

Smoldering multiple myeloma (SMM) is an asymptomatic clonal plasma cell disorder and bridges monoclonal gammopathy of undetermined significance to multiple myeloma (MM). A subset of ultra-high-risk SMM patients (∼10%) have been reclassified by the International Myeloma Working Group as MM, with a substantial risk of progression to overt MM (≥80% within 2 years). However, for most SMM patients, the standard of care remains observation until development of symptomatic MM occurs.
Keywords: Smoldering multiple myeloma, Risk factors, Progression, Treatment
Abstract
Smoldering multiple myeloma (SMM) is an asymptomatic clonal plasma cell disorder and bridges monoclonal gammopathy of undetermined significance to multiple myeloma (MM), based on higher levels of circulating monoclonal immunoglobulin and bone marrow plasmocytosis without end-organ damage. Until a Spanish study reported fewer MM-related events and better overall survival among patients with high-risk SMM treated with lenalidomide and dexamethasone, prior studies had failed to show improved survival with earlier intervention, although a reduction in skeletal-related events (without any impact on disease progression) has been described with bisphosphonate use. Risk factors have now been defined, and a subset of ultra-high-risk patients have been reclassified by the International Myeloma Working Group as MM, and thus will require optimal MM treatment, based on biomarkers that identify patients with a >80% risk of progression. The number of these redefined patients is small (∼10%), but important to unravel, because their risk of progression to overt MM is substantial (≥80% within 2 years). Patients with a high-risk cytogenetic profile are not yet considered for early treatment, because groups are heterogeneous and risk factors other than cytogenetics are deemed to weight higher. Because patients with ultra-high-risk SMM are now considered as MM and may be treated as such, concerns exist that earlier therapy may increase the risk of selecting resistant clones and induce side effects and costs. Therefore, an even more accurate identification of patients who would benefit from interventions needs to be performed, and clinical judgment and careful discussion of pros and cons of treatment initiation need to be undertaken. For the majority of SMM patients, the standard of care remains observation until development of symptomatic MM occurs, encouraging participation in ongoing and upcoming SMM/early MM clinical trials, as well as consideration of bisphosphonate use in patients with early bone loss.
Implications for Practice:
Smoldering multiple myeloma is an early stage of myeloma disease and is diagnosed before any symptoms occur. Recent research has redefined the diagnostic criteria for multiple myeloma, offering new insights into testing and classification of this malignancy. Risk factors have now been defined and three biomarkers have been validated that are able to identify patients presenting a high risk of progression toward a symptomatic disease. These biomarkers will help physicians to identify high-risk patients who may benefit from optimal treatment. This article summarizes the views of a European panel of hematologists on the implicated changes in patient care.
Introduction
Multiple myeloma (MM) is a hematological malignancy characterized by the accumulation of monoclonal plasma cells (PCs) in the bone marrow (BM). Monoclonal PC proliferation is accompanied by the secretion of a monoclonal immunoglobulin that serves as a quantitative tumor marker and can be detected in blood and/or urine [1, 2].
In 1980, Kyle and Greipp were the first to describe smoldering MM (SMM), or asymptomatic MM, as an entity fulfilling the diagnostic criteria for MM, but with a different clinical outcome [3]. They based their findings on six patients who had a medullary PC infiltration ≥10% and a serum M-spike ≥3 g/dL, but whose disease did not progress during their follow-up.
In 2003, the International Myeloma Working Group (IMWG) reviewed the diagnostic criteria for symptomatic MM, asymptomatic or smoldering myeloma (SMM), and monoclonal gammopathy of undetermined significance (MGUS) [4]. The criteria are based on biological parameters and focus on the presence of clinical symptoms. In MM, these criteria involve the presence of a monoclonal protein in blood or urine, a BMPC infiltration ≥10% (or plasmacytoma), and the presence of complications, such as hypercalcemia, radiological bone lesions, anemia, and renal failure (often referred as CRAB). Conversely, MGUS is defined by a paraprotein level that is <3 g/dL, medullar PC infiltration of <10%, and the absence of clinical complications [5]. SMM corresponds to the clinical situation in which the paraprotein level is ≥3 g/dL or medullar PC infiltration ≥ 10%, but always in the absence of complications. In 2014, the IMWG revised the diagnostic criteria of SMM and MM, a topic that is currently being discussed intensively [6].
Progression to Symptomatic Disease
The majority of SMM patients will progress to symptomatic MM and will require treatment at that time. A retrospective study by Kyle et al., including 276 cases of SMM over a period of 26 years at the Mayo Clinic, revealed a prevalence of 8% on a total MM population of 3,549 patients [7]. The median time to progression varies in different patient series between 3 and 5 years. The main objective of this study was to determine the rate of progression to symptomatic diseases (such as MM or amyloid light chain [AL] amyloidosis). Unlike patients with MGUS, the majority of SMM patients developed symptomatic disease, but the risk of progression changed over time and was found to be higher in the early years after diagnosis and decreased after 5 years. The median time to progression was 4.8 years, and the risk of progression was estimated at 10% per year for the first 5 years, 3% for the next 5 years, and 1% thereafter. The cumulative progression rate was 51% at 5 years, 66% at 10 years, and 73% at 15 years. Recently, the Heidelberg group prospectively studied 248 cases of SMM (among 2,085 cases of MM) [8]: They found that 83 (33.5%) patients progressed to active MM and 5 (2%) patients to AL-amyloidosis. Median time of progression was calculated as 5.6 years, with a cumulative progression rate of 46% over 5 years, thus well in line with prior Mayo Clinic data [8]. The SWOG S0120 prospectively followed patients with MGUS and SMM [9]: The authors found that 49 of 179 SMM patients (24%) progressed to an active MM within 2 years from diagnosis, with osteolysis being the most prominent sign of progression [9].
The main challenge of the different prognostic studies has been the identification of patients at high risk of progression who could potentially benefit from an earlier therapeutic strategy. Within this high-risk patient group, a small subgroup of patients has been defined that present an ultra-high risk of progression to symptomatic MM (≥80% within 2 years). These ultra-high-risk SMM patients are now reclassified as MM and offered earlier treatment intervention—this being performed to prevent serious complications occurring during progression or even alter the natural course of MM [10].
