiogenesis.10 A key advantage of this model is its simplicity, i.e., the tissue is easy to obtain, self-contained, and does not need to be embedded. The mesentery's thinness (20-40 μm) allows for observation of intact networks down to a single cell level and makes it an ideal tissue for printing exogenous cells. Using LDW printing technology, human breast cancer cells and fibroblasts were deposited in spatially-defined patterns onto the vascularized rat mesentery tissue. After printing, cells remained viable, proliferative, and migratory. Our results demonstrate, for the first time, cell printing onto live tissues for tracking short-term cancer cell dynamics within intact microvascular networks. Heterogeneous cell printing, quantification of cancer cell influence on angiogenesis, and observation of cancer cell integration into blood and lymphatic vessels support the feasibility of making specific spatial and temporal measurements, which are not possible in other in vitro systems. We envision this new ex vivo model platform will enable high content investigation of cancer cell behavior in a real tissue environment and future studies focused on the systematic probing of the reciprocal cellular interactions between cancer cells, fibroblasts, blood vessels, and lymphatic vessels.
Results
MDA-MB-231 breast cancer cells were successfully printed onto ex vivo mesentery tissue using laser direct-write (LDW) (Fig. 2). The real-time video feed on the LDW system allowed for the selection of a desired print area on the mesentery tissue. Ejecting a single droplet of cell suspension established a local group, or `spot,' of MDA-MB-231 cells on the mesentery tissue. After one hour of incubation, a combination of round and spindle-shaped cellular morphologies indicated various states of cell attachment. To demonstrate the deposition of cells into spatially-defined pattern positions, additional groups of cancer cells were printed to form 4×4 matrix arrays of 16 spots (0.8 mm center-to-center spot spacing, print area < 16 mm2) (Fig. 2A). Cells were printed onto microvascular networks containing blood and lymphatic vessels. Blood versus lymphatic vessel identity was distinguishable by relative diameter, vessel morphology, and lectin and PECAM fluorescence labeling intensity.10–12 Lymphatic identity based on these features is supported by our previous co-labeling of lymphatic vessels with lectin or PECAM and typical lymphatic markers: LYVE-1,8,10–12 Prox1,10–12 and podoplanin.10–12 Arterioles, venules, and capillaries were identified by position in the network, vessel morphology, and labeling intensity as previously described.8 Following printing, MDA-MB-231 cells attached to the tissue substrate and maintained their initially deposited positions. Cells displayed adherent morphology two hours after the printing process. The average spot diameter was 401 μm (SD = 68 μm) and the average number of cells per spot was 40 (SD = 19) (n = 8 completed arrays, over 2 batches). By decreasing the center-to-center spacing in the 4×4 array definition, the same CAD/CAM algorithm was capable of forming a nearly-contiguous area of overlapping cancer cell spots (Fig. 2B). In addition, arrays containing single MDA-MB-231 cancer cells were generated by lowering the cell-loading concentration on the ribbon to assure that the transferred droplets contained only single cells (Fig. 2C). At concentrations >5 × 105 cells/mL, trypsinized cells tended to cluster in groups, such that the transfer area contained several neighboring clusters (Fig. 2D). Conversely, at a loading concentration of 1 × 105 cells/mL, the diffuse areal distribution of cells enabled the transfer of single cancer cells, without affecting neighboring cells (Fig. 2D).
Fig. 2. Spatially-defined patterns of localized cell groups on mesentery tissue and single-cell printing capability.

(A) 4×4 array pattern (center-to-center spacing, dspacing = 0.8 mm) of circular 'spots' of CellTracker Red (CTR)-labeled MDA-MB-231 cancer cells on vascularized mesentery tissue, imaged two hours following LDW. Mesenteric arterioles (A), venules (V), and lymphatic vessels (L) were readily identifiable by lectin labeling. (B) Contiguous 4×4 array of MDA-MB-231 cancer cells generated by reducing dspacing to 0.4 mm. (C) 4×4 array (dspacing = 0.8 mm) of nearly-single cancer cell groups. (D) Phase contrast images of trypsinized MDA-MB-231 cells on the print ribbon at various loading concentrations (black text) and corresponding areal density (white text). Circle borders indicated cell groups selected for printing under typical transfer area.
