

Abstract
Sex differences in brain neuroanatomy and neurophysiology underpin considerable physiological and behavioural differences between females and males. Sexual differentiation of the brain is regulated by testosterone secreted by the testes predominantly during embryogenesis in humans and the neonatal period in rodents. Despite huge advances in understanding how testosterone, and its metabolite oestradiol, sexually differentiate the brain, little is known about the mechanism that actually generates the male-specific neonatal testosterone surge. This review examines the evidence for the role of the hypothalamus, and particularly the gonadotropin-releasing hormone (GnRH) neurons, in generating the neonatal testosterone surge in rodents and primates. Kisspeptin–GPR54 signalling is well established as a potent and critical regulator of GnRH neuron activity during puberty and adulthood, and we argue here for an equally important role at birth in driving the male-specific neonatal testosterone surge in rodents. The presence of a male-specific population of preoptic area kisspeptin neurons that appear transiently in the perinatal period provide one possible source of kisspeptin drive to neonatal GnRH neurons in the mouse.
Keywords: GnRH, kisspeptin, GPR54, sexual differentiation, testosterone
1. Introduction
Sexually differentiated brain circuits are a feature of the central nervous system of many species from Drosophila to humans [1–3] and are important for the proper expression of sexually differentiated physiological processes and behaviours. The field of sexual differentiation arose from experiments by Pfeiffer in the 1930s [4] and Phoenix et al. in the 1950s [5]. The work of Pfeiffer demonstrated that transplanted ovaries could exhibit cyclical activity in adult male rats that were castrated in the neonatal period, but not in those castrated as adults [4]. Phoenix et al., on the other hand, demonstrated that sexually differentiated sex behaviours of female guinea pigs were irreversibly masculinized and defeminized following the administration of testosterone during the neonatal period [5]. These results and many since have demonstrated that testosterone secreted from the testes during the early postnatal period is critical for establishing sexually dimorphic gonadotropin secretion and reproductive behaviour in rodents.
The neonatal testosterone surge is one of several significant elevations in testosterone that occur throughout life in male mammals (figure 1a). During embryogenesis, the Leydig cells of the testis secrete testosterone shortly after differentiation to help ensure, alongside other testicular hormones, that the reproductive organs undergo complete sexual differentiation and growth [7]. This begins around embryonic day (E) 13 in mice and the seventh week of gestation in humans, and gradually declines leading up to birth (figure 1a). The neonatal testosterone surge occurs in the hours following birth, and is responsible for establishing sexually dimorphic brain circuitry that controls sexually differentiated behaviours and reproductive physiological processes in several species [8]. Following the neonatal testosterone surge, circulating testosterone levels drop to low levels, where they remain until the onset of puberty [9] (figure 1a). At puberty, circulating testosterone levels rise again causing the development of secondary sex characteristics and subsequently regulate reproductive function throughout adulthood.
Figure 1.

Changes in testosterone concentrations throughout embryonic, neonatal and postnatal life. (a) Schematic of plasma testosterone levels in males during embryonic and postnatal life. (b) The mean serum testosterone levels obtained from male and female rat pups sampled at different hours following birth. 0 in (utero) represents data from rats delivered by caesarean section immediately after the appearance of the first spontaneously delivered pup. 0 ex (utero) represents data from pups collected immediately after spontaneous delivery. (c) The mean serum testosterone levels obtained from male and female mouse pups at different hours following birth. (d) The mean serum testosterone levels from human newborns of both sexes at different hours following birth. Data modified from Corbier et al. [6].
The profile of the neonatal testosterone surge has been characterized in several different species including rat (figure 1b), mouse (figure 1c), horse, sheep, primates and humans (figure 1d) [6,10–13]. In most species, the testosterone surge is relatively transient with levels declining within hours [6]; however, in some species such as non-human primates and humans, the elevations in testosterone may persist for many hours to weeks following birth [6,10,11]. The elevation in plasma testosterone quickly increases testosterone levels in the hypothalamus [14,15]. In rats, castration shortly after birth prevents the elevation of testosterone levels in the hypothalamus, indicating that the testes are the primary source of the rise in plasma testosterone [14]. Through the aromatization of testosterone by the enzyme P450 aromatase (CYP19A1), oestradiol levels within the hypothalamus are elevated [15]. As such, it is the testosterone metabolite, oestradiol, which exerts its actions within the brain to cause sexual differentiation of structure and function [16].
In rodents it is very clear that the neonatal testosterone surge is responsible for establishing brain sexual dimorphisms that underpin sexually dimorphic physiology and behaviour [8]. However, in primates, including humans, the role of the neonatal testosterone surge in the establishment of brain sexual dimorphism is less straightforward. Evidence suggests that the majority of brain sexual differentiation in primates is established by exposure to androgens prenatally rather than neonatally, though this differs among primates. Neonatal castration or blockade of the neonatal testosterone surge has little effect on the expression of sex behaviours in rhesus monkeys [17,18], but results in decreased aggression and decreased mounting behaviour in tamarins and marmosets [19–21]. It has also been noted that blockade of the neonatal testosterone surge in male rhesus monkeys can delay puberty onset and alter the response of the central nervous system to glutamate receptor activation [22]. This suggests that the neonatal surge permanently differentiates the neural circuitry underlying the hypothalamic-pituitary-gonadal (HPG) axis in monkeys. In humans, the neonatal testosterone surge is often referred to as ‘mini-puberty’ in male babies and while its function is unknown, it could conceivably be involved in some aspects of brain sexual differentiation. For example, certain sex differences in the human brain do not become apparent until after birth [23]. Furthermore, recent investigations correlating neonatal levels of testosterone with behaviour suggest that the neonatal activation of the HPG axis may be associated with gender-linked social development [24], though causal relationships are difficult to establish in human studies. In this context, it is interesting to note that males with congenital hypogonadotropic hypogonadism that do not have a neonatal testosterone surge experience significant psychosexual disorders despite treatment with testosterone as adults [25]. Thus, although fetal testosterone secretion is critical for generating sex differences in human brain function, there is some indirect evidence that the neonatal testosterone surge may also have a role in primates.
Following the discovery that neonatal exposure to testosterone and its metabolite oestradiol cause permanent and irreversible changes to the rodent central nervous system, considerable effort has been devoted to investigating the molecular and cellular mechanisms through which these hormones act to organize the sexual differentiation of the brain [26,27]. Other authors within this special issue will address several of the mechanisms by which testosterone and oestradiol sexually differentiate the brain. While not an exhaustive list, cell death, cell survival, glial cell regulation and epigenetic changes have all been shown to play a role in the establishment of sex differences in the brain [28–33].
With the importance of sex differences in the biology and treatment of disease coming to the fore [34,35], it is somewhat surprising that relatively little is known about the mechanism that actually generates the neonatal testosterone surge. Scattered evidence in recent decades has implicated various components of the HPG axis in the generation of the neonatal testosterone surge. Findings from the work of others and ourselves has led us to propose the existence of a male-specific neonatal kisspeptin → GnRH neuron signalling mechanism that drives the neonatal testosterone surge, which initiates sexual differentiation of the rodent brain (figure 2). This review will examine, in turn, the evidence for the role of gonadotropins, GnRH neurons and kisspeptin neurons in the generation of the male-specific neonatal testosterone surge.