Prognostic Factors
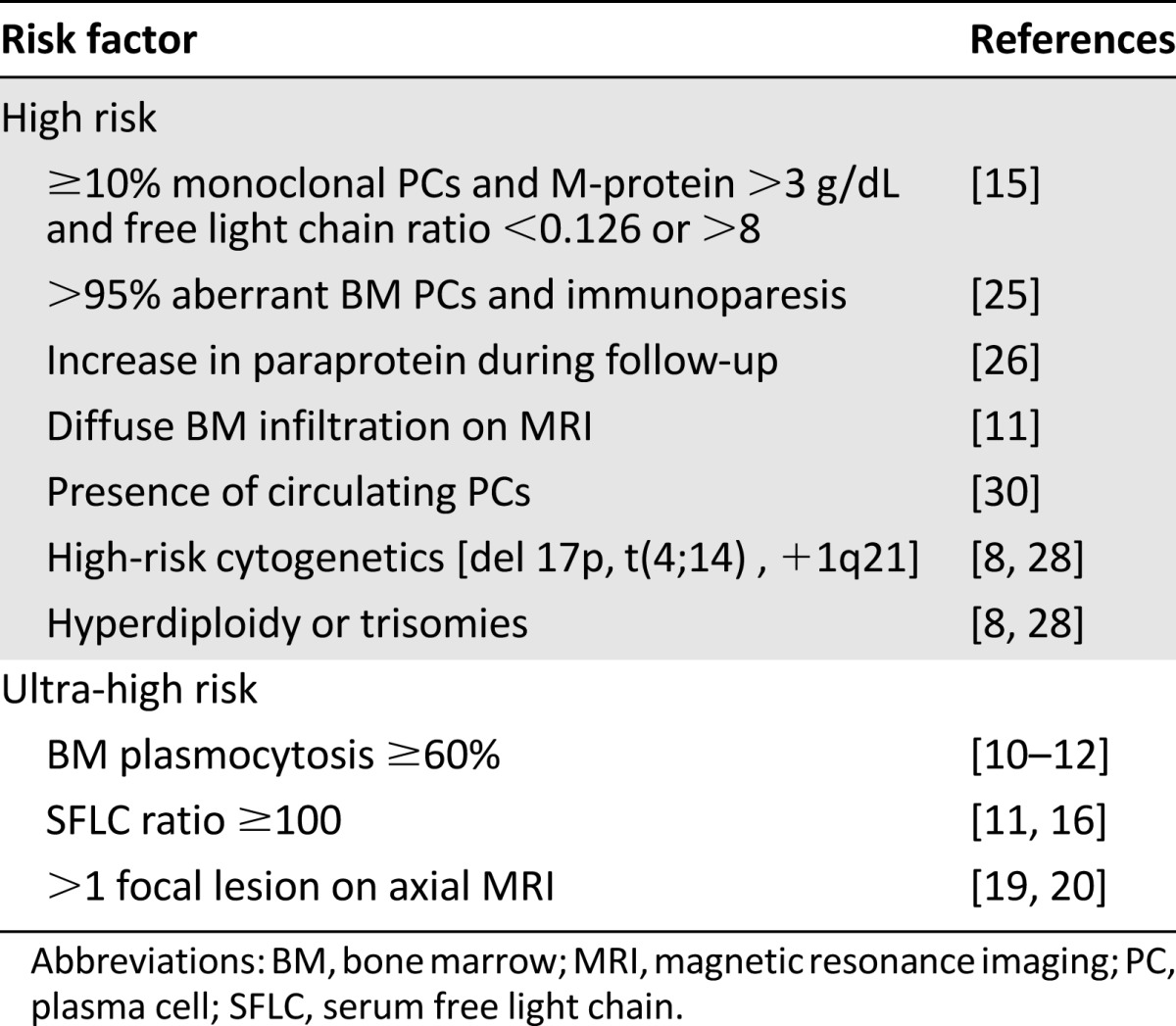
Many risk factors for progression have been proposed since the first definition of SMM (Table 1). To be clinically relevant, these prognostic factors should be accessible to all centers around the world, preferentially being performed on a routine basis and being reproducible and externally validated in different series of patients. Several of the described techniques require expertise in the realization and interpretation of results (e.g., imaging, flow cytometry, and cytogenetics) and should be confined to MM reference centers to actually use them as a reliable risk factor.
Table 1.
Risk factors for progression of smoldering multiple myeloma to active multiple myeloma
Tumor Load
A high tumor load, as assessed by the percentage of PC in the BM or the size of the serum monoclonal component, is a significant risk factor in most reported series of patients with SMM. The Mayo Clinic demonstrated that the combination of these two factors could differentiate three risk groups [7]. The highest risk group was defined by the presence of a medullary PC infiltration ≥10% and a monoclonal protein ≥3 g/dL; the other groups were defined by the presence of only one of these two criteria. The cumulative probability of progression (at 15 years) was 87%, 70%, and 39% in groups 1, 2, and 3, respectively.
This group recently identified a group of patients at ultra-high risk of progression characterized by BMPC infiltration ≥60% on BM biopsy: 95% of patients with a BM infiltration ≥60% by conventional morphology became symptomatic within 2 years after the diagnosis of SMM, with a median time to progression of 7 months (Fig. 1A) [11]. Two other studies confirmed this observation that these patients display a high risk for imminent progression [12, 13]. Such a high tumor infiltration is very infrequent (∼3% of SMM cases); nevertheless, if observed, both publications suggest considering those patients as candidates for treatment, rather than waiting until myeloma-related symptoms develop [11, 12, 14]. These criteria have been incorporated in the new IMWG recommendations [6].
Figure 1.

Risk factors that identified ultra-high-risk smoldering multiple myeloma (SMM). (A): Risk of progression for SMM patients with bone marrow plasma cells >60% (adapted and reprinted with permission from [11]). (B): Risk of progression for SMM patients with a free light chain ratio ≥100 (adapted and reprinted with permission from [17]). (C): Incidence of progression in SMM patients according to the number of focal lesions on magnetic resonance imaging (adapted and reprinted with permission from [20]).
Abbreviations: BM, bone marrow; MRI, magnetic resonance imaging; sFLC, serum free light chain ratio.
Serum Free Light Chain Ratio
The risk of progression of MGUS is significantly higher in patients with an abnormal serum free light chain (SFLC) ratio at presentation [15]. Dispenzieri et al. confirmed this observation in SMM [16]: In a cohort of 273 patients, the authors found a progression rate of 5% per year for individuals with normal or close to normal SFLC κ/λ ratios (0.26–1.65) versus 8.1% per year in patients with a severely abnormal SFLC ratio (<0.031 or >32). In multivariate analysis, the most significant threshold for an abnormal κ/λ ratio was <0.125 or >8. In these patients, the relative risk of progression to MM was 2.3 [16].
A second study on the prognostic contribution of the SFLC analysis demonstrated that the progression rate increased linearly with the SFLC ratio [17]. Of interest, 15% of the overall cohort of 586 SMM patients had an involved-to-uninvolved SFLC ratio of ≥100. The percentage of progressive patients among those with a ratio ≥100 was estimated as 43% after 1 year, 72% after 2 years, and 87% after 3 years (Fig. 1B) [17]. Also of note, 27% had renal failure as a myeloma-defining event.