The printing protocol exposed tissues to ambient surroundings (~20°C/50% RH) for 5–7 mins, during which groups of cells were printed. Based on live/dead staining, following the LDW transfer period, endogenous mesentery cells remained viable (Fig. 3A). In the co-culture systems consisting of MDA-MB-231 cancer cells and vascularized connective tissue, we investigated the influence of serum-rich (10% FBS) and reduced-serum (1% FBS) media on cancer cell attachment and viability after two hours in culture following LDW (Fig. 3B). In both 1% and 10% FBS culture conditions, the mesenteric cells directly underneath the printed spot areas remained viable, indicating cell survival upon droplet impact. The number of live CTR-labeled cancer cells was greater in 10% FBS vs. 1% FBS media (n = 20 spots per serum condition) (Fig. 3C). For the 10% FBS group, the fraction of viable breast cancer cells following LDW was 77% (SE = 3%). For the 1% FBS reduced serum group, the fraction was 35% (SE = 5%). Ribbon cell viability at time of printing was equal to 92% (SE = 5%). The proliferative capability of the printed MDA-MB-231 cells cultured in 10% FBS media after transfer onto mesentery tissue is supported by the observation of BrdU+ cells in the spot area 24 and 72 hours after printing (Fig. 4A). BrdU colocalization with MDA-MB-231 cells was confirmed by high magnification imaging (Fig. 4B).
Fig. 3. Mesenteric and printed cell viability following deposition by LDW and culture in 1% or 10% FBS media.

(A) Fluorescent live/dead staining of mesenteric cells in tissue that was prepared for LDW printing or control tissue that was suspended in MEM and not processed after harvesting. (B) Live staining of printed CTR-labeled MDA-MB-231 cells on mesentery tissue. Co-localization of CTR and live dye indicated printed cell viability. (C) Quantification of viability of printed breast cancer cells immediately following transfer (n = 20 spots per serum condition, unpaired Student's t-test, p < 0.01).
Fig. 4. Breast cancer cell proliferation following LDW printing onto mesentery tissue.

(A) BrdU labeling identified proliferating MDA-MB-231 cells 24 and 72 hours after printing. (B) Colocalization of BrdU+ nuclei with CTR-labeled MDA-MB-231 cells was confirmed with 60× magnification imaging.
Similar to MDA-MB-231 cells, printed fibroblasts and MCF-7 breast cancer cells attached to the mesentery tissue within two hours and maintained the initial pattern registry following transfer (Fig. 5A). The 4×4 arrays provided up to 16 spots for cell movement tracking. Over a culture period of two days, a fraction of fibroblasts and MDA-MB-231 cells laterally invaded the connective tissue window. Over two days in culture, both cell lines migrated outward from the Day 0 circular spot border and became distributed across the mesentery tissue. The time-window of two days was chosen to easily track individual cells before they infiltrated into invasion zones of neighboring spots. In contrast, MCF-7 cells displayed negligible migration, and by Day 2, the cells were still localized to their initial printed spots.
Fig. 5. Breast cancer cell invasion into mesentery tissue.

(A) Representative images of CTR-labeled fibroblasts and MDA-MB-231 and MCF-7 cancer cells printed onto mesentery tissue and observed over two days. The defined spots of printed cells diffused as cells infiltrate the tissue in the XY plane. White circles represent the initial border of a printed spot for reference at each time point. (B) Averaged invasion distances of cancer cells, normalized to initial-spot radius (n = 43 spots over 6 tissues, two-way repeated measures ANOVA, * = p < 0.05, ** = p < 0.01, ns = no significance). (C) Quantification of cells inside and outside of initial-spot border to determine whether the continual loss of printed cells remaining within initial-spot border was compensated by the number of invading cells (n = 43 spots, paired Student's t-test, **= p < 0.01). (D) Normalized cell counts on Day 2 in spots printed over vascularized or avascular areas (n = 43 spots, paired Student's t-test, ns = no significance). (E) 20× YZ plane projection of Day 5 printed tissue containing CTR-labeled MDA-MB-231 cells and PECAM-labeled blood vessel structure (Z-stack, 27-μm height, 0.5-μm interval, Zeiss Apotome structured illumination).
The ability to print patterns of localized breast cancer cell groups onto mesentery tissues enabled the characterization of cell movement within viable, intact microvascular networks. MDA-MB-231 cells, on average, traveled 0.40 fraction of the initial spot radius after 24 hours and 0.54 after 48 hours in culture, respectively (n = 43 spots over 6 tissues, p < 0.01) (Fig. 5B). As a control, MDA-MB-231 cells were also printed and tracked on “deactivated” tissues (i.e., non-viable tissues) incubated at 60°C for 45 mins. MDA-MB-231 cells displayed insignificant changes in migration from their printed location at Day 0 by Days 1 and 2. Cancer cells printed onto viable mesentery tissues migrated significantly farther than cells printed onto deactivated tissues (Day 1: p < 0.05; Day 2: p < 0.01). As another indicator of cell migratory behavior, the number of cells remaining within the initial-spot borders decreased by 27% by Day 2 (Fig. 5C) and the number of cancer cells outside of the printed spot area increased. (p < 0.01, n = 43 spots). The overall increase in the number of cells inside and outside the original spot area further supports the observation of BrdU labeling (Fig. 4) and the proliferative capability of the cells. No difference was observed between the normalized cell counts on Day 2 for spots printed over vascularized versus avascular areas (Fig. 5D). Importantly, MDA-MB-231 cells printed on the top of the mesenteric tissue became integrated and migrated in the z-direction past the upper mesothelial layer. Following culture, the cancer cells occupied the same tissue space as the microvascular networks (Fig. 5E). By Day 5, CTR-labeled breast cancer cells were located above, below, and in the plane of microvascular networks.
