

Abstract
Currently, the concept of ‘neuroinflammation’ includes inflammation associated with neurodegenerative diseases, in which there is little or no infiltration of blood-derived immune cells into the brain. The roles of brain-resident and peripheral immune cells in these inflammatory settings are poorly understood, and it is unclear whether neuroinflammation results from immune reaction to neuronal dysfunction/degeneration, and/or represents cell-autonomous phenotypes of dysfunctional immune cells. Here, we review recent studies examining these questions in the context of Huntington’s disease (HD), where mutant Huntingtin (HTT) is expressed in both neurons and glia. Insights into the cellular and molecular mechanisms underlying neuroinflammation in HD may provide a better understanding of inflammation in more complex neurodegenerative disorders, and of the contribution of the neuroinflammatory component to neurodegenerative disease pathogenesis.
Keywords: neuroinflammation, Huntington’s disease, microglia, astrocytes, macrophages
A comprehensive concept of neuroinflammation
The concept of ‘neuroinflammation’ was used originally to describe inflammatory settings of the central nervous system (CNS) characterized by infiltration of peripheral immune cells, such as viral and bacterial infection, ischemic stroke, HIV encephalopathy, and multiple sclerosis (MS) [1,2]. Currently, the same term has expanded to include neurodegenerative diseases that do not attract inflammatory cells from the blood. Alzheimer’s disease (AD), Parkinson’s disease (PD), and HD are characterized by cellular and molecular features of inflammation (cytokine expression and microglia activation), but lack the signs of classic ‘neuroinflammation’, such as immune cell infiltration from the blood stream [1–5]. Nevertheless, the absence of immune cell infiltration from the periphery does not rule out the potential contribution of these cells to neuroinflammation, for example, via a chronic increase of systemic pro-inflammatory cytokine production. Furthermore, whether inflammation is the response of surrounding cells to a neuron-autonomous degenerative process and/or due to cell-autonomous immune activation remains an area of active investigation.
Glial cells are non-neuronal cells in the brain that play diverse roles in tissue homeostasis and support of neuronal function. For the purposes of this review, we focus on two types of glial cell: microglia and astrocytes (see Glossary). Under conditions of infection or injury, these cells become ‘activated’, a process characterized by their production of numerous mediators that promote inflammation, changes in morphology, and, in some cases, cell division, resulting in increased cell numbers or ‘gliosis’. Regardless of the mechanisms responsible for glia activation, the contribution of inflammation to neurodegenerative diseases pathogenesis remains poorly understood. In contrast to PD and AD, which are complex multifactorial pathologies related to a spectrum of genetic mutations and environmental factors [3,4], HD or Huntington’s chorea, is a neurodegenerative disorder caused by a single mutation: a specific expansion of the PolyQ tract in the ubiquitously expressed HTT protein [6]. Interestingly, even though HTT protein is constitutively and ubiquitously expressed throughout the body, HTT mRNA expression in immune cells is on average higher than that observed in most organs (Genomics Institute of Novartis Research Foundation, transcript 202389_s_at) [7]. In recent years, several efforts have been made to understand whether mutant HTT expression could trigger cell-autonomous activation of the immune cells of the brain and periphery, and whether these, in turn, could negatively impact HD pathogenesis. Here, we review recent evidence on the impact of mutant HTT on microglia, astrocytes, and macrophages. We place these findings in the context of the current understanding of inflammation in HD, and discuss the potential contributions of these cells to HD pathogenesis.
Clinical features of neuroinflammation in HD
Accumulation of reactive microglia and astrocytes has been observed in brains from HD patients [8]. PET imaging showed that microglia activation correlates with the pathology in HD patients [9–11]. Activation of microglia is evident in presymptomatic HD gene carriers, and can be detected up to 15 years before predicted age of onset [10], approximately the same time frame when increased levels of interleukin (IL)-6 are observed in the plasma [12]. Microglia activation in tissue specimens is typically characterized by increased numbers of microglia and morphological changes, in which the extensive cytoplasmic ramifications characteristic of resting microglia are retracted, resulting in an ameboid appearance. These morphological changes are associated with increased production of cytokines, such as IL-6. Intriguingly, the plasma level of IL-6 is correlated with disease severity based on a scale of functional capacity [12]. In patients’ striatum and cortex, reactive microglia accumulate in relation to the degree of neuronal loss [10]. Reactive microglia are clearly seen even in low-grade HD human brains, suggesting an early microglia response to changes in axons [10]. Interestingly, it has been reported that activated microglia proliferate at neurites of mutant HTT-expressing neurons in vitro. Significant microglia activation in regions related to cognitive function in HD patients has recently been suggested to predict disease onset [13].
The cerebrospinal fluid of HD patients exhibits evidence of immune activation, with upregulation of IL-6, IL-8 and tumor necrosis factor (TNF)-α [12]. Significant signs of oxidative stress have been reported in postmortem brain specimens from HD individuals. Early studies reported a decrease in several mitochondrial enzymes involved in respiration [14], as well as loss of aconitase activity in caudate and putamen in symptomatic patients with striatum atrophy [15]. Protein carbonyls, markers of oxidative stress, appear to be increased in HD brains, indicating that reactive oxygen species (ROS) are overproduced [16]. In addition, increased levels of 3-nitrotyrosine, a sign of reactive nitrogen species (RNS), have been observed in HD cortex and striatum [17]. By contrast, antioxidant defense proteins, such as peroxiredoxins 1, 2, and 6, as well as glutathione peroxidases 1 and 6, are strongly induced [16]. Furthermore, iron, a metal involved in mitochondria metabolism as well as in ROS generation, appears to accumulate in the brains of HD patients [18].
Patients with HD also show multiple systemic changes [19], including alterations in the function of the peripheral immune system. Increases in expression of genes that are produced by innate immune cells have been observed, such as the gene encoding immediate early response 3 mRNA (IER3) [20]. In addition, blood levels of several proteins produced by innate immune cells [21] correlate with disease progression [21]. In particular, a significant elevation of chemokines Chemokine (C-C motif) ligand (CCL)-2, CCL4, CCL11, CCL13, CCL26, matrix metalloprotease (MMP)-9, vascular endothelial growth factor (VEGF), and transforming growth factor (TGF)-1β have been detected in the plasma from HD patients [22,23]. By contrast, plasma levels of IL-18 were significantly reduced in HD patients in comparison with controls [22]. Also the level of thioredoxin reductase-1 and thioredoxin-1 appeared to be decreased in plasma and erythrocytes from HD individuals [24]. Of note, it has been reported that the active form of signal transducer and activator of transcription (STAT)-5, a transcription factor commonly used by several cytokines, is increased in monocytes from HD patients at baseline [25]. Collectively, these observations suggest that expression of mutant HTT in peripheral immune cells results in cell-autonomous effects on their gene expression patterns and function. Whether these alterations contribute to disease severity is unknown at present, but serum levels of cytokines could potentially serve as biomarkers to assess efficacy of anti-inflammatory interventions.
The most interesting biomarker for HD-associated inflammation may be mutant HTT itself. In monocytes and T cells, mutant HTT levels were significantly associated with disease burden score and caudate atrophy rates in HD patients [26]. However, no infiltration of circulating innate or adaptive immune cells has been reported in postmortem HD brain samples [5]. At present, only two studies have reported the presence of autoantibodies in HD. In the first report, autoantibodies directed against gliadin, a protein component of gluten, were observed in 44.2% of HD patients [27]. The second report showed alteration in the titers of an antibody to angiotensin II type 1 receptors (AT1R); this antibody promoting dysfunction of the adaptive immune system [28]. MS patients show increased titers of anti-AT1R in comparison with matching controls. HD individuals showed the presence of anti-AT1R more frequently than in controls and in MS patients as well. In 37.9% of HD patients, titers were >20 U/ml. The presence of autoantibodies suggests the possibility of a failure in immune tolerance in HD. Further studies are needed to determine whether dysfunction in the adaptive immune compartment is present in HD.
