

Abstract
Bacterial diversity within animals is emerging as an essential component of health, but it is unknown how stress may influence the microbiome. We quantify a proximate link between the oral microbiome and hypothalamic–pituitary–adrenal (HPA) axis activity using faecal glucocorticoid metabolites (FGM) in wild red squirrels (Tamiasciurus hudsonicus). Not only was bacterial diversity lower at higher levels of FGM, but also between capture periods a change in bacterial relative abundance was related to an increase in FGM. These linkages between the HPA axis and microbiome communities represent a powerful capacity for stress to have multi-dimensional effects on health.
Keywords: hypothalamic–pituitary–adrenal axis, illumina sequencing, microbiome diversity
1. Introduction
The microbiome is a fundamental and dynamic dimension of an organism's physiology. Microbiome bacteria typically outnumber host cells by an order of magnitude, and recent evidence suggests a positive relationship between bacterial diversity and host health [1,2]. Oral microbiomes that are beneficial to hosts are diverse and abundantly populated with mutualistic taxa [3]. Conversely, low diversity can be correlated with a high abundance of pathogenic bacteria or increased susceptibility to low abundance opportunistic pathogens, and poor health [4,5]. Such pathogenesis of the oral (buccal and respiratory) microbiome has been connected to epizootic events in wildlife populations [6].
The bacterial microbiome is linked to its host, in part, through the hypothalamic–pituitary–adrenal (HPA) axis [7]. The HPA axis regulates the stress response through the release of glucocorticoids and is a cornerstone of the ‘brain-gut axis' [8]. Until now, the relationship between stress and the microbiome has only been studied in captivity with qualitative stressors [9,10]. We examine this relationship in the wild and quantify a proximate link between stress and the microbiome by measuring faecal glucocorticoid metabolites (FGM: an integrated measure of HPA axis activity) and the oral bacterial community in free-living North American red squirrels, for which the ecology and stress physiology have been well studied [11,12]. We examined the hypothesis that high HPA axis activity would disrupt the bacterial microbiome by testing two predictions: (i) FGM concentrations would be negatively correlated with bacterial diversity and (ii) there would be a positive relationship between change in FGM concentrations over time and change in bacterial abundance.
2. Material and methods
(a). Field protocol
A grid of 80 tomahawk live traps (Tomahawk Live Trap Co., WI, USA) was assembled in a 0.1 km2 site dominated by mixed-wood forest located within Algonquin Provincial Park (45o54′N, 78o26′W). Traps were spaced at 20 m intervals and affixed to trees 1.5 m above the ground to reduce non-target captures.
Traps were set 06.00–19.00 h, baited with an oatmeal–peanut butter mixture (10 g) and checked in less than or equal to 2 h intervals, partly to ensure trapping stress did not influence interpretation of basal FGM concentrations [11]. Squirrels (n = 12 males, 12 females; post-breeding) were captured twice for repeated sampling to examine how within-individual changes in a measure of HPA axis covary with oral bacterial communities (first capture: 17–19 July, second capture: 28–30 July). Individuals were given unique alphanumeric ear tags and, at both first and second capture, sex, reproductive condition and mass were recorded. Oral microbiome samples, collected using sterile cotton-tipped swabs circulated in the mouth for 10 s, and faecal samples, collected directly from traps, were stored in 1.5 ml microcentrifuge tubes on ice before transfer to a −20°C freezer.
(b). Faecal glucocorticoid metabolite analysis
Faecal samples were lyophilized, frozen with liquid nitrogen and pulverized with mortar and pestle. FGM extraction from 0.05 g was performed with a 0.5 ml volume of 80% methanol and analysed via enzyme immunoassay (EIA) [11,13].
(c). DNA extraction, 16S rRNA gene library preparation and sequencing
Bacterial DNA extraction was performed using a modified QIAamp DNA Mini kit (Qiagen) protocol (see the electronic supplementary material). Oral bacterial DNA purity was evaluated via spectrophotometry (Nanodrop, ThermoScientific, Waltham, MA) and stored at −20°C. Extracted DNA was processed at MetagenomBio Inc. (Toronto, Ontario), following an Illumina MiSeq paired-end sequencing method (see the electronic supplementary material). Sequences have been deposited in NCBI under the SRA accession number SRP064395.
(d). Bioinformatics and statistical methods
DNA sequences were assembled, binned and analysed using PANDAseq [14], USearch (v. 5.2.236, [15]) and QIIME (v. 1.8.0, [16]). Shannon diversity, a commonly used metric for assessing taxa richness and evenness, was calculated from the Operational Taxonomic Units (OTUs) identified from the full oral dataset [17,18] (see the electronic supplementary material for formulation); diversity was characterized at the phylum level which yielded the most reliable assignment of OTUs. The relationship between FGM and bacterial diversity was examined using linear models (GLMs) both within and between capture periods, including sex and capture period as fixed effects.