The LDW printing mechanism works independently of cell type and solely depends on the light-material interaction to actuate ejection of trypsinized cells from the ribbon to the substrate. By mixing multiple cell suspensions (total concentration = 2 × 106 cells/mL) during ribbon preparation, we demonstrated the ability to print heterogeneous spots (Fig. 6). Arrays of MDA-MB-231/fibroblasts and MDA-MB-231/MCF-7 cells printed onto mesentery tissue support the feasibility of comparing cell type-specific migratory behavior (Fig. 6). In MDA-MB-231/fibroblast constructs, fibroblasts quickly infiltrated the connective tissue ahead of breast cancer cells. On average, fibroblasts comprised 84% (SD = 14%) of the invasive cell counts beyond initial spot-borders at Day 1 (n = 36 spots over 6 tissues). In the MDA-MB-231/MCF-7 co-culture constructs, MDA-MB-231 cells invaded the tissue, whereas MCF-7 cells remained within spot borders. On average, MDA-MB-231 cancer cells comprised 90% (SD= 22%) of the invasive cell (n = 36 spots over 6 tissues).
Fig. 6. Fibroblast-directed and collective cell migration models.

Heterogeneous 4×4 arrays containing combined MDA-MB-231/fibroblast or MDA-MB-231/MCF-7 populations printed onto live, vascularized mesentery tissues. Cell behavior was monitored over two days.
In mesenteric tissues with printed cells, microvascular networks displayed robust angiogenesis over the time course of five days. Angiogenic networks were characterized by dramatic increases in vessel density and capillary sprouting (Fig. 7A). Quantification of vascular network growth in mesentery tissues with printed MDA-MB-231 cells revealed significant increases in capillary sprouts, branch points, and vascular length over five days (Fig. 7B–D). By Day 5, vascularized areas under printed cancer cell spots exhibited a greater than two-fold increase in capillary sprouting (Fig. 7B) (p < 0.01, n = total of 37 spots over 3 tissues), but not the number of branch points or vascular length, when compared to control tissue areas (Fig. 7C and D). The observation of angiogenesis within printed tissues supports the value of this novel construct for investigating cancer cell dynamics during microvascular network growth and remodeling (Fig. 7). The potential novelty of such experiments is also supported by the observations of apparent interactions between the MDA-MB-231 cells with both blood and lymphatic vessels (Fig. 8A). Sub-0.5 μm confocal images supported the observation of interactions between MDA-MB-231 cells and PECAM-positive endothelial cells (Fig. 8B). Cells were also observed to be capable of integrating into the PECAM-positive endothelial cells of microvessels (Fig. 8B).
Fig. 7. Microvascular growth in mesentery tissues containing printed cancer cells.

(A) Arrays of MDA-MB-231 cells printed on mesentery tissue and tracked over five days. By Day 5, evidence of angiogenesis in microvascular networks was observed in areas containing printed MDA-MB-231 cells. (B–D) Quantification of capillary sprouts, branch points, and vascular length, normalized to the initial-spot area (n = 37 spots over 3 tissues, two-way repeated measures ANOVA; * = p < 0.05, ** = p < 0.01, ns = no significance).
Fig. 8. Interactions between cancer cells and microvascular networks.

(A) MDA-MB-231 cells were observed possibly interacting with blood and lymphatic vessels. “L” indicates a lymphatic vessel. (B) MDA-MB-231 cells were identified interacting and integrating with blood vessels by using sub-micron confocal microscopy.
Discussion
In this study, we demonstrate the ability to print exogenous cells onto a live, 3-D tissue. The novel combination of the laser direct-write bioprinting technology and the rat mesentery culture model enabled the spatial patterning of single cells, cells clusters, and heterogeneous cell populations onto intact microvascular networks containing blood vessels and lymphatic vessels. A novelty of the ex vivo platform is the capability for quantification of cancer cell dynamics and effects within a biologically relevant tissue environment at temporal and spatial resolutions not possible with other models.
