

SUMMARY
Bacteria are central to human health and disease, but existing tools to edit microbial consortia are limited. For example, broad-spectrum antibiotics are unable to accurately manipulate bacterial communities. Bacteriophages can provide highly specific targeting of bacteria, but assembling well-defined phage cocktails solely with natural phages can be a time-, labor- and cost-intensive process. Here, we present a synthetic-biology strategy to modulate phage host ranges by engineering phage genomes in Saccharomyces cerevisiae. We used this technology to redirect Escherichia coli phage scaffolds to target pathogenic Yersinia and Klebsiella bacteria, and conversely, Klebsiella phage scaffolds to target E. coli by modular swapping of phage tail components. The synthetic phages achieved efficient killing of their new target bacteria and were used to selectively remove bacteria from multi-species bacterial communities with cocktails based on common viral scaffolds. We envision that this approach will accelerate phage-biology studies and enable new technologies for bacterial population editing.
Graphical Abstract
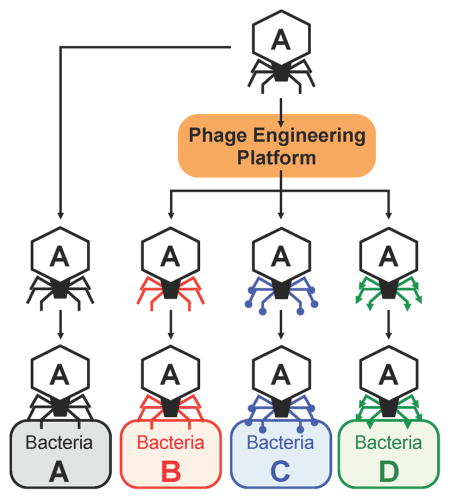
INTRODUCTION
Bacteriophages (phages) are natural biological nanomachines that have evolved to infect host bacteria with exquisite specificity and efficacy. Phages constitute the most abundant type of biological particles on Earth (Hendrix, 2003) and reproduce at the expense of their host bacteria. Thus, phages have been explored as a means of controlling pathogenic bacteria (d’Herelle, 1931), but poor understanding of the molecular relationships between bacteria and their phages can lead to highly variable treatment outcomes (Brussow, 2012; Lu and Koeris, 2011). With the rise of drug-resistant bacterial infections and the sharp decline in antibiotic discovery and development (Fischbach and Walsh, 2009), phage therapy is regaining attention after years of declining interest (Carlton, 1999; Lu and Collins, 2007, 2009). Furthermore, despite the important role that microbial populations play in regulating human health and disease (Grice and Segre, 2012), strategies for precisely manipulating complex microbial communities are lacking. With their ability to kill or deliver DNA into specific bacteria, phages constitute a promising platform for editing microbial populations (Bikard et al., 2014; Citorik et al., 2014). However, the limited host range of most naturally isolated phages means that phage cocktails are often needed to address real-world applications (Sulakvelidze et al., 2001), but the high diversity of natural phages poses a challenge for the engineering, manufacturing, and regulatory approval of phage products. Thus, enhancing the range of targetable bacteria by adding natural phages to cocktails is counterbalanced with the challenge of producing and testing well-defined multi-component mixtures for regulatory approval.
Creating phage-based therapeutics and diagnostics is also limited by the difficulty of engineering phages. Lytic phage DNA is often too large for in vitro manipulation and does not reside for very long inside of bacteria making it difficult to modify phage genomes during their reproductive cycle. Phage genome engineering is classically carried out with allele replacement methods, whereby a piece of the phage genome is cloned and manipulated in bacteria, which is then infected with the phage, leading homologous recombination of the modified DNA segment into the phage genome. This process is inefficient because many phages degrade resident DNA upon entry and time-consuming due to the need to screen for recombinant phages but the lack of phage selectable markers. Recently, CRISPR-Cas systems have been used for counter-selection against phage T7 and the heavily modified genomes of a few members of the T-even family (Kiro et al., 2014; Martel and Moineau, 2014; Yaung et al., 2014). We have used the Streptococcus pyogenes CRISPR-Cas9 system to select for mutants in phage T7 but with variable efficiencies (data not shown). Furthermore, large stretches of phage DNA can encode products toxic to bacteria, thus preventing their manipulation within bacterial hosts. Existing approaches are also limited in their ability to simultaneously engineer multiple non-contiguous loci in phage genomes. Here, we demonstrate a high-throughput phage-engineering platform that leverages the tools of synthetic biology to overcome these challenges and use this platform to engineer phages with tunable host ranges for microbial population editing.
RESULTS
Yeast platform for bacteriophage genome engineering
We used an efficient yeast-based platform (Jaschke et al., 2012; Lu et al., 2013) to create phages with novel host ranges based on common viral scaffolds. Inspired by gap-repair cloning in yeast and the work of Gibson and co-workers (Gibson et al., 2008), we captured phage genomes into Saccharomyces cerevisiae, thus enabling facile genetic manipulation of modified genomes that can be subsequently re-activated or “rebooted” into functional phages after transformation of genomic DNA into bacteria (Figure 1A). First, the entirety of the viral genome to be assembled in yeast is amplified by PCR so that each adjacent fragment has homology over ≥30 base pairs (bp). The first and last fragments of the phage genome are amplified with primers that carry “arms” that have homology with a yeast artificial chromosome (YAC) fragment, which may be obtained by PCR or any other suitable method. Upon transformation of all viral genome fragments and the YAC into yeast, gap-repair joins each fragment to the adjacent one templated by the homology regions at the end of each fragment, yielding a full phage genome cloned into a replicative yeast plasmid. Yeast transformants are then enzymatically disrupted to extract the YAC-phage DNA, which is transformed into bacterial host cells that can restart the viral life cycle. Plaques, if obtained, are then picked, amplified, and sequenced to verify proper introduction of the desired mutations. If no plaques are obtained, it is still possible to amplify parts of the YAC-phage genome from the yeast clones to verify proper DNA assembly, to eliminate the possibility of unwanted mutations, and to help determine potential reasons for the failure of the synthetic phage genome to produce viable offspring.
Figure 1. Yeast platform for phage engineering.

(A) Schematic illustrating the workflow to capture and reboot phages using our yeast platform. An entire phage genome or PCR products spanning an entire phage genome are transformed into yeast cells along with a linearized yeast replicon fragment from the yeast artificial chromosome, pRS415. In yeast, the phage genome is assembled and captured in the YAC by gap-repair cloning. The resulting YAC-phage DNA is extracted and transformed into host bacteria. Active phages are produced from the YAC-phage DNA and generate plaques on a lawn of host bacteria. (B) High-titer phage lysates (>109 PFU/ml) were spotted onto E. coli 10G lawns, which were sensitive only to T7 and T3 phages. (C) Rebooting T7 and T3 phages from purified phage genomes (10 ng) after electroporation into 10G, recovery in LB, chloroform sterilization, and plating with E. coli BL21. (D) All tested phage genomes (10–200 ng), including non-E. coli phages, could be rebooted by electroporation into E. coli 10G cells and plating supernatants with host bacteria. Host bacteria: IJ1668 K-12 hybrid; K1 capsule for K1E, K1F, and K1-5 phages, IJ612 Salmonella typhimurium LT2 for SP6 phage, Pseudomonas aeruginosa PAO1 for LUZ19 phage, Pseudomonas putida C1S for gh-1 phage, and Klebsiella sp. 390 for K11 phage. (E) An example of capturing and rebooting a phage through the yeast platform. An excised YAC pRS415 amplicon and the T7 genome were co- transformed in yeast cells. The T7 genome was captured in the YAC by gap-repair cloning in yeast. Progeny phages were produced from YAC-T7 DNA via E. coli 10G and generated plaques on E. coli BL21.