Figure 2.

Schematic of the proposed neonatal kisspeptin–GnRH neuron signalling mechanism controlling the male-specific testosterone surge that initiates the sexual differentiation of the brain.
2. Gonadotropin involvement in the generation of the neonatal testosterone surge
The first hint that gonadotropins may be involved in generating the neonatal testosterone surge came from the observation that the testes of fetal rats could be stimulated to produce testosterone by the application of luteinizing hormone (LH) in an organ culture model [36]. LH was able to stimulate the synthesis and secretion of testosterone from rat testes as young as E14.5. Cultured testes from E18 mice also increase testosterone secretion in response to LH application; moreover, co-culture of the testes with pituitary glands increased testosterone secretion [37]. Furthermore, the pituitary glands of male but not female rats were shown to have elevated concentrations of LH between E17 and E20 [38]. This evidence that LH can stimulate the testes of late embryonic rodents to secrete testosterone, alongside male-specific elevations in pituitary gland LH content, suggested a role for gonadotropins in the generation of the neonatal testosterone surge in rodents.
In humans, much of the evidence that implicates the role of gonadotropins in the generation of the neonatal testosterone surge is correlative with positive associations between LH and testosterone elevations in the neonatal period. The levels of LH in the hours following birth are elevated only in male babies and this occurs at a similar time to the rising levels of testosterone [11,39]. However, the same correlation appears less consistent in the male rat. While Corbier et al. [40] showed a strong temporal relationship between the neonatal testosterone surge and the rise in serum LH levels, others have reported plasma LH levels to be higher in female than male rat pups in the hours–days following birth [41,42]. Studies in transgenic and knockout mice have also been helpful but, again, are not entirely consistent. Male mice lacking a pituitary gland due to a deletion of the thyroid-specific enhancer binding protein (T/ebp/Nkx2.1), exhibit very reduced levels of intratesticular testosterone compared with wild-type littermates on E18.5 [43]. Nevertheless, conflicting results originate from different groups studying mice lacking the LH receptor (LuRKO mice). O'Shaughnessy et al. [44] observed that intratesticular levels of testosterone are markedly reduced in the LuRKO mice on postnatal day 1 compared with the wild-type controls. By contrast, Zhang et al. [45] report that intratesticular testosterone levels were comparable with wild-type controls in 1-day-old mice [45]. While the reasons for discrepancies between these different groups are not known, the timing of testes collection following birth will dramatically affect the levels of testosterone (figure 1c). For example, it is not clear from either study how long after birth samples were collected, or even the relationship of ‘postnatal day 1’ to the day of birth.
A study in rats using an antiserum against the LH receptor to interfere with LH receptor signalling around the time of birth has produced equivocal results. Goldman et al. [46] administered an LH antibody to male rat pups on days 1, 3 and 5 of life and examined the expression of sexually differentiated behaviours in adulthood, and the ability of ovarian grafts to ovulate. This treatment regimen resulted in sex behaviours that were neither masculinized nor defeminized, indicating that the neural circuits controlling sex behaviours had probably not been exposed to androgens during the early neonatal period. The ovarian grafts implanted within the males were not able to support corpus luteum formation indicating that the neural circuits controlling ovarian function had not been fully feminized either. However, the authors suggest that a small amount of androgen may have been produced following birth and that this could have masculinized the circuitry controlling ovarian function. Nevertheless, the authors conclude that pituitary function is important for the sexual differentiation of neuroendocrine control processes.
Considering the data at hand across different species, there appears to be sufficient positive evidence supporting a role for gonadotropins in generating the neonatal testosterone surge. (i) The neonatal testis is able to respond to LH stimulation by secreting testosterone, (ii) there is a positive relationship between LH levels and the neonatal testosterone surge, (iii) mice lacking a pituitary gland or LH receptors can fail to show an elevation in testosterone after birth, and (iv) interfering with LH receptor signalling after birth can disrupt sexual differentiation of sex behaviour and control of ovulation.
3. The role of gonadotropin-releasing hormone neurons in driving the neonatal testosterone surge
Studies in several species, including primates, indicate that the GnRH neurons begin to control pituitary gonadotropin secretion from mid- to late-embryogenesis [47,48]. As such, it is not surprising that GnRH neurons control the postnatal testosterone surge in primates. Experiments in monkeys using a GnRH receptor antagonist to block endogenous GnRH-dependent gonadotropin secretion clearly demonstrate that the neonatal testosterone surge is dependent on GnRH signalling. In non-human primates, the neonatal elevation in plasma testosterone levels persists for approximately three months. Peripheral administration of the GnRH receptor antagonist, Antide, on days 0, 3, 7, and then weekly from birth to 98 days of age in male marmoset monkeys completely abolished the neonatal rise in testosterone [10]. Furthermore, in humans, male babies with defective or absent GnRH secretion fail to exhibit the postnatal testosterone rise [47].
Investigations in mice demonstrate that communication between the hypothalamus and pituitary gland is active during the later stages of embryonic development. Using an organ culture technique, Pointis & Mahoudeau [49] demonstrated that pituitary glands from E18 mice respond to GnRH treatment by secreting LH. Furthermore Wen et al. [50] used genetically manipulated mice to show that gonadotropes in the anterior pituitary gland express GnRH receptors by E16.5, and that GnRH signalling at this time not only occurs, but is necessary for the proper development of the gonadotropes in the male mouse [50]. Taken together, these data support the idea that the GnRH neurons are active and can stimulate gonadotropes to secrete LH in the late embryonic male mouse.
However, one study using GnRH receptor antagonists to probe the role of GnRH signalling in the neonatal testosterone surge has suggested no strong relationship between plasma LH levels and testosterone levels [51]. Treatment of male rat pups 5 min after birth with a GnRH receptor antagonist had little effect on the neonatal testosterone surge observed 120 min following birth. The GnRH receptor antagonist caused a modest approximately 25% reduction in LH levels 60 min following birth compared with controls; however, this reduction was relatively short-lived with LH levels returning to normal 60 min later. It is not known whether this moderate level of suppression of LH secretion is sufficient to interfere with the neonatal testosterone surge. Studies in male mice have demonstrated that normal fertility can be achieved with as few as 12% of the normal number of GnRH neurons [52], indicating that the HPG axis has a significant safety margin before function becomes compromised. Taking this into account, it is possible that a transient 25% reduction in LH levels would not be sufficient to perturb the neonatal testosterone surge.
Investigations in the hypogonadal (hpg) mouse, which lacks GnRH peptide, also demonstrate that GnRH is a critical regulator of pituitary and gonadal function around the time of birth in mice [53]. During late embryogenesis, the levels of LH in the pituitary gland increase, and this increase is markedly reduced in the hpg mice. The levels of intratesticular testosterone are similar between control and hpg mice through late embryogenesis; however, on the day of birth there is a trend towards lower levels in the hpg mice, and this difference in testosterone level reaches significance by day 5. The exact time at which Leydig cell regulation switches from being independent to dependent upon circulating gonadotropins is not clear from this work, but indicates that it is proximate to birth. Moreover, the data show a decline in mRNA levels for important steroidogenic enzymes, particularly P450scc and P45017, prior to or on the day of birth, which supports the hypothesis that around the time of birth, Leydig cell activity and steroid production are dependent on gonadotropins.