The role of microglia in HD neuroinflammation
Microglia, accounting for less then 10% of the total brain cells [29], represent the major population of resident immune cells of the CNS [30]. In healthy brains, microglia are characterized by a small cell body and ramified processes. Such ‘patrolling-mode’ [31] microglia contribute to brain homeostasis through phagocytosis, scavenging activity, secretion of homeostatic factors, such as TGFβ, and synaptic pruning [30,32]. In response to infection or tissue damage, microglia rapidly alter their morphology to an ‘ameboid’ appearance, increase phagocytic activity, and initiate an innate immune response by secreting various inflammatory molecules, including IL-6, and TNFα [30,32]. Once the inciting stimuli have been eradicated, microglia take part in the regenerative activity of the damaged tissue. When the inciting insult cannot be eradicated, persistent expression of mediators, such as IL-6, IL-8, and TNFα, drive chronic inflammation, and can contribute to tissue damage and disease progression. It has been shown that substances released from necrotic cells within the brain trigger microglia activation, leading to consequent changes of gene expression and reorganization of the cell activity (reviewed in [33]). Microglia are thus implicated in several acute and chronic neurological disorders.
Several lines of evidence indicate altered microglia activation states in the context of HD. Using a genome-wide approach, our group provided evidence that the expression of mutant HTT solely in microglia is sufficient to confer a cell-autonomous increase in pro-inflammatory gene expression [34]. In particular, expression of mutant HTT in microglia increased the expression and transcriptional activities of the myeloid lineage-determining factors PU.1 and CCAAT/enhancer-binding protein (C/EBP)-α,β, compared with normal microglia. Binding sites for PU.1 and C/EBPs were highly enriched in enhancers and promoters associated with the genes exhibiting constitutive upregulation in mutant HTT-expressing microglia. Expression of PU.1 itself was increased at both the mRNA and protein levels in these cells, and ChIP-Seq analysis demonstrated enhanced binding of PU.1 at thousands of genomic locations. Increased binding of PU.1 was associated with enhanced co-occupancy by C/EBPs, and the combination of enhanced PU.1 and C/EBP binding was highly correlated with increased expression of nearby genes, such as Il6 and Tnfα (Figure 1). These findings therefore reveal a molecular mechanism resulting in both moderate increases in basal gene expression and enhanced responses to exogenous stimuli. This mechanism may thus explain ‘priming’ as a state of enhanced basal pro-inflammatory activation that has been defined recently based on morphological evidence [35]. Intriguingly, effects of mutant HTT on PU.1-dependent programs of gene expression appeared to be microglia specific, because we found no differences in PU.1 levels or pro-inflammatory gene expression in bone marrow-derived macrophages (BMDMs) obtained from R6/2 and HdhzQ175/175 mice (expressing human mutant HTT exon 1, and 175CAG triplet repeats inserted in the mouse endogenous Htt gene, respectively) and monocytes from HD individuals [34]. The basis for specific effects of mutant HTT on microglia gene expression are unclear, but recent lineage tracing experiments provide evidence that microglia are derived from fetal yolk sac progenitors very early in development and represent a self-renewing population of cells that is independent of BMDMs [36,37]. Furthermore, the brain environment confers to microglia a distinct phenotype that differentiates these cells from other circulating myeloid cells [38,39].
Figure 1.
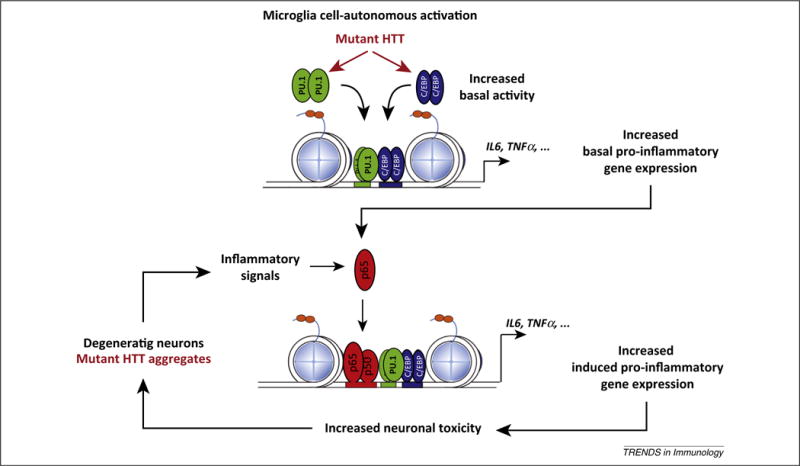
A model for mutant Huntingtin (HTT) microglia cell-autonomous activation and reactive microglia responses to neurodegeneration. In the presence of mutant HTT, increasing PU.1 expression and PU.1- CCAAT/enhancer-binding protein (C/EBP) promoter binding leads to increased enhancer activity under basal conditions that results in increased expression of basal pro-inflammatory and neurotoxic genes. This phenomenon increases the sensitivity to pro-inflammatory signals. In fact, under conditions of sterile inflammation, mutant HTT-expressing microglia appear to be more efficient in inducing neuronal death. We hypothesize that components of dead neurons or mutant HTT aggregates could trigger sterile inflammation, and this, in turn, could lead to further microglia activation, resulting in increased neuronal death and the activation of a chronic ‘feed-forward loop’. Adapted from [34]. Abbreviations: IL, interleukin; TNF, tumor necrosis factor.
The presence of a neuroinflammatory component in HD as well as in AD and PD has prompted the study of cannabinoid receptors and their agonists as potential immunomodulators to counteract microglia activation. Mouse microglia specifically expresses cannabinoid receptor 2 (CB2) and genetic ablation of this receptor in R6/2 mice enhanced microglia activation, aggravated the disease symptomatology, and reduced mice lifespan [40]. Administration of CB2 receptor-selective agonists to a chemical mouse model of HD reduced inflammation, brain edema, striatal neuronal loss, and motor symptoms [40]. Similar observations have been reported in regard to CB2 receptor signaling in peripheral immune cells [41]. Pharmacological treatments targeting the CB2 receptor have been proven protective in malonate-lesioned rats model of HD [42,43]. Recently, it has been shown that CB2 is not expressed on human microglia or astrocytes, and instead appears to be localized on CD31-positive blood vessel endothelium and vascular smooth muscle in postmortem samples from HD individuals [44]. Also, no significant difference was measured in CB2 expression in HD brains compared with controls [44]. This discrepancy between mouse and human microglia challenges the role of cannabinoid receptors as potential modulators of microglia activity in HD pathogenesis.
In the striatum of HD individuals, neurons, astrocytes, and myelin show deposition of C1q, C4, and C3, iC3b and C9 on their surface [45]. The classical complement pathway components (i.e., C1q C chain, C1r, C3, and C4), as well as complement regulators [i.e., C1 inhibitor, methyl-accepting chemotaxis protein (MCP), decay-accelerating factor (DAF) and CD59] are expressed at higher levels in HD brains compared with controls [45]. Recently, to test the hypothesis that increased levels of complement in HD brains could contribute to disease progression, mice deficient in complement C3, a key component in the complement pathway, were crossed with the R6/2 mice [46]. No alterations in multiple behavioral assays, weight, or survival in R6/2 mice lacking C3 were observed [46], demonstrating that C3 deficiency does not alter disease progression in the R6/2 model of HD. However, this observation does not completely rule out the role of the complement system in HD. The classical complement cascade is implicated in microglia-mediated synaptic pruning [47,48]. Synaptic pruning is the process of synapses elimination that occurs during development to allow the formation of mature neuronal circuits. In this process are involved complement proteins, such as C1q and/or C3, which target inappropriate synaptic connections that require a selective elimination [47,49]. In the context of HD, excessive complement protein production could lead to the elimination of useful synapses during development, and this in turn could impact the proper functionality of the brain in adult life.
