

Abstract
Increasing evidence indicates the high heterogeneity of cancer cells. Recent studies have revealed distinct subtypes of DNA methylation in colorectal cancer (CRC); however, the mechanism of heterogeneous methylation remains poorly understood. Gene expression is a natural, intermediate quantitative trait that bridges genotypic and phenotypic features. In this work, we studied the role of heterogeneous DNA methylation in tumorigenesis via gene expression analyses. Specifically, we integrated methylation and expression data in normal and tumor tissues, and examined the perturbations in coexpression patterns. We found that the heterogeneity of methylation leads to significant loss of coexpression connectivity in CRC; this finding was validated in an independent cohort. Functional analyses showed that the lost coexpression partners participate in important cancer-related pathways/networks, such as ErbB and mitogen-activated protein kinase (MAPK) signaling pathways. Our analyses suggest that the loss of coexpression connectivity induced by methylation heterogeneity might play an important role in CRC. To our knowledge, this is the first study to interpret methylation heterogeneity in cancer from the perspective of coexpression perturbation. Our results provide new perspectives in tumor biology and may facilitate the identification of potential biomedical therapies for cancer treatment.
INTRODUCTION
The initiation of cancer has long been recognized and attributed to the successive accumulation of genetic and epigenetic changes in tumor suppressor genes and oncogenes, which provides cancer cells the capacity to grow and metastasize unrestrainedly. The recent advances in next-generation sequencing and high-throughput technologies have greatly boosted our understanding of epigenetic alterations in cancer. For example, it is now well accepted that epigenetic alteration is another crucial contributor to tumorigenesis (Hon et al., 2012; Selamat et al., 2012). The epigenetic machinery and interplay between epigenetic factors play an important role in regulating many DNA-based molecular activities, such as transcriptional repression and activation, DNA repair, and DNA replication (Portela and Esteller, 2010; Hatzimichael and Crook, 2013). Consequently, epigenetic variations and their resultant abnormal gene expression profiles together may have critical effects in cancer initiation and progression (Selamat et al., 2012). Unlike genetic mutations, epigenetic changes are largely considered to be reversible (Selamat et al., 2012; Hatzimichael and Crook, 2013), and thus epigenetic therapy can open a new avenue for cancer treatment via the reversal of epigenetic effects (Dawson and Kouzarides, 2012; Hatzimichael and Crook, 2013). For example, two DNA demethylation agents, decitabine and azacitidine, have been recently shown to exert durable antitumor effects on hematological and epithelial tumor cells and have gained Food and Drug Administration approval as therapy for myelodysplastic syndrome (Fenaux et al., 2009; Tsai et al., 2012).
DNA methylation is the most studied epigenetic event in cancer (Baylin and Jones, 2011). Hypermethylation in the promoter CpG islands is commonly observed in various cancer genomes (Esteller, 2008; Dawson and Kouzarides, 2012): it facilitates cancer induction and progression by inhibiting the expression of tumor suppressor genes and can cooperate with other genetic lesions to affect key cancer pathways critical to tumorigenesis (Dai et al., 2013). In addition, DNA methylation can be treated as an important molecular feature characterizing the heterogeneity of cancer (Hinoue et al., 2012; The Cancer Genome Atlas Network, 2012). For colorectal cancer (CRC), Hinoue et al. found that CRC samples could be clustered into four subgroups based on the top 10% of loci with the highest DNA methylation variability (Hinoue et al., 2012), including a CpG island methylator phenotype-high subgroup, a CpG island methylator phenotype-low subgroup, and two other CRC subtypes. Interestingly, each subgroup shows distinctive clinical and genetic characteristics, implying the underlying biological mechanism of each subtype. Subsequently, The Cancer Genome Atlas (TCGA) consortium validated the four subgroups in their samples (The Cancer Genome Atlas Network, 2012).
The two previous reports (Hinoue et al., 2012; The Cancer Genome Atlas Network, 2012) directly stated that the highly variable DNA methylation (HVM) is strongly associated with distinctive biological features in CRC samples; however, the authors did not investigate the cellular mechanism behind methylation heterogeneity that contributes to tumorigenesis. Cancer usually occurs due to alterations of gene groups involving the biochemical and signal transduction processes that drive normal cells to malignant cells (Dreesen and Brivanlou, 2007; Yu et al., 2013; Cheng et al., 2014). Analysis of gene expression perturbation is a valid approach to investigate the alterations of molecular functions in biochemical and signaling networks (Masica and Karchin, 2011; Zhu et al., 2013). For instance, Zhu et al. (2013) studied the heterogeneity in CRC at the transcriptome level. The authors characterized three transcriptional subtypes and demonstrated how the unique combinations of key cancer pathways prevail in different CRC subtypes. Accordingly, methylation heterogeneity may contribute to tumorigenesis by perturbing the transcriptome in CRC.
In the present study, we developed an integrative framework to study the gene expression perturbations caused by DNA methylation and the resultant cellular and molecular consequences in CRC. We performed our analyses in a discovery dataset and a validation dataset, both of which have methylation and gene expression data available for the same patients.
MATERIALS AND METHODS
Data Acquisition
Discovery cohort
We obtained both DNA methylation and gene expression data for TCGA CRC samples (The Cancer Genome Atlas Network, 2012) from TCGA’s website (https://tcga-data.nci.nih.gov/docs/publications/coadread_2012/). DNA methylation data were generated on the Illumina Infinium human methylation (HM) 27 platform and were available for 236 colorectal tumors and 42 normal colorectal tissues. Notably, the Illumina Infinium HM 27 platform is designed to screen DNA methylation at gene promoter regions (The Cancer Genome Atlas Network, 2012) and is thus appropriate for the following analyses involving gene expression. The DNA methylation level is measured by the β-value, which is the ratio of the methylated probe intensity to the overall intensity (sum of methylated and unmethylated probe intensities; Du et al., 2010). RNA sequencing (RNA-seq)-based gene expression data were initially generated using the Illumina Genome Analyzer IIX (The Cancer Genome Atlas Network, 2012). The raw reads were first aligned against the reference genome using BWA (Li and Durbin, 2009) and then summarized to gene level quantification (please refer to the original paper for more details). We downloaded the expression data from TCGA for 270 colorectal tumors. Accordingly, we took the intersection of tumors for which both expression and methylation data were available and obtained a total of 231 tumors for the following analyses (Table 1). In addition to the 231 colorectal tumors, we also downloaded the RNA-seq data for 26 available normal samples from patients with “colon adenocarcinoma” and “rectum adenocarcinoma,” using the TCGA data portal (https://tcga-data.nci.nih.gov/tcga/dataAccessMatrix.htm). In summary, we obtained 231 tumors containing both gene expression and methylation data, 42 normal samples with methylation data, and 26 normal samples with gene expression data for our downstream analyses (Table 1).
TABLE 1.