A very recent study has demonstrated that GnRH neurons are activated in male but not female mice shortly after birth [54]. We exploited the fact that GnRH neurons only express cFos after periods of intense activation, such as during the preovulatory LH surge in females [55], to examine whether the GnRH neurons are active around the time of the neonatal testosterone surge in mice. Using wild-type mice collected 2–3 h after birth (at the time of the neonatal testosterone surge), we found a male-specific activation of the preoptic area GnRH neurons. Approximately 13% of GnRH neurons were activated in males in the hour following the neonatal testosterone surge, whereas virtually none of the GnRH neurons were active in female littermates [54]. These results differ from a previous report, which failed to detect a sex difference in cFos mRNA expression by GnRH neurons [56] using a wider sampling period and dual-label in situ hybridization.
Taken together, these studies in primates and rodents indicate that GnRH neurons regulate gonadotrope functioning during late embryonic and early postnatal life, with testosterone synthesis at the time of birth being dependent on GnRH neuron activity. If GnRH neurons are indeed required for the testosterone surge and sexual differentiation of the rodent brain, then manipulations affecting these neurons should generate abnormal sexually dimorphic features. In this vein, studies in the hpg mouse have been very insightful. Livne et al. [57] first demonstrated that the brains of the hpg mice are not fully masculinized during the neonatal period. The implantation of fetal hypothalamic tissue into the brains of male hpg mice was able to support normal reproductive behaviours in adulthood only when mice had been treated with testosterone to masculinize their brains on postnatal day 2. A similar rescue of male sex behaviour was obtained in hpg mice following testosterone treatment on postnatal day 2 [58]. This absence of sexually differentiated sex behaviours, and rescue by neonatal testosterone treatment in hpg mice, provide strong support for GnRH neurons in the generation of the neonatal testosterone surge. Subsequent studies by Gill et al. [59] have demonstrated that the sexual differentiation of kisspeptin neurons in the rostral periventricular area of the third ventricle (RP3V) is disrupted in the hpg mouse (figure 3). Normally, neonatal testosterone exposure acts to reduce the number of kisspeptin-expressing neurons in the RP3V of the male to approximately 10% of that observed in females [60,61]. In the hpg mouse, the number of kisspeptin neurons in the male RP3V is not masculinized, with a greater number of neurons compared with wild-type (figure 3), resulting in similar numbers of kisspeptin neurons in the RP3V of male and female hpg mice. Using in situ hybridization, Kim et al. [62] have also found a disruption of RP3V Kiss1 mRNA sexual differentiation. These studies clearly show that loss of GnRH signalling results in abnormal sexual differentiation of the RP3V kisspeptin neurons. Mice lacking the GnRH receptor have also been used to examine the role of GnRH signalling in establishing brain sexual differentiation [63]. As GnRH receptor knockout mice do not go through puberty and are hypogonadotropic, mice were gonadectomized around the time of puberty and supplemented with testosterone to ensure that post-pubertal steroid levels were normalized across all experimental groups. Under these conditions, the sexual differentiation of both the vasopressin innervation of the lateral septum and number of RP3V tyrosine hydroxylase (TH) neurons was disrupted, being female-like in male GnRH receptor-deleted mice [54].
Figure 3.
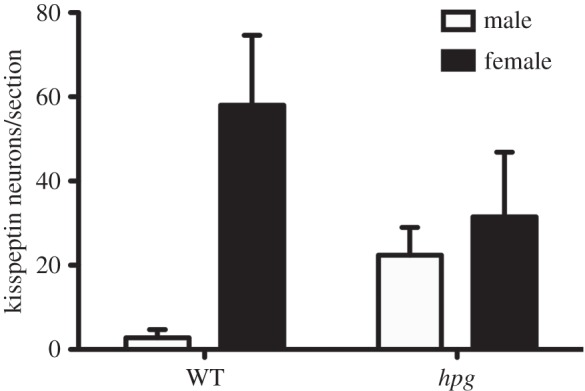
Disrupted sexual differentiation of the RP3V kisspeptin neurons in the hpg mouse. Quantification of the number of kisspeptin neurons per section in the RP3V of female and male wild-type and hpg mice at 45 days of age. WT, wild-type; hpg, hypogonadal; RP3V, rostral periventricular area of the third ventricle. Modified with permission from Gill et al. [59].
The disrupted brain sexual differentiation in the male mice lacking GnRH or GnRH receptors, combined with the male-specific activation of the GnRH neurons around the time of the neonatal testosterone surge, provides strong support for the notion that GnRH neurons play an important role in the generation of the neonatal testosterone surge. One obvious question that emerges from these findings is ‘What is responsible for activating the GnRH neurons at the time of birth?’
4. Kisspeptin neurons as orchestrators of the neonatal testosterone surge
Kisspeptin signalling through its receptor GPR54 is critical for puberty and adult fertility [64–68]. We became interested in a potential role for kisspeptin neurons in the neonatal testosterone surge because of the growing evidence that kisspeptin neurons may regulate the activity of the GnRH neurons prior to birth. Embryonic GnRH neurons express GPR54 [69,70] and can respond to kisspeptin [69,71], and kisspeptin neurons project to GnRH neurons in embryonic male and female mice [72,73]. Moreover, a male-biased sex difference in the percentage of GnRH neurons expressing GPR54 is present on the day of birth in mice [74]. In addition, studies by Kauffman et al. [75] using a global GPR54 knockout mouse demonstrated that the GPR54 is necessary for normal sexual differentiation of the male brain and behaviour. Male mice lacking the kisspeptin receptor had a greater number of TH-immunoreactive, and Kiss1 mRNA expressing cells in the RP3V than wild-type littermates, and had a feminized number of neurons in the spinal nucleus of the bulbocavernosus. Additionally, the sexually differentiated olfactory preference index was feminized in the male mice lacking GPR54. The fact that the sexual differentiation of brain and behaviour was disrupted in mice lacking GPR54 suggests that kisspeptin–GPR54 signalling plays an important role in this process.