We envision that this new ex vivo model platform offers a high-content, real tissue environment that will motivate future studies focused on the systematic probing of the reciprocal cellular interactions between cancer cells, fibroblasts, blood vessels, and lymphatic vessels. For example, consider the need to distinguish or evaluate breast cancer subtypes, which at the molecular level are characterized by unique signaling, transcriptome and epigenome signatures that define their phenotype.13,14 Application of our model will allow researchers to evaluate whether or not a molecular treatment has cell type-selective effects on capillary sprouting, cell migration, and tissue invasion. Our results also motivate future studies to investigate fundamental questions regarding whether cancer cells preferentially metastasize to blood versus lymphatic vessels or whether cells are passively or actively directed to the microvessels. And, as another example, the multi-cell printing capability motivates studies to evaluate whether the presence of fibroblasts enhances cancer cell migration.
The ability to answer such questions within one model supports the potential for our cancer cell-microvascular network model to be an intermediary platform between whole organisms and reductionist-based assays. A challenge is to maintain in vivo complexity while achieving cell level resolution. Promising techniques for cancer studies with micron resolution include cell encapsulation in hydrogels1 and embedded tumor spheroids.15 For example, recent work from Dr. Steven George's laboratory has provided an in vitro vascularized tumor model that integrates cancer cells, endothelial cells, and fibroblasts, enabling the observation of angiogenesis and tumor cell intravasation.2 Another cutting edge platform is microfluidic devices that integrate fluid flow, endothelial cell-lined channels, and cancer cell types.4,5,16,17 Exemplified by work in Dr. Roger Kamm's laboratory, use of microfluidic devices to investigate cancer cell migration into endothelial cell lined channels has highlighted the value of more realistic 3-D environments.18,19 Yet as is the case with “bottom up” approaches, microfluidic devices and the other models do not quite match the complexity across scales inherent to a real tissue microenvironment. We suggest that the combination of LDW printing and the rat mesentery culture model offers an alternative approach that enables observation of cancer cell dynamics in a multi-cell/system microvascular network scenario.
The thinness of the mesentery (20-40 μm) also allows for observation of intact networks down to a single cell level and makes it an ideal tissue for printing exogenous cells. As the tissue volume height was greater than one cell thick but less than 40 μm in thickness, printed cells have the room to invade into the 3-D tissue volume and their movements can be imaged with epifluorescent and confocal microscopy without sectioning. Immediately following LDW, cells remained viable and maintained their initial pattern positions, most likely through interactions with the mesenteric extracellular matrix. Furthermore, harvesting tissues from transgenic or aged rat strains provides the potential to study tissue dynamics in pathological scenarios. An alternative source for mesenteric tissues can be obtained from mice. Unfortunately, mouse mesentery tissues are generally avascular20 and were not used in this study. However, the future use of mouse mesentery tissues derived from transgenic mice could be useful for investigating extracellular matrix-dependent cell migration behaviors or related scientific questions that do not necessitate the presence of a microvasculature.
LDW was chosen as the primary printing technique over other cell deposition methods, such as ink-jet printing, quill-pen direct-write, and laser-induced forward transfer (LIFT). Ink-jet printing mechanisms rely on thermal expansion,21 hydraulic pressure,22 or piezoelectric-induced23 shearing of the liquid volume at the nozzle opening to eject droplets of cell suspension down to the single-cell level.24 Quill-pen direct-write techniques modify atomic-force microscopy cantilever tips with the addition of built-in reservoirs containing cell suspensions, and dispense cells where the cantilever tip contacts the substrate.25 LIFT, a precursor to LDW, similarly relies on laser-induced bubble formation at the hydrogel-ribbon interface, but requires an opaque metal energy-transduction layer to eject cells onto the substrate.26 Although these techniques may be faster at printing cells than LDW, they lack critical features required for optimal printing onto live tissue substrates. LDW integrates controls for environmental temperature and humidity management, which allows for favorable droplet formation, minimizes printed droplet evaporation, and maintains mesentery tissue hydration. In addition, the fully optical technique allows for real-time monitoring of cells on the ribbon in order to select desired cell groups, as well as single cells, for printing. More importantly, the video feed also allows for the ability to visually locate vascularized areas on the mesentery tissue. Thus, the novelty of our technique lies in the ability to localize cells onto microvascular networks to resolve micro-scale cellular interactions at the cell-vessel interface. While we chose invasive MDA-MB-231 breast cancer cells as the model cell line to demonstrate the feasibility of our model, LDW is a cell type-independent process. In addition to the MDA-MB-231 cells, we also demonstrated the ability to print MCF-7 cells and fibroblasts. The ability to print heterogeneous cell groups could allow for the generation of intratumor patterns to study fibroblast-directed events, cancer cell type-dependent behavior, and collective cell migration. We feel LDW offers several advantages that render it essential for precise deposition of cells onto live tissue substrates. Future envisioned applications could include utilizing patient-derived tumor cells or other specific cell populations for personalized drug screening strategies.