The presence of mutant HTT is a trigger of oxidative stress. Mutant HTT aggregates have been shown to cause free radical production [50] in neuronal and non-neuronal cells [51]. Interestingly, mutant HTT can be oxidized itself [52]. Furthermore, mutant HTT-induced defects in mitochondria metabolism would increase the generation of ROS; ROS and RNS are the ‘ultimate weapons’ of pro-inflammatory activated innate immune cells [53]. When the inflammatory response is not tightly regulated and becomes chronic, oxidative stress can affect endogenous DNA, lipids, and proteins [54]. These phenomena have been observed in HD [55], in particular in neurons that are highly susceptible to oxidative stress because of their oxygen consumption rate, their dependence on aerobic carbohydrate metabolism, and the high polyunsaturated fatty acid content of their membranes. On the same line of evidence, abnormal accumulation of iron has been shown in HD brains [18]. Iron is required for proper mitochondrial function, but an excess of it can lead to uncontrollable generation of ROS. HTT is an iron-regulated protein [56], and mutant HTT inclusions are iron-dependent centers of oxidative stress [57].
Several lines of evidence suggest that the overexpression of antioxidant genes that have been observed in HD are an adaptive response to cope with the unbalanced generation of ROS [16]. The iron storage protein ferritin has an antioxidant activity. Ferritin accumulates in microglia cells from R6/2 mice where mutant Htt forms aggregates, and this process increases with disease progression [58]. Accumulation of ferritin seems to be a protective mechanism to counteract the accumulation of iron. In support of this hypothesis is the observation that a mutation in ferritin causes adult-onset basal ganglia disease with extrapyramidal features, such as chorea and dystonia, which are similar to the movement disorders characteristic of HD [59]. Peroxiredoxins 1 and 6 are two other antioxidant proteins highly expressed in HD. In normal conditions, they are mainly expressed in astrocytes and microglia [60]. Their increased levels observed in HD could be due to the increased proliferation of astrocytes [8] or to the increased gene expression observed in microglia [34]. Finally, it has been recently reported that glutathione peroxidase activity is neuroprotective in animal models of HD [61].
Recent findings have revealed involvement of the kynurenine pathway in HD, which in turn may also be linked to oxidative stress. The kynurenine pathway for the metabolism of L-tryptophan produces several molecules with neuroactive properties [62]. Kynurenic acid has a neuroprotective effect through modulation of mitochondrial function [63], while 3-hydroxykynurenine (3-HK), a generator of free radicals, and quinolinic acid, an agonist of NMDA receptors, are neurotoxic. Interestingly, mutant HTT toxicity has been found to be suppressed by the genetic deletion of kynurenine 3-monooxygenase (KMO), the enzyme that converts kynurenine into 3-HK, in a yeast suppressor screen [64]. In the brain, KMO is mainly expressed by microglia but not by neurons [65]. A successive study showed that microglia from the R6/2 mice produced an increased level of neurotoxic metabolites synthesized by the kynurenine pathway. This increase can be reduced by treatment with histone deacetylase (HDAC) inhibitors [66]. These drugs have a broad effect in modulating gene expression by inhibiting deacetylation of histones and, as a consequence, alter chromatin accessibility by transcription factors. This observation supports the use of HDAC inhibitors as potential therapeutic agents to treat HD [67].
All these data taken together suggest that cell-autonomous pro-inflammatory activation of microglia due to the expression of mutant HTT could contribute to the progression of HD pathogenesis. A schematic summary of cellular and molecular features of microglia and astrocytes involved in neuroinflammation in HD is presented in Figure 2.
Figure 2.
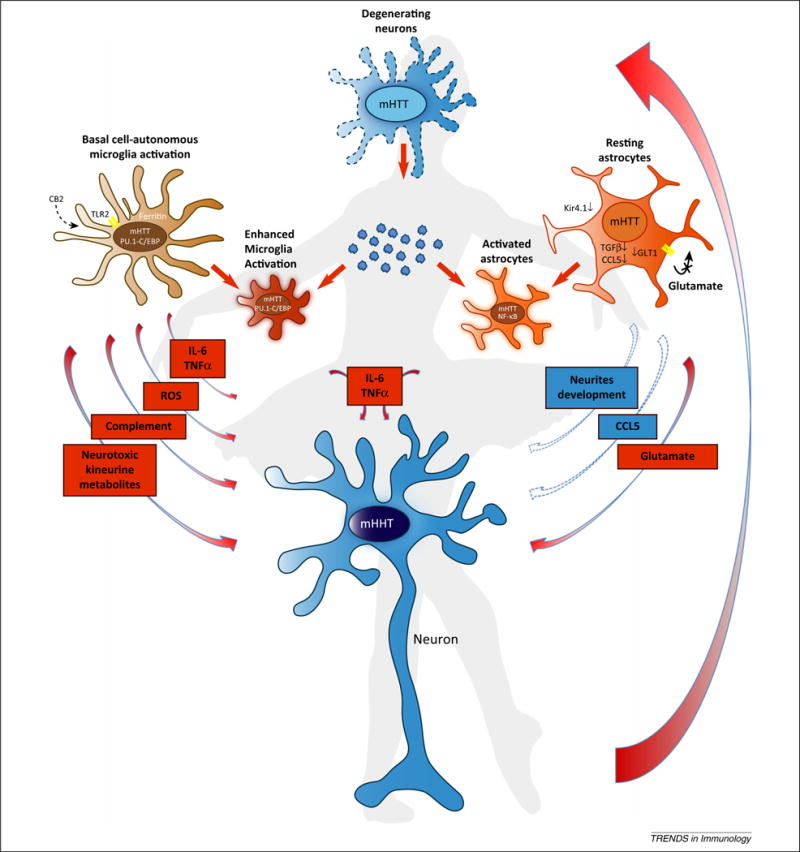
The choreography of neuroinflammation in Huntington’s disease. Expression of mutant Huntingtin (mHTT) in neurons triggers cell-autonomous neuronal degeneration and apoptosis (top center). In parallel, expression of mHTT in microglia triggers cells autonomous pro-inflammatory activation characterized by the release of pro-inflammatory cytokines, reactive oxygen species (ROS), and neurotoxic metabolites (center left). At the same time, expression of mHTT in astrocytes induces the cell-autonomous repression of several factors involved in the support of neurons wellbeing, such as, for example, Chemokine (C-C motif) ligand (CCL)-5, transforming growth factor (TGF)-β, and so on (center right). With the progression of neurodegeneration, endogenous molecules, such as components of dead neurons, protein aggregates, and potentially extracellular mHTT could be detected as DAMPs, inducing an innate immune response similar to what happens when amyloid (A)-β is phagocytosed by macrophages (center). Thus, enhanced pro-inflammatory microglia activation supported by pro-inflammatory cytokine-triggered astrocyte activation will result in further damage to neurons and the activation of a chronic ‘feed-forward loop’ of neurodegeneration. Abbreviations: IL, interleukin; NF, nuclear factor; TNF, tumor necrosis factor.
The role of astrocytes in HD neuroinflammation
Astrocytes constitute 90% of cells in the brain and are in charge of several different tasks [68]. First, astrocytes provide physical support to neurons and microglia by forming a matrix-type structure. This matrix is required to isolate synapses and limit the dispersion of neurotransmitters. Astrocytes are also required to remove toxic materials, such as for example an excess of extracellular glutamate. Furthermore, astrocytes provide neurons, microglia, and endothelial cells with factors necessary for proper function and help maintain the extracellular ion balance [69]. Finally, astrocytes provide neurons with nourishment in form of lactate [69]. In the presence of an insult, astrocytes amplify the inflammatory process initiated by microglia [70], clean up debris [71,72], and take part in repairing the damaged tissue [68].