In order to determine if kisspeptin–GPR54 signalling at the GnRH neuron itself was important in driving the neonatal testosterone surge in and sexual differentiation of the brain, we recently generated a mouse model in which GPR54 is deleted only from GnRH neurons [67]. As the GnRH–GPR54 knockout mice do not go through puberty and are hypogonadotropic, mice were gonadectomized around puberty and post-pubertal steroid levels were normalized across experimental groups using testosterone. This revealed that the vasopressin innervation of the lateral septum and the number of neurons expressing TH in the RP3V was feminized in the males lacking GPR54 in their GnRH neurons [54]. To confirm a role for GPR54 signalling in GnRH neurons in the generation of the neonatal testosterone surge plasma, testosterone was assayed in GnRH–GPR54 knockout mouse pups 1–2 h following their birth. We found that the GnRH–GPR54 knockout males had a greatly attenuated neonatal testosterone surge compared with the male wild-type littermates, and these levels were not statistically different from those of the female littermates of either genotype (figure 4). These observations differed, however, from a study in the global GPR54 knockout mouse, which showed no change in testosterone levels compared with wild-type animals when samples were collected any time between 0 and 4 h following birth [56]. As noted above (figures 1c and 5a), the neonatal testosterone surge is relatively brief in the mouse (1–3 h) and the timing of blood collection is critical. As such, we precisely determined the time of birth and took samples at 1–2 h following birth. This narrower sampling window afforded us a greater opportunity to capture the peak elevation in testosterone levels, producing an approximate sixfold sex difference in testosterone levels (figure 5a), very similar to previous results in the mouse [6]. This may explain the discrepancy with the global GPR54 knockout study in which a much smaller sex difference in testosterone levels (approx. 1.5-fold) was detected, probably due to the wide sampling window employed in that study.
Figure 4.
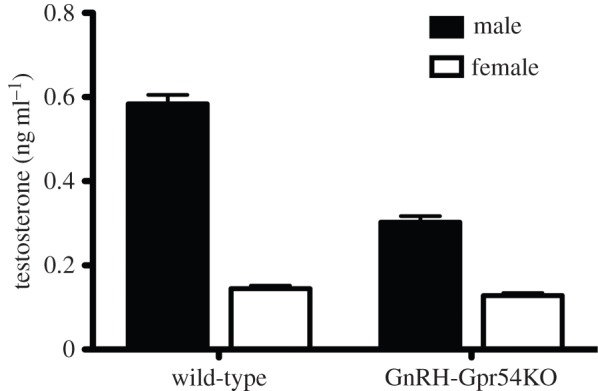
Kisspeptin–GPR54 signalling at the GnRH neuron is necessary for the perinatal testosterone surge. Quantitative analysis of the level of plasma testosterone 1–2 h after birth in male and female wild-type and GnRH–Gpr54KO mice.
Figure 5.
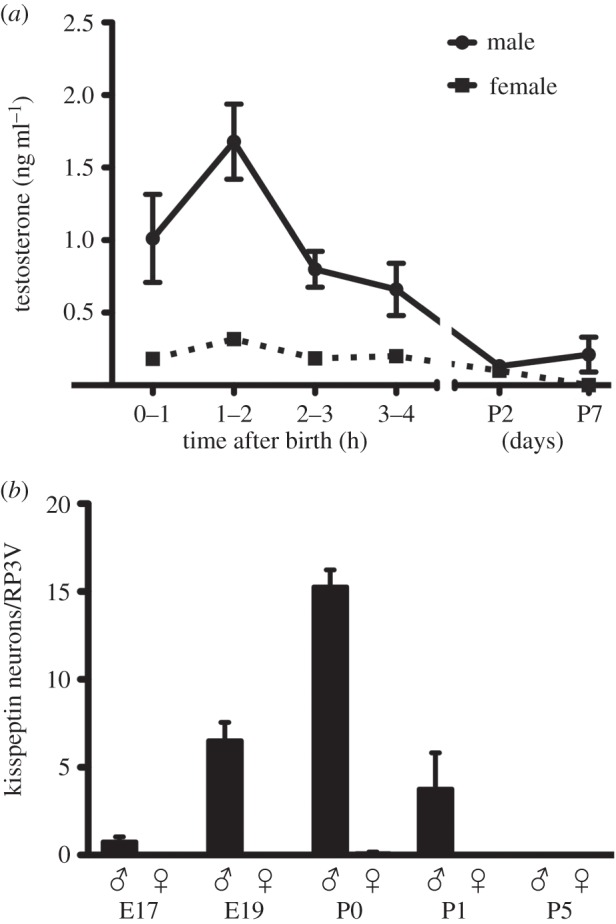
The neonatal testosterone surge correlates with the sexually dimorphic expression of kisspeptin in the RP3V. (a) Plasma testosterone levels in male and female mice killed at hourly intervals after birth on P0 and on P2 and P7. (b) Kisspeptin expression in the RP3V is sexually dimorphic in perinatal mice. Quantitative analyses of the number of kisspeptin immunoreactive neurons in the RP3V in male and female mice from E17 to P5. All values for females are zero. RP3V, rostral periventricular area of the third ventricle; P, postnatal day; E, embryonic day.
With kisspeptin–GPR54 signalling at the GnRH neuron playing an important role in the generation of the neonatal testosterone surge, it becomes important to establish where the kisspeptin innervation originates from. Given the sexually dimorphic nature of GnRH neuron activation at birth, it seemed that a sexually differentiated population of kisspeptin neurons might be involved. Kisspeptin peptide or Kiss1 mRNA is present in the arcuate nucleus (ARN) from E11.5 in the rat [76] and E13 in the mouse [70,72,73]; however, no consistent sex difference has been detected in this population at any age [70,76,77]. The other kisspeptin population in the RP3V is sexually differentiated but only begins to appear in the second postnatal week [78]. Hence, we made the decision to carefully re-characterize kisspeptin peptide expression in the neonatal mouse brain and, surprisingly, detected a small population of RP3V kisspeptin neurons only in the perinatal male mouse brain. These neurons were first detected on E17, increasing in number over E19 to reach a peak on the day of birth before declining to zero on postnatal day 5 (figure 5b). It is possible that a similar population may also exist in the embryonic rat brain [76].
We next asked what might be driving the expression of kisspeptin in the RP3V neurons prior to birth. In postnatal life, the RP3V kisspeptin neurons are positively regulated by gonadal steroids [79–82], and there is evidence of gonadotropin-independent secretion of testosterone during mid–late embryogenesis (figure 1a) in the rodent [37,53,83]. We therefore hypothesized that the transient expression of kisspeptin in the male RP3V may be driven by testosterone secretion during mid–late embryogenesis. We found that testosterone (but not dihydrotestosterone) given to pregnant dams on E18 induced the appearance of kisspeptin neurons in the RP3V of female pups. This indicates that following aromatization to oestradiol, embryonic testosterone acts to induce kisspeptin peptide expression in a small population of RP3V neurons just prior to birth [54].
The data obtained to date provide tantalizing evidence that male-specific elevations in embryonic testosterone levels give rise to RP3V kisspeptin neurons that then activate GnRH neurons to evoke the neonatal testosterone surge and, hence, brain sexual differentiation (figure 2). The weakest link in this scenario is evidence that it is indeed the neonatal RP3V kisspeptin neurons that innervate and activate GnRH neurons. Although inputs from the RP3V to GnRH neurons are established prior to birth [84], we cannot be sure that they are kisspeptinergic in nature. If not the RP3V kisspeptin neurons, then the only other possibility is that the kisspeptin input arises from the ARN kisspeptin population, and this is entirely possible. Despite the number of ARN kisspeptin neurons and embryonic innervation of GnRH neurons being the same in males and females [72,73], sex differences may well exist in their neuronal activity, as has been found in adults [85]. It has previously been speculated that the postnatal activation of GnRH neurons may arise from the sudden removal of placental gonadal steroid negative feedback at birth [86]. Whether sex differences exist in the steroid sensitivity of newborn ARN kisspeptin neurons is unknown.
