

Abstract
Amsacrine analog is a novel chemotherapeutic agent that provides potentially broad antitumor activity when compared to traditional amsacrine. However, the major limitation of amsacrine analog is that it is highly lipophilic, making it nonconductive to intravenous administration. The aim of this study was to utilize solid lipid nanoparticles (SLN) to resolve the delivery problem and to investigate the biodistribution of amsacrine analog-loaded SLN. Physicochemical characterizations of SLN, including particle size, zeta potential, entrapment efficiency, and stability, were evaluated. In vitro release behavior was also measured by the dialysis method. In vivo pharmacokinetics and biodistribution behavior of amsacrine analog were investigated and incorporated with a non invasion in vivo imaging system to confirm the localization of SLN. The results showed that amsacrine analog-loaded SLN was 36.7 nm in particle size, 0.37 in polydispersity index, and 34.5±0.047 mV in zeta potential. More than 99% of amsacrine analog was successfully entrapped in the SLN. There were no significant differences in the physicochemical properties after storage at room temperature (25°C) for 1 month. Amsacrine analog-loaded SLN maintained good stability. An in vitro release study showed that amsacrine analog-loaded SLN sustained a release pattern and followed the zero equation. An in vivo pharmacokinetics study showed that amsacrine analog was rapidly distributed from the central compartment to the tissue compartments after intravenous delivery of amsacrine analog-loaded SLN. The biodistribution behavior demonstrated that amsacrine analog mainly accumulated in the lungs. Noninvasion in vivo imaging system images also confirmed that the drug distribution was predominantly localized in the lungs when IR-780-loaded SLN was used.
Keywords: amsacrine analog, solid lipid nanoparticle, pharmacokinetics, biodistribution, water insolubility
Introduction
Amsacrine is a potential antineoplastic agent used to treat liquid cancers or blood cancers, including refractory acute leukemia, nonlymphocytic leukemias, Hodgkin’s disease, non-Hodgkin’s lymphomas, and head and neck cancers. Amsacrine is now approved in Canada for treating refractory acute adult leukemia.1–3 However, amsacrine carries a high risk of cardio toxicity in patients, and nausea, vomiting, and diarrhea are common side effects.4 In addition, the short half-life of amsacrine is about 20–30 minutes, which limits its clinical application.5
Chen et al successfully synthesized the structure–activity relationships (SARs) of amsacrine analog (Figure 1). These relationships significantly increase the potential antitumor activity, avoid certain disadvantages, and prolong the half-life.6 The IUPAC name of amsacrine analog is 1-[4-(furo[2,3-b]quinolin-4-ylamino)phenyl]ethanone. In terms of the structure–activity relationships, acridine, the backbone of amsacrine’s structure, is related to its antitumor activity and increases longer half-lives.7 Moreover, previous study demonstrated that amsacrine analog was certainly effective not only against blood cancers but also against solid cancers, including human non-small-cell lung carcinoma (NCI-H460), breast cancer (MCF-7), human glioma and astrocytoma cell (SF-268), human kidney carcinoma cell (UO-31), and human melanoma cell (UACC-257, UACC-62). According to the cytotoxicity of amsacrine analog, it exerts a 50% growth inhibition (GI50) of 0.025 μM, which shows that it has powerful antitumor activity compared to the original amsacrine (GI50 =0.42 μM).8
Figure 1.

The chemical structure of (A) amsacrine and (B) amsacrine analog.
The newly synthesized amsacrine analog demonstrates powerful antitumor activities. Nevertheless, amsacrine analog has a highly lipophilic property that causes the drug to dissolve only minimally in the biocompatible media for intravenous injection. Traditionally, the water-insoluble drugs may, in the same way as vegetable oils, have an oil-based formulation. However, administration of the oil-based formulation via intravenous injections makes fat emboli.9 Therefore, several approaches were developed to effectively deliver the water-insoluble drug.
Over the past few decades, a considerable number of studies have been conducted on the nanoparticulate field, and nanoparticles have been widely applied as controlled and site-specific drug-delivery systems.10–12 Among them, solid lipid nanoparticles (SLNs) have attractive properties in comparison to traditional drug-delivery carriers.13 SLNs have unique properties, including high drug loading, a large surface area, protection from drug degradation from the environment, ease of modulating the release behavior, and increased bioavailability.14 Based on the Biopharmaceutical Classification System (BCS) guide, amsacrine analog has lower solubility in aqueous conditions (BCS class II), making it a promising candidate for preparation in SLNs. Moreover, lipophilic drug–loaded SLNs would effectively avoid hepatic first-pass metabolism.15 The aim of the study was to utilize amsacrine analog-loaded SLNs to improve the poor solubility problem and to establish the characterization and pharmacokinetics behavior.
Materials and methods
Chemicals and reagents
Amsacrine analog was synthesized in the laboratory of the School of Medicinal and Applied Chemistry (Kaohsiung Medical University, Kaohsiung, Taiwan). Pluronic® F 68 (F-68), stearylamine, and para-phenyl phenol were purchased from Sigma-Aldrich (St Louis, MO, USA). Trimyristin (TM) was purchased from Tokyo Chemical Industry Co., Ltd. (Tokyo, Japan). Hydrogenated soybean phosphatidylcholine (97%) was purchased from Nippon Oil & Fats Co., Ltd. (Tokyo, Japan). Gelucire® 53/10 was obtained from Gattefossé (Gennevilliers, France). N,N-dimethylacetamide was obtained from Mallinckrodt (St Louis, MO, USA). Methanol, acetonitrile, dichloromethane, and other chemicals were of analytical reagent grade.