We confirmed that purified phage DNA from various phages could be transformed into bacterial hosts to generate functional phages. We targeted phages from the T7-family because their life cycle is largely host independent (Qimron et al., 2010) and there is a relatively large number of family members for which genomic sequences are publicly available. These include coliphages T7 (40 kbp), T3 (38 kbp), K1E (K1-capsule-specific, 45 kbp), K1F (K1-capsule-specific, 40 kbp), K1-5 (K1- or K5-capsule-specific, 44 kbp), Salmonella phage SP6 (44 kbp), Pseudomonas phages LUZ19 (44 kbp), gh-1 (37 kbp), and Klebsiella phage K11 (41 kbp). We used E. cloni 10G® (10G) cells (Durfee et al., 2008) as a one-time phage propagation host. Except for T7 and T3, all phages used in this study (K1E, K1F, K1-5, SP6, LUZ19, gh-1, and K11) cannot infect 10G (Figure 1B). Each purified phage genome was electroporated into 10G directly. After incubation and chloroform treatment, supernatants were mixed with overnight cultures of each natural host bacteria for each phage in soft agar, poured onto agar plates, and scored for plaque formation (Figure 1C and D). All the phages tested could be rebooted from purified DNA into functional phages through one-step propagation in 10G, even if their natural target species was not E. coli (Figure 1D and Table S1).
Rebooting bacteriophages from PCR products via the yeast platform
To determine whether phage genomes assembled in yeast can be used to create viable phages, we first captured and rebooted T7 (Figure 1E), T3, and LUZ19 phages. We used PCR to amplify the YAC pRS415 and add arms homologous to the ends of the phage genomes. We co-transformed the YAC amplicons with phage genomic DNA into yeast. Confirmed YAC-phage DNA was extracted from yeast and transformed into E. coli 10G. These cells were then chloroform treated and the lysates assessed for plaque-forming units (PFU) on the natural bacterial hosts of the phages (see Figure 1E for capturing and rebooting T7). All 3 phage genomes yielded yeast clones that could be rebooted to viable phages using this strategy.
Next, we successfully captured and rebooted eight different phages that target E. coli, Salmonella, Pseudomonas, and Klebsiella (T7, T3, K1E, K1F, K1-5, SP6, gh-1, and K11) by assembling overlapping 3.8–12 kbp-long PCR products spanning the phage genomes with the linearized YAC in yeast (Figure 2A upper panel illustrates this process with T7 as an example, data not shown for the others). This data demonstrates the efficient assembly and instantiation of functional recombinant phages via our yeast-engineering platform, which can potentially create any desired genotype in one step from PCR products.
Figure 2. Creation of synthetic phages with engineered host range.

(A) We prepared multiple PCR fragments encoding the wild-type T7 phage genome (T7WT) (PCR fragments 1 – 4; 10.0, 10.0, 10.0, and 10.1 kbp, respectively), T7 phage with the entire T3 phage tail fiber (T7T3(gp17)) (PCR fragments 1 – 3 and 5 – 7; 10.0, 10.0, 10.0, 4.7, 1.7, and 3.7 kbp, respectively), and T7 phage with a hybrid T7-T3 tail fiber (T7T3(C-gp17)) (PCR fragments 1 – 3, 8, 9, and 7; 10.0, 10.0, 10.0, 5.2, 1.3, and 3.7 kbp, respectively). All fragments were co-transformed and assembled in yeast along with YAC DNA (3.0 kbp). (B) Phage A with its primary host determinant, gene a, infects bacteria A, but cannot infect bacteria B. Phage B with its primary host determinant, gene b, infects bacteria B, but cannot infect bacteria A. We hypothesized that swapping these host determinants between phages would switch their respective host ranges. (C) Creation of synthetic T7 phage with phage 13a tail fiber (encoded by gene 17). We synthesized 13a’s gene 17 and assembled it with the rest of the T7 genome via overlapping PCR products in yeast (PCR fragments 1 – 6 and YAC amplicon; 10.0, 10.0, 10.0, 4.8, 1.7, 3.7, and 3.0 kbp, respectively). The YAC-phage DNA was extracted and used for transformation.
Swapping tail fibers enables modulation of phage host range
We explored the utility of our phage-engineering platform by engineering the host range of phages. We first selected two model phages, T7 and T3, which are well known obligate lytic phages that propagate on E. coli B (Demerec and Fano, 1945). T7 and T3 have linear genomes that share high homology with each other, in which the primary host determinant is the product of gene 17 (gp17), the tail fiber (Dunn and Studier, 1983; Pajunen et al., 2002). Alterations in the gp17 sequence have been linked to the recognition of different host receptors and shifting host ranges (Molineux, 2006). Thus, we hypothesized that exchanging gene 17 or fragments of gene 17 between T7, T3, and their relatives could be used to tune their host specificities (Figure 2B). This is supported by previous data on naturally occurring hybrids between T7 and T3 whose host range was mostly dictated by which gp17 they harbored (Lin et al., 2012).
We examined the host range of T7 and T3 phage on diverse bacterial panels that were differentially targeted by the two phages. T3 is described as incapable of targeting many common lab E. coli K-12 strains (Molineux, 2006), so we performed plaque formation assays with four K-12 strains and a B strain (BL21) as a control. As shown in Figure S1A, T7 plaqued efficiently on all strains, while T3 did not produce plaques on BW25113 and MG1655 at a detectable frequency (Figure 3). T3 exhibited ~3–4 orders-of-magnitude reductions in adsorption efficiency on BW25113 and MG1655 compared with the permissive BL21 strain (Figure S1B). Thus, we can differentiate between T7 and T3 using BW25113 or MG1655.
Figure 3. Efficiency of plating (EOP) of natural, reconstructed wild-type, and synthetic phages.

EOP was determined with respect to a reference bacterium. Klebsiella sp. 390 was used as a reference for T7K11(gp11-12-17), K11, and K11WT. Y. ptb IP2666 was used for T3R(gp17). For other phages, BL21 was used as the reference host. EOP data were log10-transformed and are presented as the mean of three independent experiments. Error bars represent SD. The “0” symbol indicates an EOP = 1, which marks the reference strain used for the EOP calculations within each phage. * indicate that the EOP was below the detection limit (EOP <10−8). Strain abbreviations: B, E. coli BL21; G, E. coli 10G; D, E. coli DH5α; W, E. coli BW25113; M, E. coli MG1655; 4, E. coli ECOR4; 13, E. coli ECOR13; 16, E. coli ECOR16; K, Klebsiella sp. 390; I, Y. ptb IP2666; Y, Y. ptb YPIII; N, E. coli Nissle 1917.