Another interesting aspect of this proposed pathway is that it effectively results in male neonatal RP3V kisspeptin neurons sealing their own demise through a ‘kiss of death’. The neonatal testosterone surge drives programmed cell death [31] to generate the sexual dimorphic features of the RP3V. Thus, in generating the testosterone surge, the neonatal RP3V kisspeptin neurons initiate a sequence of events that leads to their own demise as potential kisspeptin neurons are killed in the male to generate the 10-fold female-dominant number of kisspeptin neurons within the RP3V [61,78].
5. Conclusion
Male-specific testosterone secretion drives a variety of genetic and epigenetic mechanisms to sexually differentiate the brain [35,87,88]. While embryonic testosterone is the predominant influence in primates, the neonatal testosterone surge is the key sexually differentiating event in rodents. Although conflicting data exist, the consensus of evidence indicates that GnRH neurons are responsible for generating the postnatal testosterone surge in all species examined, including both rodents and primates. The mechanisms through which GnRH neurons become activated at the time of birth are not established but kisspeptin–GPR54 signalling is critical, at least in mice. A newly discovered population of sexually dimorphic kisspeptin neurons that appear over the perinatal period in male mice may provide the kisspeptin input necessary for GnRH neuron activation at this time (figure 2). The importance of this kisspeptin → GnRH neuron → neonatal testosterone secretion pathway in other species awaits investigation.
Authors' contributions
J.C. and A.E.H. wrote the paper.
Competing interests
We declare we have no competing interests.
Funding
This work was supported by the New Zealand Health Research Council.
References
- 1.Kimura K. 2011. Role of cell death in the formation of sexual dimorphism in the Drosophila central nervous system. Dev. Growth Differ. 53, 236–244. ( 10.1111/j.1440-169X.2010.01223.x) [DOI] [PubMed] [Google Scholar]
- 2.Luders E, Toga AW. 2010. Sex differences in brain anatomy. Prog. Brain Res. 186, 3–12. ( 10.1016/B978-0-444-53630-3.00001-4) [DOI] [PubMed] [Google Scholar]
- 3.Simerly RB. 2002. Wired for reproduction: organization and development of sexually dimorphic circuits in the mammalian forebrain. Annu. Rev. Neurosci. 25, 507–536. ( 10.1146/annurev.neuro.25.112701.142745) [DOI] [PubMed] [Google Scholar]
- 4.Pfeiffer CA. 1936. Sexual differences of the hypophyses and their determination by the gonads. Am. J. Anat. 58, 197–225. ( 10.1002/aja.1000580112) [DOI] [Google Scholar]
- 5.Phoenix CH, Goy RW, Gerall AA, Young WC. 1959. Organizing action of prenatally administered testosterone propionate on the tissues mediating mating behavior in the female guinea pig. Endocrinology 65, 369–382. ( 10.1210/endo-65-3-369) [DOI] [PubMed] [Google Scholar]
- 6.Corbier P, Edwards DA, Roffi J. 1992. The neonatal testosterone surge: a comparative study. Arch. Int. Physiol. Biochim. Biophys. 100, 127–131. ( 10.3109/13813459209035274) [DOI] [PubMed] [Google Scholar]
- 7.Klonisch T, Fowler PA, Hombach-Klonisch S. 2004. Molecular and genetic regulation of testis descent and external genitalia development. Dev. Biol. 270, 1–18. ( 10.1016/j.ydbio.2004.02.018) [DOI] [PubMed] [Google Scholar]
- 8.Lenz KM, McCarthy MM. 2010. Organized for sex-steroid hormones and the developing hypothalamus. Eur. J. Neurosci. 32, 2096–2104. ( 10.1111/j.1460-9568.2010.07511.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Plant TM. 1985. A study of the role of the postnatal testes in determining the ontogeny of gonadotropin secretion in the male rhesus monkey (Macaca mulatta). Endocrinology 116, 1341–1350. ( 10.1210/endo-116-4-1341) [DOI] [PubMed] [Google Scholar]
- 10.Lunn SF, Recio R, Morris K, Fraser HM. 1994. Blockade of the neonatal rise in testosterone by a gonadotrophin-releasing hormone antagonist: effects on timing of puberty and sexual behaviour in the male marmoset monkey. J. Endocrinol. 141, 439–447. ( 10.1677/joe.0.1410439) [DOI] [PubMed] [Google Scholar]
- 11.de Zegher F, Devlieger H, Veldhuis JD. 1992. Pulsatile and sexually dimorphic secretion of luteinizing hormone in the human infant on the day of birth. Pediatr. Res. 32, 605–607. ( 10.1203/00006450-199211000-00025) [DOI] [PubMed] [Google Scholar]
- 12.Forest MG, Cathiard AM. 1975. Pattern of plasma testosterone and delta4-androstenedione in normal newborns: evidence for testicular activity at birth. J. Clin. Endocrinol. Metab. 41, 977–980. ( 10.1210/jcem-41-5-977) [DOI] [PubMed] [Google Scholar]
- 13.Yu HK, Cabalum T, Jansen CA, Buster JE, Nathanielsz PW. 1983. Androstenedione, testosterone, and estradiol concentrations in fetal and maternal plasma in late pregnancy in the sheep. Endocrinology 113, 2216–2220. ( 10.1210/endo-113-6-2216) [DOI] [PubMed] [Google Scholar]
- 14.Rhoda J, Corbier P, Roffi J. 1983. Hypothalamic testosterone increase in the male rat at birth. Int. J. Dev. Neurosci. 1, 187–190. ( 10.1016/0736-5748(83)90213-7) [DOI] [PubMed] [Google Scholar]
- 15.Rhoda J, Corbier P, Roffi J. 1984. Gonadal steroid concentrations in serum and hypothalamus of the rat at birth: aromatization of testosterone to 17 beta-estradiol. Endocrinology 114, 1754–1760. ( 10.1210/endo-114-5-1754) [DOI] [PubMed] [Google Scholar]
- 16.McCarthy MM. 2008. Estradiol and the developing brain. Physiol. Rev. 88, 91–124. ( 10.1152/physrev.00010.2007) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brown GR, Dixson AF. 1999. Investigation of the role of postnatal testosterone in the expression of sex differences in behavior in infant rhesus macaques (Macaca mulatta). Horm. Behav. 35, 186–194. ( 10.1006/hbeh.1999.1512) [DOI] [PubMed] [Google Scholar]
- 18.Wallen K, Maestripieri D, Mann DR. 1995. Effects of neonatal testicular suppression with a GnRH antagonist on social behavior in group-living juvenile rhesus monkeys. Horm. Behav. 29, 322–337. ( 10.1006/hbeh.1995.1023) [DOI] [PubMed] [Google Scholar]