Amsacrine analog analysis by high-performance liquid chromatography
Apparatus and chromatographic conditions
The high-performance liquid chromatography (HPLC) system consisted of a Hitachi model L-7100 pump (Tokyo, Japan), a BAS LC-4C electrochemical detector (BAS, Tokyo, Japan), a Jasco 855-AS autosampler (Tokyo, Japan), and a Lichrocart® RP-18e column (125 mm ×4 mm, internal diameters, 5 μm) (Merck, Darmstadt, Germany). The mobile phase was a mixture of acetonitrile and 0.01 M sodium salt of a 1-pentansulfonic acid solution (pH 3.0) (40:60, v/v), and the flow rate was 1.0 mL/min. The effluent from the column was monitored by an electron capture detector at 1.0 V and 20 nA. A BAS MF 1000 dual glassy carbon working electrode and a DAS RE6 Ag/AL reference electrode were used in the electrochemical detector system (Bioanalytical Systems Inc., West Lafayette, IN, USA). Para-phenylphenol was prepared as the internal standard (IS). The retention time of amsacrine analog and IS was 6.4 and 9.4 minutes.
Liquid chromatographic method validation
The limit of detection and limit of quantitation of amsacrine analog were determined by dissolving it at decreasing concentrations in methanol until the signal-to-noise ratios were 3 and 10, respectively. The recovery of amsacrine analog from plasma was assessed by comparing the peak area obtained from spiked amsacrine analog samples with those standard samples in a dichloromethane solution that was processed in the same way as the samples. Standard samples of rat plasma were prepared and analyzed during method validation. The linearity of the standard curves and the intra-day and inter-day precision and accuracy were determined from seven concentrations of amsacrine analog of 0.005, 0.01, 0.05, 0.1, 0.5, 1, and 2 μg/mL under the conditions previously described. The standard calibration curves were analyzed by a regression analysis to obtain the equation and correlation coefficients, which were y=0.4938x+0.0022 and 0.9990.
Preparation of SLNs
SLNs formulation was prepared by using hot homogenization followed by an ultrasonication method. The oil phase consisted of amsacrine analog, hydrogenated soybean phosphatidylcholine, TM, Glyceryl Monosterate, and Gelucire 53/10; and the aqueous phase with water consisted of 1% F-68. The oil phase was dissolved in 3 mL of a chloroform and methanol mixture (1:1, v/v) in order to ensure it was soluble. Organic solvents were completely removed by evaporator under a vacuum, and the residual organic solvent was then removed by nitrogen for 30 minutes. The two phases were preheated separately to 80°C for 20 minutes. The aqueous phase was added to the lipid phase and initially mixed by using a high-shear homogenizer (Pro 250, Pro Scientific, Monroe, CT, USA) at 10,500 rpm for 3 minutes. The mixture was further treated using a probe-type sonicator (Geprüfte Sicherheit UP50H, Teltow, Germany) for 20 minutes at 50 W.
Particle size and zeta potential
The mean particle size (z-average), polydispersity index (PDI), and zeta potential of the SLNs were measured by photon correlation spectroscopy (Zetasizer 3000HS; Malvern Instruments, Worcestershire, UK) using a helium–neon laser with a wavelength of 633 nm at 25°C. The size values are given as a volume distribution. All vesicles were diluted 150-fold with double-distilled water to achieve the count rate for the measurements. The determination was repeated three times per sample for three samples.
Entrapment efficiency
The entrapment efficiency of amsacrine analog in SLNs was determined by an ultracentrifugation method. The systems were centrifuged at 120,000 rpm (649,000× g) for 2 hours at 4°C in a Hitachi CS150 GXL ultracentrifuge (Japan) to separate the incorporated drug from the free form. The supernatant was analyzed by HPLC to determine the unentrapped parts. Moreover, 900 μL of tetrahydrofuran was added to 100 μL of SLNs to destroy the shell of SLNs, and then centrifuged at 120,000 rpm at 4°C for 10 minutes. A total of 200 μL of supernatants and 50 μL of IS (5 μg/mL) were mixed to determine the total drug load. The drug entrapment efficiency was calculated by the following equation:
Storage stability
The prepared SLN colloids were stored at room temperature (25°C) for 1 month. An aliquot of a sample was taken at predetermined time intervals to investigate the particle size, PDI, zeta potential, and entrapment efficiency. The mean size was determined by photon correlation spectroscopy as described in the previous section. The concentration of amsacrine analog was determined by the ultracentrifugation method to understand changes in the drug entrapment efficiency.
Transmission electron microscopy morphology
The morphology of the SLNs was observed by transmission electron microscopy (TEM) to characterize the microstructure. An SLN suspension (200 μL) was added to a carbon film-covered copper grid (Ted Pella Inc., Redding, CA, USA), which was stained with 0.5% phosphotungstic acid for 10 minutes, washed with double-distilled water, and placed in a vacuum overnight. The sample was then examined using a JEOL JEO-2000EX II transmission electron microscope (Tokyo, Japan).