The predictive value for progression to overt myeloma of an abnormal involved/uninvolved SFLC ratio was confirmed by a Greek study based on different biological and radiological parameters [12]. Multivariate analysis retained only a SFLC ≥ 100 and BMPC ≥ 60% as prognostic factors [12]. These patients also showed a higher risk for developing AL-amyloidosis.
Imaging Studies
Although the detection of bone lesions on the standard radiologic skeletal survey defines symptomatic myeloma, other techniques are more sensitive to detect bone lesions [18]. Moulopoulos et al. described the presence of BM abnormalities on magnetic resonance imaging (MRI) of the dorso-lumbar spine of 38 patients with SMM and reported an abnormal MRI in 50% of them, which was associated with an increased risk of progression [19]. More recently, the same group described the prognostic value of spinal MRI. In their study, the hazard ratio estimating the risk of progression in patients with an abnormal MRI was calculated as 5.8, compared with patients with normal MRI. Five of the eight patients with an abnormal MRI progressed to symptomatic MM within 18 months. In another series of 149 SMM patients, Hillengass et al. showed that the presence of ≥2 focal lesions on whole-body MRI (wb-MRI) was a prognostic factor for progression to MM (Fig. 1C) [20]. The median time to progression was not reached in patients with 0 or 1 focal bone lesions versus a time to progression of 13 months in those who had at least 2 lesions [20]. A smaller study on 69 patients conducted by the Greek Myeloma Group indicated that the presence of ≥1 focal lesion in spine MRI was also associated with a short time to symptomatic disease (median 9 months vs. >5 years) [21]. The Heidelberg group retrospectively analyzed the outcome of 63 patients with SMM that received at least two MRIs during their follow-up [22]. Radiological progression was defined by the appearance of new focal lesions or a new diffuse infiltration of previously unaffected regions. Progressive disease was identified in 31 patients (49%) and was associated with a 16.5-fold risk of progression into symptomatic MM compared with patients with a stable MRI [22] It is important to note that a focal lesion on MRI reflects the presence of tumor infiltration and is insufficient for the identification of an osteolytic process. Although the results on MRI are consistent between different groups and currently incorporated in the IMWG criteria of treatment-requiring events, standardization in interpretation and reporting of MRI results is required in subsequent studies. Standardization is also required for the use and implication of positron emission tomography (PET)/computed tomography (CT). In their study on 120 patients with SMM, the Bologna group reported that PET/CT identified either focal lesions or a diffuse infiltration in 19 (16%) patients [23]. Patients presenting small osteolytic lesions were excluded from the study. The probability of progression within 2 and 3 years for patients with positive PET/CT was 58% and 66%, respectively, as compared with 33% and 42% for negative patients [23] The Mayo Clinic reported retrospective results on 122 patients with SMM that underwent PET/CT, finding that 75% of patients with a positive PET/CT progressed to active myeloma within 2 years versus 30% with a negative PET/CT [24].
Surface Molecules Expression by Flow Cytometry
Immunophenotyping by flow cytometry has been developed to diagnose various hematological malignancies and can be used to detect monoclonal PCs in patients with MM. Ocqueteau et al. showed that the proportion of phenotypically abnormal PCs to normal PCs (>95%) was one of the most important criteria for the differential diagnosis between MGUS and myeloma [25]. Pérez-Persona et al. showed that this proportion could have an impact on the risk of MGUS or SMM progression [26]. They reported on a series of 407 patients with MGUS and 93 patients with SMM and found a significant impact of the preponderance of aberrant PC on the risk of progression. Patients with ≥95% aberrant BMPC had a median time to symptomatic progression of 34 months versus progression not reached in the case of ≥5% of normal BMPC [26]. This definition of high-risk SMM was used to identify patients at high risk for progression to symptomatic MM in the prospective Programa Español de Tratamientos en Hematología (PETHEMA) study of lenalidomide and dexamethasone (Rd). Unfortunately, flow cytometry to identify aberrant PC is not uniformly and widely used, but is eagerly sought to be standardized at various MM centers worldwide.
Serum Paraprotein Progression
The evolution of disease parameters (such as serum paraprotein or urine paraprotein) after the initial diagnosis could be an important element in estimating the risk of progressive SMM. Rosiñol et al. followed 53 patients with SMM and defined two subpopulations: a population of 22 patients with progressive or “evolving” SMM (defined as a progressive increase of ≥10% in the paraprotein level at two consecutive follow-up visits) and another population of 26 patients whose serum M-spike remained stable [27]. As expected, the median time to progression was shorter in patients with evolving SMM (1.3 vs. 3.9 years). Similarly, Landgren et al. retrospectively evaluated 71 patients who developed MM and were able to detect the presence of a monoclonal gammopathy in the years preceding the diagnosis of symptomatic MM, showing a progressive increase in the SFLC levels and the size of the monoclonal peak [28]. However, the definition of progressive M-spikes differs among the reported studies, and small fluctuations of the M-spike (i.e., ±5%–10%) are frequently seen and may also depend on the techniques used to evaluate the M-spike (e.g., agarose vs. capillary serum protein electrophoresis). Thus, a continuous trend over time, rather than a single or few measurements, should be considered in the evaluation of evolving SMM patients. An increasing M-component should be taken into account when one considers starting treatment and may be used as an additional argument in patients who present with other relevant risk factors. However, watchful waiting—with attentive, close follow-up—may also be a possible option and should be discussed with the SMM patient, especially with slowly evolving disease, because these patients rarely present with suddenly explosive progression and devastating complications. Subsequent analyses are needed to specifically and even better define these subgroups by incorporating other valuable biomarkers and to distinguish slowly evolving patients from those patients requiring an early treatment.
