

Abstract
The importance of eukaryotic DNA methylation [5-methylcytosine (5mC)] in transcriptional regulation and development was first suggested almost 40 years ago. However, the molecular mechanism underlying the dynamic nature of this epigenetic mark was not understood until recently, following the discovery that the TET proteins, a family of AlkB-like Fe(II)/α-ketoglutarate-dependent dioxygenases, can oxidize 5mC to generate 5-hydroxymethylcytosine (5hmC), 5-formylcytosine (5fC), and 5-carboxylcytosine (5caC). Since then, several mechanisms that are responsible for processing oxidized 5mC derivatives to achieve DNA demethylation have emerged. Our biochemical understanding of the DNA demethylation process has prompted new investigations into the biological functions of DNA demethylation. Characterization of two additional AlkB family proteins, FTO and ALKBH5, showed that they possess demethylase activity toward N6-methyladenosine (m6A) in RNA, indicating that members of this subfamily of dioxygenases have a general function in demethylating nucleic acids. In this review, we discuss recent advances in this emerging field, focusing on the mechanism and function of TET-mediated DNA demethylation.
Keywords: DNA/RNA demethylation, 5-methylcytosine oxidation, Fe(II)/α-ketoglutarate-dependent dioxygenases, Tet, N6-methyladenosine
INTRODUCTION
Cellular DNA and RNA are subjected to various forms of methylation. Methylation has two forms: damage and modifications. Methylation damage, such as N1-methyladenosine (m1A) and N3-methylcytosine (m3C), which are introduced by endogenous or exogenous methylation agents, is considered cytotoxic and/or mutagenic through blocking or altering Watson–Crick base-pairing. Methylation modifications, notably in the forms of 5-methylcytosine (5mC) in DNA and N6-methyladenosine (m6A) in messenger RNA (mRNA), are generated by S-adenosylmethionine (SAM)-dependent methyltransferases (for a review, see References 1 and 2). These modifications do not interfere with Watson–Crick base-pairing; instead, they perform important regulatory functions in mammalian development, and their dysregulation can lead to various human diseases, including cancer (3–6).
The AlkB family of Fe(II)/α-ketoglutarate (αKG)-dependent dioxygenases are important mediators in mammalian nucleic acid methylation (7). AlkB is the Escherichia coli prototype that repairs DNA methylation damage. Human AlkB homologs, such as ALKBH2 and ALKBH3, have long been known to repair various types of methylation damage in DNA and RNA, guarding the genome from methylation agents (Figure 1a) (8). Recent studies have identified members of dioxygenase family that can modulate DNA and RNA demethylation in mammalian systems. On the DNA level, the TET (ten-eleven translocation) family proteins mediate the reversal of 5mC methylation through iterative oxidation of 5mC to the newly discovered 5-hydroxymethylcytosine (5hmC), 5-formylcytosine (5fC), and 5-carboxylcytosine (5caC) (9–12). The complete reversal of cytosine methylation can be achieved by either replication-dependent dilution of 5mC oxidation derivatives or thymine-DNA glycosylase (TDG)-mediated base excision repair (BER), a pathway involving removal of the entire 5fC or 5caC base and its subsequent repair to replace the residue with unmodified cytosine (Figure 1b) (12–14). On the RNA level, the human AlkB homologs FTO (fat mass– and obesity-associated protein) (2) and ALKBH5 (15) participate in removal of the methyl group of m6A through oxidation (Figure 1c). These newly identified Fe(II)/αKG-dependent dioxygenases and nucleic acid modifications play pivotal roles in regulating development and diseases (16–19). This review highlights the mechanisms and function of the oxidative reversal of DNA and RNA methylation, which is mediated by these newly characterized enzymes. We begin by discussing the current mechanistic understanding of TET-mediated DNA demethylation and its regulation. We then compare methods for the detection of 5hmC, 5fC, and 5caC and review functions of TET-mediated demethylation in diverse biological processes. Finally, we briefly review recent discoveries involving RNA demethylation.
Figure 1.