Summary of Datasets
| Discovery cohort
|
Validation cohort
|
|||
|---|---|---|---|---|
| # Tumor samplesa | # Normal samples | # Tumor samplesa | # Normal samples | |
| DNA methylation | 231 | 42 | 25 | 29 |
| Gene expression | 231 | 26 | 25 | 26 |
Note that we required the DNA methylation and gene expression data from the same tumors for the following analyses.
Validation cohort
We downloaded both DNA methylation and gene expression data used in an independent study (Hinoue et al., 2012) for validation from the NCBI Gene Expression Omnibus (GEO) database (access IDs are GSE25062 and GSE25070, respectively). The DNA methylation dataset was comprised of 125 colorectal tumors and 29 adjacent nontumor colorectal tissues and was generated on the same platform used in the discovery dataset, that is, Illumina Infinium HM 27. Gene expression assays were performed using the Illumina Ref-8 v3.0 whole-genome BeadChip, and the dataset comprised 26 colorectal tumors and 26 adjacent nontumor colorectal tissues. Similarly, we only retained the 25 tumor samples for which both types of data were available (Table 1).
Data Preprocessing and Filtering
The CRC expression dataset of the discovery cohort was comprised of 20,531 genes. Expression levels were measured in “reads per kilo base per million” (RPKM). To filter genes with insignificant expression, we adopted the strategy used in a previous study (Imielinski et al., 2012). We used the log2 transformed RPKM as an expression index in the discovery dataset. We estimated the distribution of medians for the transformed RPKM and then used it to define a threshold to exclude the genes with insignificant expression (Supporting Information Fig. S1). This step resulted in 15,749 expressed genes. Among them, 10,908 genes had valid methylation data. For the 26 normal tissues collected from TCGA, the insignificant expression values were removed following the same procedure. In summary, we had four data matrices in the discovery cohort: a gene expression profile of 10,908 genes across 231 CRC tumors; a DNA methylation panel of 18,948 CpG sites covering the same 10,908 genes across the same 231 tumors; and two datasets from normal tissues (gene expression of 26 samples and DNA methylation of 42 samples) for comparison.
The preprocessing of gene expression and methylation data in the validation cohort was well performed in the original study (Hinoue et al., 2012). A total of 11,269 genes displayed both expression and DNA methylation values. Likewise, we had four data matrices in our validation cohort: an expression profile of 11,269 genes across 25 CRC tumors; a DNA methylation panel of 19,459 CpG sites covering the same 11,269 genes across the same 25 tumors; and two datasets from normal tissues (gene expression of 26 samples and DNA methylation of 29 samples) for comparison.
Coexpression Analyses
Gene coexpression was measured based on the Pearson correlation coefficient (PCC). To determine coexpressed genes, we calculated the pairwise PCCs among all genes in tumors and in normal samples, respectively. For each dataset, we ranked the gene pairs according to their PCC values and selected those having a PCC greater than the upper 2.5% quantile or less than the lower 2.5% quantile of the overall distribution as coexpression. In this way, no arbitrary or hard threshold was defined for either tumor or normal dataset. Rather, the sample size (231 vs. 26 in discovery cohort and 25 vs. 26 in validation cohort) was appropriately adjusted, and coexpressed gene pairs were selected according to the PCC distribution in the corresponding cohort.
Functional Enrichment Analyses
Functional enrichment analyses were performed using the Ingenuity Pathway Analysis (IPA, http://www.ingenuity.com/) system. The original P-values of enriched pathways from the Fisher’s exact test were adjusted using the Benjamini & Hochberg method (Benjamini and Hochberg, 1995) for multiple testing correction.
RESULTS
A schematic overview of our strategy is shown in Figure 1. We used gene expression and methylation data of the TCGA cohort (The Cancer Genome Atlas Network, 2012) as our discovery dataset and an independent cohort (Hinoue et al., 2012) as the validation dataset. To accurately characterize the relationship between DNA methylation and gene expression, we required the availability of both expression and methylation data from the same tumor samples. Starting from methylation data, we first identified the HVM loci based on the fluctuation of the DNA methylation β-values across the entire TCGA CRC tumor panel. Next, we probed the expression profile to explore how HVM perturbs gene expression and identified the ones whose expression patterns are strongly affected by the HVM status. We refer to these genes as “methylation-perturbed (MP) genes” hereafter. Then, we identified the differential coexpression patterns of MP genes between tumors and normal tissues. Meanwhile, we validated the HVM sites, MP genes, and differential coexpression patterns from the discovery data in an independent cohort. Lastly, we conducted functional analyses of MP genes and their perturbed coexpression partners, aiming to explore the underlying roles heterogeneous methylation plays in tumorigenesis.
Figure 1.
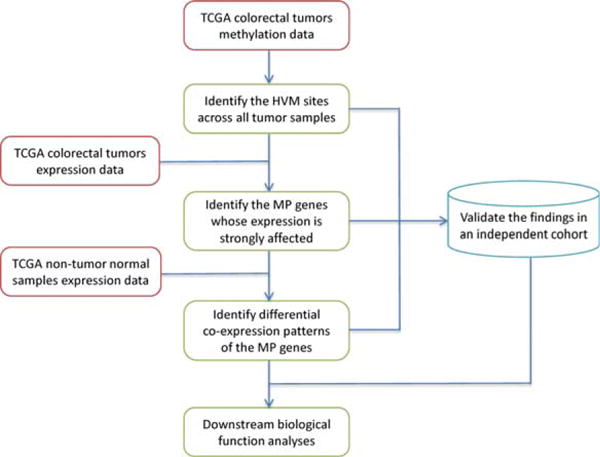
Schematic overview of this study. HVM: highly variable DNA methylation (standard deviation of β-values > 0.2). MP genes: MP genes. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Identification of HVM Sites and MP Genes in Discovery Cohort
We adopted the same criterion as in previous studies (Hinoue et al., 2012; The Cancer Genome Atlas Network, 2012) to define HVM sites, that is, the standard deviation of DNA methylation β-values across all the tumors > 0.2. Based on this threshold, we identified 1,157 HVM sites across the 231 CRC samples. Of note, almost all the HVM sites had a stationary methylation state in normal samples (Hinoue et al., 2012; The Cancer Genome Atlas Network, 2012). However, to make our analyses more stringent, we further excluded the sites that had a standard deviation of β-values > 0.2 across 42 normal tissues, reducing the HVM sites from 1,157 to 1,013.
To measure methylation impact on expression, we calculated the PCCs between the methylation status of 1,013 HVM sites and their target genes” expression. A majority of these sites (84.2%) had a negative PCC with gene expression, consistent with previous findings that DNA methylation at promoter regions generally inhibits gene expression (Portela and Esteller, 2010; Dawson and Kouzarides, 2012). Overall, the 1,013 HVM sites mapped to 802 unique target genes. Among the 802 targets, 629 genes had only one HVM site in their promoter regions; 155 genes had two corresponding sites; and 18 genes had three or more corresponding sites. To determine whether the multiple HVM sites covered by the same target gene play a consistent role in regulation, we regarded a positive PCC as methylation up-regulating gene expression and a negative one as down-regulating expression. Among the 173 (155 + 18) genes with multiple HVM sites, only 12 genes carried inconsistent directions. We manually inspected these 12 genes and found that all inconsistent PCCs showed a very small deviation from zero. For instance, gene CTSA covered two HVM sites, and the PCCs were 0.022 and −0.008, respectively. These small absolute values showed weak evidence of association between methylation and expression, and thus we removed these genes from further analysis. Next, we focused on the genes whose expression was strongly affected by methylation. We retained the PCCs less than −0.5 and referred to the corresponding genes as MP genes. Note that for the genes with more than one HVM site, we only kept the smallest PCC. The threshold −0.5 is indicated by a previous study (Lokk et al., 2014). A total of 118 MP genes were identified in this way (Supporting Information Table S1).