Several reports indicate that mutant HTT-expressing astrocytes are defective in performing functions that support neuronal wellbeing. Expression of glutamate transporter GLT1 was lower in astrocytes from R6/2 mice compared with nontransgenic controls [73], and a subsequent report showed that mutant HTT suppresses glutamate uptake in primary astrocytes. These phenomena contribute to neuronal excitotoxicity observed in HD [74]. In fact, striatal neurons are connected by glutamatergic neurons from the cortex, and are for this reason particularly susceptible to an excess of extracellular glutamate [75]. Transgenic mice expressing mutant Htt exclusively in astrocytes present with evident body weight loss, motor function deficits, and shorter lifespans compared with nontransgenic littermates [76]. Furthermore, it has been reported that mutant HTT-expressing astrocytes reduce the availability of CCL5 to neurons [77]. On the one hand, mutant HTT reduced the transcription of the CCL5 gene in an nuclear factor (NF)-κB-dependent manner (in microglia? astrocytes?); on the other hand, it prevents the secretion of CCL5 protein by astrocytes in basal conditions. The lack of CCL5 could produce detrimental effects on the functionality of neurons [78,79]. A recent study reported that the onset of the symptoms in R6/2 and HdhzQ175/175 HD mouse models was not associated with classical astrogliosis, but rather with decreased expression of the potassium channel Kir4.1 [80]. This phenomenon led to elevated in vivo striatal extracellular potassium, which increased medium-spiny projection neurons (MSNs) excitability in vitro [80]. Viral delivery of Kir4.1 channels in the striatum ameliorated MSNs dysfunction, attenuated motor phenotypes, and prolonged the survival of R6/2 mice [80].
Astrocytes expressing mutant HTT are more prone to support pro-inflammatory activation. In the presence of an inflammatory stimulation, such as lipopolysaccharide (LPS), an enhanced activation of p65 was observed in astrocytes from R6/2 mice compared with astrocytes from nontransgenic littermates [81]. Primary R6/2 astrocytes exhibited higher IkB kinase (IKK) activity, resulting in prolonged NF-κB activation and the expression of pro-inflammatory factors, such as TNFα and IL-1β. R6/2 astrocytes also produced more damage on primary R6/2 neurons in comparison to control astrocytes during inflammation in vitro. The role of TNFα in HD pathogenesis was examined using the dominant negative inhibitor XPro1595 [82]. XPro1595 is a selective inhibitor of soluble TNFα that interferes with its binding to TNFα receptors. Intracranial infusion of XPro1595 decreased the elevated levels of TNFα in the cortex and striatum, improved motor function, and reduced caspase activation in R6/2 mice in vivo. Furthermore, a diminished amount of mutant Htt aggregates, an increased neuronal density, and decreased gliosis in brains of R6/2 mice were observed after XPro1595 infusion. In partial contrast with these observations is the fact that striatal injection of adeno-associated virus (AAV)-encoding dominant negative TNF did not rescue MSNs in the YAC128 model of HD (i.e., containing a yeast artificial chromosome expressing the full-length human HTT gene containing 128 CAG repeat expansion in exon-1) [83]. Using a lentivirus system to deliver control or mutant Htt N terminal proteins to all cells of the brain, the Janus kinase (JAK)/STAT3 pathway was found to play a prominent role in astrocyte activation in mice and monkeys [84] JAK/STAT3 signaling is commonly triggered by IL-6, a cytokine produced in HD brains by microglia [5]. These findings are consistent with the possibility that astrocytes amplify pro-inflammatory signaling initiated by microglia.
Monocytes and macrophages in HD neuroinflammation
Monocytes and monocyte-derived macrophages obtained from blood collected from HD patients express mutant HTT but do not show increased pro-inflammatory cytokines expression and/or production compared with monocytes obtained from healthy donors [12,34,85]. However, these cells are hyperactive in response to an external pro-inflammatory stimulation [12,85]. Upon LPS stimulation in vitro, mutant HTT in myeloid cells binds IKKγ and results in increased NF-κB activity, likely explaining the altered transcription of NF-κB target genes, and RNAi of HTT reverses the effects associated with NF-κB dysregulation [85]. Monocytes from HD individuals display phenotypic heterogeneity in terms of M1–M2 polarization throughout the clinical course of the disease [86]. Pre-HD subjects and early-stage HD patients showed a preferential pro-inflammatory M1 phenotype. Intriguingly, macrophages from HD patients in the late stage of the disease displayed changes in the expression of surface markers in favor of an anti-inflammatory M2 phenotype [86]. Furthermore, NF-κB–p65 protein expression is increased in macrophages from pre-HD subjects [86]. This predominant M1 activation observed in pre-HD subjects and early-stage HD patients is associated with a reduction of the number of TGFβ-producing cells, which is likely the explanation of the reduced level of this anti-inflammatory cytokine in the patients’ serum [86].
Mutant HTT is also associated with defective macrophage migration. Macrophages isolated from the BACHD mouse model of HD (i.e., containing a bacterial artificial chromosome expressing the full-length human HTT gene containing 97 mixed CAA-CAG repeat expansion in exon 1) exhibit impaired migration to an inflammatory stimulus, and this defect was also seen in blood monocytes from HD patients [87]. It is possible that the observed defect in migration of mutant HTT-expressing macrophages could help explain the absence of innate immune cells infiltration detected in HD brains. This same group recently reported that CD11b+ bone marrow (BM) myeloid cells as well as BMDM from R6/2, HdhQ150, and YAC128 HD mice fail to show increased pro-inflammatory cytokine production or phagocytosis before as well as after pro-inflammatory stimulation in vitro [88]. Instead, blood-derived CD11b+ myeloid cells showed increased pro-inflammatory cytokines secretion after a substantial pro-inflammatory stimulation, such as interferon (IFN)-γ priming and stimulation with LPS for 24 h. We also observed a lack of differential pro-inflammatory gene expression between BMDM from R6/2 and HdhzQ175/175 mice and nontransgenic littermates [34]. These observations show that peripheral immune cells expressing mutant HTT are defective rather than hyperactive. Thus, they seem to be poorly involved in triggering inflammatory processes in the CNS in HD.
Impact of neuroinflammation on HD pathogenesis
Striatal microglia in YAC128 mice show age-dependent changes in morphology, including a decrease in microglia ramifications that is associated with a reduction in blood vessel diameter and an increase in density [35]. Based on the idea that systemic challenge with LPS is known to induce a pro-inflammatory activation of immune cells in the periphery as well as in the CNS [89], YAC128 mice were challenged with peripheral injection of LPS [35]. Although LPS caused an increase in microglia activation, the phenotypic hallmarks of HD in YAC128 mice, such as motor coordination deficits and decreased striatal volume, were not exacerbated by chronic peripheral LPS exposure [35]. In another study, wild type (WT) BM cells were transplanted into two lethally irradiated transgenic mouse models of HD that ubiquitously express mutant Htt [90]. BM transplantation resulted in increased levels of synapses in the cortex, but conferred only modest benefits in terms of hypokinetic and motor deficits. This cannot be attributed to a decreased efficiency of the transplantation, because reconstitution of HD mice was comparable and/or superior to that of WT mice. Importantly, transplantation of HD BM into WT mice was stated to not cause behavioral or neuropathological deficits [90], but these data were not shown and it is unclear whether the efficiency of transplantation in this experiment was comparable between the two genotypes. In addition, this result could be partially explained by the fact that macrophages from mouse models of HD showed defective migratory capacities [87]. Thus, it is possible that the transplanted cells efficiently reconstituted the recipient mice, but did not reach the brain. Finally, it has been observed that neither CD11b+ BM myeloid cells nor BMDM of three different HD mice models show increased cytokines production or phagocytosis after strong pro-inflammatory stimulation [88]. These observations provide additional evidence that peripheral immune cells expressing mutant HTT are not major contributors to HD neuroinflammation.
Microglia and macrophages represent two different myeloid cell populations, due to their different origins and environmental influences [36,38,39]. For these reasons, we considered the possibility that sterile inflammation specifically in the brain could be triggered in HD patients by endogenous molecules, such as components of dead neurons and protein aggregates, referred to as damage-associated molecular patterns (DAMPs). In addition, it is possible that extracellular mutant HTT could be detected as a DAMP, inducing an immune response similar to what happens to amyloid (A)-β when phagocytosed by macrophages [91]. In fact, it has been shown that polyQ peptides can be phagocytosed by Cos7 and neuronal PC-12 cells in culture [92]. A recent study reported the presence of mutant HTT aggregates within intracerebral allografts of striatal tissue in three HD individuals who received transplants approximately a decade earlier and subsequently died secondary to the progression of their disease [93]. The mutant HTT aggregates were observed in the extracellular matrix of the transplanted tissue, while in the host brain they were seen in neurons, neuropil, extracellular matrix, and blood vessels. Furthermore, transneuronal propagation of mutant HTT has been observed ex vivo and in vivo in three different neural network models [94]. Given these findings, it is plausible that extracellular mutant HTT fragments could trigger sterile inflammation, which in turn could lead to a further microglia activation resulting in an increased neuronal death and the activation a chronic ‘feed forward loop’ (Figures 1 and 2). Consistent with this possibility, stereotactic injection of LPS (as an inducer of sterile inflammation) into the striatum of mice expressing mutant Htt exclusively in microglia in the CNS resulted in enhanced incidence of neuronal death in comparison to nontransgenic littermates [34]. These results support a role of mutant HTT-expressing microglia in initiating and/or amplifying endogenous pro-inflammatory signals that result in neurotoxicity.