Overview of the oxidative reversal of mammalian DNA and RNA methylation. (a) DNA methylation damage, such as N1-methyladenosine (m1A) and N3-methylcytosine (m3C), can be repaired by the AlkB family dioxygenases ALKBH2 and ALKBH3 through an unstable hemiaminal intermediate (brackets). (b) DNA methylation at the 5-position of cytosine is deposited by DNA methyltransferases (DNMTs). The Fe(II)/α-ketoglutarate (αKG)-dependent dioxygenases TET1, TET2, and TET3 can oxidize 5-methylcytosine (5mC) to the chemically stable intermediates 5-hydroxymethylcytosine (5hmC), 5-formylcytosine (5fC), and 5-carboxylcytosine (5caC) in a stepwise manner. 5mC, 5hmC, 5fC, and 5caC can be passively converted back to cysteine during DNA replication, when DNMTs fail to methylate the newly synthesized strand (dashed arrows). In addition, 5fC and 5caC can be excised by thymine-DNA glycosylase (TDG) and repaired through base excision repair (BER). (c) RNA methylation modification in the form of N6-methyladenosine (m6A) is generated from adenine by RNA methyltransferase. The AlkB family dioxygenases FTO (fat mass– and obesity-associated protein) and ALKBH5 remove the methyl group of m6A by oxidation. FTO oxidizes m6A to form metastable products of N6-hydroxymethyladenosine (hm6A) and N6-formyladenosine (f 6A), which decompose back to adenosine.
BIOCHEMICAL AND MOLECULAR MECHANISMS OF TET-MEDIATED OXIDATIVE DNA DEMETHYLATION
Although AlkB family dioxygenases vary significantly in terms of domain structure and function (Figure 2a), their oxidation chemistry and catalytic domain are similar to those of the E. coli prototype AlkB (20). In particular, all AlkB family dioxygenases use a base-flipping mechanism to flip their target base out of the double-stranded DNA (dsDNA) helix into their catalytic pocket (Figure 2b), and the core of their catalytic domain contains a double-stranded β-helix (DSBH) fold that is conserved among Fe(II)/αKG-dependent dioxygenases (Figure 2c). The AlkB homologs and TET proteins contain the same His–Xaa–Asp–(Xaa)n–His (where Xaa means any amino acid) iron-binding motif in the catalytic site for oxygen activation (21).
Figure 2.

The structural and chemical basis of methyl oxidation by Fe(II)/α-ketoglutarate (αKG)-dependent dioxygenases. (a) Domain structures of Escherichia coli AlkB, mouse FTO (fat mass– and obesity-associated protein), ALKBH5, JBPs ( J-binding proteins), and TET proteins. TET proteins harbor three conserved domains, including the CXXC zinc finger, the cysteine-rich (Cys-rich) domain, and the double-stranded β-helix (DSBH) fold. (b) Crystal structure of human TET2 catalytic domain with DNA and an αKG analog, N-oxalylglycine (NOG) (Protein Data Bank identifier 4NM6). The conserved β-strands in the DSBH fold are colored green, the α-helixes in the DSBH fold are in yellow, the large insert is in gray, and the Cys-rich domain is in brown. The red and gray spheres represent Fe(II) and Zn(II), respectively. (c) The topology diagram for the conserved DSBH fold. The locations of Fe(II) and the αKG-binding sites are indicated. (d ) The chemical mechanism underlying Fe(II)/αKG-dependent dioxygenase–mediated 5mC oxidation. In the dioxygen activation stage, Fe(II) and αKG activate dioxygen to form a highly active Fe(IV)-oxo species. In the substrate oxidation stage, Fe(IV)-oxo inserts the oxygen atom into the C–H bond of the substrate, and Fe(IV) is reduced back to Fe(II) to complete the catalytic cycle.
The oxidation reaction can be split into two stages: dioxygen activation and substrate oxidation (Figure 2d). The dioxygen activation stage is a four-electron process. At this stage, Fe(II) and αKG may each contribute two electrons to activate a dioxygen molecule first into bridged peroxo and then into the Fe(IV)-oxo intermediate (22). In the subsequent substrate oxidation stage, the highly active Fe(IV)-oxo species oxidizes the inert C–H bond of the substrate, and Fe(IV) is reduced back into Fe(II) to complete the catalytic cycle. Overall, four electrons—two from αKG and two from the substrate C–H bond—are consumed to fully reduce a dioxygen molecule. The two oxygen atoms of the dioxygen molecule are incorporated into the succinate (the oxidized and decarboxylated product of αKG) and the oxidized product (Figure 2d).