One of the limitations of our integrated model platform includes the lack of microvascular perfusion. While this is a common limitation of other ex vivo models, excluding microfluidic devices, future experiments are required to incorporate this critical factor. Another limitation is that the tissue explant is a non-cancerous connective tissue from a rat and does not mimic a tumor-stroma environment. While the results from our short term studies support the ability to observe initial cancer cell dynamics and their effects on angiogenesis, future studies will also be needed to determine longer term cancer cell behavior and whether the printed cells can form tumors. Still, for now, our characterization of cancer cell influences on angiogenic sprouts, quantification of cancer cell migration from spatially defined “spots” in a physiologically relevant extracellular environment, and observation of cancer cell interaction with both blood and lymphatic vessels supports the novelty of the model for investigating cancer cell behavior in the context of an intact microvascular network scenario – a capability which is not matched by other models.
For this initial validation study, we investigated the effect of serum-rich and reduced-serum environments on blood vessel growth and breast cancer cell behavior. As expected, higher serum levels induced greater cell viability and infiltration distance following LDW. However, improvements to the printing procedure are required to increase cell viability to 90%, as previously reported for 2-D printing.27 Printed cancer cells also retained the ability to proliferate following transfer. Even with a serum concentration of 10%, it was possible to decouple local cancer cell-directed capillary sprouting from system-wide angiogenic response due to the availability of growth factors in media. Interestingly, we observed a statistically-significant increase in capillary sprouting, but no difference in vascular density. A possible explanation for these combined results based on our previous characterization of microvascular network remodeling in the mesentery12 is that increases in vascular density temporally lag increases in sprouting. Additional experiments are required to determine the longer term effects of cancer cell presence on angiogenesis and whether the increase in sprouting at Day 5 results in associated increases in vascular density at later time points.
In summary, our work combines the latest bioprinting technology with an established angiogenesis model to generate an intermediary platform that bridges the gap between in vivo observations and in vitro co-culture systems. Taken together, our 3-D tissue-level model advances the field of biofabrication and cancer cell-driven angiogenesis research, allowing researchers to 1) print cancer cells, fibroblasts, and cancer cell/fibroblast co-populations onto live tissue substrates with intact microvascular networks, 2) track individual cell migration over the time course of angiogenesis, and 3) quantify microvascular network responses.
Materials and Methods
Cell lines and media
Human dermal fibroblasts, BJ5ta, and breast cancer cell lines, MDA-MB-231 and MCF-7, (ATCC, Manassas, VA) were cultured in Dulbecco's Modified Eagle's Medium (DMEM), supplemented with 10% fetal bovine serum (Atlas Biologicals, Fort Collins, CO; FBS) and 1% penicillin-streptomycin. “1% FBS” media utilized for reduced-serum culture conditions was made by diluting cell culture media at a 1:10 ratio in minimum essential media (MEM). Cell lines were labeled with CellTracker Red (CTR) or CellTracker Blue (CTB), according to manufacturer's protocol, one hour prior to printing. All media, supplements, and labeling agents were obtained from Life Technologies (Carlsbad, CA), unless otherwise stated.
Preparation of rat mesentery tissue for printing
All experiments were performed in accordance with the guidelines of the Tulane University Institutional Animal Care and Use Committee. Rat mesenteric tissue windows, defined as the thin connective tissue between the artery/vein pairs feeding the small intestine, were harvested from the ileum of adult male Wistar rats (Harlan, Indianapolis, IN) and incubated at 37°C/5% CO2/100% RH in MEM prior to LDW preparation. The mesentery tissues, spanning a triangular area of ~1.5 cm2 and a thickness of 20–40 μm, were then removed from media and stretched on inverted transwell inserts (CellCrown, Sigma Aldrich) with polycarbonate filters (Millipore, Billerica, MA). Surface tension interactions physically held tissues flat against the transwell filter membranes. The tissues were then rehydrated with 50 μL MEM and incubated in culture conditions for 20 mins. Excess media was removed prior to cell printing.