Concluding remarks
There is now substantial evidence that expression of mutant HTT protein results in a cell-autonomous pro-inflammatory state of activation of microglia and, to a certain extent, of astrocytes. Furthermore, astrocytes, whose main role is to support and provide nutrients to neurons, appear to be defective in HD [80]. By contrast, monocytes and macrophages expressing mutant HTT do not show differences in pro-inflammatory cytokines production under basal conditions, but seem to be hyperactive after external pro-inflammatory stimulation [12,85]. Furthermore, macrophages from mouse models of HD as well as monocytes from HD individuals exhibit defective migratory capacities [87]. Thus, monocytes and macrophages appear defective in HD.
Systemic challenge of peripheral immune cells via LPS injection in HD mice as well as transplantation of mutant Htt-expressing BMDM into WT mice did not cause behavioral or neuropathological deficits in comparison to control animals [90]. BM transplantation into a mouse model of HD induced increased levels of synapses in the cortex, but conferred only modest benefits [90]. Instead, transgenic mice expressing mutant Htt exclusively in astrocytes show several symptoms of HD with time, demonstrating that mutant HTT expression in non-neuronal cells is sufficient to cause neurological symptoms [76]. Furthermore, mice that express mutant Htt exclusively in microglia showed an enhanced neuronal death in the presence of sterile inflammation. These results suggest a potential contribution of mutant HTT-expressing brain immune cells activation and neuroinflammation to the development of HD pathogenesis. Of course, neuroinflammation and microglia activation are not the primary cause of HD. In fact, recent studies with BAC HD mice showed marked amelioration of disease with reduction of mutant Htt in both cortical and striatal neurons, without affecting its expression in microglia [95]. The selective induction and/or reduction of mutant HTT expression in specific brain cells populations, such as cortical neurons [95,96], microglia [34], and astrocytes [76], have helped define the non-cell-autonomous mechanisms as well as some of the basis for selective neuronal vulnerability underling HD pathogenesis. All of these observations support the notion that cell–cell interactions are necessary for the generation of the striatal pathogenesis typical of HD. Furthermore, these results support once more the previously defined concept that both cell-autonomous toxicity and cell–cell interactions are necessary to define HD pathogenesis [96].
Similarly to the mutation in HTT protein that confers cell-autonomous pro-inflammatory activation to microglia in the context of HD, two different mutations have recently been linked to cell-autonomous roles of microglia activation in the context of AD. The R47H substitution in triggering receptor expressed on myeloid cells 2 (TREM2) has been associated with late-onset AD [97]. TREM2 is a receptor expressed on the microglia surface and it is involved in the clearance of neural debris [98]. TREM2 signaling creates an anti-inflammatory cytokine environment, as well as mediates the clearance of apoptotic neural tissue [98]. It has been shown that the R47H substitution leads to an increased predisposition to AD through impaired cell surface transport and phagocytosis [97,99]. Recently, it has been reported that TREM2 deficiency augments Aβ accumulation due to a dysfunctional response of microglia [100]. Furthermore, the R47H mutation was shown to impair TREM2 detection of lipid ligands [100]. These results suggest that chronic inflammation coupled with dysfunction in the microglia clearance of Aβ neurodegeneration in TREM2-associated AD.
Natural genetic variants in the CD33 gene have been identified as additional risk factors for AD [101]. AD brains have increased CD33 and CD33+ microglia [101]. Mice lacking CD33 have less AD pathology, suggesting a role of microglia for Aβ clearance [101]. Recently, it has been shown that the minor allele of the CD33 single nucleotide polymorphism (SNP) rs3865444 confers protection against AD [101]. This SNP was associated with reductions in both CD33 expression and insoluble Aβ42 levels in AD brain [101]. These observations suggest the idea that suppressing CD33-mediated microglia pro-inflammatory activation could represent a novel therapeutic approach for AD. A major difference between AD and HD is the fact that, in the first, the role of neuroinflammation is still controversial [102,103]. In fact, in AD, neuroinflammation has been described as impairing as well as promoting the progression of the disease [102,103]. Instead, in HD, all the available data point toward a deleterious contribution of neuroinflammation to the disease development in all stages of progression and a worsening of this situation with aging [8–11].
From this review of the literature emerges the concept that neuroinflammation is a strong component of HD pathogenesis. The expression of mutant HTT alters specific processes in microglia and astrocytes, such as transcription, cell signaling, cytokine release, and migration. Altered functions of microglia and astrocytes may lead to a chronic pathogenic inflammatory response that contributes to the death of additional neurons, and, as consequence, more neuroinflammation, in a ‘feed-forward loop’. Also, the activities of microglia are influenced by astrocytes and neurons and vice versa through cell–cell interactions, cytokines, and neurotransmitter release, respectively. A challenge for the future will be to shed light on the complex interplay between neurons, microglia, and astrocytes, which could be defined as the ‘choreography of neuroinflammation’ (Figure 2). The increasingly well-established roles of the single ‘dancers’ in the pathogenesis of HD may also prove instructive for understanding the roles of microglia and astrocytes in other neurodegenerative disorders, such as PD and AD (Box 1).
Box 1. Focus on HD.
HD is an inherited neurodegenerative disorder characterized by abnormal involuntary jerking movements similar to a dance (chorea), muscle problems, such as rigidity and contracture (dystonia), cognitive impairment, and emotional disturbance. The cause of HD is a single mutation, a specific expansion of the CAG stretch in the HTT gene that translates into an expanded poly glutamine tract in the HTT protein. Despite being ubiquitously expressed throughout the body, mutant HTT affects specifically a population of inhibitory neurons called MSNs localized in the central part of the brain called the striatum (composed of the caudate nucleus and putamen). The second most-affected area of the brain in HD is the cortex, the external part of the brain that projects into the striatum. One of the main mechanisms by which mutant HTT affects neurons function and viability is called excitotoxicity. This is a pathological process by which neurons are damaged or killed by excessive stimulation by excitatory neurotransmitters, such as glutamate. NMDA and AMPA receptors, when activated by pathologically high levels of glutamate, allow high levels of calcium ions to enter the cells. This in turn triggers the activation of several enzymes that damage cell components. Several murine models have been generated to study HD and these can be grouped in three main categories: (i) expressing N terminus human mutant Htt (R6/2, N-171); (ii) expressing full-length human mutant Htt (YAC 128, BAC HD); and (iii) knock-in models, where an expanded CAG stretch has been inserted in the endogenous mouse Htt gene (HdhQ150, HdhzQ175). For an exhaustive review on the topic, see [104].