Similarly, we explored how change in FGM between first and second capture influenced change in the relative abundance of the most prevalent bacterial families (see the electronic supplementary material).
Samples from an antibiotic manipulation of the oral microbiome were ultimately pooled for analysis owing to a lack of evidence of an effect of treatment (see the electronic supplementary material). All FGM measures were natural log-transformed to meet assumptions of normality. All analyses were performed in R [19,20]; GLMs contained ID as a random effect.
3. Results and discussion
We provide, we believe, the first evidence for a proximate relationship between HPA axis activity and microbiome community dynamics in a free-living system. Consistent with our hypothesis, FGM were negatively correlated with bacterial diversity within and across sampling periods (first capture: R2 = 0.33, t21 = −3.447, p < 0.01; second capture: R2 = 0.24, t21 = −3.040, p < 0.01; overall: R2 = 0.26, t44 = −4.28, p < 0.001, figure 1, and the electronic supplementary material, table S1 for analysis at the class, order and family level). This was observed despite large, within-individual changes in FGM concentration between sampling periods that likely corresponds to a seasonal endocrinological shift [11]. There was no effect of sex in our models (first capture, second capture, overall: all p ≥ 0.05). Reduced microbiome diversity with increased FGM may be the result of stress-induced activation of the innate immune system [9]. The HPA axis interacts directly with immune function [21] and an acute stress response bolsters innate immunity while simultaneously diminishing adaptive immunity [22,23]. More specifically, elevated glucocorticoids can increase circulating cytokine and macrophage concentrations, which have direct antimicrobial effects [22,24]. Thus, while the interplay between the HPA axis and immune system are complex and multidirectional depending on the stressor, the HPA axis can facilitate immune system-mediated changes in the bacterial microbiome, but also increases host vulnerability to pathogenic species invasion by weakening the adaptive immune system and disrupting steady-state communities [10].
Figure 1.
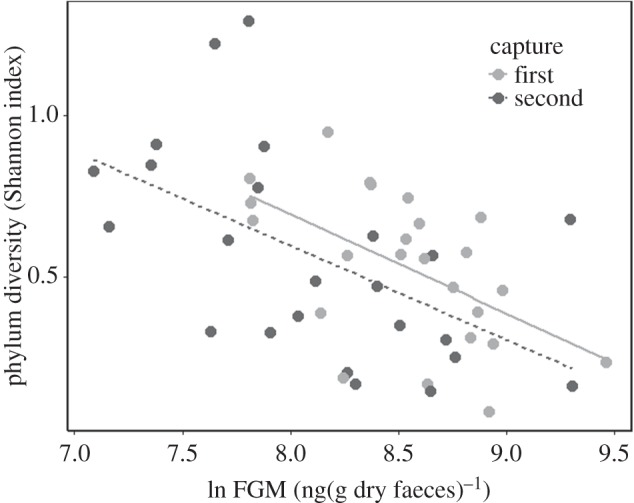
Shannon diversity of the red squirrel oral microbiome analysed at the phylum level. Diversity decreased with increasing FGM concentration (natural log-transformed FGM) during both capture periods.
Not only was bacterial diversity lower in individuals with elevated FGM, but also between capture periods a change in bacterial abundance was correlated with FGM (see the electronic supplementary material). Interestingly, the relative abundance of the most prevalent family in the oral microbiome, Pasteurellaceae, increased with increasing FGM (R2 = 0.23, t22 = 2.536, p = 0.02; figure 2). Members of this bacterial family have been linked to large epizootic events in bighorn sheep (Ovis canadensis) [6] as well as the near extinction of saiga antelope (Saiga tatarica) [25], and in both cases the proliferation of Pasteurellaceae was thought to have been enabled by environmental stress-induced reduction in host condition. These taxa-specific responses to perturbation by HPA axis activity could be due to attenuation of adaptive immunity, which, unlike innate immunity, is a non-general response [23,26]. Therefore, taxa suppressed by an adaptive immune response would be expected to increase in abundance following heightened HPA axis activity. Alternately, taxa persisting at higher abundance may possess a greater capacity for increase following HPA axis-mediated microbiome disruption.
Figure 2.