The gp17 tail fibers of T7 and T3 can be split in two domains. The N-terminal 149 residues are thought to be necessary for the tail fiber to bind to the rest of the capsid while the remaining C-terminal region forms a kinked shaft and harbors the recognition domain for host receptors at its tip (Steven et al., 1988). The N-terminal regions of T7 and T3 share 99% identity at the protein level, while the C-termini exhibit 83% identity, with the last 104 amino acids (aa) of the T3 protein showing only 62% of identity to the corresponding 99 aa of the T7 protein. Therefore, we hypothesized that swapping the C-terminal domain between the two viruses would result in exchanging the host ranges. We constructed synthetic phages, based on either the T7 or T3 viral chassis, which carried engineered gene 17 alleles composed of fragments from the other phage. Specifically, we created six synthetic phages: T7 phage with the wild-type T7 tail fiber (T7WT), T7 phage with the C-terminal 410 aa region of the T3 tail fiber (T7T3(C-gp17)), T7 phage with the entire T3 tail fiber (T7T3(gp17)), T3 phage with the wild-type T3 tail fiber (T3WT), T3 phage with the C-terminal 405 aa region of the T7 tail fiber (T3T7(C-gp17)), and T3 phage with the entire T7 tail fiber (T3T7(gp17)). T7WT and T7 phages are the same at the genetic level; however, T7WT phage was created by capturing the T7 genome in yeast and then rebooting this phage genome in bacteria and served as a control for the faithfulness of the reconstruction process, whereas T7 was obtained from ATCC. The same applies to T3WT and T3. Each phage was assembled in yeast via four or six PCR fragments and was rebooted via transformation into E. coli 10G (example schematics in Figure 2A). No unexpected mutations were found in the heterologous gp17 regions of the rebooted phages.
To examine the host specificities of our six engineered phages, we performed plaque formation assays on a range of E. coli, Klebsiella, and Yersinia pseudotuberculosis (Y. ptb) strains (Figure 3, Figure S2). T3T7(C-gp17) and T3T7(gp17) plaqued on E. coli BW25113 and E. coli MG1655 at a similar EOP as T7 and T7WT, while T3, T3WT, T7T3(C-gp17), and T7T3(gp17) had >108-fold-reduced EOPs on these strains. In addition, T3, T7T3(C-gp17), and T7T3(gp17) plaqued on E. coli ECOR16 while T7, T3T7(C-gp17), and T3T7(gp17) did not. Furthermore, we also synthesized a codon-optimized version of the tail fiber of the T7-like coliphage 13a and created synthetic T7 phages containing the entire 13a tail fiber (T713a(gp17)) or the C-terminal region of the 13a tail fiber (T713a(C-gp17)) (Figure 2C, Table S2). Although T7 and T7WT did not plaque on E. coli ECOR16, both T713a(gp17) and T713a(C-gp17) were able to do so efficiently (Figure 3, Figure S2). Thus, the C-terminal region of gp17 is a major host range determinant and new host ranges can be conferred onto T7-like phage scaffolds by engineering tail fibers. Interestingly, T713a(C-gp17) efficiently infected E. coli BW25113 and MG1655, similar to T7 and T7WT, but T713a(gp17) did not, which suggests that the N-terminus of the phage 13a tail fiber can also alter infectivity of the virus although the mechanism is still to be investigated (Figure 3). A second example of this phenomenon can be found between T7T3(C-gp17) (Figure 3 and Figure S2 lane 3) and T7T3(gp17) (Figure 3 and Figure S2 lane 4). While the former phage infected Y. ptb YPIII (albeit with a low EOP), the latter phage as well as wild-type T3 did not.
Coliphage T3 with a Yersinia phage tail fiber infects both E. coli and Y. pseudotuberculosis
We further demonstrated that gene swapping between phages can overcome species barriers by designing synthetic phage based on T7 or T3 scaffolds that can infect bacteria other than E. coli. We started with coliphage T3 and Yersinia phage R (38 kbp), since their gp17’s share 99.5% identity at the protein level and differ by only 3 nucleotides in gene 17, suggesting these could be responsible for their divergent host ranges. We introduced these three mutations in T3 gene 17 by PCR so that it would encode the same tail fiber as phage R (Figure 4A). While T3 did not plaque on Y. ptb strains IP2666 and YPIII, which are known hosts for phage R (Rashid et al., 2012), synthetic T3 phage with the R tail fiber (T3R(gp17)) was able to infect Y. ptb IP2666 and YPIII (Figure 3 and Figure S2). The adsorption efficiencies of T3WT and T3R(gp17) against Y. ptb IP2666 were found to be 0.01±0.01% and 90.05±1.1%, respectively.
Figure 4. Creation of synthetic T3 phage with Yersinia phage R tail fiber.

(A) We introduced mutations in T3 gene 17 by PCR to convert it into phage R gene 17 and assembled the resulting product with the rest of the T3 genome and YAC DNA in yeast (PCR products 1 – 6 and YAC amplicon; 10.0, 10.0, 10.0, 4.4, 0.2, 3.8, and 3.0 kbp, respectively). The YAC-phage DNA was extracted and used for transformation into E. coli. (B) Plaquing assay with T3WT and T3R(gp17) on E. coli BL21, Y. ptb IP2666, and Y. ptb YPIII shows that T3R(gp17) can infect both E. coli and Y. ptb. Ten-fold serial dilutions of phage lysates were spotted on bacterial lawns and incubated for 4 h at 37°C for E. coli BL21 or 24 h at 30°C for Y. ptb strains. These pictures were cut out from Figure S2. Bottom panels show images of individual plaques. NP, no plaque. (C) Killing curves of Y. ptb IP2666 treated with T3R(gp17). ~108 CFU/ml bacteria and ~107 PFU/ml phage were used (MOI ~0.1). The data are presented as the mean of three independent experiments. Error bars represent SD. Small error bars are obscured by symbols. The detection limit was 2.0 × 103 CFU/ml.
Interestingly, T3R(gp17) maintained the capacity to infect E. coli BL21 (Figure 4B), demonstrating that the introduced mutations conferred a host range expansion and not just a host range shift. In addition to plaquing assays, we further characterized the ability of T3WT versus T3R(gp17) to kill Y. ptb IP2666 over time. After 1.5 h of treatment, T3R(gp17) killed 99.999% of IP2666 while T3 had no effect on the bacteria (Figure 4C and Table S3A).
Redirection of host range between coliphage and Klebsiella phage by swapping whole tail components
We further overcame species barriers by engineering phages with lower similarity with one another. K11 is a Klebsiella phage that belongs to the T7-like family (Dietz et al., 1990). K11 shares gene synteny with T7 but homology between K11 and T7 genes is low, averaging only 59% among the genes that have homologs between the two viruses. For comparison, T7 and T3 share 72% identity at the genomic level between homologous genes. While T7 is a coliphage and does not infect Klebsiella, K11 infects Klebsiella, such as Klebsiella sp. 390, but not E. coli (Figure 3, Figure S2) (Bessler et al., 1973). Their respective host range determinants, gp17, are very different and do not share any homology outside of the N-terminal 150 amino acids, which is only 47% identical between the two proteins. Specifically, the T7 gp17 encodes tail fibers while the 322 aa longer K11 gp17 directs the synthesis of a tail spike, an enzymatic host range determinant that actively breaks down the capsule of Klebsiella to allow K11 phage to gain access to unknown secondary receptors located beneath the capsule (Bessler et al., 1973).