Glossary
- Astrocyte
the most abundant type of glial cell in the brain. Astrocytes play numerous roles in supporting neuronal function and establishing the blood–brain barrier
- Chemokine
a group of small proteins within the broad category of cytokines that are released from cells and result in directed migration of nearby cells. Cytokines play important roles in attracting immune cells to sites of injury and infection
- Cytokine
a broad category of small proteins that are released from cells and exert biological effects on target cells. A subset of cytokines, including molecules such as TNFα and IL-6, are released from activated microglia and astrocytes and promote inflammatory responses
- Damage associated molecular pattern (DAMP)
this term refers to endogenously derived molecules that are recognized by pattern recognition receptors as indicators of tissue injury
- Gliosis
a histological term referring to an increase in the number of glial cells in the brain accompanied by morphological changes associated with glial activation
- Microglia
the major resident macrophage population in the brain. In addition to contributing to tissue homeostasis, microglia play primary roles in sensing injury and infection through expression of pattern recognition receptors
- Monocyte
a circulating innate immune cell that can migrate into tissues in response to infection or injury and differentiate into a macrophage
References
- 1.Hensley K. Neuroinflammation in Alzheimer’s disease: mechanisms, pathologic consequences, and potential for therapeutic manipulation. J Alzheimers Dis. 2010;21:1–14. doi: 10.3233/JAD-2010-1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aguzzi A, et al. Microglia: scapegoat, saboteur, or something else? Science. 2013;339:156–161. doi: 10.1126/science.1227901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Samii A, et al. Parkinson’s disease. Lancet. 2004;363:1783–1793. doi: 10.1016/S0140-6736(04)16305-8. [DOI] [PubMed] [Google Scholar]
- 4.Schellenberg GD, Montine TJ. The genetics and neuropathology of Alzheimer’s disease. Acta Neuropathol. 2012;124:305–323. doi: 10.1007/s00401-012-0996-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Silvestroni A, et al. Distinct neuroinflammatory profile in postmortem human Huntington’s disease. Neuroreport. 2009;20:1098–1103. doi: 10.1097/WNR.0b013e32832e34ee. [DOI] [PubMed] [Google Scholar]
- 6.MacDonald ME, et al. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell. 1993;72:971–983. doi: 10.1016/0092-8674(93)90585-e. [DOI] [PubMed] [Google Scholar]
- 7.Soulet D, Cicchetti F. The role of immunity in Huntington’s disease. Mol Psychiatry. 2011;16:889–902. doi: 10.1038/mp.2011.28. [DOI] [PubMed] [Google Scholar]
- 8.Rosas HD, et al. Altered white matter microstructure in the corpus callosum in Huntington’s disease: implications for cortical ‘disconnection’. Neuroimage. 2009;49:2995–3004. doi: 10.1016/j.neuroimage.2009.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pavese N, et al. Clinical correlates of levodopa-induced dopamine release in Parkinson disease: a PET study. Neurology. 2006;67:1612–1617. doi: 10.1212/01.wnl.0000242888.30755.5d. [DOI] [PubMed] [Google Scholar]
- 10.Tai YF, et al. Microglial activation in presymptomatic Huntington’s disease gene carriers. Brain. 2007;130:1759–1766. doi: 10.1093/brain/awm044. [DOI] [PubMed] [Google Scholar]
- 11.Tai YF, et al. Imaging microglial activation in Huntington’s disease. Brain Res Bull. 2007;72:148–151. doi: 10.1016/j.brainresbull.2006.10.029. [DOI] [PubMed] [Google Scholar]
- 12.Bjorkqvist M, et al. A novel pathogenic pathway of immune activation detectable before clinical onset in Huntington’s disease. J Exp Med. 2008;205:1869–1877. doi: 10.1084/jem.20080178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sapp E, et al. Early and progressive accumulation of reactive microglia in the Huntington disease brain. J Neuropathol Exp Neurol. 2001;60:161–172. doi: 10.1093/jnen/60.2.161. [DOI] [PubMed] [Google Scholar]
- 14.Brennan WA, Jr, et al. Regional mitochondrial respiratory activity in Huntington’s disease brain. J Neurochem. 1985;44:1948–1950. doi: 10.1111/j.1471-4159.1985.tb07192.x. [DOI] [PubMed] [Google Scholar]
- 15.Tabrizi SJ, et al. Biochemical abnormalities and excitotoxicity in Huntington’s disease brain. Ann Neurol. 1999;45:25–32. doi: 10.1002/1531-8249(199901)45:1<25::aid-art6>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 16.Sorolla MA, et al. Proteomic and oxidative stress analysis in human brain samples of Huntington disease. Free Radic Biol Med. 2008;45:667–678. doi: 10.1016/j.freeradbiomed.2008.05.014. [DOI] [PubMed] [Google Scholar]
- 17.Browne SE, et al. Oxidative damage and metabolic dysfunction in Huntington’s disease: selective vulnerability of the basal ganglia. Ann Neurol. 1997;41:646–653. doi: 10.1002/ana.410410514. [DOI] [PubMed] [Google Scholar]
- 18.van den Bogaard SJ, et al. The role of iron imaging in Huntington’s disease. Int Rev Neurobiol. 2013;110:241–250. doi: 10.1016/B978-0-12-410502-7.00011-9. [DOI] [PubMed] [Google Scholar]
- 19.van der Burg JM, et al. Beyond the brain: widespread pathology in Huntington’s disease. Lancet Neurol. 2009;8:765–774. doi: 10.1016/S1474-4422(09)70178-4. [DOI] [PubMed] [Google Scholar]
- 20.Runne H, et al. Analysis of potential transcriptomic biomarkers for Huntington’s disease in peripheral blood. Proc Natl Acad Sci USA. 2007;104:14424–14429. doi: 10.1073/pnas.0703652104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dalrymple A, et al. Proteomic profiling of plasma in Huntington’s disease reveals neuroinflammatory activation and biomarker candidates. J Proteome Res. 2007;6:2833–2840. doi: 10.1021/pr0700753. [DOI] [PubMed] [Google Scholar]
- 22.Chang KH, et al. Plasma inflammatory biomarkers for Huntington’s disease patients and mouse model. Brain Behav Immunity. 2015;44:121–127. doi: 10.1016/j.bbi.2014.09.011. [DOI] [PubMed] [Google Scholar]
- 23.Wild E, et al. Abnormal peripheral chemokine profile in Huntington’s disease. PLoS Curr. 2011;3:1231. doi: 10.1371/currents.RRN1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sanchez-Lopez F, et al. Oxidative stress and inflammation biomarkers in the blood of patients with Huntington’s disease. Neurol Res. 2012;34:721–724. doi: 10.1179/1743132812Y.0000000073. [DOI] [PubMed] [Google Scholar]
- 25.Trager U, et al. JAK/STAT signalling in Huntington’s disease immune cells. PLoS Curr. 2014;5:125–134. doi: 10.1371/currents.hd.5791c897b5c3bebeed93b1d1da0c0648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Weiss A, et al. Mutant huntingtin fragmentation in immune cells tracks Huntington’s disease progression. J Clin Invest. 2012;122:3731–3736. doi: 10.1172/JCI64565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bushara KO, et al. Antigliadin antibodies in Huntington’s disease. Neurology. 2004;62:132–133. doi: 10.1212/wnl.62.1.132. [DOI] [PubMed] [Google Scholar]
- 28.Lee DH, et al. Increase of angiotensin II type 1 receptor auto-antibodies in Huntington’s disease. Mol Neurodegener. 2014;9:49. doi: 10.1186/1750-1326-9-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lawson LJ, et al. Heterogeneity in the distribution and morphology of microglia in the normal adult mouse brain. Neuroscience. 1990;39:151–170. doi: 10.1016/0306-4522(90)90229-w. [DOI] [PubMed] [Google Scholar]
- 30.Ransohoff RM, Perry VH. Microglial physiology: unique stimuli, specialized responses. Annu Rev Immunol. 2009;27:119–145. doi: 10.1146/annurev.immunol.021908.132528. [DOI] [PubMed] [Google Scholar]
- 31.Nimmerjahn A, et al. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science. 2005;308:1314–1318. doi: 10.1126/science.1110647. [DOI] [PubMed] [Google Scholar]
- 32.Weinstein JR, et al. Microglia in ischemic brain injury. Future Neurol. 2010;5:227–246. doi: 10.2217/fnl.10.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hanisch UK, Kettenmann H. Microglia: active sensor and versatile effector cells in the normal and pathologic brain. Nat Neurosci. 2007;10:1387–1394. doi: 10.1038/nn1997. [DOI] [PubMed] [Google Scholar]