In the case of N-methylated substrates, such as m1A and m3C, the initial oxidation renders the C–N bond unstable and undergoes hydrolytic deformylation, causing direct demethylation of the methylation damage. However, in the case of C-methylation substrates, such as 5mC, the oxidized 5-substituents are connected through a C–C bond to the rest of the base and are chemically stable under physiological conditions. In this case, 5hmC can be further oxidized to generate 5fC and even 5caC. Although 5hmC, 5fC, and 5caC are stable under physiological conditions, all of them can serve as intermediates for DNA demethylation, as discussed further in the section titled Potential Mechanisms of TET-Mediated DNA Demethylation, below (Figure 3). Compelling evidence suggests that replication-dependent passive dilution and TDG-mediated excision of 5fC and 5caC, and its subsequent restoration of unmodified cytosine by BER (for a review of BER, see Reference 23), may play predominant roles in completing TET-mediated oxidative DNA demethylation.
Figure 3.
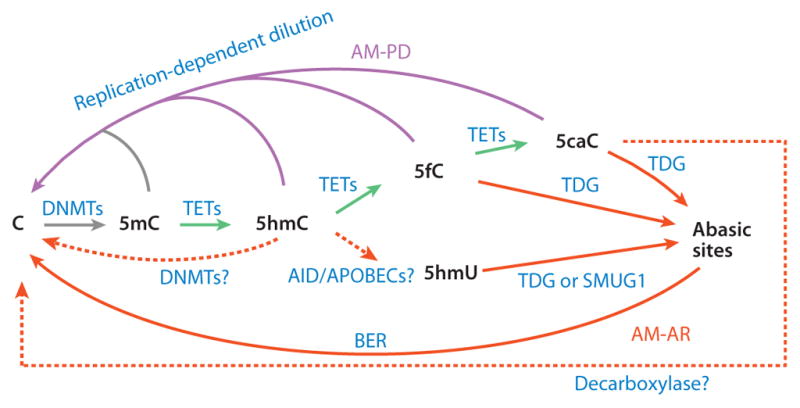
Proposed mechanisms of TET-mediated DNA demethylation. TET proteins can oxidize 5-methylcytosine (5mC) to generate 5-hydroxymethylcytosine (5hmC), which is recognized poorly by DNA methyltransferase 1 (DNMT1) and thus can be diluted during DNA replication. 5hmC can also be further oxidized by TET proteins to produce 5-formylcytosine (5fC) and 5-carboxylcytosine (5caC). Alternatively, 5hmC may be deaminated by AID and APOBECs to become 5-hydroxymethyluracil (5hmU). 5fC, 5caC, and 5hmU can be excised from DNA by glycosylases. In addition, DNMT3A and DNMT3B may directly dehydroxymethylate 5hmC to generate cytosine, and a putative decarboxylase may also directly convert 5caC to cytosine. TET-catalyzed reactions are colored green, AM-AR (active modification followed by active restoration) pathways red, and AM-PD (active modification followed by passive dilution) pathways magenta. Solid lines represent processes with strong evidence; dashed lines indicate processes that either are hypothetical or need further verification. Abbreviation: BER, base excision repair.
TET Family Dioxygenases
The names of TET genes stem from the involvement of the human TET1 gene in the ten-eleven translocation [t(10;11)(q22;q23)] in rare cases of acute myeloid leukemia (AML), which fuses the TET1 gene on chromosome 10 with the mixed-lineage leukemia gene (MLL; also known as KMT2A) on chromosome 11 (24, 25). Two TET1 paralog genes, TET2 and TET3, were identified on the basis of sequence homology. The three TET genes are conserved in all jawed vertebrates, suggesting that the ancestral TET gene underwent a triplication event in the jawed vertebrate lineage (26, 27).