In vitro release study
An in vitro release study used the dialysis method. Cellulose dialysis bags with a molecular weight cutoff of 12,000–14,000 Daltons (Orange Scientific, Houston, TX, USA) were soaked with double-distilled water for 12 hours before the experiment. The control group of amsacrine analog was dissolved in the vehicle, which contained 300 μL N,N-dimethylacetamide, 900 μL polyethylene glycol (PEG)-400, 1,200 μL tetraglycol, and 600 μL injection water due to its water-insoluble property. The control group of amsacrine analog and amsacrine analog-loaded SLN suspensions (1 mL) was put into dialysis bags and subsequently placed in flasks containing 150 mL medium (pH 7.4 phosphate buffer containing 1% Pluronic F-127 [F-127]) to maintain the solubility during the experiment’s sink conditions at 100 rpm in a 37°C water bath. At appropriate intervals, 500 μL aliquots of the medium were withdrawn from the receiver compartment through a side tube and immediately replaced with an equal volume of fresh medium. The amounts of the drug released went through a 0.45 μm filter and were analyzed by the HPLC.
Animals and drug administration
ICR, CD-1 mice, 4 weeks old and weighing 20–25 g (obtained from the Laboratory Animal Center of the National Science Council, Taipei, Taiwan) were used in the study. The animal experiment protocol was reviewed and approved by the Institutional Animal Care and Use Committee of Kaohsiung Medical University. Animals were housed and handled according to institutional guidelines. All animals were starved overnight prior to the experiments. Mice were anesthetized by an intraperitoneal injection of 25% urethane (3 mL/kg) before the experiment. The amsacrine analog control group (amsacrine analog dissolved in 300 μL N,N-dimethylacetamide, 900 μL PEG-400, 1,200 μL tetraglycol, and 600 μL of injection water) or the amsacrine analog entrapped in SLNs was administered via the tail vein at dosages of 8.4 mg/kg.
In vivo pharmacokinetics
Blood samples were collected from the heart at specified time intervals after amsacrine analog administration with and without SLNs. Blood samples were then centrifuged at 3,000 rpm for 10 minutes at 4°C and plasma was immediately separated and stored at −20°C until analysis. Aliquots of blank organs and plasma samples at 100 μL were mixed with 50 μL of 5 μg/mL of an IS solution and 4 mL dichloromethane as the extraction solvent. Mice were sacrificed after the administration of amsacrine analog with and without SLNs at specified time intervals. Tissues and organs, including the heart, lungs, liver, spleen, kidneys, whole brain, spine, and small and large intestines, were harvested and weighed on an analytical scale. The drug in the tissues and organs was extracted by a homogenization method. Normal saline was added to the excised tissue in a homogenizer at 16,800 rpm for 1 minute. Then 50 μL of 5 μg/mL IS and 4 mL dichloromethane were added for extraction. The mixture was horizontally shaken at 200 rpm for 30 minutes and centrifuged at 3,000 rpm for 10 minutes. A total of 2.5 mL of the organic layer was transferred to another tube and evaporated to dryness under a vacuum. The dry residue was reconstituted in 200 μL acetonitrile and 20 μL of clear supernatant was injected into the HPLC system.
Analysis of pharmacokinetic data
The pharmacokinetics were calculated based on each individual set of data, using the pharmacokinetic software WinNonLin (v 4.0; Pharsight Software, Mountain View, CA, USA) to measure the type of compartment model using the correlation coefficient from the observed and predicted data. The pharmacokinetic data measured included the area under the plasma concentration–time curves from time zero to infinity (AUC0−∞), the half-life of the distribution phase (t1/2α), the half-life of the elimination phase (t1/2β), and the clearance (Cl). The AUC0−∞ was calculated using the trapezoidal rule (AUC0−t) and was extrapolated to infinity by the addition of AUCt−∞. The Cl value was calculated as the dose/AUC0−∞, and the mean residence time (MRT) and the volume of distribution at a steady state were obtained by the summation of the central and tissue compartments.
Optical biodistribution by a noninvasion in vivo imaging system
Mice were anesthetized by halothane vapor with a vaporizer system and then were intravenously injected with 500 μL IR-780-loaded SLN. Mice were sacrificed 3 hours after IR-780-loaded SLN administration. The liver, kidney, lungs, heart, spleen, and gastrointestinal tract were collected. The fluorescence intensities of the tissues were recorded with a NightOWL II LB 983 NC100 image system (Berthold Technologies, Bad Wildbad, Germany).
Statistical analysis
All data were expressed as the mean ± standard derivation (SD). Statistical analyses were performed using the unpaired Student’s t-test with Winks SDA 6.0 software (Texasoft, Duncanville, TX, USA). An analysis of variance test was also used. Subgroup comparisons were made using the Newman–Keuls multiple comparisons. A 0.05 level of probability was used as the level of significance.
Results
Characterization of amsacrine analog-loaded SLN
Amsacrine analog-loaded SLN is a semitransparent yellow colloid. When amsacrine analog gets well dissolved in the matrix of SLN, the semitransparent yellow was observed due to the yellow color of amsacrine analog. Table 1 summarizes the physicochemical properties and the stability changes of amsacrine analog-loaded SLN. The particle size of amsacrine analog-loaded SLN was 36.7 nm and the PDI showed a distribution in the range 0.37±0.04. Moreover, the zeta potential ranged from 34.5±4.7 mV. SLN showed a high entrapment efficiency for carrying amsacrine analog of approximately 100%. The results also showed that physicochemical properties, including particle sizes, zeta potential, and entrapment efficiency of amsacrine analog SLN, remained relatively stable after 1 month of storage (P>0.05). Figure 2 shows the TEM micrograph of amsacrine analog-loaded SLN. The images presented nanometer-sized spherical outlines and the monodispersed distribution pattern.