To create a T7 phage with a K11 tail fiber and a K11 phage with a T7 tail fiber, we first swapped the entire gene 17, but this yielded no viable phages. We then tried to construct composite tail fibers composed of gene 17 fragments from both phages hybridized at various points along the length of the gene, but this was also unsuccessful at generating functional synthetic phages. We speculated that one possible reason for these failures could be that the K11 genome cannot create productive phages within E. coli 10G. However, the natural K11 genome produced Klebsiella-infecting virions when it was rebooted via 10G cells (Figure 1D and Table S1).
Alternatively, the gene 17 product from K11 may require a function or factor that is absent from T7. Cuervo et al. reported that the tail of T7 phage, which assembles independently of the head, is assembled from a dodecamer of gp11 (the adaptor) and a hexamer of gp12 (the nozzle) (Figure 5A, upper panel) onto which 6 trimers of gp17 attach (Cuervo et al., 2013). T7’s six tail fibers attach at the interface between the adaptor and nozzle, thus making contacts with both proteins. The adaptor ring is responsible for the attachment of the preformed tail to the prohead via interactions with the portal composed of 12 subunits of gp8. The homology between the gp8 of T7 and K11 (80% identity at the amino acid level) is much higher than the homology between the gp11 and gp12 proteins of T7 and K11 (60 and 61% identity, respectively), which led us to suspect that replacing all three tail genes of T7 with their K11 equivalents (gp11, gp12, and gp17) could be necessary to create functional virions (Figure 5A, lower panel). Indeed, both T7 with K11 tail components (T7K11(gp11-12-17)) and K11 with T7 tail components (K11T7(gp11-12-17)) were successfully created and exhibited tail-dependent host ranges. Specifically, T7K11(gp11-12-17) infected Klebsiella sp. 390 and did not target E. coli, while K11T7(gp11-12-17) infected E. coli, but did not plaque on Klebsiella (Figure 5B, Figure 3, and Figure S2). The yeast-based phage engineering platform enabled the facile construction of these phages via one-step genome construction even though gene 11 and 12 are physically separated from gene 17, a feat that would be difficult to achieve with other phage engineering methods. To further validate the ability of synthetic T7K11(gp11-12-17) to target Klebsiella, we performed a time-course experiment that showed that T7K11(gp11-12-17) killed 99.955% of Klebsiella sp. 390 after 1 hour of treatment (Figure 5C and Table S3B), but was about 100-fold less effective than K11WT (Figure 5B and Figure S3). Measured adsorption efficiencies of T7K11(gp11-12-17) and K11WT against Klebsiella sp. 390 were 98 ± 2% and 92 ± 6% after 10 minutes, respectively, suggesting that the reduced efficacy of T7K11(gp11-12-17) is not due to an adsorption defect.
Figure 5. Creation of synthetic T7 phage with Klebsiella phage K11 tail components as well as K11 phage with T7 tail components.

(A) The tail complex of T7 phage is composed of two components, a tubular structure and tail fibers. The tubular structure consists of an upper dodecameric ring made of adaptor protein gp11 and a pyramidal hexameric complex of the nozzle protein gp12. The tail fiber protein gp17 interacts with the interface between gp11 and gp12 (Cuervo et al., 2013). Schematics illustrating the construction of synthetic hybrids between phages T7 and K11. Whole genomes were amplified as overlapping PCR amplicons. PCR fragments were co-transformed and assembled in yeast (PCR fragments 1 – 8 and YAC amplicon; 10.0, 10.0, 4.3, 3.0, 2.7, 4.8, 2.7, 3.7, and 3.0 kbp, respectively). YAC-phage genomes were extracted and used for transformation. We swapped K11 genes 11, 12 and 17 into T7 to create T7K11(gp11-12-17) and T7 genes 11, 12 and 17 into K11 to create K11T7(gp11-12-17). (C) Plaquing of T7K11(gp11-12-17) and K11T7(gp11-12-17) on E. coli BL21 and Klebsiella sp. 390. Ten-fold serial dilutions of phage lysates were spotted on bacterial lawns and incubated for 4 h at 37°C. These pictures were cut from Figure S3. Bottom panels show images of individual plaques. NP, no plaque. (D) Killing curves of Klebsiella sp. 390 treated with T7K11(gp11-12-17). ~108 CFU/ml bacteria and ~107 PFU/ml phage were used (MOI ~0.1). The data are presented as the mean of three independent experiments. Error bars represent SD. Small error bars are obscured by symbols. The detection limit was 2.0 × 103 CFU/ml.
Synthetic phage cocktails efficiently remove target bacteria from mixed bacterial populations
Our results show that common phage scaffolds can be retargeted against new bacteria hosts by engineering single or multiple tail components. This capability enables the construction of defined phage cocktails that only differ in their host-range determinants and can be used to edit the composition of microbial consortia and/or treat bacterial infections. To demonstrate microbial population editing using synthetic phages derived from T7 or T3 scaffolds but with differing host specificities, we designed experiments to remove specific bacteria from mixed populations containing Klebsiella sp. 390, Y. ptb IP2666, and the probiotic E. coli strain Nissle 1917. The amount of each bacterial member in this mixed population was quantified using their differing sensitivities to chemical antimicrobials (Figure S4). After 1 h treatment of the multi-species population with T7K11(gp11-12-17) or T3R(gp17), >99.9% or >99% of their target bacteria, Klebsiella sp. 390 or Y. ptb IP2666, respectively, were removed with no detectable impact on the remaining bacterial species (Figure 6 and Table S4A). Furthermore, a phage cocktail consisting of two phages with the same chassis but different host ranges, T7WT and T7K11(gp11-12-17), resulted in >99.9% killing of Klebsiella sp. 390 and >99.9% killing of Y. ptb IP2666 after 1 hour, thus enriching for probiotic E. coli Nissle 1917 (Figure 6 and Table S4A).
Figure 6. Microbial population editing assay.

A synthetic microbial community composed of E. coli Nissle 1917, Klebsiella sp. 390, and Y. ptb IP2666 was treated with various individual synthetic phages and a pairwise combination of phages. After adding ~107 PFU/ml of each phage, the resulting samples were incubated at 30°C with shaking for 1 h. At each time point, bacteria were collected, washed in saline, serially diluted, and plated onto selective plates for viable cell counts after a 24 h incubation at 30°C. The data are presented as the mean of three independent experiments and the total numbers of cells (CFU/ml) are shown. The sizes of the pie charts reflect the total number of cells. Note that the chart does not allow the display of fractions smaller than ~1%. The detailed data and the SD are shown in Table S4A. The detection limit was 2.0 × 103 CFU/ml.
We performed similar assays with a phage cocktail consisting of two phages based on the T3 chassis (Figure S5). In this experiment, we used a mixed bacterial population containing the probiotic E. coli strain Nissle 1917, E. coli ECOR16 (sensitive to T3WT), and Y. ptb IP2666 (sensitive to T3R(gp17)). After one hour treatment with a phage cocktail consisting of two phages with the same T3 chassis but different host ranges, T3WT and T3R(gp17), >99.9% E. coli ECOR16 and >98% Y. ptb IP2666 were killed without affecting the remaining probiotic E. coli Nissle 1917 (Figure S6 and Table S4B). These results demonstrate the high efficiency and selectivity of our engineered phages in microbial consortia, and the potential of generating well-defined phage cocktails and combining them with probiotics.