Recently, the crystal structures of a catalytically active truncated human TET2 and a Naegleria Tet-like dioxygenase were reported (28, 29). Similar to all characterized Fe(II)/αKG-dependent dioxygenases, the TET catalytic domain contains a DSBH fold (also known as a jelly-roll fold) (9), which includes eight conserved antiparallel β-strands (I–VIII) and a conserved iron-binding motif (Figure 2b,c) (30). In addition to these common motifs of the DSBH fold, the catalytic domain of TET proteins also has unique characteristics including a cysteine-rich domain adjacent to the N terminus of the DSBH fold and a large, nonconserved, low-complexity region inserted between conserved β-strands IV and V (26, 27). In vitro studies suggest that the cysteine-rich domain, but not the low-complexity insert, is essential for the catalytic activity of TET proteins (30). Indeed, the crystal structure of TET2 catalytic domain reveals that the cysteine-rich domain wraps around the DSBH core and stabilizes the substrate DNA above it (28). Although the low-complexity insert is predicted to be unstructured, and no structural information about this region is available, the insert in TET1 shows sequence similarity to the C-terminal domain of Saccharomyces cerevisiae RNA polymerase II, suggesting a possible regulatory role of this region (31).
TET1 is also known as CXXC6 (CXXC zinc finger 6) and LCX (leukemia-associated CXXC protein) due to the presence of a CXXC (cysteine–X–X–cysteine) domain (24). Both TET1 and TET3, but not TET2, contain a CXXC domain at the N terminus. In vitro DNA-binding analyses and crystal structures of the Xenopus laevis Tet3 CXXC domain have revealed that this domain strongly binds to unmethylated DNA (32). However, the CXXC domain does not appear to account for all of the DNA-binding activity of TET proteins, because catalytic domains of these three TET proteins—without their CXXC domains—can still oxidize 5mC DNA in vitro and in vivo (10). It follows that the catalytic domains of the TET proteins may possess a non-sequence-specific DNA-binding capacity similar to that of most AlkB family proteins (21), whereas the CXXC domain may increase the sequence selectivity to facilitate and regulate binding of TET proteins to their genomic targets (32–34).
TET-Mediated Iterative Oxidation of 5-Methylcytosine
The biochemical function of TET proteins was not known until 2009, when Rao and colleagues (9) demonstrated that human TET1 can convert 5mC of DNA to 5hmC. This finding was inspired by an investigation of the reaction involved in the biosynthesis of base J (β-D-glucosyl-hydroxymethyluracil), which is a modified thymine base found in kinetoplastid genomes. The first step of base J synthesis involves oxidation of the 5-methyl group on thymine to 5-hydroxymethyluracil (5hmU) by J-binding proteins 1 and 2 ( JBP1 and JBP2) (35). Given the structural similarity between 5mC and thymine, mammalian homologs of JBPs were thought to have the capacity to oxidize 5mC (27). This possibility was experimentally confirmed; the mammalian homologs of JBPs—TET proteins—oxidize 5mC to 5hmC (9, 10, 36). Furthermore, the presence of TET genes in metazoans appears to depend on the presence of 5mC in the genome (26, 27), supporting the idea that the biochemical function of TET proteins is 5mC oxidation. Importantly, these studies were further supported by the demonstrations that 5hmC is relatively abundant in Purkinje neurons and mouse embryonic stem cells (ESCs) and that its presence is TET protein dependent (9, 10, 37).
Interestingly, further oxidation of 5hmC was proposed on the basis of the similarity between the chemistry of 5mC demethylation and thymine-to-uracil conversion (38). Thymine-to-uracil conversion is part of the thymidine salvage pathway and involves three thymine-7-hydroxylase [an AlkB-like Fe(II)/αKG-dependent dioxygenase]-catalyzed iterative oxidation reactions to generate isoorotate, whose carboxyl group is subsequently removed by an isoorotate decarboxylase to generate uracil [thymine → 5hmU → 5-formyluracil → 5-carboxyluracil (isoorotate) → uracil] (39). Similar to the iterative oxidation of thymine by thymine-7-hydroxylase, TET proteins can oxidize 5mC not only to 5hmC, but also to 5fC and 5caC in vitro (11, 12). This activity is mechanistically supported by the finding that the active cavity of TET2 recognizes CpG dinucleotide regardless of its methylation status. The lack of specific interactions between the active cavity and the methyl group of 5mC may allow 5mC oxidation derivatives to be oxidated further (28). Consistent with this in vitro activity, 5fC and 5caC can be reliably detected in mouse ESCs in a TET-dependent manner, supporting their biological relevance. However, 5fC and 5caC levels are two orders of magnitude lower than that of 5hmC in mouse ESCs (11, 12, 40). With synthetic oligo substrates, the initial reaction rate of purified TET2 protein toward 5hmC or 5fC is at least fivefold lower than that toward 5mC; turnover numbers of TET2 protein toward 5mC, 5hmC, and 5fC are estimated to be 0.5 min−1, 0.1 min−1, and 0.07 min−1, respectively (11). These results are consistent with the finding that 5hmC, 5fC, and 5caC levels in mammalian genomes are low. Detailed kinetics analyses of the TET-catalyzed reaction are needed for an understanding of its processivity and regulation.