- 34.Crotti A, et al. Mutant Huntingtin promotes autonomous microglia activation via myeloid lineage-determining factors. Nat Neurosci. 2014;4:513–521. doi: 10.1038/nn.3668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Franciosi S, et al. Age-dependent neurovascular abnormalities and altered microglial morphology in the YAC128 mouse model of Huntington disease. Neurobiol Dis. 2011;45:438–449. doi: 10.1016/j.nbd.2011.09.003. [DOI] [PubMed] [Google Scholar]
- 36.Ginhoux F, et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science. 2010;330:841–845. doi: 10.1126/science.1194637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schulz C, et al. A lineage of myeloid cells independent of Myb and hematopoietic stem cells. Science. 2012;336:86–90. doi: 10.1126/science.1219179. [DOI] [PubMed] [Google Scholar]
- 38.Gosselin D, et al. Environment drives selection and function of enhancers controlling tissue-specific macrophage identities. Cell. 2014;159:1327–1340. doi: 10.1016/j.cell.2014.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lavin Y, et al. Tissue-resident macrophage enhancer landscapes are shaped by the local microenvironment. Cell. 2014;159:1312–1326. doi: 10.1016/j.cell.2014.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Palazuelos J, et al. Microglial CB2 cannabinoid receptors are neuroprotective in Huntington’s disease excitotoxicity. Brain. 2009;132:3152–3164. doi: 10.1093/brain/awp239. [DOI] [PubMed] [Google Scholar]
- 41.Bouchard J, et al. Cannabinoid receptor 2 signaling in peripheral immune cells modulates disease onset and severity in mouse models of Huntington’s disease. J Neurosci. 2012;32:18259–18268. doi: 10.1523/JNEUROSCI.4008-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sagredo O, et al. Cannabinoid CB2 receptor agonists protect the striatum against malonate toxicity: relevance for Huntington’s disease. Glia. 2009;57:1154–1167. doi: 10.1002/glia.20838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Valdeolivas S, et al. Sativex-like combination of phytocannabinoids is neuroprotective in malonate-lesioned rats, an inflammatory model of Huntington’s disease: role of CB1 and CB2 receptors. ACS Chem Neurosci. 2012;3:400–406. doi: 10.1021/cn200114w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dowie MJ, et al. Cannabinoid receptor CB2 is expressed on vascular cells, but not astroglial cells in the post-mortem human Huntington’s disease brain. J Chem Neuroanat. 2014:59–60. doi: 10.1016/j.jchemneu.2014.06.004. [DOI] [PubMed] [Google Scholar]
- 45.Singhrao SK, et al. Increased complement biosynthesis by microglia and complement activation on neurons in Huntington’s disease. Exp Neurol. 1999;159:362–376. doi: 10.1006/exnr.1999.7170. [DOI] [PubMed] [Google Scholar]
- 46.Larkin PB, Muchowski PJ. Genetic deficiency of complement Component 3 does not alter disease progression in a mouse model of Huntington’s disease. J Huntingtons Dis. 2012;1:107–118. doi: 10.3233/JHD-2012-120021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Stevens B, et al. The classical complement cascade mediates CNS synapse elimination. Cell. 2007;131:1164–1178. doi: 10.1016/j.cell.2007.10.036. [DOI] [PubMed] [Google Scholar]
- 48.Schafer DP, et al. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron. 2012;74:691–705. doi: 10.1016/j.neuron.2012.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chu Y, et al. Enhanced synaptic connectivity and epilepsy in C1q knockout mice. Proc Natl Acad Sci USA. 2010;107:7975–7980. doi: 10.1073/pnas.0913449107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hands S, et al. In vitro and in vivo aggregation of a fragment of huntingtin protein directly causes free radical production. J Biol Chem. 2011;286:44512–44520. doi: 10.1074/jbc.M111.307587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wyttenbach A, et al. Heat shock protein 27 prevents cellular polyglutamine toxicity and suppresses the increase of reactive oxygen species caused by huntingtin. Hum Mol Genet. 2002;11:1137–1151. doi: 10.1093/hmg/11.9.1137. [DOI] [PubMed] [Google Scholar]
- 52.Fox JH, et al. Cysteine oxidation within N-terminal mutant huntingtin promotes oligomerization and delays clearance of soluble protein. J Biol Chem. 2011;286:18320–18330. doi: 10.1074/jbc.M110.199448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Block ML, et al. Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat Rev Neurosci. 2007;8:57–69. doi: 10.1038/nrn2038. [DOI] [PubMed] [Google Scholar]
- 54.Barja G. Free radicals and aging. Trends Neurosci. 2004;27:595–600. doi: 10.1016/j.tins.2004.07.005. [DOI] [PubMed] [Google Scholar]
- 55.Shirendeb U, et al. Abnormal mitochondrial dynamics, mitochondrial loss and mutant huntingtin oligomers in Huntington’s disease: implications for selective neuronal damage. Hum Mol Genet. 2011;20:1438–1455. doi: 10.1093/hmg/ddr024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hilditch-Maguire P, et al. Huntingtin: an iron-regulated protein essential for normal nuclear and perinuclear organelles. Hum Mol Genet. 2000;9:2789–2797. doi: 10.1093/hmg/9.19.2789. [DOI] [PubMed] [Google Scholar]
- 57.Firdaus WJ, et al. Huntingtin inclusion bodies are iron-dependent centers of oxidative events. FEBS J. 2006;273:5428–5441. doi: 10.1111/j.1742-4658.2006.05537.x. [DOI] [PubMed] [Google Scholar]
- 58.Simmons DA, et al. Ferritin accumulation in dystrophic microglia is an early event in the development of Huntington’s disease. Glia. 2007;55:1074–1084. doi: 10.1002/glia.20526. [DOI] [PubMed] [Google Scholar]
- 59.Curtis AR, et al. Mutation in the gene encoding ferritin light polypeptide causes dominant adult-onset basal ganglia disease. Nat Genet. 2001;28:350–354. doi: 10.1038/ng571. [DOI] [PubMed] [Google Scholar]
- 60.Rhee SG, et al. Peroxiredoxins: a historical overview and speculative preview of novel mechanisms and emerging concepts in cell signaling. Free Radic Biol Med. 2005;38:1543–1552. doi: 10.1016/j.freeradbiomed.2005.02.026. [DOI] [PubMed] [Google Scholar]
- 61.Mason RP, et al. Glutathione peroxidase activity is neuroprotective in models of Huntington’s disease. Nat Genet. 2013;45:1249–1254. doi: 10.1038/ng.2732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Schwarcz R, et al. Of mice, rats and men: revisiting the quinolinic acid hypothesis of Huntington’s disease. Prog Neurobiol. 2009;90:230–245. doi: 10.1016/j.pneurobio.2009.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Beal MF, et al. Kynurenine pathway measurements in Huntington’s disease striatum: evidence for reduced formation of kynurenic acid. J Neurochem. 1990;55:1327–1339. doi: 10.1111/j.1471-4159.1990.tb03143.x. [DOI] [PubMed] [Google Scholar]
- 64.Giorgini F, et al. A genomic screen in yeast implicates kynurenine 3-monooxygenase as a therapeutic target for Huntington disease. Nat Genet. 2005;37:526–531. doi: 10.1038/ng1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Guillemin GJ, et al. Expression of the kynurenine pathway enzymes in human microglia and macrophages. Adv Exp Med Biol. 2003;527:105–112. doi: 10.1007/978-1-4615-0135-0_12. [DOI] [PubMed] [Google Scholar]
- 66.Giorgini F, et al. Histone deacetylase inhibition modulates kynurenine pathway activation in yeast, microglia. and mice expressing a mutant huntingtin fragment. J Biol Chem. 2008;283:7390–7400. doi: 10.1074/jbc.M708192200. [DOI] [PubMed] [Google Scholar]
- 67.Jia H, et al. HDAC inhibition imparts beneficial transgenerational effects in Huntington’s disease mice via altered DNA and histone methylation. Proc Natl Acad Sci USA. 2015;112:E56–E64. doi: 10.1073/pnas.1415195112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sofroniew MV, Vinters HV. Astrocytes: biology and pathology. Acta Neuropathol. 2010;119:7–35. doi: 10.1007/s00401-009-0619-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chen Y, Swanson RA. Astrocytes and brain injury. J Cereb Blood Flow Metab. 2003;23:137–149. doi: 10.1097/01.WCB.0000044631.80210.3C. [DOI] [PubMed] [Google Scholar]