Table 1.
Characterization of amsacrine analog–loaded solid lipid nanoparticles (SLNs) in terms of the particle size, polydispersity index (PDI), zeta potential, and entrapment efficiency, and stability changes following storage at room temperature (25°C) for 1 month
| Formulation | Original | After 1 month |
|---|---|---|
| Particle diameter (nm) | 36.7±0.6 | 40.10±7.1 |
| PDI | 0.37±0.04 | 0.44±0.12 |
| Zeta potential (mV) | 34.5±4.7 | 37.4±3.2 |
| Entrapment efficiency (%) | 99.58±0.09 | 98.75±0.10 |
Note: Each value represents mean ± SD (n=3).
Abbreviation: SD, standard deviation.
Figure 2.
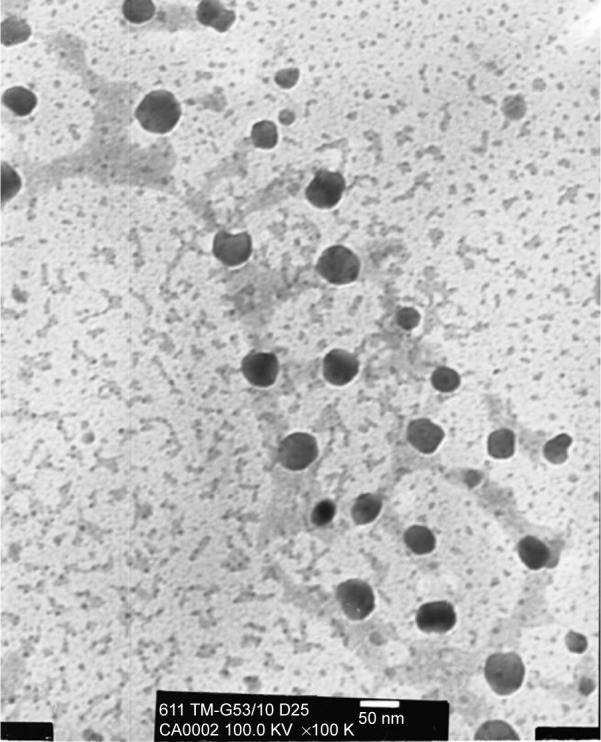
Transmission electron microscopic micrograph of amsacrine analog-loaded solid lipid nanoparticle (SLN) (original magnification ×100 K).
In vitro release behavior
The release behaviors of amsacrine analog with or without SLN were investigated as an accumulative amount of release percentage (Figure 3). The amsacrine analog control group exhibited a biphasic pattern, with an initial burst release of approximately 61.6% within 5 hours, followed by saturation at 24 hours. Moreover, the results of amsacrine analog-loaded SLN showed a sustained release trend with 27.79% in the period of 24 hours. In addition, there was a significant difference between amsacrine analog with and without being amsacrine analog loaded in the SLN group (P<0.05). On the other hand, the parameters of the release kinetics were also calculated based on the release behavior. For the calculation of the release period of the amsacrine analog, the results showed that the initial 5 hours and later 5–24 hours were fitted zero-order in the control group, and the relative coefficients (R) of the Hiquchi-order were 0.9734 and 0.9654, respectively. Moreover, amsacrine analog-loaded SLN followed zero equation (R=0.9999).
Figure 3.
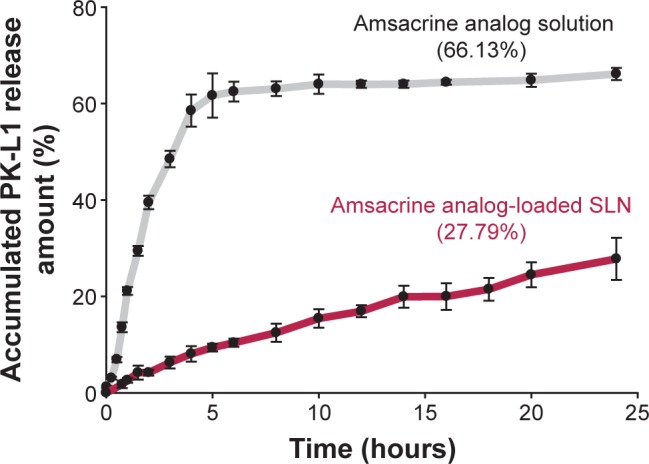
In vitro accumulative amount release versus time profiles of amsacrine analog from solid lipid nanoparticle (SLN).
Note: Each value represents mean ± SD (n=3).
Abbreviation: SD, standard deviation.
In vivo pharmacokinetics
Table 2 summarizes the pharmacokinetic parameters acquired by a two-compartment model analysis after an intravenous bolus of amsacrine analog-loaded SLN for 4 hours. The t1/2α of the amsacrine analog-loaded SLN was significantly higher than that of the amsacrine analog solution (P<0.05); however, the t1/2β of the amsacrine analog-loaded SLN was significantly lower than that of the amsacrine analog solution (P<0.05). Moreover, both the Cl and volume of distribution at steady state of SLNs were significantly lower than those of the PK-L4 solution (P<0.05). A comparison of the AUC0→∞ values for amsacrine analog with or without SLN in plasma showed the values as 0.4132±0.003 and 0.1905±0.004 h·μg/mL, respectively. In terms of MRT, the amsacrine analog solution was slightly higher than the amsacrine analog-loaded SLN.