DISCUSSION
In this study, we utilized an efficient yet simple yeast-based platform for phage engineering to modulate phage host ranges for several members of the T7 phage family. Traditional phage engineering strategies, such as in vitro manipulation, allele-exchange within bacterial hosts, and phage crossing via co-infection of bacteria (Beier et al., 1977; Garcia et al., 2003; Lin et al., 2011) have been used to modulate phage host range (Pouillot et al., 2010; Tetart et al., 1998; Trojet et al., 2011; Yoichi et al., 2005), but these strategies are inefficient and unable to achieve multiple genetic modifications in a single step. Screening for a desired mutation after classical crossing or recombination experiments can require PCR, restriction digestion, or plaque hybridization on hundreds of individual plaques, which are all costly and time-consuming methods. Conversely, our strategy rarely requires the screening of more than a few yeast clones, since we found that >25% of yeast clones contained properly assembled phage genomes (composed of up to 11 DNA fragments) that were bootable into functional phages after transformation into bacteria. Previously, a scheme for engineering phage T4 through electroporation of PCR products was devised (Pouillot et al., 2010), but it is based on a particular feature of the genetic regulation of T4 and cannot easily be applied to other phage families. Recently, the 5.4 kb filamentous coliphage ϕX174 was assembled in yeast in order to stably store the genome and aid in phage refactoring (Jaschke et al., 2012). In this approach, the majority of the genome assembly was performed in vitro and the YAC cloning was mostly used to store the resulting genome, whereas the majority of the genome engineering in our approach stems from the actual gap-repair cloning process in yeast. In addition, the phages we have cloned using this method are in the 38–45 kbp range and we have indications that it can also be used for much larger phage genomes (e.g., up to 100 kbp, data not shown). Leveraging yeast to modify phages enables the decoupling of phage genome engineering from phage fitness and viability, obviates the need for selective or screenable markers in phage genomes, reduces the risks of phage contamination during the engineering process, and permits facile one-step genetic manipulations. For example, the ability to simultaneously engineer multiple loci in a phage genome was crucial for constructing T7K11(gp11-12-17) and K11T7(gp11-12-17).
However, a challenge of yeast-based phage engineering (which is shared by in vitro engineering strategies) is the need to reboot modified phage genomes into functional phages. Here, we used high-efficiency DNA transformation to deliver phage DNA into bacterial hosts, but future work may be facilitated by in vitro transcription-translation systems capable of supporting functional phage synthesis (Shin et al., 2012). Another challenge for the approach outlined here is identifying the loci that can be manipulated to change phage host range. We anticipate that the systematic construction of phage mutants will enable more detailed mapping of host range determinants and potentially of their bacterial targets. Phages such as T7, T3 or R initiate contact with their hosts through their tail fibers encoded by gene 17 (Hu et al., 2013). On E. coli B strains, T3 recognizes the penultimate glucose residue of the LPS, while T7 binds to an undetermined site further inwards in the LPS (Molineux, 2006). The exact receptor of phage R has not been determined but may be somewhere within the LPS of Y. ptb, since the related phage phiA1122 has been shown to plaque efficiently on strains of E. coli expressing the core LPS of Yersinia (Kiljunen et al., 2011). The actual receptor bound by Klebsiella phage K11 has not been formally documented but by analogy to phage T7, we assumed that its gene 17 bore its host-determinant activity. Interestingly, K11’s gp17 is known to be a capsule depolymerase specific to the K11 capsule of Klebsiella (Bessler et al., 1973) and we suspect that it is a tail spike because known Podoviridae that bind to and degrade capsules or smooth O-antigens (e.g., K1F, SP6, P22, K1-5, K1E) display tail spikes instead of fibers.
In summary, we demonstrated that synthetic phages based on common viral scaffolds can be designed to target a range of different bacterial hosts. Furthermore, we showed that a cocktail containing multiple engineered phages could effectively remove select bacterial targets in mixed microbial populations. We anticipate that the systematic and high-throughput engineering of viral genomes will enable new applications for the editing of microbial communities and enhanced understanding of bacterial viruses. For example, the engineering of common viral scaffolds could help simplify the discovery and manufacturing of novel bacteriophages and reduce the regulatory burden required for the use of phage cocktails as human therapeutics. In future work, we plan to expand our efforts to phages that are not relatives of T3/T7 phages, and phages that target bacteria outside of the Enterobacteriaceae family, including those which are major members of the human gut microbiome (Cryan and Dinan, 2012; Gibson et al., 2014; Zeeuwen et al., 2013). Also, abundant phage sequences contained within metagenomic databases could be synthesized and booted into functional phage particles for study and use. Finally, the systematic deconstruction and manipulation of these viral nanomachines will enable a greater understanding of phage biology and may provide insights that are useful for bioinspired nanotechnologies.
EXPERIMENTAL PROCEDURES
Strains, vector, and primers
Phages T7 (ATCC BAA-1025-B2, NC_001604) and T3 (ATCC 110303-B3, AJ318471) were lab stocks. Phages K1E (NC_007637), K1F (NC_007456), K1-5 (NC_008152), SP6 (NC_004831), and K11 (EU734173) were provided by Ian Molineux (University of Texas, Austin). Phage LUZ19 (NC_010326) was provided by Rob Lavigne (KU Leuven). Phage gh-1 (ATCC 12633-B1, NC_004665) was obtained from ATCC. Synthetic phages are listed in Table S5. S. cerevisiae BY4741 (MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0) was obtained from Thermo Scientific. E. coli strains BL21 [B, F− ompT hsdSB (rB− mB−) gal dcm], DH5α [K–12, F− λ − Φ80d lacZΔM15 Δ(lacZYA-argF)U169 deoR recA1 endA1 hsdR17 (rK− mK+) phoA supE44 thi-1 gyrA96 relA1], BW25113 [K-12, F− λ − Δ(araD-araB)567 ΔlacZ4787(::rrnB-3) rph-1 Δ(rhaD-rhaB)568 hsdR514], MG1655 (K-12, F− λ − ilvG− rfb-50 rph-1), and Nissle 1917 were obtained from lab stocks. E. cloni 10G [K–12, F− λ − Δ(ara leu)7697 araD139 ΔlacX74 galU galK Φ80d lacZΔM15 recA1 endA1 nupG rpsL (StrR) Δ(mrr-hsdRMS-mcrBC) tonA] from Lucigen is an E. coli DH10B derivative and is suitable for maintaining large DNA constructs (Durfee et al., 2008). Bacterial strains IJ284 Klebsiella sp. 390 (O3:K11), IJ1668 K-12 hybrid; K1 capsule, and IJ612 Salmonella typhimurium LT2 were provided by Ian Molineux. Virulence-plasmid-less Y. pseudotuberculosis IP2666 and YPIII were provided by Joan Mecsas (Tufts University). P. aeruginosa PAO1 was obtained from a lab stock. E. coli libraries, such as the ECOR group and DECA set, were sourced from the Thomas S. Whittam STEC Center (Michigan State University). P. putida C1S (ATCC 23287) was obtained from ATCC. The pRS415 yeast centromere vector with LEU2 marker (ATCC 87520) was obtained from a lab stock. Primers used in this study are listed in Table S6.