Potential Mechanisms of TET-Mediated DNA Demethylation
DNA methylation is relatively stable compared with most histone modifications. Nevertheless, DNA demethylation, either passive or active, has been observed in various biological contexts. Passive DNA demethylation refers to the loss of 5mC during successive rounds of replication in the absence of functional DNA methylation maintenance machinery, and it has been suggested that this term be used only for replication-dependent dilution of 5mC, but not its oxidation derivatives, to avoid confusion (41). In contrast, active DNA demethylation refers to an enzymatic process that removes or modifies 5mC with regeneration of unmodified cytosine. Thus, all TET-mediated demethylation processes are best viewed as active DNA demethylation.
Once TET enzymes actively convert 5mC to 5hmC, 5fC, and 5caC [referred to as active modification (AM)], this base can be further processed through either (a) passive dilution (PD) to regenerate unmodified cytosine through DNA replication or (b) active restoration (AR) through further enzymatic modification. An AM-AR pathway may function rapidly and seems well suited for locus-specific demethylation that requires a rapid response to environmental stimuli; however, an AM-PD pathway may be well suited for global demethylation events in which DNA replication takes place (41). To date, at least five different demethylation pathways involving TET proteins have been proposed (Figure 3).
The first mechanism is direct removal of the 5-carboxyl group of 5caC by a putative decarboxylase (AM-AR). This potential mechanism is the most straightforward and is mechanistically similar to the last step of thymine-to-uracil conversion in the thymidine salvage pathway (42). Consistent with this possibility, a recent study detected weak 5caC decarboxylase activity in mouse ESC extracts (43). However, the identity of the putative decarboxylase has yet to be revealed.
The second mechanism is direct removal of the 5-hydroxymethyl group from 5hmC (AM-AR). In vitro studies have shown that mammalian de novo DNA methyltransferases (DNMTs) 3A and 3B, as well as the bacterial DNA methyltransferase M.HhaI, can remove the 5-hydroxymethyl group of 5hmC to generate unmodified cytosine in the absence of the methyl donor SAM (44, 45). However, the biological relevance of this reaction is questionable, given that SAM is a general methyl donor of many important biochemical reactions that take place in all cell types.
The third mechanism is replication-dependent dilution of 5mC oxidation products (AM-PD). Previous studies have demonstrated that DNMT1—the DNMT responsible for maintaining DNA methylation during DNA replication—is much less efficient in utilizing hemihydroxymethylated CpGs (5hmC/C) substrates versus hemimethylated CpGs (5mC/C) (46, 47), raising the possibility that 5mC-to-5hmC conversion can facilitate replication-dependent DNA demethylation. Accumulating evidence indicates that, in addition to passive demethylation (i.e., dilution of 5mC), AM-PD also plays an important role in global DNA demethylation during preim-plantation development and primordial germ cell (PGC) reprogramming (see the section titled Biological Functions of TET-Mediated Oxidation of 5-Methylcytosine, below).
The fourth mechanism is 5hmC deamination, followed by glycosylation and BER (AM-AR). One study reported that overexpression of AID/APOBEC deaminases into HEK293 cells demethylates the cotransfected 5hmC-containing reporter and generates the deamination product 5hmU (48). This report raises the possibility that DNA demethylation can be achieved through sequential actions of TET-AID/APOBEC-BER. However, this possible mechanism is challenged by the fact that AID efficiently acts only on single-stranded DNA (ssDNA), not on dsDNA (49). Furthermore, a systematic biochemical study revealed that AID/APOBEC deaminases have no detectable deamination activity on 5hmC (50). Nevertheless, a deamination/BER-dependent mechanism had previously been proposed for the removal of 5mC (whose deamination product is thymine) in various biological systems, including zebrafish embryos, mouse PGCs, and mouse ESC/human fibroblast fused heterokaryons (51–54). Thus, deamination-based DNA demethylation is more likely to act on 5mC than on 5hmC. Further studies are needed to clarify the role of AID/APOBEC in DNA demethylation.