MP Genes Undergo Loss of Coexpression Connectivity in CRC Samples
Gene expression usually acts as the intermediate phenotype bridging DNA methylation and gene functions. As a result, our next topic of exploration was the expression perturbation of MP genes and the subsequent effect in carcinogenesis. We computed the pairwise PCCs of expression for all genes in the CRC samples and their counterparts in nontumor normal samples. Comparison of the PCCs in both the tumor and normal samples showed that the coexpression of MP genes was substantially reduced in CRC samples (Figs 2A and 2B). To determine to what extent the coexpression was lost, we calculated the number of coexpression partners for each gene (Fig. 2C). The median number of coexpression partners of MP genes in normal samples was 291, while a majority of MP genes (74, 62.7%) had fewer than 5 partners in CRC samples (Supporting Information Table S1). From Figure 2C, we saw that the non-MP genes had a greater variance in CRC samples compared to normal samples. The number of coexpression partners of MP genes in CRC was significantly smaller than that in normal samples (Wilcoxon test, P-value = 1.07 × 10−19). When examined within the CRC dataset, MP genes had significantly fewer coexpression partners compared to non-MP genes (Wilcoxon test, P-value = 8.74 × 10−30). In contrast, there was no significant difference between MP genes and non-MP genes within normal samples (Wilcoxon test, P-value > 0.05). These findings confirmed the loss of coexpression partners of MP genes in CRC samples.
Figure 2.
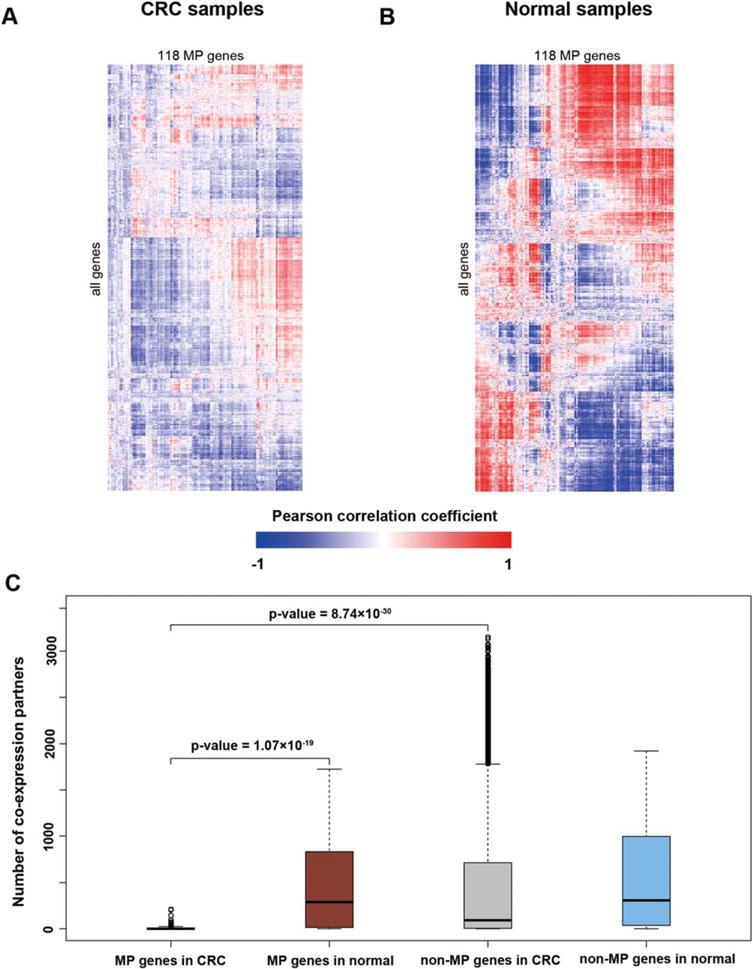
MP genes lose the co-expression connectivity in CRC. (A) Coexpression patterns of MP genes in CRC samples. (B) Coexpression patterns of MP genes in normal samples. (C) The boxplot of coexpression partners of MP genes and non-MP genes in CRC and normal dataset, respectively. The partners of MP genes in CRC are significantly less than in the normal dataset (Wilcoxon test, P-value = 1.07 × 10−19) and non-MP genes in CRC (Wilcoxon test, P-value = 8.74 × 10−30). [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Validation in an Independent Cohort
To validate our findings in the discovery cohort, we performed the same analysis in an independent cohort (Hinoue et al., 2012). Among the 1,013 HVM probes identified in the discovery cohort, 896 probes were eligible for analyses in the validation cohort with valid DNA methylation values. Strikingly, 675 of these 896 HVM sites (75.3%) were successfully replicated in the validation cohort (Fig. 3A). The hypergeometric test showed a significant overlap between HVM sites identified in discovery and in validation cohorts (P-value < 2.2 × 10−16). We next examined the MP genes in the validation cohort. Among the 118 MP genes identified in the discovery cohort, 103 had available expression values and 44 of them (42.7%) could be replicated in the validation cohort (hypergeometric test, P-value < 2.2 × 10−16).
Figure 3.