Synthesis of codon-optimized 13a gene 17
The gene was synthesized by Gen9 (Table S2).
Culture conditions
S. cerevisiae BY4741 was cultured in YPD [1% Bacto Yeast Extract (BD), 2% Bacto Peptone (BD), 2% dextrose (VWR)] at 30°C. Y. pseudotuberculosis strains and P. putida C1S were cultured in LB (BD) at 30°C. All other strains were cultured in LB at 37°C.
Preparation of phage genomes
Lysates were made by infecting 200 ml of logarithmically growing cells with the appropriate phage at a MOI of 0.1–0.01 and incubating the cultures until clearance. Cells were lysed and lysates were sterilized by adding 200 μl chloroform (Sigma). Lysates were centrifuged at 8,000 g for 5 min and then filtered through 0.22 μm filters (VWR) to remove cell debris. We added 216 μl of buffer L1 [20 mg/ml RNase A (Sigma), 6 mg/ml DNase I (NEB), 0.2 mg/ml BSA (NEB), 10 mM EDTA (Teknova), 100 mM Tris-HCl (VWR), 300 mM NaCl (VWR), pH 7.5] and incubated at 37°C for 1 h with gentle shaking. Then we added 30 ml of ice cold buffer L2 [30% polyethylene glycol (PEG) 6000 (Sigma), 3 M NaCl] and stored the samples overnight in 4°C. Samples were centrifuged at 10,000 g for 30 min at 4°C. Phage pellets were suspended in 9 ml buffer L3 (100 mM Tris-HCl, 100 mM NaCl, 25 mM EDTA, pH7.5). Then, we added 9 ml buffer L4 [4% SDS (VWR)] and incubated the samples at 70°C for 20 min. After cooling down on ice, 9 ml buffer L5 [2.55 M potassium acetate, pH4.8 (Teknova)] were added, and the samples were centrifuged at 10,000 g for 30 min at 4°C. Supernatants were passed onto Qiagen tip-100 system according to the manufacturer’s instructions to extract DNA.
Preparation of PCR products for assembling phage genomes
All PCR products were prepared with specific primer sets (Table S6) and KAPA HiFi DNA Polymerase. Five to ten 3.8–12.0 kbp PCR products including the YAC were used per reaction. Homology arms between the YAC and the phage genomes were added to the first and last phage genome fragments or to the YAC when capturing phage genomes from genomic DNA. The YAC amplicon was gel extracted to reduce background.
Preparation of yeast competent cells
S. cerevisiae BY4741 was grown in 5 ml YPD at 30°C for 24 h. Overnight cultures were added into 50 ml YPD, and incubated at 30°C for 4 h. Cells were harvested by centrifugation at 3,000 g and washed with 25 ml water and then with 1 ml of 100 mM lithium acetate (LiAc) (Alfa Aesar), and suspended in 400 μl of 100 mM LiAc. Fifty microliters were used for each transformation.
Yeast transformation
All DNA samples and a linearized pRS415 were collected in a tube (0.5 – 4.0 μg for each DNA sample and 100 ng linearized pRS415 in 50 μl water), and mixed with the transformation mixture [50 μl yeast competent cell, 240 μl 50% PEG3350 (Sigma), 36 μl 1 M LiAc, 25 μl 2 mg/ml salmon sperm DNA (Sigma)]. The mixture was incubated at 30°C for 30 min, then at 42°C for 20 min or at 42°C for 45 min, centrifuged at 8,000 g for 15 sec, and suspended in 200 μl water. Transformants were selected on complete synthetic defined medium without leucine (SD-Leu) [0.67% YNB+Nitrogen (Sunrise Science Products), 0.069% CSM-Leu (Sunrise Science Products), 2% dextrose] agar plates at 30°C for 3 days.
Extraction of captured phage genomes
Individual yeast transformants were picked into SD-Leu liquid medium and incubated at 30°C for 24 h. DNA was extracted from these cells using the YeaStar Genomic DNA Kit (Zymo Research) or Yeast Genomic DNA Purification Kit (Amresco) according to manufacturer instructions.
Rebooting of phages
E. coli 10G strain was used as a host bacterium for the initial propagation of phages. To reboot T7 and T3 phages, 3 μl of extracted DNA were electroporated into 20 – 25 μl cells in a 2 mm gap electroporation cuvette (Molecular BioProducts) at 2,500 V, 25 μF, 200 Ω using a Gene Pulser Xcell (Bio-Rad). Cells were mixed with 3 ml LB soft agar (LB + 0.6% agar) warmed at 55°C, poured onto LB plate, and incubated for 4 h at 37°C. To reboot other phages, after electroporation, cells were incubated at 37°C for 1–2 h in 1 ml LB medium. Then, we added drops of chloroform to kill the cells and release phages. After centrifugation at 12,000 g for 1 min, supernatants were mixed with 300 μl overnight cultures of host bacteria for the phages and 3 ml LB soft agar, poured onto LB plate, and incubated for 4 – 24 h at 30 or 37°C.
One-time phage propagation assays
We electroporated 10 – 200 ng of purified phage genomes into E. coli 10G. After incubation for 1 – 2 h, we added chloroform to kill the cells and release phages that may have failed to lyse cells. Then, supernatants were mixed with overnight cultures of each natural host bacteria for each phage in soft agar, poured onto agar plates, incubated for 4 – 24 h at 30 to 37°C, and analyzed for plaque formation.
Determination of Plaque Forming Units (PFUs)
We mixed serially diluted phages in 0.95% saline, 300 μl overnight culture of host bacteria, and 3 ml LB soft agar, and poured the mixture onto LB plates. After 4 – 24 h incubation at 30 or 37°C, phage plaques were counted, and PFU/ml values were calculated.
Plaque formation assays
We mixed 300 μl bacterial overnight cultures and 3 ml LB soft agar, and poured the mixtures onto LB plate. After 5 min at room temperature (RT), 2.5 μl of 10-fold serially diluted phages in 0.95% saline were spotted onto LB soft agar and incubated at 30 or 37°C. Efficiency of plating (EOP) was determined with respect to a reference bacterium for each phage. Klebsiella sp. 390 was used as the EOP reference for T7K11(gp11-12-17), K11, and K11WT. Y. ptb IP2666 was used for T3R(gp17). For other phages, BL21 was used as the reference host.
Adsorption assay
We mixed 100 μl of ~108 CFU/ml target bacterial strains and phages (MOI = 0.5), and incubated at RT for 10 min. Then, we added 700 μl of 0.95% saline and drops of chloroform to kill the cells and prevent the production of progeny phages. After centrifugation at 11,000 g for 1 min, supernatants were serially diluted and mixed with 300 μl of overnight cultures of host bacterial strains and 3 ml LB soft agar, and the mixtures were poured onto LB plates. After incubation, phage plaques were counted, and adsorption efficiencies were calculated according to the following equation:
Bacterial killing assays
Overnight cultures of Y. pseudotuberculosis IP2666 and Klebsiella sp. 390 were diluted 1:200 into LB and grown to log-phase (~108 CFU/ml), i.e., for 5 h at 30°C and for 3 h at 37°C, respectively. Bacterial cultures were mixed with phage lysates (MOI ~0.1) and incubated at 30 or 37°C. At each time point, bacteria were collected, washed twice with 0.95% saline, serially diluted, plated onto LB, and incubated at 30 or 37°C. Colonies were enumerated to calculate CFU/ml.