Cytogenetic Abnormalities and Gene Expression Profile
The presence of certain cytogenetic abnormalities has major prognostic significance in symptomatic MM. Such cytogenetic lesions, however, also may occur early in patients with MGUS or SMM. The Mayo Clinic team studied the prognostic value of cytogenetics in a cohort of 351 SMM patients and found a median time to progression of 28 months for patients with t(4;14) translocation, 34 months in patients with trisomies, and 55 months for other anomalies. The time to progression in patients with 17p deletion was 24 months [29]. Cytogenetic abnormalities also determined the OS; indeed, OS after diagnosis of SMM was 105 months for t(4;14) and 147 months for t(11;14) aberrations [29]. The Heidelberg group confirmed the prognostic importance of the presence of cytogenetic abnormalities and added gain of the 1q21 chromosome as a risk factor for SMM patients [8]. They showed that the 3-year progression rate was 45% for patients with high-risk aberrations [t(4;14), del17p, +1q21] versus 24% in the standard-risk group. Multivariate analysis indicated that high-risk cytogenetics, hyperdiploidy, the percentage of >95% aberrant PCs (identified by interphase fluorescent in situ hybridization), and a high tumor mass (based on BMPCs and paraprotein concentrations) were independent predictors of progression in SMM patients. These authors elegantly proposed a prognostic model based on paraprotein levels and unfavorable cytogenetic abnormalities and identified four groups with different progression-free survival (PFS) [8]. Regarding the prognostic value of hyperdiploidy in SMM, the Mayo Clinic assessed the OS of SMM patients: They observed that patients presenting with trisomies progressed more rapidly to symptomatic myeloma, but survived approximately as long as standard-risk patients and much longer than those with deletion 17p [14]. In the SWOG S0120 trial, gene expression profiling was introduced to identify additional predictive markers for SMM progression [9]: A gene-expression score, based on 70 genes (earlier found to be associated with a high risk in patients with symptomatic MM), together with a serum M-protein of >3 g/dL and serum FLC of >25 mg/dL, identified SMM patients with a 70% 2-year risk of progression to MM requiring therapy [9]. This list of 70 genes could be further reduced to 4 genes (RRM2 [2p25-p24], DTL [1q32], TMEM48 [1p32.3], and ASPM [1q31]) that were integrated in a prognostic score and identified a subset of patients (13%) with a 2-year progression probability of 86% [30].
Circulating Peripheral PCs
Circulating tumor cells can be detected in a small fraction of newly diagnosed MM patients (<15%) by conventional morphology, but their rate of detection increases to 50%–70% once more sensitive techniques (i.e., immunohistochemistry or flow cytometry) are used [31]. These circulating PCs disseminate through the bloodstream to distant BM sites. The Mayo Clinic group evaluated the presence and prognostic significance of peripheral blood circulating PCs in SMM patients by an immunofluorescent assay performed on fixed peripheral blood mononuclear cells [32]. Their study included 91 evaluable patients, and 14 of them (15%) were found to have high circulating PCs. The median time to progression (TTP) was 12 versus 57 months for those with high and low circulating PCs [32]. Although the proposed technique to identify circulating PCs is not standard practice (and could eventually be replaced by flow cytometry), the latter analysis showed that—depending on the amount of cells being assessed therein—approximately 75% of newly diagnosed and relapsing MM patients show circulating peripheral blood PCs [33]. The lack of broadly available techniques and clear cut-off points hamper the implication of this risk factor in daily routine. The prognostic importance of circulating PCs, however, should be validated in subsequent analyses to further assess their value to the diagnostic SMM criteria and risk of progression.
Prognostic Modeling
Based on the observations by Kyle et al. [7] and Dispenzieri et al. [16], the Mayo Clinic team proposed a classification model (Table 2) based on the following risk factors: BMPC ≥ 10%, monoclonal protein ≥3 g/dL, and abnormal SFLC ratio of <0.125 or >8 (Fig. 2A). In multivariate analysis, each of these factors independently correlated with an increased risk of progression. The cumulative probability of progression at 5 years was 25%, 51%, and 76%, depending on whether the patients had 1, 2, or 3 risk factors, with a median time to progression of 10, 5.1, and 1.9 years, respectively [16].
Table 2.
Risk of progression of smoldering multiple myeloma patients according to the Mayo Clinic and Spanish PETHEMA models

Figure 2.

Proposed algorithm for the management of smoldering multiple myeloma/multiple myeloma (MM) in 2015.
Abbreviations: BM, bone marrow; BMPC, bone marrow plasma cell; CRAB, hypercalcemia, radiological bone lesions, anemia, and renal failure; M-protein, monoclonal protein level quantified by protein electrophoresis; MRI, magnetic resonance imaging; sFLC, serum involved/uninvolved free light chain ratio.
A second classification model (Table 2) was proposed by the Spanish group PETHEMA that studied anomalies by flow cytometry and the presence of immunoparesis (defined as a decrease by >25% of the level of one of the two other uninvolved immunoglobulins). Depending on whether patients had 0, 1, or 2 risk factors, progression rates at 5 years were 4%, 46%, and 72%, respectively. The median time to progression was not reached and was reached at 73 months and 23 months, respectively [26]. Both studies indicated that the probability of progression of patients in the low-risk groups was almost equivalent to MGUS patients (1% per year).
Landgren’s team prospectively evaluated these two risk models in a series of 77 patients with SMM and was able to detect a concordant classification in 29% of cases [34], suggesting that both risk models could be used in a complementary way, rather than alternatively, and that other biomarkers (e.g., gene expression profiling, sensitive imaging and their harmonized assessment interpretation, cytogenetics, etc.) should be added to the existing parameters to further improve this risk assessment.
Diagnosis and Follow-Up
Based on these clinical findings, the following recommendations have been proposed. If SMM is suspected, standard laboratory tests searching for target-organ damage should be implemented. These include a complete blood count; renal function with determination of urea, creatinine, albumin, and total protein levels; serum calcium; serum protein electrophoresis (with immunofixation, if it has not yet been performed); and proteinuria in a 24-hour collection to search for Bence Jones protein. In addition, SFLC analysis, BM cytology and biopsy, and bone survey should be performed [35]. To detect myeloma-related bone disease, a more sensitive radiological survey is relevant, such as a wb-MRI, to determine diffuse versus focal, extramedullary lesions, and extent of BM infiltration. Depending on local expertise and availability of MRI, a low-dose whole-body CT may also be considered, although data on the use of this imaging technique in SMM are incomplete. PET/CT is eagerly used and further assessed in SMM within clinical trials.
Laboratory tests should be repeated after 2–3 months, then every 4–6 months for 1 year, and then every 6–12 months, if the SMM remains stable. A bone survey can be repeated when a progression is suspected.
Patients with >1 focal lesions on wb-MRI are now defined as active MM [36]. Ultra-high-risk SMM is now defined by the presence of either a BMPC infiltration of ≥60%, an abnormal SFLC ratio ≥ 100, and >1 focal lesions detected by wb-MRI. These patients have a >80% probability of progression to symptomatic disease within 2 years from the diagnosis of SMM. Because a symptomatic progression may present with serious complications (e.g., fractures or acute renal failure), it is recommended that these patients are defined as MM and offered antimyeloma treatment. Such a treatment aims to avoid major complications, possibly prolong OS, and improve the quality of life of MM patients [14]—issues that are currently and very eagerly evaluated in early intervention SMM clinical trials.