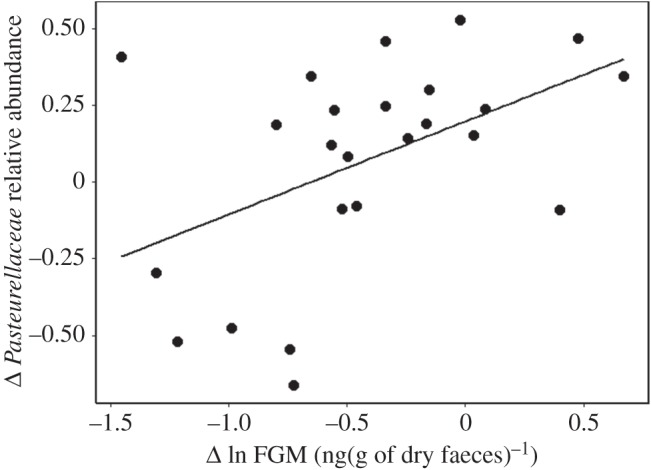
Over time, intra-individual change in FGM was correlated with change in the relative abundance of Pasteurellaceae within the red squirrel oral microbiome.
4. Conclusion
We report novel interactions between the HPA axis and oral microbiome diversity where disruption of bacterial communities may represent an ancillary cost of heightened HPA axis activity. Increased HPA axis activity resulted in bacterial communities that were lower in richness and dominated by a smaller number of taxa. These communities may have a direct negative impact on host health while simultaneously increasing susceptibility to pathogen invasion [1,27]. These findings using a free-living system lay the foundation for understanding the proximate mechanisms linking stress and the microbiome.
Supplementary Material
Acknowledgements
We thank C. Jardine for manuscript comments, C. Bosson for aid in performing the FGM assays, K. S. Brown for assistance in the field, and two anonymous reviewers for excellent feedback.
Ethics
All work was approved by the Laurentian University Animal Care Committee and conformed to Canadian Council of Animal Care requirements.
Data accessibility
The data underlying this study are available from Dryad: http://dx.doi.org/10.5061/dryad.js25t.
Authors' contributions
M.R.S. and C.B.B. conducted the field research; M.R.S., C.B.B., A.I.S., A.E.M.N. conceived and designed the study; R.B. and R.P. contributed antibodies and laboratory support for FGM EIA analysis; C.B.B., M.R.S. and N.C.S.M. interpreted the bacterial sequence data; M.R.S., C.B.B., A.I.S., R.B., R.P. and A.E.M.N. wrote the manuscript; all authors edited and gave final approval for publication. All authors agree to be accountable for all aspects of the work performed.
Competing interests
We have no competing interests.
Funding
Funding was provided by the University of Guelph and a Natural Sciences and Engineering Research Council (NSERC) Discovery grant to A.E.M.N., and by Laurentian University and NSERC Discovery grant to A.I.S. and by an NSERC Undergraduate Student Research Award to M.R.S.
References
- 1.Lozupone CA, Stombaugh JI, Gordon JI, Jansson JK, Knight R. 2012. Diversity, stability and resilience of the human gut microbiota. Nature 489, 220–230. ( 10.1038/nature11550) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cénit MC, Matzaraki V, Tigchelaar EF, Zhernakova A. 2014. Rapidly expanding knowledge on the role of the gut microbiome in health and disease. Biochim. Biophys. Acta. 1842, 1981–1992. ( 10.1016/j.bbadis.2014.05.023) [DOI] [PubMed] [Google Scholar]
- 3.Huttenhower C, et al. 2012. Structure, function and diversity of the healthy human microbiome. Nature 486, 207–214. ( 10.1038/nature11234) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Avila M, Ojcius DM, Yilmaz O. 2009. The oral microbiota: living with a permanent guest. DNA Cell Biol. 28, 405–411. ( 10.1089/dna.2009.0874) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Carroll IM, Ringel-Kulka Y, Siddle JP, Ringel Y. 2012. Alterations in composition and diversity of the intestinal microbiota in patients with diarrhea-predominant irritable bowel syndrome. Neurogastroent. Motil. 24, 521–530. ( 10.1111/j.1365-2982.2012.01891.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Besser TE, et al. 2012. Causes of pneumonia epizootics among bighorn sheep, western United States, 2008–2010. Emerg. Infect. Dis. 18, 406–414. ( 10.3201/eid1803.111554) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cryan JF, O'Mahony SM. 2011. The microbiome-gut-brain axis: from bowel to behavior. Neurogastroent. Motil. 23, 187–192. ( 10.1111/j.1365-2982.2010.01664.x) [DOI] [PubMed] [Google Scholar]