- 70.Saijo K, et al. A Nurr1/CoREST pathway in microglia and astrocytes protects dopaminergic neurons from inflammation-induced death. Cell. 2009;137:47–59. doi: 10.1016/j.cell.2009.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chung WS, et al. Astrocytes mediate synapse elimination through MEGF10 and MERTK pathways. Nature. 2013;504:394–400. doi: 10.1038/nature12776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Loov C, et al. Engulfing astrocytes protect neurons from contact-induced apoptosis following injury. PLoS ONE. 2012;7:e33090. doi: 10.1371/journal.pone.0033090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lievens JC, et al. Impaired glutamate uptake in the R6 Huntington’s disease transgenic mice. Neurobiol Dis. 2001;8:807–821. doi: 10.1006/nbdi.2001.0430. [DOI] [PubMed] [Google Scholar]
- 74.Shin JY, et al. Expression of mutant huntingtin in glial cells contributes to neuronal excitotoxicity. J Cell Biol. 2005;171:1001–1012. doi: 10.1083/jcb.200508072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Estrada-Sanchez AM, Rebec GV. Corticostriatal dysfunction and glutamate transporter 1 (GLT1) in Huntington’s disease: interactions between neurons and astrocytes. Basal Ganglia. 2012;2:57–66. doi: 10.1016/j.baga.2012.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bradford J, et al. Expression of mutant huntingtin in mouse brain astrocytes causes age-dependent neurological symptoms. Proc Natl Acad Sci USA. 2009;106:22480–22485. doi: 10.1073/pnas.0911503106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Chou SY, et al. Expanded-polyglutamine huntingtin protein suppresses the secretion and production of a chemokine (CCL5/RANTES) by astrocytes. J Neurosci. 2008;28:3277–3290. doi: 10.1523/JNEUROSCI.0116-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Spires TL, et al. Dendritic spine pathology and deficits in experience-dependent dendritic plasticity in R6/1 Huntington’s disease transgenic mice. Eur J Neurosci. 2004;19:2799–2807. doi: 10.1111/j.0953-816X.2004.03374.x. [DOI] [PubMed] [Google Scholar]
- 79.Ariano MA, et al. Striatal potassium channel dysfunction in Huntington’s disease transgenic mice. J Neurophysiol. 2005;93:2565–2574. doi: 10.1152/jn.00791.2004. [DOI] [PubMed] [Google Scholar]
- 80.Tong X, et al. Astrocyte Kir4.1 ion channel deficits contribute to neuronal dysfunction in Huntington’s disease model mice. Nat Neurosci. 2014;5:694–703. doi: 10.1038/nn.3691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hsiao HY, et al. A critical role of astrocyte-mediated nuclear factor-kappaB-dependent inflammation in Huntington’s disease. Hum Mol Genet. 2013;22:1826–1842. doi: 10.1093/hmg/ddt036. [DOI] [PubMed] [Google Scholar]
- 82.Hsiao HY, et al. Inhibition of soluble tumor necrosis factor is therapeutic in Huntington’s disease. Hum Mol Genet. 2014;23:4328–4344. doi: 10.1093/hmg/ddu151. [DOI] [PubMed] [Google Scholar]
- 83.Alto LT, et al. AAV-dominant negative tumor necrosis factor (DN-TNF) gene transfer to the striatum does not rescue medium spiny neurons in the YAC128 mouse model of Huntington’s disease. PLoS ONE. 2014;9:e96544. doi: 10.1371/journal.pone.0096544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ben Haim L, et al. The JAK/STAT3 pathway is a common inducer of astrocyte reactivity in Alzheimer’s and Huntington’s diseases. J Neurosci. 2015;35:2817–2829. doi: 10.1523/JNEUROSCI.3516-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Trager U, et al. HTT-lowering reverses Huntington’s disease immune dysfunction caused by NFkappaB pathway dysregulation. Brain. 2014;137:819–833. doi: 10.1093/brain/awt355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Di Pardo A, et al. Changes of peripheral TGF-beta1 depend on monocytes-derived macrophages in Huntington disease. Mol Brain. 2013;6:55. doi: 10.1186/1756-6606-6-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kwan W, et al. Mutant huntingtin impairs immune cell migration in Huntington disease. J Clin Invest. 2012;122:4737–4747. doi: 10.1172/JCI64484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Trager U, et al. Characterisation of immune cell function in fragment and full-length Huntington’s disease mouse models. Neurobiol Dis. 2014;73C:388–398. doi: 10.1016/j.nbd.2014.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Qin L, et al. Systemic LPS causes chronic neuroinflammation and progressive neurodegeneration. Glia. 2007;55:453–462. doi: 10.1002/glia.20467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kwan W, et al. Bone marrow transplantation confers modest benefits in mouse models of Huntington’s disease. J Neurosci. 2012;32:133–142. doi: 10.1523/JNEUROSCI.4846-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Morten IJ, et al. Investigation into the role of macrophages in the formation and degradation of beta2-microglobulin amyloid fibrils. J Biol Chem. 2007;282:29691–29700. doi: 10.1074/jbc.M705004200. [DOI] [PubMed] [Google Scholar]
- 92.Yang W, et al. Aggregated polyglutamine peptides delivered to nuclei are toxic to mammalian cells. Hum Mol Genet. 2002;11:2905–2917. doi: 10.1093/hmg/11.23.2905. [DOI] [PubMed] [Google Scholar]
- 93.Cicchetti F, et al. Mutant huntingtin is present in neuronal grafts in Huntington’s disease patients. Ann Neurol. 2014;1:31–42. doi: 10.1002/ana.24174. [DOI] [PubMed] [Google Scholar]
- 94.Pecho-Vrieseling E, et al. Transneuronal propagation of mutant huntingtin contributes to non-cell autonomous pathology in neurons. Nat Neurosci. 2014;17:1064–1072. doi: 10.1038/nn.3761. [DOI] [PubMed] [Google Scholar]
- 95.Wang N, et al. Neuronal targets for reducing mutant huntingtin expression to ameliorate disease in a mouse model of Huntington’s disease. Nat Med. 2014;20:536–541. doi: 10.1038/nm.3514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Gu X, et al. Pathological cell–cell interactions are necessary for striatal pathogenesis in a conditional mouse model of Huntington’s disease. Mol Neurodegener. 2007;2:8. doi: 10.1186/1750-1326-2-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Jonsson T, et al. Variant of TREM2 associated with the risk of Alzheimer’s disease. N Engl J Med. 2013;368:107–116. doi: 10.1056/NEJMoa1211103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Takahashi K, et al. Clearance of apoptotic neurons without inflammation by microglial triggering receptor expressed on myeloid cells-2. J Exp Med. 2005;201:647–657. doi: 10.1084/jem.20041611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Kleinberger G, et al. TREM2 mutations implicated in neurodegeneration impair cell surface transport and phagocytosis. Sci Transl Med. 2014;6:243ra86. doi: 10.1126/scitranslmed.3009093. [DOI] [PubMed] [Google Scholar]
- 100.Wang Y, et al. TREM2 lipid sensing sustains the microglial response in an Alzheimer’s disease model. Cell. 2015;160:1061–1071. doi: 10.1016/j.cell.2015.01.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Griciuc A, et al. Alzheimer’s disease risk gene CD33 inhibits microglial uptake of amyloid beta. Neuron. 2013;78:631–643. doi: 10.1016/j.neuron.2013.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Heneka MT, et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015;14:388–405. doi: 10.1016/S1474-4422(15)70016-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Mosher KI, Wyss-Coray T. Microglial dysfunction in brain aging and Alzheimer’s disease. Biochem Pharmacol. 2014;88:594–604. doi: 10.1016/j.bcp.2014.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Zuccato C, Cattaneo E. Huntington’s disease. Handb Exp Pharmacol. 2014;220:357–409. doi: 10.1007/978-3-642-45106-5_14. [DOI] [PubMed] [Google Scholar]