The fifth and final mechanism is that TDG acts robustly on 5fC and 5caC to generate abasic sites, which can be repaired through the BER pathway to restore unmodified cytosine (AM-AR). As discussed in more detail in the following section, this is the most plausible active demethylation pathway, and it has gained the most support from multiple laboratories (11–14, 50).
TDG-Mediated Excision of 5-Formylcytosine and 5-Carboxylcytosine
TDG belongs to the uracil-DNA glycosylase (UDG) superfamily, which uses a base-flipping mechanism to excise target bases from dsDNA to initiate BER. The well-established function of TDG is to remove pyrimidine moiety from guanine/uracil and guanine/thymine mismatches (55). Interestingly, the substrate specificity of TDG appears to depend on the stability of the base–sugar bond (N-glycosidic bond); TDG also efficiently excises properly base-paired cytosine bases with 5-position substituents, such as 5-fluorocytosine, that destabilize the N-glycosidic bond through electronic effects (56).
Computational analyses indicate that 5fC and 5caC destabilize the N-glycosidic bond relative to cytosine, 5mC, 5hmC, and even 5-fluorocytosine (57). TDG can consistently recognize and excise 5fC and 5caC, but not cytosine, 5mC, and 5hmC, from dsDNA when paired with guanine (12, 13). In fact, it has a slightly higher binding affinity toward guanine/5caC and guanine/5fC pairs than to guanine/uracil and guanine/thymine mismatches (13). Co-overexpression of TDG with TET proteins in HEK293 cells depletes TET-generated 5fC and 5caC (12, 50), whereas knockdown of TDG leads to a 5–10-fold increase of 5fC and 5caC in mouse ESCs (12, 58, 59), confirming the 5fC/5caC base-removal activity of TDG.
The crystal structure of human TDG in complex with 5caC-containing dsDNA clearly shows that, compared with the other UDGs [i.e., UNG, methyl-CpG-binding domain protein 4 (MBD4), and single-strand-selective monofunctional uracil-DNA glycosylase 1 (SMUG1)], the active site of TDG is uniquely configured to accommodate 5caC and facilitate its cleavage (14). One of the major functions of TDG in mammals is probably to recognize and excise 5fC and 5caC in the genome. Consistent with this idea is the finding that TDG is the only UDG protein required during mouse embryonic development (60–63). Tdg-knockout mice exhibit developmental defects and die around embryonic day (E)12.5 (60, 61). Importantly, the DNA glycosylase activity of TDG is essential for embryonic development because the Tdg catalytic mutant has the same phenotype as the Tdg null mutant (60). Thus, the unique 5fC and 5caC excision activity of TDG may be essential for DNA demethylation during embryonic development.
REGULATION OF OXIDATIVE DNA DEMETHYLATION
DNA methylation carries cell type– and developmental stage–specific epigenetic information that is critical to the maintenance of proper gene transcription and genome stability. Therefore, both DNA methylation and demethylation must be precisely controlled to avoid dysregulation of gene expression. Although the regulation of DNA methylation has been extensively studied (1, 64), how DNA demethylation is regulated is only beginning to be understood.
Regulation of TET Expression
The three TET proteins exhibit developmental stage– and tissue-specific expression patterns. Specifically, TET1 is highly expressed in the inner cell mass (ICM) of mouse blastocysts (10) and mouse E10.5–13.5 PGCs (65); TET2 is broadly expressed in various mouse tissues (10); and TET3, although exhibiting a broad expression pattern in adult mouse tissues, is the only TET protein that is highly expressed in mouse oocytes and zygotes (66, 67). In line with the upregulation of TET1 in mouse E10.5 PGCs (65), 5mC is actively converted to 5hmC at this time point (68, 69), indicating that regulation of TET expression is a primary way to control 5hmC production.