- 19.Dixson AF. 1993. Effects of testosterone propionate upon the sexual and aggressive behavior of adult male marmosets (Callithrix jacchus) castrated as neonates. Horm. Behav. 27, 216–230. ( 10.1006/hbeh.1993.1016) [DOI] [PubMed] [Google Scholar]
- 20.Epple G, Alveario MC, Belcher AM. 1990. Copulatory behavior of adult tamarins (Saguinus fuscicollis) castrated as neonates or juveniles: effect of testosterone treatment. Horm. Behav. 24, 470–483. ( 10.1016/0018-506X(90)90036-W) [DOI] [PubMed] [Google Scholar]
- 21.Epple G, Alveario MC, St. Andre E. 1987. Sexual and social behavior of adult saddle-back tamarins (Saguinus fascicollis), castrated as neonates. Am. J. Primatol. 13, 37–49. ( 10.1002/ajp.1350130106) [DOI] [PubMed] [Google Scholar]
- 22.Mann DR, Akinbami MA, Gould KG, Tanner JM, Wallen K. 1993. Neonatal treatment of male monkeys with a gonadotropin-releasing hormone agonist alters differentiation of central nervous system centers that regulate sexual and skeletal development. J. Clin. Endocrinol. Metab. 76, 1319–1324. ( 10.1210/jcem.76.5.8496324) [DOI] [PubMed] [Google Scholar]
- 23.Swaab DF, Hofman MA. 1988. Sexual differentiation of the human hypothalamus: ontogeny of the sexually dimorphic nucleus of the preoptic area. Brain Res. Dev. Brain Res. 44, 314–318. ( 10.1016/0165-3806(88)90231-3) [DOI] [PubMed] [Google Scholar]
- 24.Alexander GM. 2014. Postnatal testosterone concentrations and male social development. Front. Endocrinol. (Lausanne) 5, 15 ( 10.3389/fendo.2014.00015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dwyer AA, Quinton R, Pitteloud N, Morin D. 2015. Psychosexual development in men with congenital hypogonadotropic hypogonadism on long-term treatment: a mixed methods study. Sex. Med. 3, 32–41. ( 10.1002/sm2.50) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Forger NG, de Vries GJ. 2010. Cell death and sexual differentiation of behavior: worms, flies, and mammals. Curr. Opin. Neurobiol. 20, 776–783. ( 10.1016/j.conb.2010.09.006) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lenz KM, Nugent BM, McCarthy MM. 2012. Sexual differentiation of the rodent brain: dogma and beyond. Front. Neurosci. 6, 26 ( 10.3389/fnins.2012.00026) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Amateau SK, McCarthy MM. 2002. Sexual differentiation of astrocyte morphology in the developing rat preoptic area. J. Neuroendocrinol. 14, 904–910. ( 10.1046/j.1365-2826.2002.00858.x) [DOI] [PubMed] [Google Scholar]
- 29.Amateau SK, McCarthy MM. 2004. Induction of PGE2 by estradiol mediates developmental masculinization of sex behavior. Nat. Neurosci. 7, 643–650. ( 10.1038/nn1254) [DOI] [PubMed] [Google Scholar]
- 30.Davis EC, Popper P, Gorski RA. 1996. The role of apoptosis in sexual differentiation of the rat sexually dimorphic nucleus of the preoptic area. Brain Res. 734, 10–18. ( 10.1016/0006-8993(96)00298-3) [DOI] [PubMed] [Google Scholar]
- 31.Forger NG, Rosen GJ, Waters EM, Jacob D, Simerly RB, de Vries GJ. 2004. Deletion of Bax eliminates sex differences in the mouse forebrain. Proc. Natl Acad. Sci. USA 101, 13 666–13 671. ( 10.1073/pnas.0404644101) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Murray EK, Hien A, de Vries GJ, Forger NG. 2009. Epigenetic control of sexual differentiation of the bed nucleus of the stria terminalis. Endocrinology 150, 4241–4247. ( 10.1210/en.2009-0458) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Simerly RB, Zee MC, Pendleton JW, Lubahn DB, Korach KS. 1997. Estrogen receptor-dependent sexual differentiation of dopaminergic neurons in the preoptic region of the mouse. Proc. Natl Acad. Sci. USA 94, 14 077–14 082. ( 10.1073/pnas.94.25.14077) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Clayton JA, Collins FS. 2014. Policy: NIH to balance sex in cell and animal studies. Nature 509, 282–283. ( 10.1038/509282a) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McCarthy MM, Arnold AP, Ball GF, Blaustein JD, De Vries GJ. 2012. Sex differences in the brain: the not so inconvenient truth. J. Neurosci. 32, 2241–2247. ( 10.1523/JNEUROSCI.5372-11.2012) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Picon R, Ktorza A. 1976. Effect of LH on testosterone production by foetal rat testes in vitro. FEBS Lett. 68, 19–22. ( 10.1016/0014-5793(76)80394-8) [DOI] [PubMed] [Google Scholar]
- 37.Pointis G, Mahoudeau JA. 1976. Demonstration of a pituitary gonadotrophin hormone activity in the male foetal mouse. Acta Endocrinol. (Copenh.) 83, 158–165. ( 10.1530/acta.0.0830158) [DOI] [PubMed] [Google Scholar]
- 38.Chowdhury M, Steinberger E. 1976. Pituitary and plasma levels of gonadotrophins in foetal and newborn male and female rats. J. Endocrinol. 69, 381–384. ( 10.1677/joe.0.0690381) [DOI] [PubMed] [Google Scholar]
- 39.Corbier P, Dehennin L, Castanier M, Mebazaa A, Edwards DA, Roffi J. 1990. Sex differences in serum luteinizing hormone and testosterone in the human neonate during the first few hours after birth. J. Clin. Endocrinol. Metab. 71, 1344–1348. ( 10.1210/jcem-71-5-1344) [DOI] [PubMed] [Google Scholar]
- 40.Corbier P, Kerdelhue B, Picon R, Roffi J. 1978. Changes in testicular weight and serum gonadotropin and testosterone levels before, during, and after birth in the perinatal rat. Endocrinology 103, 1985–1991. ( 10.1210/endo-103-6-1985) [DOI] [PubMed] [Google Scholar]
- 41.Pang SF, Caggiula AR, Gay VL, Goodman RL, Pang CS. 1979. Serum concentrations of testosterone, oestrogens, luteinizing hormone and follicle-stimulating hormone in male and female rats during the critical period of neural sexual differentiation. J. Endocrinol. 80, 103–110. ( 10.1677/joe.0.0800103) [DOI] [PubMed] [Google Scholar]
- 42.Slob AK, Ooms MP, Vreeburg JT. 1980. Prenatal and early postnatal sex differences in plasma and gonadal testosterone and plasma luteinizing hormone in female and male rats. J. Endocrinol. 87, 81–87. ( 10.1677/joe.0.0870081) [DOI] [PubMed] [Google Scholar]