Table 2.
Average pharmacokinetic parameters after intravenous administration of amsacrine analog solution and amsacrine analog-loaded SLN to mice with a dose of 8.4 mg/kg for 4 hours
| Parameters | Amsacrine analog solution | Amsacrine analog-loaded SLN |
|---|---|---|
| t1/2α (hour) | 0.1955±0.002 | 0.3469±0.007 |
| t1/2β (hour) | 1.4375±0.067 | 0.8026±0.087 |
| Cl (L/h/kg) | 20.3287±1.254 | 44.0903±3.501 |
| Vdss (L/kg) | 23.1743±1.563 | 34.0650±4.184 |
| AUC0→∞ (h·μg/mL) | 0.4132±0.003 | 0.1905±0.004 |
| MRT (hour) | 1.1400±0.006 | 0.7726±0.009 |
Note: Each value represents mean ± SD (n=6).
Abbreviations: SLN, solid lipid nanoparticle; t1/2α, half-life of the distribution phase; t1/2β, half-life of the elimination phase; Cl, clearance; Vdss, steady-state volume of distribution; AUC, area under tissue concentration–time curve; MRT, mean residence; SD, standard deviation.
Biodistribution
The biodistribution of amsacrine analog of mice was determined after intravenous administration (8.4 mg/kg) of amsacrine analog with and without SLN. Amsacrine analog amounts of the tissues and organs showed quantification, and the linear range was 0.025–10 μg/g (R2=0.9945–0.9995). Figure 4 shows the concentration of amsacrine analog in the organs and time profile. Table 3 presents the relative organ distribution values. The results showed that the amsacrine analog amounts with SLN in the lungs, liver, spleen, and kidney were significantly higher than those without SLN (P<0.05). Moreover, the highest deposition of amsacrine analog-loaded SLN was mainly found in the lungs followed by the liver, spleen, and kidney. There was no significant accumulation in the heart, intestines, and brain compared to the SLN control group (P>0.05). Among the organs’ accumulation, lung tissue showed a high deposition presented in AUC, MRT, and relative bioavailability showing about 120.
Figure 4.

Concentration–time profiles in organs after intravenous administration of amsacrine analog with or without SLN at dose 8.4 mg/kg in mice.
Note: Each value represents mean ± SD (n=6).
Abbreviations: SLN, solid lipid nanoparticle; SD, standard deviation.
Table 3.
AUC and mean residence time of amsacrine analog in various organs upon administration of amsacrine analog with or without SLN
| Parameters | Formulations | Organs
|
||||||
|---|---|---|---|---|---|---|---|---|
| Heart | Liver | Spleen | Lung | Kidney | Brain | Intestine | ||
| AUC0−∞ (h·μg/mL) | Amsacrine analog solution | 0.3143 | 0.6913 | 0.2117 | 0.5915 | 1.0669 | 0.4015 | 0.4222 |
| Amsacrine analog-loaded SLN | 0.2164 | 2.3000 | 1.7113 | 71.1698 | 1.0826 | 0.0588 | 0.0636 | |
| MRT (hour) | Amsacrine analog solution | 0.6883 | 0.7727 | 0.9841 | 0.7681 | 0.7991 | 0.8547 | 0.8785 |
| Amsacrine analog-loaded SLN | 0.8075 | 1.2507 | 1.2821 | 2.7101 | 1.2685 | 0.6088 | 0.6572 | |
| Fr | 0.6885 | 3.3270 | 8.0836 | 120.3208 | 1.0147 | 0.1465 | 0.1506 | |
Note: Fr = AUCSLN/AUCsolution.
Abbreviations: SLN, solid lipid nanoparticle; Fr, relative bioavailability; AUC, area under tissue concentration–time curve; MRT, mean residence time.
In vivo optical images of IR-780-loaded SLN
In order to confirm where the SLNs could selectively target in vivo, the mice were intravenously injected with IR-780-loaded SLNs and observed by a noninvasion in vivo imaging system (IVIS). The IVIS image as shown in Figure 5, the left side was a bright field. The right side shows the relative fluorescent intensities of individual organs. Fluorescence was a predominant observation in the lungs and liver.
Figure 5.

In vivo biodistribution images of IR-780-loaded SLN in mice by using IVIS technique. The left side is a bright field; the right side is the intensity of IR-780 in various organs. The color bar presents the signal efficiency of the fluorescence emission coming out from the violet.
Abbreviations: SLN, solid lipid nanoparticle; Min, minimum; Max, maximum; IVIS, noninvasion in vivo imaging system.
Discussion
Poor water solubility of amsacrine analog obstructed the drug’s administration by intravenous delivery. The aim of the study was to focus on resolving the delivery problem. The present work demonstrated amsacrine analog-loaded SLN improving the intravenous-injected delivery problem.