Preparation of ribbon for printing
A UV-transparent quartz disk, termed “ribbon,” was subjected to spin-coating at 2,000 RPM to generate a ~30 μm thick layer of liquefied 20% w/v gelatin (Sigma Aldrich; Type A porcine, 300 Bloom). A 1 mL suspension of trypsinized cancer or fibroblast cells at 2 × 106/mL was pipetted onto the coated ribbon surface. Cells were incubated for 7 mins at 37°C/5% CO2/100% RH to partially embed cells in the liquefied gelatin layer, followed by another 5 mins of incubation at room temperature for the gelatin to solidify. Excess media was removed prior to cell printing. A single ribbon could be used to print cell patterns onto three successive mesentery tissues. Fresh ribbons were prepared for each batch printing.
Laser Direct-Write (LDW)27,28
Prepared ribbons were inverted to face the flattened mesentery tissues for cell transfer, with a 0.7-mm gap-spacing between the print and receiving surfaces. Both the ribbon and transwell supports were loaded onto parallel holders with independently-motorized XY translation for computer-aided design/manufacturing ability. The working space environmental conditions were set at 21°C and 45% RH. A single pulse of 193 nm ArF excimer laser (Precision Microfab; near-Gaussian distribution, pulse width = ~8 ns, fluence = 0.42 ± 0.01 J/cm2) interacts with the sacrificial gelatin layer on the ribbon, forming a local vapor pocket that ejects a single droplet of desired cells onto the tissue. Subsequent groups of cells, targeted in real-time with the aid of an in situ camera, were additively deposited into computer-programmable positions to complete the array patterns. The minimum turnaround time for this technique is one hour, with a printing resolution of 5 μm (single-cell). Following the printing procedure, the cells were incubated for 12 mins at 37°C/5% CO2/100% RH to initialize cell attachments to the tissue. Afterwards, 50 μL of media supplemented with 1% or 10% FBS was pipetted onto the tissues and incubated for another 20 mins to further cell attachment. Finally, the transwell insert was inverted and inserted into a 6-well plate, such that the tissues were wedged between the membrane of the insert and the well bottom for the duration of the culture period. Tissues were cultured for up to five days.
Viability of mesenteric and printed cells
A live/dead staining kit (BioVision; calcein and propidium iodide-based) was utilized according to manufacturer's protocol to determine mesenteric cell viability in tissues. As printed cells were labeled with CTR, co-localization with of CTR with the live dye produced a positive count for cell viability. Image-based counts were utilized to quantify live and dead cells.
Proliferation potential of printed cells
To evaluate the effect of LDW on the proliferation potential of deposited cells, mesentery tissues containing printed MDA-MB-231 cell patterns were labeled with BrdU (Sigma Aldrich; 5-bromo-2'-deoxyuridine, 10 μM). BrdU was added with 10% FBS cell culture media to the tissues immediately following printing or after two days and incubated for two hours. Tissues were then fixed in 100% methanol at −20°C, washed in PBS, and labeled with 1:100 mouse anti-BrdU antibody (Dako, Carpinteria, CA) and Alexa Fluor 488-conjugated goat anti-mouse secondary antibody (Jackson ImmunoResearch, West Grove, PA). Proliferating MDA-MB-231 cells were identified by colabeling of BrdU with CTR.
Lectin labeling and antibody staining
To identify blood and lymphatic vessels during culture, tissues were incubated with 1:40 FITC-conjugated BS-I lectin (Sigma Aldrich), as previously described.8,9 Tissues were labeled with lectin for imaging at Day 0 and Day 2 or Day 5. Whole tissues were then mounted on glass slides, fixed in 100% methanol, and washed and permeabilized with 0.1% saponin in PBS. At the designated endpoint, tissues were stained with 1:200 biotinylated anti-rat PECAM antibody (BD Pharmingen, San Diego, CA) and 1:500 Cy2-conjugated streptavidin (Jackson ImmunoResearch) to identify endothelial cells and with 1:3000 DAPI (Life Technologies) to identify cell nuclei.
Image analysis for invasion and vascular growth
Circle borders (termed “initial-spot borders”), which encompassed transferred cells within a printed spot on Day 0, were overlaid on respective pattern spots in images taken at Days 0–2. The number of cells within and outside of the initial-spot border was counted over the time period. Invasion events were defined as events in which individual cells migrated beyond the circle border. Positions of labeled cells were recorded relative to the spot center, and invasion distances were calculated as the difference between circle radius and cell position, and finally normalized to the radius length. Quantification of microvascular remodeling was performed as described previously.8,10 The number of capillary sprouts, number of branch points, and vessel length were quantified on Days 0 and 5 within the initial-spot areas of printed cells and in equivalently-sized control areas of the same tissue that did have printed cells. Capillary sprouts were defined as blind-ended blood vessel segments extending from existing vessels. Branch points were defined as the intersection of three or more blood vessel segments. Vessel length was defined as the total length of blood vessels. All metrics were normalized to their corresponding area, either the printed initial-spot area or the defined control area. Patterns in which cell spots were printed over avascular areas were not included in the analysis.