Microbial population editing assays
Overnight cultures of E. coli Nissle 1917, Y. pseudotuberculosis IP2666, and Klebsiella sp. 390 or E. coli ECOR16 were diluted 1:200 into LB and grown to log-phase (~108 CFU/ml), i.e., for 3 h at 37°C, for 5 h at 30°C, for 3 h at 37°C, and for 3 h at 37°C respectively. Cultures were mixed and treated with phage lysates (MOI ~0.1) and incubated at 30°C. At each time point, bacteria were collected, washed twice with 0.95% saline, serially diluted, plated onto LB, LB containing 25 μg/ml carbenicillin (VWR), and LB containing 1 μg/ml triclosan (VWR), and incubated at 30°C. Colonies were enumerated to calculate CFU/ml.
Statistical analysis
For all data points in all experiments, three samples were collected. The data are presented as the mean, and the error bars represent the SD. In the “Bacterial killing assays” and the “Microbial population editing assays”, all CFU data were log10-transformed before analysis. EOPs were log10-transformed before analysis (Figure 3).
Supplementary Material
Highlights.
A generalizable and efficient strategy for phage genome engineering was established
Synthetic phages with tunable host ranges were created
Engineered phages enacted efficient killing of their new target bacteria
Phage cocktails with engineered host ranges edited mixed bacterial populations
Acknowledgments
Strains IJ284 Klebsiella sp. 390, IJ1668 K-12 hybrid; K1 capsule, IJ612 S. typhimurium LT2, K1E, K1F, K1-5, SP6, and K11 were kindly provided by Ian Molineux (University of Texas at Austin). LUZ19 was kindly provided by Rob Lavigne (KU Leuven). Y. pseudotuberculosis IP2666 and YPIII were kindly provided by Joan Mecsas (Tufts University). We thank Oliver Purcell and Jennifer Henry for critical reading. This work was supported by grants from the Defense Threat Reduction Agency (HDTRA1-14-1-0007), the National Institutes of Health (1DP2OD008435, 1P50GM098792, 1R01EB017755), and the U.S. Army Research Laboratory / Army Research Office via the Institute for Soldier Nanotechnologies, under contract number W911NF-13-D-0001. H.A. was supported by fellowships from the Japan Society for the Promotion of Science and the Naito Foundation. D.P.P. was supported by the Portuguese Foundation for Science and Technology (SFRH/BD/76440/2011).
Footnotes
AUTHOR CONTRIBUTIONS
H.A., S.L., and T.K.L. designed the study, wrote the manuscript, and filed a provisional application on this work. H.A., S.L., and D.P.P. performed experiments. All authors analyzed the data and discussed results.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Beier H, Golomb M, Chamberlin M. Isolation of recombinants between T7 and T3 bacteriophages and their use in vitro transcriptional mapping. Journal of virology. 1977;21:753–765. doi: 10.1128/jvi.21.2.753-765.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bessler W, Freund-Molbert E, Knufermann H, Rduolph C, Thurow H, Stirm S. A bacteriophage-induced depolymerase active on Klebsiella K11 capsular polysaccharide. Virology. 1973;56:134–151. doi: 10.1016/0042-6822(73)90293-6. [DOI] [PubMed] [Google Scholar]
- Bikard D, Euler CW, Jiang W, Nussenzweig PM, Goldberg GW, Duportet X, Fischetti VA, Marraffini LA. Exploiting CRISPR-Cas nucleases to produce sequence-specific antimicrobials. Nature biotechnology. 2014;32:1146–1150. doi: 10.1038/nbt.3043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brussow H. What is needed for phage therapy to become a reality in Western medicine? Virology. 2012;434:138–142. doi: 10.1016/j.virol.2012.09.015. [DOI] [PubMed] [Google Scholar]
- Carlton RM. Phage therapy: past history and future prospects. Archivum immunologiae et therapiae experimentalis. 1999;47:267–274. [PubMed] [Google Scholar]
- Citorik RJ, Mimee M, Lu TK. Sequence-specific antimicrobials using efficiently delivered RNA-guided nucleases. Nature biotechnology. 2014;32:1141–1145. doi: 10.1038/nbt.3011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cryan JF, Dinan TG. Mind-altering microorganisms: the impact of the gut microbiota on brain and behaviour. Nat Rev Neurosci. 2012;13:701–712. doi: 10.1038/nrn3346. [DOI] [PubMed] [Google Scholar]
- Cuervo A, Pulido-Cid M, Chagoyen M, Arranz R, Gonzalez-Garcia VA, Garcia-Doval C, Caston JR, Valpuesta JM, van Raaij MJ, Martin-Benito J, et al. Structural characterization of the bacteriophage T7 tail machinery. The Journal of biological chemistry. 2013;288:26290–26299. doi: 10.1074/jbc.M113.491209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- d’Herelle F. Bacteriophage as a Treatment in Acute Medical and Surgical Infections. Bulletin of the New York Academy of Medicine. 1931;7:329–348. [PMC free article] [PubMed] [Google Scholar]
- Demerec M, Fano U. Bacteriophage-Resistant Mutants in Escherichia Coli. Genetics. 1945;30:119–136. doi: 10.1093/genetics/30.2.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietz A, Weisser HJ, Kossel H, Hausmann R. The gene for Klebsiella bacteriophage K11 RNA polymerase: sequence and comparison with the homologous genes of phages T7, T3, and SP6. Molecular & general genetics : MGG. 1990;221:283–286. doi: 10.1007/BF00261733. [DOI] [PubMed] [Google Scholar]
- Dunn JJ, Studier FW. Complete nucleotide sequence of bacteriophage T7 DNA and the locations of T7 genetic elements. Journal of molecular biology. 1983;166:477–535. doi: 10.1016/s0022-2836(83)80282-4. [DOI] [PubMed] [Google Scholar]
- Durfee T, Nelson R, Baldwin S, Plunkett G, 3rd, Burland V, Mau B, Petrosino JF, Qin X, Muzny DM, Ayele M, et al. The complete genome sequence of Escherichia coli DH10B: insights into the biology of a laboratory workhorse. Journal of bacteriology. 2008;190:2597–2606. doi: 10.1128/JB.01695-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischbach MA, Walsh CT. Antibiotics for emerging pathogens. Science. 2009;325:1089–1093. doi: 10.1126/science.1176667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia E, Elliott JM, Ramanculov E, Chain PS, Chu MC, Molineux IJ. The genome sequence of Yersinia pestis bacteriophage phiA1122 reveals an intimate history with the coliphage T3 and T7 genomes. Journal of bacteriology. 2003;185:5248–5262. doi: 10.1128/JB.185.17.5248-5262.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson DG, Benders GA, Andrews-Pfannkoch C, Denisova EA, Baden-Tillson H, Zaveri J, Stockwell TB, Brownley A, Thomas DW, Algire MA, et al. Complete chemical synthesis, assembly, and cloning of a Mycoplasma genitalium genome. Science. 2008;319:1215–1220. doi: 10.1126/science.1151721. [DOI] [PubMed] [Google Scholar]