- 43.Pakarinen P, Kimura S, El-Gehani F, Pelliniemi LJ, Huhtaniemi I. 2002. Pituitary hormones are not required for sexual differentiation of male mice: phenotype of the T/ebp/Nkx2.1 null mutant mice. Endocrinology 143, 4477–4482. ( 10.1210/en.2002-220052) [DOI] [PubMed] [Google Scholar]
- 44.O'Shaughnessy PJ, Morris ID, Huhtaniemi I, Baker PJ, Abel MH. 2009. Role of androgen and gonadotrophins in the development and function of the Sertoli cells and Leydig cells: data from mutant and genetically modified mice. Mol. Cell. Endocrinol. 306, 2–8. ( 10.1016/j.mce.2008.11.005) [DOI] [PubMed] [Google Scholar]
- 45.Zhang FP, Pakarainen T, Zhu F, Poutanen M, Huhtaniemi I. 2004. Molecular characterization of postnatal development of testicular steroidogenesis in luteinizing hormone receptor knockout mice. Endocrinology 145, 1453–1463. ( 10.1210/en.2003-1049) [DOI] [PubMed] [Google Scholar]
- 46.Goldman BD, Quadagno DM, Shryne J, Gorski RA. 1972. Modification of phallus development and sexual behavior in rats treated with gonadotropin antiserum neonatally. Endocrinology 90, 1025–1031. ( 10.1210/endo-90-4-1025) [DOI] [PubMed] [Google Scholar]
- 47.Bouvattier C, Maione L, Bouligand J, Dode C, Guiochon-Mantel A, Young J. 2012. Neonatal gonadotropin therapy in male congenital hypogonadotropic hypogonadism. Nat. Rev. Endocrinol. 8, 172–182. ( 10.1038/nrendo.2011.164) [DOI] [PubMed] [Google Scholar]
- 48.Foster DL, Hileman SM. 2015. Puberty in the sheep. In Knobil and Neill's physiology of reproduction (eds Plant TM, Zeleznik AJ), pp. 1441–1477, 4th edn Amsterdam, The Netherlands: Elsevier. [Google Scholar]
- 49.Pointis G, Mahoudeau JA. 1976. Release of immuno-reactive and biologically active LH from fetal mouse pituitary in response to synthetic gonadotropin releasing factor (LRF). Experientia 32, 1347–1348. ( 10.1007/BF01953132) [DOI] [PubMed] [Google Scholar]
- 50.Wen S, Ai W, Alim Z, Boehm U. 2010. Embryonic gonadotropin-releasing hormone signaling is necessary for maturation of the male reproductive axis. Proc. Natl Acad. Sci. USA 107, 16 372–16 377. ( 10.1073/pnas.1000423107) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.McGivern RF, Hermans RH, Handa RJ, Longo LD. 1995. Plasma testosterone surge and luteinizing hormone beta (LH-beta) following parturition: lack of association in the male rat. Eur. J. Endocrinol. 133, 366–374. ( 10.1530/eje.0.1330366) [DOI] [PubMed] [Google Scholar]
- 52.Herbison AE, Porteous R, Pape JR, Mora JM, Hurst PR. 2008. Gonadotropin-releasing hormone neuron requirements for puberty, ovulation, and fertility. Endocrinology 149, 597–604. ( 10.1210/en.2007-1139) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.O'Shaughnessy PJ, Baker P, Sohnius U, Haavisto AM, Charlton HM, Huhtaniemi I. 1998. Fetal development of Leydig cell activity in the mouse is independent of pituitary gonadotroph function. Endocrinology 139, 1141–1146. ( 10.1210/endo.139.3.5788) [DOI] [PubMed] [Google Scholar]
- 54.Clarkson J, Busby ER, Kirilov M, Schutz G, Sherwood NM, Herbison AE. 2014. Sexual differentiation of the brain requires perinatal kisspeptin-GnRH neuron signaling. J. Neurosci. 34, 15 297–15 305. ( 10.1523/JNEUROSCI.3061-14.2014) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hoffman GE, Smith MS, Verbalis JG. 1993. c-Fos and related immediate early gene products as markers of activity in neuroendocrine systems. Front. Neuroendocrinol. 14, 173–213. ( 10.1006/frne.1993.1006) [DOI] [PubMed] [Google Scholar]
- 56.Poling MC, Kauffman AS. 2012. Sexually dimorphic testosterone secretion in prenatal and neonatal mice is independent of kisspeptin-Kiss1r and GnRH signaling. Endocrinology 153, 782–793. ( 10.1210/en.2011-1838) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Livne I, Silverman AJ, Gibson MJ. 1992. Reversal of reproductive deficiency in the hpg male mouse by neonatal androgenization. Biol. Reprod. 47, 561–567. ( 10.1095/biolreprod47.4.561) [DOI] [PubMed] [Google Scholar]
- 58.Nwagwu MO, Baines H, Kerr JB, Ebling FJ. 2005. Neonatal androgenization of hypogonadal (hpg) male mice does not abolish estradiol-induced FSH production and spermatogenesis. Reprod. Biol. Endocrinol. 3, 48 ( 10.1186/1477-7827-3-48) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gill JC, Wang O, Kakar S, Martinelli E, Carroll RS, Kaiser UB. 2010. Reproductive hormone-dependent and -independent contributions to developmental changes in kisspeptin in GnRH-deficient hypogonadal mice. PLoS ONE 5, e11911 ( 10.1371/journal.pone.0011911) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Homma T, et al. 2009. Significance of neonatal testicular sex steroids to defeminize anteroventral periventricular kisspeptin neurons and the GnRH/LH surge system in male rats. Biol. Reprod. 81, 1216–1225. ( 10.1095/biolreprod.109.078311) [DOI] [PubMed] [Google Scholar]
- 61.Kauffman AS, et al. 2007. Sexual differentiation of Kiss1 gene expression in the brain of the rat. Endocrinology 148, 1774–1783. ( 10.1210/en.2006-1540) [DOI] [PubMed] [Google Scholar]
- 62.Kim J, Tolson KP, Dhamija S, Kauffman AS. 2013. Developmental GnRH signaling is not required for sexual differentiation of kisspeptin neurons but is needed for maximal kiss1 gene expression in adult females. Endocrinology 154, 3273–3283. ( 10.1210/en.2013-1271) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wu S, Wilson MD, Busby ER, Isaac ER, Sherwood NM. 2010. Disruption of the single copy gonadotropin-releasing hormone receptor in mice by gene trap: severe reduction of reproductive organs and functions in developing and adult mice. Endocrinology 151, 1142–1152. ( 10.1210/en.2009-0598) [DOI] [PubMed] [Google Scholar]
- 64.d'Anglemont de Tassigny X, et al. 2007. Hypogonadotropic hypogonadism in mice lacking a functional Kiss1 gene. Proc. Natl Acad. Sci. USA 104, 10 714–10 719. ( 10.1073/pnas.0704114104) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.de Roux N, Genin E, Carel JC, Matsuda F, Chaussain JL, Milgrom E. 2003. Hypogonadotropic hypogonadism due to loss of function of the KiSS1-derived peptide receptor GPR54. Proc. Natl Acad. Sci. USA 100, 10 972–10 976. ( 10.1073/pnas.1834399100) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Funes S, Hedrick JA, Vassileva G, Markowitz L, Abbondanzo S, Golovko A, Yang S, Monsma FJ, Gustafson EL. 2003. The KiSS-1 receptor GPR54 is essential for the development of the murine reproductive system. Biochem. Biophys. Res. Commun. 312, 1357–1363. ( 10.1016/j.bbrc.2003.11.066) [DOI] [PubMed] [Google Scholar]