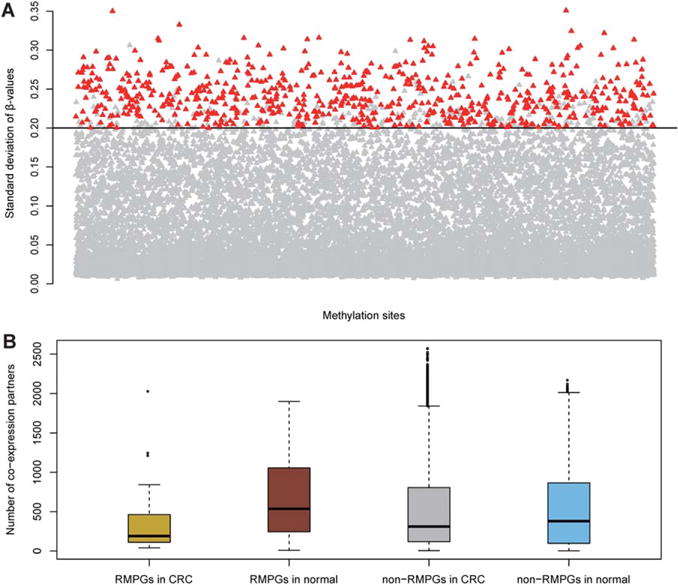
Validation in an independent cohort. (A) The replication of HVM sites. Total of 675 (75.33%) HVM sites (highlighted) identified in discovery cohort could be replicated in validation cohort. (B) The boxplot of co-expression partners of RMPGs and non-RMPGs in CRC and normal categories in the validation cohort. Similarly, the loss of co-expression connectivity can be observed, confirming the reliability of connectivity loss identified in the discovery cohort. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
In addition, we also investigated the loss of coexpression partners of MP genes in the validation cohort. Here, we focused on the 44 replicated MP genes (RMPGs). We computed the number of coexpression partners of RMPGs and the remaining genes (i.e., non-RMPGs) in CRC and in normal samples, respectively. As shown in Figure 3B, the patterns of coexpression connectivity were well replicated in the discovery cohort (Fig. 2C). Non-RMPGs had a more divergent range in terms of partner numbers in CRC samples compared to normal. As expected, the coexpression partners of RMPGs in CRC were significantly fewer than in the normal dataset (Wilcoxon test, P-value = 7.37 × 10−4). Within the CRC dataset, RMPGs had significantly fewer coexpression partners compared with non-RMPGs (Wilcoxon test, P-value = 0.046). In contrast, RMPGs had more co-expression partners than non-RMPGs in the normal dataset (Wilcoxon test, P-value = 0.019), which is a discrepancy between the discovery and validation cohorts. In the discovery cohort, there was no significant difference between MP genes and non-MP genes within normal samples (Wilcoxon test, P-value > 0.05). It is worth noting that this small discrepancy supports, rather than undermines, the previous findings that MP genes lose coexpression connectivity in CRC.
In summary, most of the HVM sites and a moderate proportion of MP genes identified in the discovery cohort were successfully replicated in the independent validation cohort. Moreover, the coexpression pattern of RMPGs in the validation cohort showed the same trend as in the discovery cohort. Of note, we performed the validation process within an independent cohort with much fewer samples, especially for the tumors. The high consistency between the two cohorts indicated the reliability of our findings in the discovery cohort.
Functional Enrichment Analyses of Lost Coexpression Partners
To investigate the possible functions of MP genes in tumorigenesis, we performed literature mining by searching NCBI PubMed titles/abstracts for the co-occurrence of a gene symbol with several CRC-related keywords (“colorectal cancer,” “colon cancer,” or “rectum cancer”). However, automated literature mining may be prone to false positives; therefore, we manually checked the interesting genes gathered from the literature mining results. Some MP genes had previously been identified as playing important roles in CRC, and several of them even promise to be targets of epigenetic therapy. We highlighted six well-studied genes in Table 2 and collected evidence showing that these genes are frequently observed with loss of function in tumors, mainly due to DNA methylation.
TABLE 2.
Well-Studied Methylated Genes in CRC and the Relevant References
| Gene symbol | Description | References |
|---|---|---|
| MLH1 | MutL Homolog 1 |
Kane, et al., 1997 Cunningham, et al., 1998 Weisenberger, et al., 2006 |
| BNIP3 | BCL2/Adenovirus E1B 19kDa Interacting Protein 3 |
Murai, et al., 2005 Shimizu, et al., 2010 Liu, et al., 2011 |
| CHFR | Checkpoint with forkhead and ring finger domains, E3 ubiquitin protein ligase |
Morioka, et al., 2006 Tanaka, et al., 2011 |
| MGMT | O-6-methylguanine-DNA methyltransferase |
Nakamura, et al., 2001 Gerson 2004 Ogino, et al., 2007 Nagasaka, et al., 2008 Hibi, et al., 2009 Hawkins, et al., 2009 Lee, et al., 2009 |
| HLTF | Helicase-like transcription factor |
Moinova, et al., 2002 Herbst, et al., 2009 Sandhu, et al., 2012 |
| RBP1 | Retinol binding protein 1, cellular |
Esteller, et al., 2002 Toki, et al., 2010 Peralta, et al., 2012 |
We next performed pathway and network enrichment analyses to explore the cellular and molecular functions in which the lost coexpression partners of the six MP genes are involved. We used Ingenuity Pathway Analysis (IPA, http://www.ingenuity.com/) to perform the pathway and network analyses. Supporting Information Table S2 summarizes the top five significantly enriched IPA canonical pathways [adjusted P-value < 0.1, Benjamini & Hochberg multiple testing correction (Benjamini and Hochberg, 1995)] for the lost coexpression partners for each of the six MP genes. In Supporting Information Table S2, we used the six MP genes as column names to represent their corresponding lost partners. The most recurrently enriched pathway, “Signaling by Rho Family GTPases,” was observed in three genes (adjusted P-value = 1.17 × 10−6 for BNIP3, adjusted P-value = 2.64 × 10−5 for CHFR, and adjusted P-value = 7.53 × 10−8 for HLTF), and the pathway “RhoGDI Signaling” was enriched in BNIP3 (adjusted P-value = 1.14 × 10−5) and HLTF (adjusted P-value = 7.53 × 10−8). Other pathways of interest included “Molecular Mechanisms of Cancer” (adjusted P-value = 0.0648 for MLH1), “EIF2 Signaling” (adjusted P-value = 6.35 × 10−23 for MGMT), and “mTOR Signaling” (adjusted P-value = 1.78 × 10−5 for MGMT) (See Supporting Information Table S2 for details). Overall, these are well-known pathways involved in tumor biology, suggesting the loss of coexpression connectivity might play a role in cancer initiation and development.
Connectivity Loss of BNIP3 Disturbs ErbB and MAPK Pathways
We illustrated the promise of MP genes in tumorigenesis using BNIP3 as an example (Fig. 4). A total of 1,503 genes were coexpressed with BNIP3 in the normal samples, all of which lost the coexpression relationship in CRC. We then explored the enriched networks of 637 lost partners that harbored negative coexpression PCCs with BNIP3 using IPA. As shown in Figure 4A, ErbB and MAPK signaling pathway members were frequently observed in one of the top-enriched networks reported by IPA. ERBB2 and ERBB3 are two members of the epidermal growth factor receptor (EGFR) family of receptor tyrosine kinases. Their function is mainly to bind to other EGFR members to form a heterodimer, enhancing the activation of downstream signaling pathways which lead to cell proliferation or differentiation. Overexpression of both genes has been widely reported in many cancer types, including CRC (Maurer et al., 1998; Braun et al., 2001; Holbro et al., 2003; Zhou et al., 2004). The MAPK pathway is a common downstream target of the ErbB signaling pathway. Interestingly, five MAPK members (MAPK3, MAP2K2, FGFR4, FGFR4, and MAP3K11) form a small module in the same network (Fig. 4A). We specifically examined the expression pattern of these seven ErbB/MAPK pathway members in CRC and in normal samples. As shown in Figures 4B and C, we found that the negative correlations between each of them and BNIP3 in normal samples were strongly disrupted, due to the highly variable methylation status of BNIP3 in CRC samples. Collectively, these results support the previous findings that BNIP3 is a proapoptotic protein and that the methylation of BNIP3 contributes to the dysfunction of apoptotic pathways, suggesting our strategy is effective to explore cancer pathogenesis from the perspective of DNA methylation.