Therapeutic Strategies
Earlier intervention studies comparing an imminent with a delayed treatment with oral melphalan and prednisone for SMM patients could never show any benefit in terms of response rate, PFS, or OS [37, 38]. After the first reports of its activity in MM, thalidomide was also evaluated in SMM patients, and treatment with this agent resulted in partial responses in ∼35% of the cases, but was associated with a significant toxicity [39, 40]. The Mayo Clinic recently reported the results of a randomized trial that compared thalidomide plus zoledronate versus zoledronate alone in SMM patients [41]. The response rate was 37% in the thalidomide arm (whereas no responses were seen in the zoledronate arm), but there were no significant differences in the TTP to symptomatic MM (4.3 vs. 3.3 years) or in OS (74% vs. 73% at 5 years) [41]. Earlier trials of bisphosphonate treatment demonstrated the absence of clear antitumoral effects, whereas a strong effect on the bone metabolism was uniformly described with a significant reduction in the incidence of skeletal-related events [42–45]. Given the anabolic effects on bone metabolism, bisphosphonates are recommended for those SMM patients with osteoporosis identified by dual-energy x-ray absorptiometry scan in doses used for osteoporosis [46]. For ultra-high-risk SMM, the treating physician should consider using bisphosphonate doses and schedules as with symptomatic MM [46].
Trying to incorporate agents targeting novel pathways, anakinra (IL1-β receptor antagonist) was studied in 47 patients with SMM and resulted in a prolonged PFS [47]. Given the limited side effects and the lenalidomide-sparing approach, this agent seems interesting, but further trials are required.
The lack of a clear benefit in terms of PFS and OS in early clinical trials in SMM may be explained by the fact that most included limited and unselected SMM patients and did not focus on (ultra) high-risk patients who may benefit most from early interventions. Furthermore, the limited activity and poor tolerability of either melphalan/prednisone or thalidomide (without dexamethasone) were significant limitations of these prior studies. Therefore, the Spanish group PETHEMA recently published the results of a randomized study comparing treatment with Rd with no treatment (observational arm) in 125 high-risk patients [48]. In the treatment group, patients received nine cycles of Rd, followed by lenalidomide maintenance up to 2 years. The average follow-up period was 40 months. At the end of the induction phase, 79% of patients had a partial response or better, and 28% achieved at least a very good partial response. After an average of 15 maintenance cycles, response rates increased up to 90% in the Rd treatment group. The median time to progression in the Rd arm was not reached versus 21 months in the control group. A total of 13 patients (23%) progressed to symptomatic disease in the Rd treatment group versus 47 (76%) in the observational group. Treatment also prolonged OS, with a projected 5-year OS of 94% in patients with Rd versus 78% in the control group. This prominent study was the first to demonstrate the benefit of a therapeutic intervention compared with a watch-and-wait strategy, suggesting that not treating subsets of high-risk SMM may be more detrimental than treatment, because renal failure, skeletal-related events, severe infections, and other adversities may occur. The results and implications for daily practice were discussed worldwide because of the limited patient number (however, a larger patient population would have given similar—or even more significant—differences) and the addition of dexamethasone during lenalidomide maintenance in patients presenting an asymptomatic biochemical progression. The latter was given to fully exploit the therapeutic possibilities of the initiated lenalidomide treatment, because the duration of the maintenance was limited to 2 years after the warnings of secondary primary malignancies (SPMs). Results of the other randomized trials being conducted in high-risk SMM patients will yield additional information on early treatment in this patient population.
Of further interest and in line with the PETHEMA study, Ola Langren’s group recently reported the results obtained with an induction treatment containing eight cycles with carfilzomib, lenalidomide, and dexamethasone (KRD), followed by 2 years of lenalidomide maintenance [49]. This phase 2 study included 12 patients with SMM and 45 patients with symptomatic MM. One SMM patient discontinued treatment after 6 months due to occurrence of a congestive heart failure, possibly related to carfilzomib. The remaining patients all achieved complete response with disappearance of minimal residual disease (assessed by multiparametric flow cytometry and/or detection of clonal rearrangements by next-generation sequencing). Deeper responses were observed in patients with SMM than in patients with symptomatic MM (complete remission rates of 100% vs. 62%, respectively), and the reported toxicity was mostly hematological (lymphopenia and thrombocytopenia) and gastrointestinal, whereas two patients developed nonmelanoma skin cancers as SPMs. These responses are impressive, but longer follow-up and randomized studies with this regimen are needed. In addition, its financial burden may hamper the introduction of this regimen as a treatment option for SMM. Other studies that are currently ongoing in SMM are listed in Table 3.
Table 3.
Current ongoing trials in smoldering myeloma

IMWG Definition of Early Myeloma
The earlier definition of symptomatic MM required the presence of overt clinical manifestations of serious end-organ damage, such as osteolytic bone lesions and renal failure. This was acceptable in an era of limited treatment options and less sensitive techniques for early detection of organ damage. However, this approach seems less justifiable with improved treatment schedules and potentially devastating complications of the MM (e.g., renal failure with dialysis, vertebral fracture, or neurological complications). The IMWG therefore aimed to identify valid biomarkers that were associated with a ≥80% probability of progression to MM within 2 years [6]. This threshold would identify a small cohort of SMM patients (10%–15%) with a median time to development of end-organ damage of ∼12 months in whom delaying therapy was not associated with any tangible benefit—and conversely increased significantly the risk of severe debilitating myeloma-related complications at the moment myeloma would become symptomatic. The IMWG has thus defined the ultra-high-risk SMM (also called early myeloma) now as MM with one of the following biomarkers: a clonal BM plasmocytosis ≥60%, an involved/uninvolved SFLC ratio ≥ 100 with a minimum involved SFLC level of ≥100 mg/L, and the presence of >1 focal lesions on MRI studies (Table 4; Fig. 2) [6].
Table 4.
Revised definition of MM by the International Myeloma Working Group

Traditionally, bone disease has been identified on the basis of conventional skeletal radiography. The current disease definition also includes the presence of osteolytic bone destruction (at least 5 mm or more in size) seen on CT (including low-dose wb-CT) and/or (18)F-fluorodeoxyglucose PET combined with CT (PET-CT). In case of doubt (equivoqual lesions or tiny lesions), the radiological study should be repeated in 3–6 months before making a diagnosis of symptomatic MM.
Comments on the SMM Practice Change
According to the IMWG, all SMM patients should undergo, in addition to the conventional assessments, a BM aspiration and biopsy to quantify malignant PCs, determination of SFLCs, and wb-MRI. The results and implications for therapy should then be carefully interpreted and discussed with the patient. Potential harmful organ complications with significant long-term morbidity need to be avoided; therefore, the current SMM-risk definition is a step forward because it allows the initiation of earlier antimyeloma therapy. Because the evidence for improved survival with immediate start of therapy presently is based on one study only, we encourage physicians to offer these patients participation in one of the ongoing trials. Immediate start of therapy is also an option, but the decision to treat should include an evaluation of the patient’s general health status, and, whenever possible, these patients should be referred to centers specialized in MM therapy and discussed in a multidisciplinary MM tumor board [50]. Patients should be correctly informed about the expected benefits and possible risks of starting an antimyeloma treatment. Some patients might decide to delay immediate treatment, arguing that being without therapy for 1 or more years is an attractive alternative. In case of delay, it is important to assess the dynamics of the MM disease with close follow-up of the laboratory parameters (initially every month and gradually less frequent, if nonevolving). Evolving paraprotein levels, additional lesions on follow-up MRI, or even moderate changes in renal or BM function should prompt treatment initiation, whereas their lack may comfort both physicians and patients.