Particle size is an important concern in the development of a colloid delivery system, which influences the organ biodistribution, particularly in the intravenous injection administration route.16 Generally, the blood capillary diameter average was approximately 3.7 μm; the design of the pharmaceutically acceptable particle size require limited less than 3.7 μm in diameter for injection usage.17,18
Our past evidence demonstrated that TM and Gelucire 53/10 in the ratio 1:1 presented a smaller particle size. When a high amount of TM is incorporated (TM:Gelucire 53/10=9:1), the particle size would increase to 110 nm and broad distribution would occur. However, without the incorporation of Gelucire 53/10, precipitation of SLN colloidal was found to occur (data not shown). In this work, amsacrine analog-loaded SLN was less than 40 nm in terms of particle size and had a well distributed PDI of 0.37. The TEM image also confirmed that it was monodispersed.
Several factors would influence the entrapment efficiency of drug-loaded SLN. Amsacrine analog is a lipophilic compound with a marked solubility and a miscibility of melted lipid. We assumed that increased entrapment efficiency was contributed by amsacrine analog melted in matrix SLN. Previous study demonstrated that glyceride matrix would produce less-ordered crystal lattices.19 The phenomena were also relevant with TM, owing to the long chain of fatty acids attached to the triglyceride, resulting in increased accommodation of lipophilic drugs.20
Stability is a crucial factor for practical use of any pharmaceutical formulation.21 After 1 month, amsacrine analog-loaded SLN showed marked stability, with the particle size, zeta potential, and entrapment efficiency remaining similar to the original ranges. The colloidal system can be stabilized by two different mechanisms including electrostatic stabilization and steric stabilization.22 The composition of amsacrine analog-loaded SLN contains F68, which provides steric stabilization, and stearylamine enabled the acquisition of the positive particle surface charge that produces electric repulsion to help prevent particle aggregation.19,23
Traditional SLN existed with several problems, such as a rapid burst effect, making for an unstable plasma concentration and high toxicity. Hence, in this work, we focus on the formulation design to minimize the aforementioned problem. According to the result of the release study, the amsacrine analog control group exhibited an initial rapid release of 60% drug within 5 hours, followed by a slow release until 24 hours. The results indicated a biphasic pattern. The early burst-release phase of amsacrine analog contributed to the drug solutes’ rapid dissolution to the release medium. The phenomenon contributed to the vehicle itself and the passive diffusion force. The diffusion process in most of the drug was by concentration gradient.24 The marked initial burst-release effect would make the drug quickly enter the blood or organs. The phenomenon is dangerous clinically, especially in the case of low-therapeutic window drugs. On the other hand, amsacrine analog-loaded SLN followed the zero equation. An ideal drug-delivery system would follow zero-order kinetics, particularly for some medicines such as antibiotics, pain control, and chemotherapy.25 The literature describes the solutes’ transport process in which the materials’ release requires more time than the characteristic solvent diffusion time that follows Fick’s law. Hence, the composition of SLN plays the major role in the release rate and pattern.26 Generally, the drug is released from excipients by diffusion and by using endogenous enzymes that degrade excipients (particularly lipids). Hence, selection of a suitable lipid type is an important consideration in the formulation design that influences the degradation process.27,28 Our past evidence demonstrated that the solid lipid core of SLN composed of TM and Gelucire 53/10 presented good sustained release behavior.29 The sustained release behavior was mainly attributed to medium-chain triglycerides of TM and PEG glyceryl stearate derivatives of Gelucire 53/10. Although previous studies pointed out that traditional SLNs exhibited a marked burst effect, this study did not present this phenomenon. It contributed the observation that the low surfactant amount used in the formulation made no significant burst effect to occur.30 Moreover, the high relative coefficient of a zero-order release pattern was fitted with amsacrine analog-loaded SLN within 24 hours. This phenomenon also indicates that the sustained release of amsacrine analog from all SLNs was achieved in this work.
The area under the curve of plasma from the control group was significantly higher than that of the SLN group (P<0.05). The relative pharmacokinetic parameters presented that the t1/2α of SLNs was higher than those of the control group, and, to the contrary, the t1/2β of SLNs was less than those of the control group (P<0.05). These phenomena mean that amsacrine analog was rapidly distributed from the central compartment to the tissue compartments and disappeared quickly from the blood. Moreover, SLN also showed a higher volume of distribution at steady state and Cl when compared with the control group. MRT of SLN was less than in the control. In the case of the study, all the evidence pointed out that amsacrine analog-loaded SLN rapidly left the plasma. Actually, the composition and surface properties of amsacrine analog-loaded SLN were the contribution factors influencing the pharmacokinetics parameter and biodistribution characteristics.
Gelucire 53/10 is a PEG-32 glyceryl stearate. PEG glyceryl stearates are a family of lipid-based excipients composed of glycerides and esters of PEG, which offer hydrophobic and hydrophilic properties to the vehicle. Moreover, F68 also provides a hydrophilic environment on the outside of SLN. Past studies have also revealed that the biodistribution behavior of SLN can be decorated with nonionic surfactants such as polysorbates, poloxamers, and poloxamines.31 In terms of the biodistribution, amsacrine analog amounts in organs were measured to be mainly accumulated in the lungs, followed by the liver, spleen, and kidney. We also measured the in vivo organs’ distribution image by IVIS with the expectation of the drug being localized. The results also indicated that the lungs, liver, spleen, and kidneys had a high localization intensity. All the pharmacokinetics and IVIS demonstrated that amsacrine analog SLN was localized in the reticuloendothelial system organs. Similar results were reported when carrier intravenous administration was rapidly taken up by the reticuloendothelial system in the liver and spleen.32,33 Indeed, with intravenously administered colloid carriers, the biodistributions behavior of amsacrine analog would be changed. We assumed that this was mainly contributed to by SLNs adsorbing the surface protein of blood (opsonins). Hence, the relationship between SLN and phagocytes was changed by opsonization.34
Conclusion
In summary, the aim of the study was to utilize SLN to resolve the hydrophobic property of amsacrine analog application for intravenous injection. We established the physicochemical characterization and biodistribution behavior of amsacrine analog-loaded SLN. Due to the lipid ingredients of SLN, amsacrine analog was successfully entrapped in SLN and presented marked stability. An in vivo pharmacokinetics study presented that amsacrine analog-loaded SLN was rapidly distributed from the plasma to the organs. The main organ distribution of amsacrine analog accumulated in the lungs and a large number of SLNs were also localized in the lungs by IVIS. The present result indicated successful delivery of amsacrine analog by SLN and the pharmacokinetics and biodistribution demonstrated the potential of lung targeting. The preliminary results encouraged us to develop lung-cancer targeting in the future.