Figure 4.
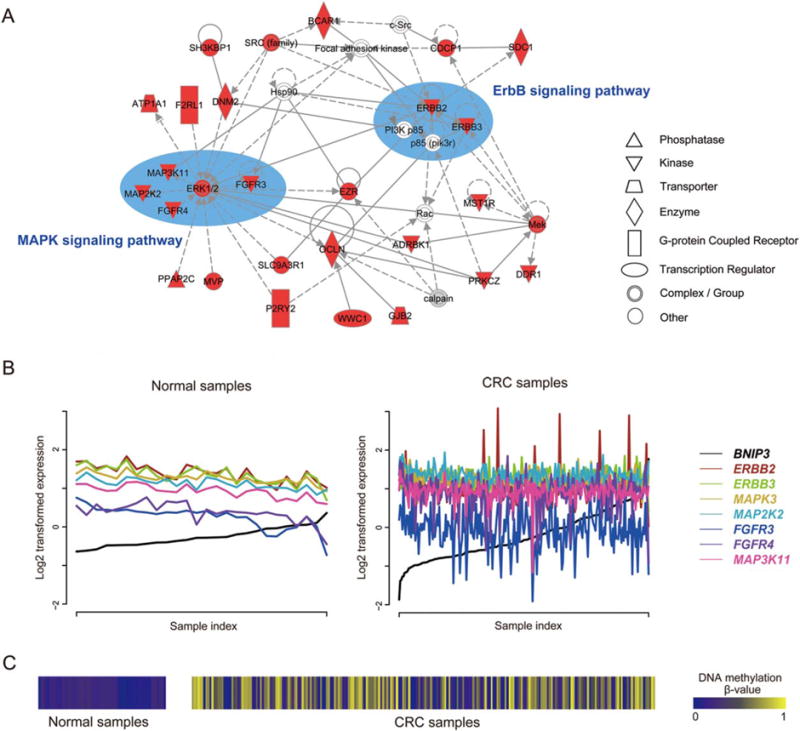
Network analyses of the lost co-expression partners of MP gene BNIP3 by Ingenuity Pathway Analysis (IPA) tool. (A) One of the top networks reported by IPA, using 637 lost partners that harbor negative co-expression PCCs with BNIP3 as input. The highlighted nodes are co-expression partners of MP genes in the normal dataset but are lost in CRC. The key components were manually depicted within ellipses and labelled according to the KEGG pathway database (Kanehisa and Goto, 2000). Prominent in the network are seven kinases (ERBB2, ERBB3, MAPK3, MAP2K2, FGFR4, FGFR4, and MAP3K11) from the ErbB/MAPK signaling pathways. Notably, ERK1/2 in the network is a dimer consisting of MAPK1 and MAPK3, as reported by IPA, and only MAPK3 is in our input list. Figure 4B shows that the distinct negative correlations between the seven kinases and BNIP3 in the normal panel are disrupted in CRC samples. See the text for more details. (B) The co-expression pattern between seven ErbB/MAPK pathway member genes (ERBB2, ERBB3, MAPK3, MAP2K2, FGFR4, FGFR4, and MAP3K11) and BNIP3 in the normal dataset (left) and CRC (right). To clarify the co-expression pattern, we rearranged the sample index according to the ascending order of BNIP3 expression values. (C) The DNA methylation pattern of BNIP3 across normal samples (left) and CRC samples (right). [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
DISCUSSION
The initiation and development of cancer has been proven to be highly heterogeneous, and different molecular and cellular alterations may lead to the similar or same phenotype (Burrell et al., 2013; Marte, 2013). The effort to characterize genetic diversity both between and within tumors has continued for a long time (Burrell et al., 2013; Marte, 2013). So far, the heterogeneity in epigenetics and how it influences tumorigenic processes have been barely investigated. Recently, two independent groups investigated the heterogeneity of DNA methylation in CRC (Hinoue et al., 2012; The Cancer Genome Atlas Network, 2012). Using the top variable methylation sites as molecular features, both groups characterized four distinctive subtypes in CRC samples; however, the authors performed little research regarding the downstream effects of heterogeneous methylation in tumor biology. Generally, promoter methylation induces variation within the expression of target genes, and gene expression is a legitimate intermediate bridging genotypic and phenotypic features. Interestingly, another recent publication focused on the heterogeneity of CRC at the transcriptome level (Zhu et al., 2013). The authors identified three distinctive transcriptional subtypes in a CRC cohort and validated the findings in an independent group of patients. Moreover, the authors found that subtype-specific cancer-related pathways or signaling networks prevail in different subtypes.
Motivated by these findings, we attempted to build a link between methylation heterogeneity and gene expression perturbation. In this study, we first identified the HVM sites in CRC samples and then focused on the MP genes whose expression profiles were strongly affected by the heterogeneous methylation. As a result, the expression level of MP genes was more diverse in CRC compared to normal samples. Next, we found that the diverse expression patterns of MP genes led to a loss of coexpression connectivity. The underlying hypothesis in this study is that the loss of coexpression connectivity of MP genes arising from heterogeneous methylation promotes the transformation of tumor cells. We further assessed this hypothesis through a combination of literature data, pathway and network enrichment analyses of the lost coexpression partners, and replication in an independent cohort.
Among the 118 MP genes, we highlighted six of them through literature search and manual curation (Table 2). The roles of methylation in these six genes have been well studied in CRC. Specifically, the promoter methylation of MLH1 has long been studied in CRC since 1997 (Kane et al., 1997). Nowadays it is well established that promoter hypermethylation, accompanied by decreased expression, is one of the major mechanisms that inactivates MLH1 in CRC, especially in sporadic CRC with microsatellite instability (Cunningham et al., 1998; Weisenberger et al., 2006). BNIP3, a Bcl-2 family proapoptotic protein, can induce cell death. The methylation and silencing of the BNIP3 gene has been observed to occur in ~60% of CRC patients, subsequently contributing to the dysfunction of the apoptotic pathway and leading to drug resistance (Murai et al., 2005; Shimizu et al., 2010). More promisingly, BNIP3 is a potential chemotherapeutic target. Previous studies implicated that Verticillin A might induce apoptosis by increasing BNIP3 expression through demethylation of the BNIP3 promoter, showing great promise to eventually overcome drug resistance in human cancer therapy (Liu et al., 2011). Similarly, methylated CHFR was also found in ~60% of CRC patients (Tanaka et al., 2011). CHFR is a tumor suppressor gene, and the silencing of this gene due to aberrant methylation has been reported in several primary tumors, including CRC (Morioka et al., 2006). MGMT is a ubiquitously expressed DNA mismatch repair protein that removes alkylating lesions at the O6 of guanine residues (Nakamura et al., 2001). Failure in this step can undermine cancer therapies and result in neoplastic transformation (Gerson, 2004). The low expression of MGMT due to methylation has been frequently observed in all stages of CRC (Ogino et al., 2007; Nagasaka et al., 2008; Hibi et al., 2009). In addition, MGMT methylation has also been associated with the mutations of some cancer genes, such as KRAS and TP53 (Nakamura et al., 2001; Hawkins et al., 2009). In consideration of its importance, MGMT methylation has been suggested to be a valuable biomarker for early detection of CRC (Lee et al., 2009). Another gene, HLTF, encodes a DNA helicase protein involved in the regulation of gene expression by altering the chromatin structure. The epigenetic inactivation of HLTF is a common event in CRC, promoting carcinogenesis by inducing genomic instability (Sandhu et al., 2012). Moreover, the functional loss of HLTF was found to be associated with poor survival outcome and a relatively high risk of disease recurrence after curative surgery (Herbst et al., 2009). These findings suggested that HLTF is a tumor suppressor gene in CRC (Moinova et al., 2002). The last gene in Table 2, RBP1, encodes the carrier protein involved in the intracellular transport of retinol. Methylation-based functional loss of RBP1 has been observed in multiple cancers, and it may have potential for cancer prevention and therapy (Esteller et al., 2002; Toki et al., 2010; Peralta et al., 2012).