Ideally, high-risk SMM patients should be included in clinical trials to answer questions on timing, optimal treatment regimen, and identification of patients who would benefit most from these interventions. Existing clinical trials that investigate single-agent treatments might prove insufficient in patients presenting with high-risk SMM, whereas specifically studies on the immune surveillance within the BM microenvironment and those on the molecular monitoring are of great interest, especially if these follow the effect of treatment on different PC clones that are present at diagnosis versus follow-up. Ideally, upcoming clinical trials for newly diagnosed MM patients will incorporate the new IMWG definition, allowing subgroup analyses for patients presenting with one of the biomarkers of ultra-high-risk SMM/early MM.
Of note, these new IMWG criteria do not affect the majority of SMM patients because 85%–90% do not present with ultra-high-risk features. These SMM patients are currently monitored without antimyeloma treatment until progression to MM occurs. Patients who do not present any risk factor should be followed every 3 months and—if stable and without symptoms—gradually less frequently. For all SMM patients, it is important to follow the evolution of the different MM parameters to have insight into the dynamics of the disease. Low-risk SMM remains a challenging entity, and further research should enable us to determine subgroups of patients with a very low likelihood of progression (∼1% per year), similar to MGUS patients in which follow-up can be further delayed. A management algorithm for patients with a suspected MM is presented in Fig. 2.
Conclusion
The introduction of new therapeutic agents and the identification of SMM patients at high risk and ultra-high risk of progression are major advances in the management of myeloma precursor diseases. With the new IMWG recommendations, ultra-high-risk SMM patients are considered as MM, allowing us to consider initiation of active antimyeloma treatment. High-risk SMM patients should be offered clinical trials testing early interventions, with prudent clinical judgment being employed in these patients. The regimens that are currently tested consist of single-agent and multiagent treatment options. For low-risk SMM patients, the standard care remains observation until development of symptomatic MM. In patients with signs of osteoporosis and in ultra-high-risk patients, treatment with bisphosphonates should be considered. In the future, prospective studies are critical for validating, but also improving, current risks, biomarkers, and prognostic models and to even better allow us to correctly identify individuals who would receive the utmost benefit from early interventions.
Acknowledgments
This work was supported by grants from the Belgian Foundation Against Cancer, Fonds de la Recherche Scientifique Médicale, the Fonds National de la Recherche Scientifique (Belgium), and the Fonds spéciaux de la Recherche, Université de Liège (to J.C., R.H., and Y.B.). The work was also supported through Deutsche Krebshilfe Grant 111424 (to M.E. and R.W.).
Author Contributions
Conception/Design: Jo Caers, Monika Engelhardt
Collection and/or assembly of data: Carlos Fernández de Larrea, Xavier Leleu, Niklas Zojer, Olivier Decaux, Efstathios Kastritis, Monique Minnema, Maria Victoria Mateos, Heinz Ludwig, Monika Engelhardt
Data analysis and interpretation: Jo Caers, Carlos Fernández de Larrea, Xavier Leleu, Roy Heusschen, Niklas Zojer, Olivier Decaux, Efstathios Kastritis, Monique Minnema, Antonio Palumbo, Maria Victoria Mateos, Heinz Ludwig, Monika Engelhardt
Manuscript writing: Jo Caers, Carlos Fernández de Larrea, Xavier Leleu, Roy Heusschen, Niklas Zojer, Olivier Decaux, Efstathios Kastritis, Monique Minnema, Artur Jurczyszyn, Yves Beguin, Ralph Wäsch, Antonio Palumbo, Meletios Dimopoulos, Maria Victoria Mateos, Heinz Ludwig, Monika Engelhardt
Final approval of manuscript: Jo Caers, Carlos Fernández de Larrea, Xavier Leleu, Roy Heusschen, Niklas Zojer, Olivier Decaux, Efstathios Kastritis, Monique Minnema, Artur Jurczyszyn, Yves Beguin, Ralph Wäsch, Antonio Palumbo, Meletios Dimopoulos, Maria Victoria Mateos, Heinz Ludwig, Monika Engelhardt
Disclosures
Carlos F. de Larrea: Celgene, Janssen (C/A); Niklas Zojer: Celgene, Bristol-Myers Squibb, Takeda, Novartis, Amgen, Janssen (C/A, H), Celgene, Takeda, Amgen (RF), Amgen (Other); Efstathios Kastritis: Takeda, Janssen (H); Monique Minnema: Amgen, Celgene, Janssen Cilag (C/A); Yves Beguin: Celgene (C/A), Celgene, Janssen (RF); Antonio Palumbo: Amgen, BMS, Genmab, Celgene, Janssen-Cilag, Millennium, Onyx (C/A), Amgen, BMS, Genmab, Celgene, Janssen-Cilag, Millennium, Onyx, Novartis, Sanofi (H); Meletios Dimopoulos: Celgene, Janssen, Novartis, Amgen (H); Maria Victoria Mateos: Janssen, Takeda, Celgene, Onyx, Novartis (C/A); Heinz Ludwig: Böhringer Ingelheim, Janssen Cilag, Amgen, Roche (C/A), Janssen Cilag, Böhringer Ingelheim, Bristol Myers Squibb, Amgen, Novartis, Onyx (H), Takeda (RF); The other authors indicated no financial relationships.