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Arlin ZA, Feldman E, Kempin S, et al. Amsacrine with high-dose cytarabine is highly effective therapy for refractory and relapsed acute lymphoblastic leukemia in adults. Blood. 1988;72:433–435. [PubMed] [Google Scholar]
- 2.Legha SS, Keating MJ, McCredie KB, Bodey GP, Freireich EJ. Evaluation of AMSA in previously treated patients with acute leukemia results of therapy in 109 adults. Blood. 1982;60:484–490. [PubMed] [Google Scholar]
- 3.Jehn U, Heinemann V. New drugs in the treatment of acute and chronic leukemia with some emphasis on m-AMSA. Anticancer Res. 1991;11:705–711. [PubMed] [Google Scholar]
- 4.Fenaux P, Tertian G, Castaigne S, et al. A randomized trial of amsacrine and rubidazone in 39 patients with acute promyelocytic leukemia. J Clin Oncol. 1991;9:1556–1561. doi: 10.1200/JCO.1991.9.9.1556. [DOI] [PubMed] [Google Scholar]
- 5.Paxton JW, Kim SN, Whitfield LR. Pharmacokinetic and toxicity scaling of the antitumor agents amsacrine and CI-921, a new analogue, in mice, rats, rabbits, dogs, and humans. Cancer Res. 1990;50:2692–2697. [PubMed] [Google Scholar]
- 6.Chen IL, Chen YL, Tzeng CC, Chen IS. Synthesis and cytotoxic evaluation of some 4-anilinofuro[2,3-b]quinoline derivatives. Helv Chim Acta. 2002;85:2214–2221. [Google Scholar]
- 7.Finlay GJ, Riou JF, Baguley BC. From amsacrine to DACA (N-[2-(dimethylamino)ethyl]acridine-4-carboxamide): selectivity for topoisomerases I and II among acridine derivatives. Eur J Cancer. 1996;32A:708–714. doi: 10.1016/0959-8049(95)00604-4. [DOI] [PubMed] [Google Scholar]
- 8.Chen YL, Chen IL, Wang TC, Han CH, Tzeng CC. Synthesis and anticancer evaluation of certain 4-anilinofuro[2,3-b]quinoline and 4-anilinofuro[3,2-c]quinoline derivatives. Eur J Med Chem. 2005;40:928–934. doi: 10.1016/j.ejmech.2005.04.003. [DOI] [PubMed] [Google Scholar]
- 9.Desai A, Lee M. Gibaldi’s Drug Delivery Systems in Pharmaceutical Care. Bethesda, Maryland: American Society of Health-System Pharmacist; 2007. p. 111. [Google Scholar]
- 10.Tammam SN, Azzazy HM, Lamprecht A. Biodegradable particulate carrier formulation and tuning for targeted drug delivery. J Biomed Nanotechnol. 2015;11:555–577. doi: 10.1166/jbn.2015.2017. [DOI] [PubMed] [Google Scholar]
- 11.Yohan D, Chithrani BD. Applications of nanoparticles in nanomedicine. J Biomed Nanotechnol. 2014;10:2371–2392. doi: 10.1166/jbn.2014.2015. [DOI] [PubMed] [Google Scholar]
- 12.Li J, Wang Y, Liang R, et al. Recent advances in targeted nanoparticles drug delivery to melanoma. Nanomedicine. 2015;11:769–794. doi: 10.1016/j.nano.2014.11.006. [DOI] [PubMed] [Google Scholar]
- 13.Müller RH, Radtke M, Wissing SA. Solid lipid nanoparticles (SLN) and nanostructured lipid carriers (NLC) in cosmetic and dermatological preparations. Adv Drug Deliv Rev. 2002;54(Suppl 1):S131–S155. doi: 10.1016/s0169-409x(02)00118-7. [DOI] [PubMed] [Google Scholar]
- 14.Ezzati Nazhad Dolatabadi J, Valizadeh H, Hamishehkar H. Solid lipid nanoparticles as efficient drug and gene delivery systems: recent breakthroughs. Adv Pharm Bull. 2015;5:151–159. doi: 10.15171/apb.2015.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kamboj S, Bala S, Nair AB. Solid lipid nanoparticles: an effective lipid based technology for poorly water soluble drugs. Int J Pharm Sci Rev Res. 2010;5:78–90. [Google Scholar]
- 16.Kanke M, Simmons GH, Weiss DL, Bivins BA, DeLuca PP. Clearance of 141C3-labeled microspheres from blood and distribution in specific organs following intravenous and intraarterial administration in beagle dogs. J Pharm Sci. 1980;69:755–762. doi: 10.1002/jps.2600690703. [DOI] [PubMed] [Google Scholar]
- 17.Wiedeman MP. Dimensions of blood vessels from distributing artery to collecting vein. Circ Res. 1963;12:375–378. doi: 10.1161/01.res.12.4.375. [DOI] [PubMed] [Google Scholar]