We next performed the pathway and network enrichment analyses of the lost coexpression partners of the six well-studied MP genes to assess our hypothesis. IPA analyses summarized the top five enriched canonical pathways for the lost coexpression partners of the six MP genes. All of the enriched lists carry well-known cancer-related pathways except RBP1, including “Molecular Mechanisms of Cancer,” “Signaling by Rho Family GTPases,” “RhoGDI Signaling,” “EIF2 Signaling,” and “mTOR Signaling” (Supporting Information Table S2). In addition, we also attempted to exemplify in detail how aberrant methylation contributes to tumorigenesis through IPA network analyses of the lost partners of MP gene BNIP3 (Fig. 4). Interestingly, ErbB/MAPK pathway members act prominently in Figure 4A. ERBB2 and ERBB3 are two important members of the EGFR family. Their main function is to induce the activation of downstream signaling pathways which then lead to cell proliferation and differentiation. Importantly, the MAPK signaling pathway, which is often involved in cell proliferation, differentiation, and migration, is a common target of all ErbB receptors. In normal samples, the expression level between BNIP3 and the seven ErbB/MAPK member genes was negatively correlated, but the coexpression connectivity was substantially disrupted in CRC patients (Fig. 4B). The aberrant methylation status of BNIP3 in tumors might be the reason for this coexpression connectivity loss (Fig. 4C). Collectively, these findings enhance the previous understanding that the methylation of the proapoptotic gene BNIP3 contributes to the dysfunction of apoptotic pathways, which subsequently leads to cell proliferation and differentiation (Murai et al., 2005; Shimizu et al., 2010; Liu et al., 2011).
Another piece of encouraging evidence regarding the loss of coexpression connectivity in tumor biology has recently been published (Anglani et al., 2014): Anglani et al. collected expression data for five distinct cancer types (colorectal, lung, gastric, pancreatic, and cervical) and investigated the topological features of coexpression networks for tumor and normal phenotypes. They found that coexpression connectivity underwent dramatic losses in all five studied cancer types compared to normal controls and concluded that coexpression loss is common in cancer and should be taken into account in carcinogenesis studies.
To confirm the reliability of our findings, we repeated the same analysis procedures in an independent CRC dataset. Despite the far fewer samples in the validation cohort, we could replicate 72.3% of HVM sites and 42.7% of MP genes. The hypergeometric test indicated that these overlap rates were unlikely by chance (both P-values < 2.2 × 10−16). More importantly, we also observed the significant loss of the coexpression connectivity of MP genes in the validation cohort. It is worth noting that the validation was performed in a totally independent cohort with much fewer samples. The high level of consistency between the discovery and validation cohorts indicated the reliability of our findings and supported our hypothesis.
Supplementary Material
Acknowledgments
The authors thank Drs. Xingyi Guo and Wei Jiang at Vanderbilt University for their valuable discussion. We thank Ms. Christen Parzych for proofreading and polishing the manuscript.
Supported by: National Institutes of Health, Grant numbers: R01LM011177, P50CA095103, P50CA098131, and P30CA068485; Ingram Professorship Funds (Z.Z.).
Footnotes
Additional Supporting Information may be found in the online version of this article.
References
- Anglani R, Creanza TM, Liuzzi VC, Piepoli A, Panza A, Andriulli A, Ancona N. Loss of connectivity in cancer co-expression networks. PLoS One. 2014;9:e87075. doi: 10.1371/journal.pone.0087075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baylin SB, Jones PA. A decade of exploring the cancer epigenome – biological and translational implications. Nat Rev Cancer. 2011;11:726–734. doi: 10.1038/nrc3130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y. Controlling the false discovery rate – a practical and powerful approach to multiple testing. J RStat Soc Ser B-Methodol. 1995;57:289–300. [Google Scholar]
- Braun S, Schlimok G, Heumos I, Schaller G, Riethdorf L, Riethmuller G, Pantel K. ErbB2 overexpression on occult metastatic cells in bone marrow predicts poor clinical outcome of stage I–III breast cancer patients. Cancer Res. 2001;61:1890–1895. [PubMed] [Google Scholar]
- Burrell RA, McGranahan N, Bartek J, Swanton C. The causes and consequences of genetic heterogeneity in cancer evolution. Nature. 2013;501:338–345. doi: 10.1038/nature12625. [DOI] [PubMed] [Google Scholar]
- Cheng F, Jia P, Wang Q, Lin CC, Li WH, Zhao Z. Studying tumorigenesis through network evolution and somatic mutational perturbations in the cancer interactome. Mol Biol Evol. 2014;31:2156–2169. doi: 10.1093/molbev/msu167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham JM, Christensen ER, Tester DJ, Kim CY, Roche PC, Burgart LJ, Thibodeau SN. Hypermethylation of the hMLH1 promoter in colon cancer with microsatellite instability. Cancer Res. 1998;58:3455–3460. [PubMed] [Google Scholar]
- Dai W, Zeller C, Masrour N, Siddiqui N, Paul J, Brown R. Promoter CpG island methylation of genes in key cancer pathways associates with clinical outcome in high-grade serous ovarian cancer. Clin Cancer Res. 2013;19:5788–5797. doi: 10.1158/1078-0432.CCR-13-1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson MA, Kouzarides T. Cancer epigenetics: From mechanism to therapy. Cell. 2012;150:12–27. doi: 10.1016/j.cell.2012.06.013. [DOI] [PubMed] [Google Scholar]
- Dreesen O, Brivanlou AH. Signaling pathways in cancer and embryonic stem cells. Stem Cell Rev. 2007;3:7–17. doi: 10.1007/s12015-007-0004-8. [DOI] [PubMed] [Google Scholar]