- 67.Kirilov M, Clarkson J, Liu X, Roa J, Campos P, Porteous R, Schutz G, Herbison AE. 2013. Dependence of fertility on kisspeptin-Gpr54 signaling at the GnRH neuron. Nat. Commun. 4, 2492 ( 10.1038/ncomms3492) [DOI] [PubMed] [Google Scholar]
- 68.Seminara SB, et al. 2003. The GPR54 gene as a regulator of puberty. N. Engl. J. Med. 349, 1614–1627. ( 10.1056/NEJMoa035322) [DOI] [PubMed] [Google Scholar]
- 69.Constantin S, Caligioni CS, Stojilkovic S, Wray S. 2009. Kisspeptin-10 facilitates a plasma membrane-driven calcium oscillator in GnRH-1 neurons. Endocrinology 150, 3221–3227. ( 10.1210/en.2008-1711) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Knoll JG, Clay CM, Bouma GJ, Henion TR, Schwarting GA, Millar RP, Tobet SA. 2013. Developmental profile and sexually dimorphic expression of kiss1 and kiss1r in the fetal mouse brain. Front. Endocrinol. (Lausanne) 4, 140 ( 10.3389/fendo.2013.00140) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Fiorini Z, Jasoni CL. 2010. A novel developmental role for kisspeptin in the growth of gonadotrophin-releasing hormone neurites to the median eminence in the mouse. J. Neuroendocrinol. 22, 1113–1125. ( 10.1111/j.1365-2826.2010.02059.x) [DOI] [PubMed] [Google Scholar]
- 72.Kumar D, Freese M, Drexler D, Hermans-Borgmeyer I, Marquardt A, Boehm U. 2014. Murine arcuate nucleus kisspeptin neurons communicate with GnRH neurons in utero. J. Neurosci. 34, 3756–3766. ( 10.1523/JNEUROSCI.5123-13.2014) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kumar D, Periasamy V, Freese M, Voigt A, Boehm U. 2015. In utero development of kisspeptin/GnRH neural circuitry in male mice. Endocrinology 156, 3084–3090. ( 10.1210/EN.2015-1412) [DOI] [PubMed] [Google Scholar]
- 74.Herbison AE, de Tassigny X, Doran J, Colledge WH. 2010. Distribution and postnatal development of Gpr54 gene expression in mouse brain and gonadotropin-releasing hormone neurons. Endocrinology 151, 312–321. ( 10.1210/en.2009-0552) [DOI] [PubMed] [Google Scholar]
- 75.Kauffman AS, et al. 2007. The kisspeptin receptor GPR54 is required for sexual differentiation of the brain and behavior. J. Neurosci. 27, 8826–8835. ( 10.1523/JNEUROSCI.2099-07.2007) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Desroziers E, Droguerre M, Bentsen AH, Robert V, Mikkelsen JD, Caraty A, Tillet Y, Duittoz A, Franceschini I. 2012. Embryonic development of kisspeptin neurones in rat. J. Neuroendocrinol. 24, 1284–1295. ( 10.1111/j.1365-2826.2012.02333.x) [DOI] [PubMed] [Google Scholar]
- 77.Walker DM, Kirson D, Perez LF, Gore AC. 2012. Molecular profiling of postnatal development of the hypothalamus in female and male rats. Biol. Reprod. 87, 129 ( 10.1095/biolreprod.112.102798) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Clarkson J, Herbison AE. 2006. Postnatal development of kisspeptin neurons in mouse hypothalamus: sexual dimorphism and projections to gonadotropin-releasing hormone neurons. Endocrinology 147, 5817–5825. ( 10.1210/en.2006-0787) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Clarkson J, Boon WC, Simpson ER, Herbison AE. 2009. Postnatal development of an estradiol-kisspeptin positive feedback mechanism implicated in puberty onset. Endocrinology 150, 3214–3220. ( 10.1210/en.2008-1733) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Clarkson J, Shamas S, Mallinson S, Herbison AE. 2012. Gonadal steroid induction of kisspeptin peptide expression in the rostral periventricular area of the third ventricle during postnatal development in the male mouse. J. Neuroendocrinol. 24, 907–915. ( 10.1111/j.1365-2826.2012.02294.x) [DOI] [PubMed] [Google Scholar]
- 81.Smith JT, Cunningham MJ, Rissman EF, Clifton DK, Steiner RA. 2005. Regulation of Kiss1 gene expression in the brain of the female mouse. Endocrinology 146, 3686–3692. ( 10.1210/en.2005-0488) [DOI] [PubMed] [Google Scholar]
- 82.Smith JT, Dungan HM, Stoll EA, Gottsch ML, Braun RE, Eacker SM, Clifton DK, Steiner RA. 2005. Differential regulation of KiSS-1 mRNA expression by sex steroids in the brain of the male mouse. Endocrinology 146, 2976–2984. ( 10.1210/en.2005-0323) [DOI] [PubMed] [Google Scholar]
- 83.Weisz J, Ward IL. 1980. Plasma testosterone and progesterone titers of pregnant rats, their male and female fetuses, and neonatal offspring. Endocrinology 106, 306–316. ( 10.1210/endo-106-1-306) [DOI] [PubMed] [Google Scholar]
- 84.Polston EK, Simerly RB. 2006. Ontogeny of the projections from the anteroventral periventricular nucleus of the hypothalamus in the female rat. J. Comp. Neurol. 495, 122–132. ( 10.1002/cne.20874) [DOI] [PubMed] [Google Scholar]
- 85.de Croft S, Piet R, Mayer C, Mai O, Boehm U, Herbison AE. 2012. Spontaneous kisspeptin neuron firing in the adult mouse reveals marked sex and brain region differences but no support for a direct role in negative feedback. Endocrinology 153, 5384–5393. ( 10.1210/en.2012-1616) [DOI] [PubMed] [Google Scholar]
- 86.Kaplan SL, Grumbach MM, Aubert ML. 1976. The ontogenesis of pituitary hormones and hypothalamic factors in the human fetus: maturation of central nervous system regulation of anterior pituitary function. Recent Prog. Horm. Res. 32, 161–243. ( 10.1016/b978-0-12-571132-6.50015-4) [DOI] [PubMed] [Google Scholar]
- 87.McCarthy MM, Arnold AP. 2011. Reframing sexual differentiation of the brain. Nat. Neurosci. 14, 677–683. ( 10.1038/nn.2834) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Bakker J, Baum MJ. 2008. Role for estradiol in female-typical brain and behavioral sexual differentiation. Front. Neuroendocrinol. 29, 1–16. ( 10.1016/j.yfrne.2007.06.001) [DOI] [PMC free article] [PubMed] [Google Scholar]