Microscopy
Epifluorescent images were acquired using an Olympus IX70 inverted microscope (Olympus America, Center Valley, PA) with 10× and 60× objectives or Carl Zeiss Cell Observer inverted microscope (Carl Zeiss, Thornwood, NY) with 10× and 20× objectives. High resolution z-stack images were acquired using the Carl Zeiss Cell Observer microscope in Apotome structured illumination mode or a Nikon A1 confocal microscope (Nikon Instruments, Melville, NY) using a 60× objective (oil, NA = 1.4) at an optical section thickness of 0.47 μm.
Fig. 1. Schematic of laser direct-write (LDW) set up, adapted for the transfer of targeted cells from the ribbon onto the ex vivo mesentery tissue.
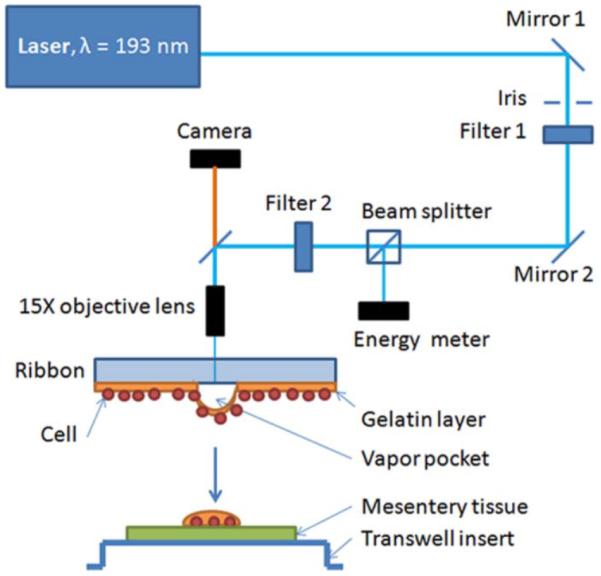
An inline camera provided real-time video feed for the selection of desired cells for printing. Through CAD/CAM control, cell groups were additively printed into spatially-defined patterns onto thin mesentery tissue, flattened on an inverted transwell insert.
Insight Statement.
Biomimetic models are extremely valuable for the investigation of underlying cell and tissue mechanisms and the pre-clinical development of therapies. The current interdisciplinary study integrates two approaches – laser direct write cell printing and the rat mesentery culture model – to create an innovative ex vivo experimental platform that enables time-lapse evaluation of cancer cell dynamics during angiogenesis within a real microvascular network scenario. Our results advance the field of biofabrication by demonstrating the efficient printing of cancer cells, with high precision as single cells or as groups of cells, onto live rat connective tissues with intact microvascular networks. This platform will allow for the systematic probing of the complex, reciprocal cellular interactions between cancer cells, fibroblasts, blood vessels, and lymphatic vessels.
Acknowledgements
Research work was supported by the National Institutes of Health - CA125806 (MEB) and 5-P20GM103629 (WLM), Tulane Center for Aging (WLM), and the National Science Foundation - 1258536 (DBC).
References
- 1.Delort L, Lequeux C, Dubois V, Dubouloz A, Billard H, Mojallal A, Damour O, Vasson MP, Caldefie-Chezet F. PLoS One. 2013;8:e66284. doi: 10.1371/journal.pone.0066284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ehsan SM, Welch-Reardon KM, Waterman ML, Hughes CC, George SC. Integr. Biol. (Camb) 2014;6:603–610. doi: 10.1039/c3ib40170g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Salameh TS, Le TT, Nichols MB, Bauer E, Cheng J, Camarillo IG. Int. J. Cancer. 2013;132:288–296. doi: 10.1002/ijc.27672. [DOI] [PubMed] [Google Scholar]
- 4.Shin Y, Han S, Chung E, Chung S. Integr. Biol. (Camb) 2014;6:654–661. doi: 10.1039/c4ib00022f. [DOI] [PubMed] [Google Scholar]
- 5.Moya ML, Hsu YH, Lee AP, Hughes CC, George SC. Tissue Eng. Part C. Methods. 2013;19:730–737. doi: 10.1089/ten.tec.2012.0430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lee SL, Rouhi P, Dahl Jensen L, Zhang D, Ji H, Hauptmann G, Ingham P, Cao Y. Proc. Natl. Acad. Sci. U. S. A. 2009;106:19485–19490. doi: 10.1073/pnas.0909228106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Beerling E, Ritsma L, Vrisekoop N, Derksen PW, van Rheenen J. J. Cell. Sci. 2011;124:299–310. doi: 10.1242/jcs.072728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stapor PC, Azimi MS, Ahsan T, Murfee WL. Am. J. Physiol. Heart Circ. Physiol. 2013;304:H235–45. doi: 10.1152/ajpheart.00552.