- Du P, Zhang X, Huang CC, Jafari N, Kibbe WA, Hou L, Lin SM. Comparison of Beta-value and M-value methods for quantifying methylation levels by microarray analysis. BMC Bioinformatics. 2010;11:587. doi: 10.1186/1471-2105-11-587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esteller M. Epigenetics in cancer. N Engl J Med. 2008;358:1148–1159. doi: 10.1056/NEJMra072067. [DOI] [PubMed] [Google Scholar]
- Esteller M, Guo M, Moreno V, Peinado MA, Capella G, Galm O, Baylin SB, Herman JG. Hypermethylation-associated Inactivation of the Cellular Retinol-Binding-Protein 1 Gene in Human Cancer. Cancer Res. 2002;62:5902–5905. [PubMed] [Google Scholar]
- Fenaux P, Mufti GJ, Hellstrom-Lindberg E, Santini V, Finelli C, Giagounidis A, Schoch R, Gattermann N, Sanz G, List A, Gore SD, Seymour JF, Bennett JM, Byrd J, Backstrom J, Zimmerman L, McKenzie D, Beach C, Silverman LR, International Vidaza High-Risk MDSSSG Efficacy of azacitidine compared with that of conventional care regimens in the treatment of higher-risk myelodysplastic syndromes: a randomised, open-label, phase III study. Lancet Oncol. 2009;10:223–232. doi: 10.1016/S1470-2045(09)70003-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerson SL. MGMT: its role in cancer aetiology and cancer therapeutics. Nat Rev Cancer. 2004;4:296–307. doi: 10.1038/nrc1319. [DOI] [PubMed] [Google Scholar]
- Hatzimichael E, Crook T. Cancer epigenetics: new therapies and new challenges. J Drug Deliv. 2013;2013:529312. doi: 10.1155/2013/529312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkins NJ, Lee JH, Wong JJ, Kwok CT, Ward RL, Hitchins MP. MGMT methylation is associated primarily with the germline C>T SNP (rs16906252) in colorectal cancer and normal colonic mucosa. Mod Pathol. 2009;22:1588–1599. doi: 10.1038/modpathol.2009.130. [DOI] [PubMed] [Google Scholar]
- Herbst A, Wallner M, Rahmig K, Stieber P, Crispin A, Lamerz R, Kolligs FT. Methylation of helicase-like transcription factor in serum of patients with colorectal cancer is an independent predictor of disease recurrence. Eur J Gastroenterol Hepatol. 2009;21:565–569. doi: 10.1097/MEG.0b013e328318ecf2. [DOI] [PubMed] [Google Scholar]
- Hibi K, Goto T, Mizukami H, Kitamura Y, Sakata M, Saito M, Ishibashi K, Kigawa G, Nemoto H, Sanada Y. MGMT gene is aberrantly methylated from the early stages of colorectal cancers. Hepatogastroenterology. 2009;56:1642–1644. [PubMed] [Google Scholar]
- Hinoue T, Weisenberger DJ, Lange CP, Shen H, Byun HM, Van Den Berg D, Malik S, Pan F, Noushmehr H, van Dijk CM, Tollenaar RA, Laird PW. Genome-scale analysis of aberrant DNA methylation in colorectal cancer. Genome Res. 2012;22:271–282. doi: 10.1101/gr.117523.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holbro T, Beerli RR, Maurer F, Koziczak M, Barbas CF, 3rd, Hynes NE. The ErbB2/ErbB3 heterodimer functions as an oncogenic unit: ErbB2 requires ErbB3 to drive breast tumor cell proliferation. Proc Natl Acad Sci USA. 2003;100:8933–8938. doi: 10.1073/pnas.1537685100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hon GC, Hawkins RD, Caballero OL, Lo C, Lister R, Pelizzola M, Valsesia A, Ye Z, Kuan S, Edsall LE, Camargo AA, Stevenson BJ, Ecker JR, Bafna V, Strausberg RL, Simpson AJ, Ren B. Global DNA hypomethylation coupled to repressive chromatin domain formation and gene silencing in breast cancer. Genome Res. 2012;22:246–258. doi: 10.1101/gr.125872.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imielinski M, Berger AH, Hammerman PS, Hernandez B, Pugh TJ, Hodis E, Cho J, Suh J, Capelletti M, Sivachenko A, Sougnez C, Auclair D, Lawrence MS, Stojanov P, Cibulskis K, Choi K, de Waal L, Sharifnia T, Brooks A, Greulich H, Banerji S, Zander T, Seidel D, Leenders F, Ansen S, Ludwig C, Engel-Riedel W, Stoelben E, Wolf J, Goparju C, Thompson K, Winckler W, Kwiatkowski D, Johnson BE, Janne PA, Miller VA, Pao W, Travis WD, Pass HI, Gabriel SB, Lander ES, Thomas RK, Garraway LA, Getz G, Meyerson M. Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell. 2012;150:1107–1120. doi: 10.1016/j.cell.2012.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kane MF, Loda M, Gaida GM, Lipman J, Mishra R, Goldman H, Jessup JM, Kolodner R. Methylation of the hMLH1 promoter correlates with lack of expression of hMLH1 in sporadic colon tumors and mismatch repair-defective human tumor cell lines. Cancer Res. 1997;57:808–811. [PubMed] [Google Scholar]
- Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28:27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee BB, Lee EJ, Jung EH, Chun HK, Chang DK, Song SY, Park J, Kim DH. Aberrant methylation of APC, MGMT, RASSF2A, and Wif-1 genes in plasma as a biomarker for early detection of colorectal cancer. Clin Cancer Res. 2009;15:6185–6191. doi: 10.1158/1078-0432.CCR-09-0111. [DOI] [PubMed] [Google Scholar]
- Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F, Liu Q, Yang D, Bollag WB, Robertson K, Wu P, Liu K. Verticillin A overcomes apoptosis resistance in human colon carcinoma through DNA methylation-dependent upregulation of BNIP3. Cancer Res. 2011;71:6807–6816. doi: 10.1158/0008-5472.CAN-11-1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lokk K, Modhukur V, Rajashekar B, Martens K, Magi R, Kolde R, Kolt Ina M, Nilsson TK, Vilo J, Salumets A, Tonisson N. DNA methylome profiling of human tissues identifies global and tissue-specific methylation patterns. Genome Biol. 2014;15:R54. doi: 10.1186/gb-2014-15-4-r54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marte B. Tumour heterogeneity. Nature. 2013;501:327. doi: 10.1038/501327a. [DOI] [PubMed] [Google Scholar]
- Masica DL, Karchin R. Correlation of somatic mutation and expression identifies genes important in human glioblastoma progression and survival. Cancer Res. 2011;71:4550–4561. doi: 10.1158/0008-5472.CAN-11-0180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurer CA, Friess H, Kretschmann B, Zimmermann A, Stauffer A, Baer HU, Korc M, Buchler MW. Increased expression of erbB3 in colorectal cancer is associated with concomitant increase in the level of erbB2. Hum Pathol. 1998;29:771–777. doi: 10.1016/s0046-8177(98)90444-0. [DOI] [PubMed] [Google Scholar]