- 18.Kostera VS, Kuks PF, Lange R, Talsma H. Particle size in parenteral fat emulsions, what are the true limitations? Int J Pharm. 1996;134:235–238. [Google Scholar]
- 19.Nair R, Arun Kumar KS, Vishnu Priya K, Sevukarajan M. Recent advances in solid lipid nanoparticle based drug delivery systems. J Biomed Sci Res. 2011;3:368–384. [Google Scholar]
- 20.Kumar VV, Chandrasekar D, Ramakrishna S, Kishan V, Rao YM, Diwan PV. Development and evaluation of nitrendipine loaded solid lipid nanoparticles: Influence of wax and glyceride lipids on plasma pharmacokinetics. Int J Pharm. 2007;335:167–175. doi: 10.1016/j.ijpharm.2006.11.004. [DOI] [PubMed] [Google Scholar]
- 21.Doktorovova S, Shegokar R, Rakovsky E, et al. Cationic solid lipid nanoparticles (cSLN): structure, stability and DNA binding capacity correlation studies. Int J Pharm. 2011;420:341–349. doi: 10.1016/j.ijpharm.2011.08.042. [DOI] [PubMed] [Google Scholar]
- 22.Fritz G, Schädler V, Willenbacher N, Wagner NJ. Electrosteric stabilization of colloidal dispersions. Langmuir. 2002;18:6381–6390. [Google Scholar]
- 23.Müller RH, Mäder K, Gohla S. Solid lipid nanoparticles (SLN) for controlled drug delivery – a review of the state of the art. Eur J Pharm Biopharm. 2000;50:161–177. doi: 10.1016/s0939-6411(00)00087-4. [DOI] [PubMed] [Google Scholar]
- 24.Fu Y, Kao WJ. Drug release kinetics and transport mechanism of non-degradable and degradable polymeric delivery system. Expert Opin Drug Deliv. 2010;7:429–444. doi: 10.1517/17425241003602259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Singhvi G, Singh M. Review: in-vitro drug release characterization models. Int J Pharm Studies Res. 2011;2:77–84. [Google Scholar]
- 26.Grassi M, Grassi G. Mathematical modeling and controlled drug delivery: matrix system. Current Drug Deliv. 2005;2:97–116. doi: 10.2174/1567201052772906. [DOI] [PubMed] [Google Scholar]
- 27.Nunthanid J, Huanbutta K, Luangtana-anan M, Sriamornsak P, Limmatvapirat S, Puttipipatkhachorn S. Development of time-, pH-, and enzyme-controlled colonic drug delivery using spray-dried chitosan acetate and hydroxypropyl methylcellulose. Eur J Pharm Biopharm. 2008;68:253–259. doi: 10.1016/j.ejpb.2007.05.017. [DOI] [PubMed] [Google Scholar]
- 28.Fang YP, Wu PC, Tzeng CC, Chen YL, Lin HL, Tsai YH. A new antitumor agent, (3-chloro-7-methoxyfuro[2,3-b]-quinolin-4-yl)-(4-methoxyphenyl)amine, loaded in solid lipid nanoparticles: characterization and pharmacokinetics. Curr Nanosci. 2012;8:266–273. [Google Scholar]
- 29.Fang YP, Wu PC, Huang YB, et al. Modification of polyethylene glycol onto solid lipid nanoparticles encapsulating a novel chemotherapeutic agent (PK-L4) to enhance solubility for injection delivery. Int J Nanomedicine. 2012;7:4995–5005. doi: 10.2147/IJN.S34301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ekambaram P, Abdul Hasan Sathali A, Priyanka K. Solid lipid nanoparticles – a review. Int J App Pharm. 2013;5:8–18. [Google Scholar]
- 31.Liu M, Li H, Luo G, Liu Q, Wang Y. Pharmacokinetics and biodistribution of surface modification polymeric nanoparticles. Arch Pharm Res. 2008;31:547–554. doi: 10.1007/s12272-001-1191-8. [DOI] [PubMed] [Google Scholar]
- 32.Manjunath K, Venkateswarlu V. Pharmacokinetics, tissue distribution and bioavailability of clozapine solid lipid nanoparticles after intravenous and intraduodenal administration. J Control Release. 2005;107:215–228. doi: 10.1016/j.jconrel.2005.06.006. [DOI] [PubMed] [Google Scholar]
- 33.Harivardhan Reddy L, Sharma RK, Chuttani K, Mishra AK, Murthy RS. Influence of administration route on tumor uptake and biodistribution of etoposide loaded solid lipid nanoparticles in Dalton’s lymphoma tumor bearing mice. J Control Release. 2005;105:185–198. doi: 10.1016/j.jconrel.2005.02.028. [DOI] [PubMed] [Google Scholar]
- 34.Bagul R, Mahajan V, Dhakenew A. New approaches in nanoparticulate drug delivery system – a review. Int J Curr Pharm Res. 2012;4:29–38. [Google Scholar]