(C/A) Consulting/advisory relationship; (RF) Research funding; (E) Employment; (ET) Expert testimony; (H) Honoraria received; (OI) Ownership interests; (IP) Intellectual property rights/inventor/patent holder; (SAB) Scientific advisory board
References
- 1.Engelhardt M, Terpos E, Kleber M, et al. European Myeloma Network recommendations on the evaluation and treatment of newly diagnosed patients with multiple myeloma. Haematologica. 2014;99:232–242. doi: 10.3324/haematol.2013.099358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Röllig C, Knop S, Bornhäuser M. Multiple myeloma. Lancet. 2015;385:2197–2208. doi: 10.1016/S0140-6736(14)60493-1. [DOI] [PubMed] [Google Scholar]
- 3.Kyle RA, Greipp PR. Smoldering multiple myeloma. N Engl J Med. 1980;302:1347–1349. doi: 10.1056/NEJM198006123022405. [DOI] [PubMed] [Google Scholar]
- 4.International Myeloma Working Group Criteria for the classification of monoclonal gammopathies, multiple myeloma and related disorders: A report of the International Myeloma Working Group. Br J Haematol. 2003;121:749–757. [PubMed] [Google Scholar]
- 5.Caers J, Vekemans M-C, Bries G, et al. Diagnosis and follow-up of monoclonal gammopathies of undetermined significance; information for referring physicians. Ann Med. 2013;45:413–422. doi: 10.3109/07853890.2013.801562. [DOI] [PubMed] [Google Scholar]
- 6.Rajkumar SV, Dimopoulos MA, Palumbo A, et al. International Myeloma Working Group updated criteria for the diagnosis of multiple myeloma. Lancet Oncol. 2014;15:e538–e548. doi: 10.1016/S1470-2045(14)70442-5. [DOI] [PubMed] [Google Scholar]
- 7.Kyle RA, Remstein ED, Therneau TM, et al. Clinical course and prognosis of smoldering (asymptomatic) multiple myeloma. N Engl J Med. 2007;356:2582–2590. doi: 10.1056/NEJMoa070389. [DOI] [PubMed] [Google Scholar]
- 8.Neben K, Jauch A, Hielscher T, et al. Progression in smoldering myeloma is independently determined by the chromosomal abnormalities del(17p), t(4;14), gain 1q, hyperdiploidy, and tumor load. J Clin Oncol. 2013;31:4325–4332. doi: 10.1200/JCO.2012.48.4923. [DOI] [PubMed] [Google Scholar]
- 9.Dhodapkar MV, Sexton R, Waheed S, et al. Clinical, genomic, and imaging predictors of myeloma progression from asymptomatic monoclonal gammopathies (SWOG S0120) Blood. 2014;123:78–85. doi: 10.1182/blood-2013-07-515239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rajkumar SV, Landgren O, Mateos M-V. Smoldering multiple myeloma. Blood. 2015;125:3069–3075. doi: 10.1182/blood-2014-09-568899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rajkumar SV, Larson D, Kyle RA. Diagnosis of smoldering multiple myeloma. N Engl J Med. 2011;365:474–475. doi: 10.1056/NEJMc1106428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kastritis E, Terpos E, Moulopoulos L, et al. Extensive bone marrow infiltration and abnormal free light chain ratio identifies patients with asymptomatic myeloma at high risk for progression to symptomatic disease. Leukemia. 2013;27:947–953. doi: 10.1038/leu.2012.309. [DOI] [PubMed] [Google Scholar]
- 13.Rago A, Grammatico S, Za T, et al. Prognostic factors associated with progression of smoldering multiple myeloma to symptomatic form. Cancer. 2012;118:5544–5549. doi: 10.1002/cncr.27657. [DOI] [PubMed] [Google Scholar]
- 14.Dispenzieri A, Stewart AK, Chanan-Khan A, et al. Smoldering multiple myeloma requiring treatment: Time for a new definition? Blood. 2013;122:4172–4181. doi: 10.1182/blood-2013-08-520890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rajkumar SV, Kyle RA, Therneau TM, et al. Serum free light chain ratio is an independent risk factor for progression in monoclonal gammopathy of undetermined significance. Blood. 2005;106:812–817. doi: 10.1182/blood-2005-03-1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dispenzieri A, Kyle RA, Katzmann JA, et al. Immunoglobulin free light chain ratio is an independent risk factor for progression of smoldering (asymptomatic) multiple myeloma. Blood. 2008;111:785–789. doi: 10.1182/blood-2007-08-108357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Larsen JT, Kumar SK, Dispenzieri A, et al. Serum free light chain ratio as a biomarker for high-risk smoldering multiple myeloma. Leukemia. 2013;27:941–946. doi: 10.1038/leu.2012.296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Caers J, Withofs N, Hillengass J, et al. The role of positron emission tomography-computed tomography and magnetic resonance imaging in diagnosis and follow up of multiple myeloma. Haematologica. 2014;99:629–637. doi: 10.3324/haematol.2013.091918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Moulopoulos LA, Dimopoulos MA, Smith TL, et al. Prognostic significance of magnetic resonance imaging in patients with asymptomatic multiple myeloma. J Clin Oncol. 1995;13:251–256. doi: 10.1200/JCO.1995.13.1.251. [DOI] [PubMed] [Google Scholar]
- 20.Hillengass J, Fechtner K, Weber MA, et al. Prognostic significance of focal lesions in whole-body magnetic resonance imaging in patients with asymptomatic multiple myeloma. J Clin Oncol. 2010;28:1606–1610. doi: 10.1200/JCO.2009.25.5356. [DOI] [PubMed] [Google Scholar]
- 21.Kastritis E, Moulopoulos LA, Terpos E, et al. The prognostic importance of the presence of more than one focal lesion in spine MRI of patients with asymptomatic (smoldering) multiple myeloma. Leukemia. 2014;28:2402–2403. doi: 10.1038/leu.2014.230. [DOI] [PubMed] [Google Scholar]
- 22.Merz M, Hielscher T, Wagner B, et al. Predictive value of longitudinal whole-body magnetic resonance imaging in patients with smoldering multiple myeloma. Leukemia. 2014;28:1902–1908. doi: 10.1038/leu.2014.75. [DOI] [PubMed] [Google Scholar]
- 23.Zamagni E, Nanni C, Gay F, et al. 18F-FDG PET/CT focal, but not osteolytic, lesions predict the progression of smoldering myeloma to active disease. Leukemia. 2015 doi: 10.1038/leu.2015.291. [Epub ahead of print]. doi:10.1038/leu.2015.291. [DOI] [PubMed] [Google Scholar]
- 24.Siontis B, Kumar S, Dispenzieri A, et al. Positron emission tomography-computed tomography in the diagnostic evaluation of smoldering multiple myeloma: Identification of patients needing therapy. Blood Cancer J. 2015;5:e364. doi: 10.1038/bcj.2015.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ocqueteau M, Orfao A, Almeida J, et al. Immunophenotypic characterization of plasma cells from monoclonal gammopathy of undetermined significance patients. Implications for the differential diagnosis between MGUS and multiple myeloma. Am J Pathol. 1998;152:1655–1665. [PMC free article] [PubMed] [Google Scholar]
- 26.Pérez-Persona E, Vidriales M-B, Mateo G, et al. New criteria to identify risk of progression in monoclonal gammopathy of uncertain significance and smoldering multiple myeloma based on multiparameter flow cytometry analysis of bone marrow plasma cells. Blood. 2007;110:2586–2592. doi: 10.1182/blood-2007-05-088443. [DOI] [PubMed] [Google Scholar]