- Moinova HR, Chen WD, Shen L, Smiraglia D, Olechnowicz J, Ravi L, Kasturi L, Myeroff L, Plass C, Parsons R, Minna J, Willson JK, Green SB, Issa JP, Markowitz SD. HLTF gene silencing in human colon cancer. Proc Natl Acad Sci USA. 2002;99:4562–4567. doi: 10.1073/pnas.062459899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morioka Y, Hibi K, Sakai M, Koike M, Fujiwara M, Kodera Y, Ito K, Nakao A. Aberrant methylation of the CHFR gene is frequently detected in non-invasive colorectal cancer. Anticancer Res. 2006;26:4267–4270. [PubMed] [Google Scholar]
- Murai M, Toyota M, Suzuki H, Satoh A, Sasaki Y, Akino K, Ueno M, Takahashi F, Kusano M, Mita H, Yanagihara K, Endo T, Hinoda Y, Tokino T, Imai K. Aberrant methylation and silencing of the BNIP3 gene in colorectal and gastric cancer. Clin Cancer Res. 2005;11:1021–1027. [PubMed] [Google Scholar]
- Nagasaka T, Goel A, Notohara K, Takahata T, Sasamoto H, Uchida T, Nishida N, Tanaka N, Boland CR, Matsubara N. Methylation pattern of the O6-methylguanine-DNA methyltransferase gene in colon during progressive colorectal tumorigenesis. Int J Cancer. 2008;122:2429–2436. doi: 10.1002/ijc.23398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura M, Watanabe T, Yonekawa Y, Kleihues P, Ohgaki H. Promoter methylation of the DNA repair gene MGMT in astrocytomas is frequently associated with G:C –> A:T mutations of the TP53 tumor suppressor gene. Carcinogenesis. 2001;22:1715–1719. doi: 10.1093/carcin/22.10.1715. [DOI] [PubMed] [Google Scholar]
- Ogino S, Kawasaki T, Kirkner GJ, Suemoto Y, Meyerhardt JA, Fuchs CS. Molecular correlates with MGMT promoter methylation and silencing support CpG island methylator phenotype-low (CIMP-low) in colorectal cancer. Gut. 2007;56:1564–1571. doi: 10.1136/gut.2007.119750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peralta R, Valdivia A, Alvarado-Cabrero I, Gallegos F, Apresa T, Hernandez D, Mendoza M, Romero P, Paniagua L, Ibanez M, Cabrera L, Salcedo M. Correlation between expression of cellular retinol-binding protein 1 and its methylation status in larynx cancer. J Clin Pathol. 2012;65:46–50. doi: 10.1136/jclinpath-2011-200304. [DOI] [PubMed] [Google Scholar]
- Portela A, Esteller M. Epigenetic modifications and human disease. Nat Biotechnol. 2010;28:1057–1068. doi: 10.1038/nbt.1685. [DOI] [PubMed] [Google Scholar]
- Sandhu S, Wu X, Nabi Z, Rastegar M, Kung S, Mai S, Ding H. Loss of HLTF function promotes intestinal carcinogenesis. Mol Cancer. 2012;11:18. doi: 10.1186/1476-4598-11-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selamat SA, Chung BS, Girard L, Zhang W, Zhang Y, Campan M, Siegmund KD, Koss MN, Hagen JA, Lam WL, Lam S, Gazdar AF, Laird-Offringa IA. Genome-scale analysis of DNA methylation in lung adenocarcinoma and integration with mRNA expression. Genome Res. 2012;22:1197–1211. doi: 10.1101/gr.132662.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu S, Iida S, Ishiguro M, Uetake H, Ishikawa T, Takagi Y, Kobayashi H, Higuchi T, Enomoto M, Mogushi K, Mizushima H, Tanaka H, Sugihara K. Methylated BNIP3 gene in colorectal cancer prognosis. Oncol Lett. 2010;1:865–872. doi: 10.3892/ol_00000153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka M, Chang P, Li Y, Li D, Overman M, Maru DM, Sethi S, Phillips J, Bland GL, Abbruzzese JL, Eng C. Association of CHFR promoter methylation with disease recurrence in locally advanced colon cancer. Clin Cancer Res. 2011;17:4531–4540. doi: 10.1158/1078-0432.CCR-10-0763. [DOI] [PubMed] [Google Scholar]
- The Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330–337. doi: 10.1038/nature11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toki K, Enokida H, Kawakami K, Chiyomaru T, Tatarano S, Yoshino H, Uchida Y, Kawahara K, Nishiyama K, Seki N, Nakagawa M. CpG hypermethylation of cellular retinol-binding protein 1 contributes to cell proliferation and migration in bladder cancer. Int J Oncol. 2010;37:1379–1388. doi: 10.3892/ijo_00000789. [DOI] [PubMed] [Google Scholar]
- Tsai HC, Li H, Van Neste L, Cai Y, Robert C, Rassool FV, Shin JJ, Harbom KM, Beaty R, Pappou E, Harris J, Yen RW, Ahuja N, Brock MV, Stearns V, Feller-Kopman D, Yarmus LB, Lin YC, Welm AL, Issa JP, Minn I, Matsui W, Jang YY, Sharkis SJ, Baylin SB, Zahnow CA. Transient low doses of DNA-demethylating agents exert durable antitumor effects on hematological and epithelial tumor cells. Cancer Cell. 2012;21:430–446. doi: 10.1016/j.ccr.2011.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisenberger DJ, Siegmund KD, Campan M, Young J, Long TI, Faasse MA, Kang GH, Widschwendter M, Weener D, Buchanan D, Koh H, Simms L, Barker M, Leggett B, Levine J, Kim M, French AJ, Thibodeau SN, Jass J, Haile R, Laird PW. CpG island methylator phenotype underlies sporadic microsatellite instability and is tightly associated with BRAF mutation in colorectal cancer. Nat Genet. 2006;38:787–793. doi: 10.1038/ng1834. [DOI] [PubMed] [Google Scholar]
- Yu H, Lin CC, Li YY, Zhao Z. Dynamic protein interaction modules in human hepatocellular carcinoma progression. BMC Syst Biol. 2013;7(Suppl 5):S2. doi: 10.1186/1752-0509-7-S5-S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Tan M, Stone Hawthorne V, Klos KS, Lan KH, Yang Y, Yang W, Smith TL, Shi D, Yu D. Activation of the Akt/mammalian target of rapamycin/4E-BP1 pathway by ErbB2 overexpression predicts tumor progression in breast cancers. Clin Cancer Res. 2004;10:6779–6788. doi: 10.1158/1078-0432.CCR-04-0112. [DOI] [PubMed] [Google Scholar]
- Zhu J, Wang J, Shi Z, Franklin JL, Deane NG, Coffey RJ, Beauchamp RD, Zhang B. Deciphering genomic alterations in colorectal cancer through transcriptional subtype-based network analysis. PLoS One. 2013;8:e79282. doi: 10.1371/journal.pone.0079282. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.