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Azimi MS, Myers L, Lacey M, Stewart SA, Shi Q, Katakam PV, Mondal D, Murfee WL. PLoS One. 2015;10:e0119227. doi: 10.1371/journal.pone.0119227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sweat RS, Sloas DC, Murfee WL. Microcirculation. 2014;21:532–540. doi: 10.1111/micc.12132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Robichaux JL, Tanno E, Rappleye JW, Ceballos M, Stallcup WB, Schmid-Schonbein GW, Murfee WL. Anat. Rec. (Hoboken) 2010;293:1629–1638. doi: 10.1002/ar.21195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sweat RS, Stapor PC, Murfee WL. Lymphat Res. Biol. 2012;10:198–207. doi: 10.1089/lrb.2012.0014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brenton JD, Carey LA, Ahmed AA, Caldas C. J. Clin. Oncol. 2005;23:7350–7360. doi: 10.1200/JCO.2005.03.3845. [DOI] [PubMed] [Google Scholar]
- 14.Lehmann BD, Bauer JA, Chen X, Sanders ME, Chakravarthy AB, Shyr Y, Pietenpol JA. J. Clin. Invest. 2011;121:2750–2767. doi: 10.1172/JCI45014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wiercinska E, Naber HP, Pardali E, van der Pluijm G, van Dam H, ten Dijke P. Breast Cancer Res. Treat. 2011;128:657–666. doi: 10.1007/s10549-010-1147-x. [DOI] [PubMed] [Google Scholar]
- 16.Chan JM, Zervantonakis IK, Rimchala T, Polacheck WJ, Whisler J, Kamm RD. PLoS One. 2012;7:e50582. doi: 10.1371/journal.pone.0050582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Song JW, Munn LL. Proc. Natl. Acad. Sci. U. S. A. 2011;108:15342–15347. doi: 10.1073/pnas.1105316108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zervantonakis IK, Hughes-Alford SK, Charest JL, Condeelis JS, Gertler FB, Kamm RD. Proceedings of the National Academy of Sciences. 2012;109:13515–13520. doi: 10.1073/pnas.1210182109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Aref AR, Huang RY, Yu W, Chua KN, Sun W, Tu TY, Bai J, Sim WJ, Zervantonakis IK, Thiery JP, Kamm RD. Integr. Biol. (Camb) 2013;5:381–389. doi: 10.1039/c2ib20209c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Norrby K. J. Cell. Mol. Med. 2006;10:588–612. doi: 10.1111/j.1582-4934.2006.tb00423.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cui X, Boland T, D'Lima DD, Lotz MK. Recent. Pat. Drug Deliv. Formul. 2012;6:149–155. doi: 10.2174/187221112800672949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chang R, Nam J, Sun W. Tissue Eng. Part A. 2008;14:41–48. doi: 10.1089/ten.a.2007.0004. [DOI] [PubMed] [Google Scholar]
- 23.Xu T, Jin J, Gregory C, Hickman JJ, Boland T. Biomaterials. 2005;26:93–99. doi: 10.1016/j.biomaterials.2004.04.011. [DOI] [PubMed] [Google Scholar]
- 24.Yamaguchi S, Ueno A, Akiyama Y, Morishima K. Biofabrication. 2012;4 doi: 10.1088/1758-5082/4/4/045005. 045005-5082/4/4/045005. Epub 2012 Oct 17. [DOI] [PubMed] [Google Scholar]
- 25.Hynes WF, Doty NJ, Zarembinski TI, Schwartz MP, Toepke MW, Murphy WL, Atzet SK, Clark R, Melendez JA, Cady NC. Biosensors (Basel) 2014;4:28–44. doi: 10.3390/bios4010028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gruene M, Pflaum M, Hess C, Diamantouros S, Schlie S, Deiwick A, Koch L, Wilhelmi M, Jockenhoevel S, Haverich A, Chichkov B. Tissue Eng. Part C. Methods. 2011;17:973–982. doi: 10.1089/ten.tec.2011.0185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schiele NR, Chrisey DB, Corr DT. Tissue Eng. Part C. Methods. 2011;17:289–298. doi: 10.1089/ten.tec.2010.0442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Phamduy TB, Raof NA, Schiele NR, Yan Z, Corr DT, Huang Y, Xie Y, Chrisey DB. Biofabrication. 2012;4 doi: 10.1088/1758-5082/4/2/025006. 025006-5082/4/2/025006. Epub 2012 May 4. [DOI] [PubMed] [Google Scholar]