- 27.Rosiñol L, Bladé J, Esteve J, et al. Smoldering multiple myeloma: Natural history and recognition of an evolving type. Br J Haematol. 2003;123:631–636. doi: 10.1046/j.1365-2141.2003.04654.x. [DOI] [PubMed] [Google Scholar]
- 28.Landgren O, Kyle RA, Pfeiffer RM, et al. Monoclonal gammopathy of undetermined significance (MGUS) consistently precedes multiple myeloma: A prospective study. Blood. 2009;113:5412–5417. doi: 10.1182/blood-2008-12-194241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rajkumar SV, Gupta V, Fonseca R, et al. Impact of primary molecular cytogenetic abnormalities and risk of progression in smoldering multiple myeloma. Leukemia. 2013;27:1738–1744. doi: 10.1038/leu.2013.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Khan R, Dhodapkar M, Rosenthal A, et al. Four genes predict high risk of progression from smoldering to symptomatic multiple myeloma (SWOG S0120) Haematologica. 2015;100:1214–1221. doi: 10.3324/haematol.2015.124651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Paiva B, Paino T, Sayagues J-M, et al. Detailed characterization of multiple myeloma circulating tumor cells shows unique phenotypic, cytogenetic, functional, and circadian distribution profile. Blood. 2013;122:3591–3598. doi: 10.1182/blood-2013-06-510453. [DOI] [PubMed] [Google Scholar]
- 32.Bianchi G, Kyle RA, Larson DR, et al. High levels of peripheral blood circulating plasma cells as a specific risk factor for progression of smoldering multiple myeloma. Leukemia. 2013;27:680–685. doi: 10.1038/leu.2012.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Billadeau D, Van Ness B, Kimlinger T, et al. Clonal circulating cells are common in plasma cell proliferative disorders: A comparison of monoclonal gammopathy of undetermined significance, smoldering multiple myeloma, and active myeloma. Blood. 1996;88:289–296. [PubMed] [Google Scholar]
- 34.Cherry BM, Korde N, Kwok M, et al. Modeling progression risk for smoldering multiple myeloma: Results from a prospective clinical study. Leuk Lymphoma. 2013;54:2215–2218. doi: 10.3109/10428194.2013.764419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kyle RA, Durie BG, Rajkumar SV, et al. Monoclonal gammopathy of undetermined significance (MGUS) and smoldering (asymptomatic) multiple myeloma: IMWG consensus perspectives risk factors for progression and guidelines for monitoring and management. Leukemia. 2010;24:1121–1127. doi: 10.1038/leu.2010.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rajkumar SV, Merlini G, San Miguel JF. Haematological cancer: Redefining myeloma. Nat Rev Clin Oncol. 2012;9:494–496. doi: 10.1038/nrclinonc.2012.128. [DOI] [PubMed] [Google Scholar]
- 37.Hjorth M, Hellquist L, Holmberg E, et al. Initial versus deferred melphalan-prednisone therapy for asymptomatic multiple myeloma stage I—a randomized study. Eur J Haematol. 1993;50:95–102. doi: 10.1111/j.1600-0609.1993.tb00148.x. [DOI] [PubMed] [Google Scholar]
- 38.Riccardi A, Mora O, Tinelli C, et al. Long-term survival of stage I multiple myeloma given chemotherapy just after diagnosis or at progression of the disease: A multicentre randomized study. Br J Cancer. 2000;82:1254–1260. doi: 10.1054/bjoc.1999.1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Detweiler-Short K, Hayman S, Gertz MA, et al. Long-term results of single-agent thalidomide as initial therapy for asymptomatic (smoldering or indolent) myeloma. Am J Hematol. 2010;85:737–740. doi: 10.1002/ajh.21821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Barlogie B, van Rhee F, Shaughnessy JD, Jr, et al. Seven-year median time to progression with thalidomide for smoldering myeloma: Partial response identifies subset requiring earlier salvage therapy for symptomatic disease. Blood. 2008;112:3122–3125. doi: 10.1182/blood-2008-06-164228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Witzig TE, Laumann KM, Lacy MQ, et al. A phase III randomized trial of thalidomide plus zoledronic acid versus zoledronic acid alone in patients with asymptomatic multiple myeloma. Leukemia. 2013;27:220–225. doi: 10.1038/leu.2012.236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.D’Arena G, Gobbi PG, Broglia C, et al. Pamidronate versus observation in asymptomatic myeloma: Final results with long-term follow-up of a randomized study. Leuk Lymphoma. 2011;52:771–775. doi: 10.3109/10428194.2011.553000. [DOI] [PubMed] [Google Scholar]
- 43.Martín A, García-Sanz R, Hernández J, et al. Pamidronate induces bone formation in patients with smouldering or indolent myeloma, with no significant anti-tumour effect. Br J Haematol. 2002;118:239–242. doi: 10.1046/j.1365-2141.2002.03549.x. [DOI] [PubMed] [Google Scholar]
- 44.Musto P, Falcone A, Sanpaolo G, et al. Pamidronate for early-stage, untreated myeloma. J Clin Oncol. 2003;21:3177–3178; author reply 3178. doi: 10.1200/JCO.2003.99.248. [DOI] [PubMed] [Google Scholar]
- 45.Musto P, Petrucci MT, Bringhen S, et al. A multicenter, randomized clinical trial comparing zoledronic acid versus observation in patients with asymptomatic myeloma. Cancer. 2008;113:1588–1595. doi: 10.1002/cncr.23783. [DOI] [PubMed] [Google Scholar]
- 46.Terpos E, Morgan G, Dimopoulos MA, et al. International Myeloma Working Group recommendations for the treatment of multiple myeloma-related bone disease. J Clin Oncol. 2013;31:2347–2357. doi: 10.1200/JCO.2012.47.7901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lust JA, Lacy MQ, Zeldenrust SR, et al. Induction of a chronic disease state in patients with smoldering or indolent multiple myeloma by targeting interleukin 1β-induced interleukin 6 production and the myeloma proliferative component. Mayo Clin Proc. 2009;84:114–122. doi: 10.4065/84.2.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mateos M-V, Hernández M-T, Giraldo P, et al. Lenalidomide plus dexamethasone for high-risk smoldering multiple myeloma. N Engl J Med. 2013;369:438–447. doi: 10.1056/NEJMoa1300439. [DOI] [PubMed] [Google Scholar]
- 49.Korde N, Roschewski M, Zingone A, et al. Treatment with carfilzomib-lenalidomide-dexamethasone with lenalidomide extension in patients with smoldering or newly diagnosed multiple myeloma. JAMA Oncol. 2015;1:746–754. doi: 10.1001/jamaoncol.2015.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Selder R, Pandurevic M, Möller M-D, et al. First results of the Interdisciplinary Comprehensive Cancer Center Freiburg Tumor Board Multiple Myeloma assessing questions, advice adherence, satisfaction of patients, participants and reffering physicians, inclusion of difficult-to-treat-MM pts in clinical trials (CT) and patient outcome. Paper presented at 56th American Society of Hematology Annual Meeting and Exposition; December 6–9, 2014, San Francisco, CA. [Google Scholar]