- 8.Grenham S, Clarke G, Cryan JF, Dinan TG. 2011. Brain-gut-microbe communication in health and disease. Front. Phys. 2, 1–15. ( 10.3389/fphys.2011.00094) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bailey MT, Dowd SE, Galley JD, Hufnagle AR, Allen RG, Lyte M. 2011. Exposure to a social stressor alters the structure of the intestinal microbiota: implications for stressor-induced immunomodulation. Brain Behav. Immun. 25, 397–407. ( 10.1016/j.bbi.2010.10.023) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bailey MT, Dowd SE, Parry NMA, Galley JD, Schauer DB, Lyte M. 2010. Stressor exposure disrupts commensal microbial populations in the intestines and leads to increased colonization by Citrobacter rodentium. Infect. Immun. 78, 1509–1519. ( 10.1128/IAI.00862-09) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dantzer B, McAdam AG, Palme R, Fletcher QE, Boutin S, Humphries MM, Boonstra R. 2010. Fecal cortisol metabolite levels in free-ranging North American red squirrels: assay validation and the effects of reproductive condition. Gen. Comp. Endocr. 167, 279–286. ( 10.1016/j.ygcen.2010.03.024) [DOI] [PubMed] [Google Scholar]
- 12.Dantzer B, Newman AEM, Boonstra R, Palme R, Boutin S, Humphries MM, McAdam AG. 2013. Density triggers maternal hormones that increase adaptive offspring growth in a wild mammal. Science 340, 1215–1217. ( 10.1126/science.1235765) [DOI] [PubMed] [Google Scholar]
- 13.Palme R, Touma C, Arias N, Dominchin MF, Lepschy M. 2013. Steroid extraction: get the best out of faecal samples. Wiener Tierärztl. Mschrift. 100, 238–246. [Google Scholar]
- 14.Masella AP, Bartram AK, Truszkowski JM, Brown DG, Neufeld JD. 2012. PANDAseq: paired-end assembler for illumine sequences. BMC Bioinform. 13, 1–7. ( 10.1186/1471-2105-13-31) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Edgar RC. 2010. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461. ( 10.1093/bioinformatics/btq461) [DOI] [PubMed] [Google Scholar]
- 16.Caporaso JG, et al. 2010. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7, 335–336. ( 10.1038/nmeth.f.303) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hill CJ, Walsh KA, Harris JA, Moffett BF. 2003. Using ecological diversity measures with bacterial communities. FEMS Microbiol. Ecol. 43, 1–11 ( 10.1111/j.1574-6941.2003.tb01040.x) [DOI] [PubMed] [Google Scholar]
- 18.Shannon CE, Weaver W. 1949. The mathematical theory of communication, vol. 117 Urbana, IL: University of Illinois Press. [Google Scholar]
- 19.R Core Team. 2014. R: a language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing; See http://www.R-project.org/. [Google Scholar]
- 20.McMurdie PJ, Holmes S. 2013. phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 8, e61217 ( 10.1371/journal.pone.0061217) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bateman A, Singh A, Kral T, Solomon S. 1989. The immune-hypothalamic-pituitary-adrenal axis. Endocr. Rev. 10, 92–112. ( 10.1210/edrv-10-1-92) [DOI] [PubMed] [Google Scholar]
- 22.Bailey MT, Kinsey SG, Padgett DA, Sheridan JF, Leblebicioglu B. 2009. Social stress enhances IL-1β and TNF-α production by Porphyromonas gingivalis lipopolysaccharide-stimulated CD11b+ cells. Physiol. Behav. 98, 351–358. ( 10.1016/j.physbeh.2009.06.013) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fleshner M, Bellgrau D, Watkins LR, Laudenslager ML, Maier SF. 1995. Stress-induced reduction in the rat mixed lymphocyte reaction is due to macrophages and not to changes in T cell phenotypes. J. Neuroimmunol. 56, 45–52. ( 10.1016/0165-5728(94)00132-8) [DOI] [PubMed] [Google Scholar]
- 24.Moloney RD, Desbonnet L, Clarke G, Dinan TG, Cryan JF. 2014. The microbiome: stress, health and disease. Mamm. Genome 25, 49–74. ( 10.1007/s00335-013-9488-5) [DOI] [PubMed] [Google Scholar]
- 25.Milner-Gullan EJ. 2015. Catastrophe and hope for the saiga. Oryx 49, 577 ( 10.1017/S0030605315000824) [DOI] [Google Scholar]
- 26.Zhang H, Sparks JB, Karyala SV, Settlage R, Luo XM. 2015. Host adaptive immunity alters gut microbiota. ISME J. 9, 770–781. ( 10.1038/ismej.2014.165) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dillon RJ, Vennard CT, Buckling A, Charnley AK. 2005. Diversity of locust gut bacteria protects against pathogen invasion. Ecol. Lett. 8, 1291–1298. ( 10.1111/j.1461-0248.2005.00828.x) [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data underlying this study are available from Dryad: http://dx.doi.org/10.5061/dryad.js25t.