- Gibson MK, Pesesky MW, Dantas G. The yin and yang of bacterial resilience in the human gut microbiota. J Mol Biol. 2014;426:3866–3876. doi: 10.1016/j.jmb.2014.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grice EA, Segre JA. The human microbiome: our second genome. Annual review of genomics and human genetics. 2012;13:151–170. doi: 10.1146/annurev-genom-090711-163814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendrix RW. Bacteriophage genomics. Current opinion in microbiology. 2003;6:506–511. doi: 10.1016/j.mib.2003.09.004. [DOI] [PubMed] [Google Scholar]
- Hu B, Margolin W, Molineux IJ, Liu J. The bacteriophage t7 virion undergoes extensive structural remodeling during infection. Science. 2013;339:576–579. doi: 10.1126/science.1231887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaschke PR, Lieberman EK, Rodriguez J, Sierra A, Endy D. A fully decompressed synthetic bacteriophage oX174 genome assembled and archived in yeast. Virology. 2012;434:278–284. doi: 10.1016/j.virol.2012.09.020. [DOI] [PubMed] [Google Scholar]
- Kiljunen S, Datta N, Dentovskaya SV, Anisimov AP, Knirel YA, Bengoechea JA, Holst O, Skurnik M. Identification of the lipopolysaccharide core of Yersinia pestis and Yersinia pseudotuberculosis as the receptor for bacteriophage phiA1122. Journal of bacteriology. 2011;193:4963–4972. doi: 10.1128/JB.00339-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiro R, Shitrit D, Qimron U. Efficient engineering of a bacteriophage genome using the type I-E CRISPR-Cas system. RNA biology. 2014;11:42–44. doi: 10.4161/rna.27766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin A, Jimenez J, Derr J, Vera P, Manapat ML, Esvelt KM, Villanueva L, Liu DR, Chen IA. Inhibition of bacterial conjugation by phage M13 and its protein g3p: quantitative analysis and model. PloS one. 2011;6:e19991. doi: 10.1371/journal.pone.0019991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin TY, Lo YH, Tseng PW, Chang SF, Lin YT, Chen TS. A T3 and T7 recombinant phage acquires efficient adsorption and a broader host range. PloS one. 2012;7:e30954. doi: 10.1371/journal.pone.0030954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu TK, Collins JJ. Dispersing biofilms with engineered enzymatic bacteriophage. Proceedings of the National Academy of Sciences. 2007;104:11197–11202. doi: 10.1073/pnas.0704624104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu TK, Collins JJ. Engineered bacteriophage targeting gene networks as adjuvants for antibiotic therapy. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:4629–4634. doi: 10.1073/pnas.0800442106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu TK, Koeris MS. The next generation of bacteriophage therapy. Current opinion in microbiology. 2011;14:524–531. doi: 10.1016/j.mib.2011.07.028. [DOI] [PubMed] [Google Scholar]
- Lu TK, Koeris MS, Chevalier BS, Holder JW, Mckenzie GJ, Brownell DR. Recombinant phage and methods. WO 2013049121 A2 Patent. 2013
- Martel B, Moineau S. CRISPR-Cas: an efficient tool for genome engineering of virulent bacteriophages. Nucleic acids research. 2014;42:9504–9513. doi: 10.1093/nar/gku628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molineux IJ. The T7 Group. In: Calendar R, editor. In The Bacteriophages. New York: Oxford Univ. Press; 2006. pp. 277–301. [Google Scholar]
- Pajunen MI, Elizondo MR, Skurnik M, Kieleczawa J, Molineux IJ. Complete nucleotide sequence and likely recombinatorial origin of bacteriophage T3. Journal of molecular biology. 2002;319:1115–1132. doi: 10.1016/S0022-2836(02)00384-4. [DOI] [PubMed] [Google Scholar]
- Pouillot F, Blois H, Iris F. Genetically engineered virulent phage banks in the detection and control of emergent pathogenic bacteria. Biosecurity and bioterrorism : biodefense strategy, practice, and science. 2010;8:155–169. doi: 10.1089/bsp.2009.0057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qimron U, Tabor S, CCR . In Microbe magazine. 2010. New details about bacteriophage T7-host interactions. [Google Scholar]
- Rashid MH, Revazishvili T, Dean T, Butani A, Verratti K, Bishop-Lilly KA, Sozhamannan S, Sulakvelidze A, Rajanna C. A Yersinia pestis-specific, lytic phage preparation significantly reduces viable Y. pestis on various hard surfaces experimentally contaminated with the bacterium. Bacteriophage. 2012;2:168–177. doi: 10.4161/bact.22240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin J, Jardine P, Noireaux V. Genome replication, synthesis, and assembly of the bacteriophage T7 in a single cell-free reaction. ACS Synth Biol. 2012;1:408–413. doi: 10.1021/sb300049p. [DOI] [PubMed] [Google Scholar]
- Steven AC, Trus BL, Maizel JV, Unser M, Parry DA, Wall JS, Hainfeld JF, Studier FW. Molecular substructure of a viral receptor-recognition protein. The gp17 tail-fiber of bacteriophage T7. Journal of molecular biology. 1988;200:351–365. doi: 10.1016/0022-2836(88)90246-x. [DOI] [PubMed] [Google Scholar]
- Sulakvelidze A, Alavidze Z, Morris JG., Jr Bacteriophage therapy. Antimicrob Agents Chemother. 2001;45:649–659. doi: 10.1128/AAC.45.3.649-659.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tetart F, Desplats C, Krisch HM. Genome plasticity in the distal tail fiber locus of the T-even bacteriophage: recombination between conserved motifs swaps adhesin specificity. Journal of molecular biology. 1998;282:543–556. doi: 10.1006/jmbi.1998.2047. [DOI] [PubMed] [Google Scholar]
- Trojet SN, Caumont-Sarcos A, Perrody E, Comeau AM, Krisch HM. The gp38 adhesins of the T4 superfamily: a complex modular determinant of the phage’s host specificity. Genome biology and evolution. 2011;3:674–686. doi: 10.1093/gbe/evr059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaung SJ, Esvelt KM, Church GM. CRISPR/Cas9-mediated phage resistance is not impeded by the DNA modifications of phage T4. PloS one. 2014;9:e98811. doi: 10.1371/journal.pone.0098811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoichi M, Abe M, Miyanaga K, Unno H, Tanji Y. Alteration of tail fiber protein gp38 enables T2 phage to infect Escherichia coli O157:H7. Journal of biotechnology. 2005;115:101–107. doi: 10.1016/j.jbiotec.2004.08.003. [DOI] [PubMed] [Google Scholar]
- Zeeuwen PL, Kleerebezem M, Timmerman HM, Schalkwijk J. Microbiome and skin diseases. Curr Opin Allergy Clin Immunol. 2013;13:514–520. doi: 10.1097/ACI.0b013e328364ebeb. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.