The regulation of TET expression has been reported at different levels. The upstream region of the mouse Tet1 gene contains a large cluster of binding sites for core pluripotency transcription factors (70), which explains why TET1 is highly expressed in mouse ESCs but are rapidly downregulated after differentiation (10, 71). TET expression is also regulated at the posttranscriptional level. For example, the oncogenic microRNA (miRNA) miR-22 targets TET2 by directly interacting with its 3′ untranslated region (3′UTR) in breast cancer cells and hematopoietic stem cells (72, 73). Interestingly, it appears that both human and mouse TET genes carry predicted miRNA recognition elements of miR-22 within their 3′UTRs (73), implicating a conserved miRNA regulatory mechanism. Furthermore, TET expression can also be regulated at the protein level. For example, one study showed that the CXXC domain–containing protein IDAX (inhibition of the Dvl and Axin complex; also known as CXXC4) directly interacts with the catalytic domain of TET2 to downregulate the TET2 protein through caspase-mediated degradation (34).
Regulation by TET-Interacting Proteins and 5-Hydroxymethylcytosine-Binding Proteins
In addition to IDAX, many other TET-interacting proteins, such as O-linked N-acetylglucosamine (O-GlcNAc) transferase (OGT) and the SIN3A complex (74–78), have been identified. OGT can GlcNAcylate TET proteins; mutation of the putative O-GlcNAcylation site decreases the level of TET1 (74). Although OGT does not directly regulate the enzymatic activity of TET proteins, knockdown of Ogt in mouse ESCs decreases the association of TET1 with chromatin and alters 5hmC enrichment at certain loci (74, 78). Nevertheless, how other TET-interacting partners regulate TET proteins remains to be determined.
In addition to TET-interacting proteins, some 5hmC-binding proteins can also regulate TET-catalyzed 5mC oxidation. For example, one study showed that knockdown of methyl-CpG-binding domain protein 3 (MBD3), which binds to both 5mC and 5hmC, strongly reduces global 5hmC levels in mouse ESCs (79). In another study, UHRF2 was identified as a 5hmC-specific binding protein in neuronal progenitor cells. Surprisingly, UHRF2 can stimulate the processivity of TET1 when co-overexpressed with the catalytic domain of TET1 in HEK293T cells (80). However, the molecular mechanisms underlying these observations, as well as their biological significance, remain to be determined.
Regulation by Metabolites and Cofactors
As discussed above, TET-mediated reaction requires Fe(II) and αKG as cofactors. Because αKG is an intermediate of the tricarboxylic acid cycle, it is reasonable to suppose that cells’ metabolic state, which affects intracellular αKG levels, may influence TET activity. Consistent with this idea, oncometabolite 2-hydroxyglutarate (2HG) inhibits TET proteins by competing with αKG (81, 82). Indeed, the five-carbon dicarboxylic acids 2HG and αKG are chemical analogs, and the substitution of the keto group on αKG to a hydroxyl group on 2HG could interfere with Fe(II) binding and stabilize the reaction intermediate. Remarkably, cellular accumulation of 2HG is often caused by tumor-associated mutations in the NADP+-dependent isocitrate dehydrogenase genes (IDH1 and IDH2), which encode enzymes that normally produce αKG. These tumor-associated IDH1 and IDH2 mutations (affecting R132 of IDH1 or R140 and R172 of IDH2) impair αKG production and confer a neomorphic enzyme activity to convert αKG to 2HG (83, 84), inhibiting TET activity. Co-expression of mutant IDH enzymes with TET consistently inhibits TET-mediated 5mC-to-5hmC conversion (81, 82).
In addition to 2HG, two other metabolites, fumarate and succinate, also share structural similarity with αKG. Both function as competitive inhibitors of Fe(II)/αKG-dependent dioxygenases and accumulate in a subset of human cancers with inactivation mutations of fumarate hydratase (FH) and succinate de-hydrogenase (SDH), respectively (85). Thus, multiple intracellular metabolites may regulate TET-mediated oxidative DNA demethylation, at least under certain pathological conditions.
Recently, three groups reported that ascorbate (vitamin C) enhances the catalytic activity of TET proteins both in vitro and in vivo (86–88). In wild-type (but not Tet1- or Tet2-deficient) mouse ESCs, ascorbate significantly increases the levels of all 5mC oxidation products (86, 88), particularly 5fC and 5caC, by more than an order of magnitude, leading to a global loss of 5mC (~40%) (86). Mechanistically, ascorbate interacts with the catalytic domain of TET and probably facilitates its folding (86). Interestingly, ascorbate-induced demethylation has a stronger effect on DNA sequences that gain methylation in cultured ESCs compared with blastocysts (i.e., DNA sequences that normally gain methylation after implantation) (88). Taken together, these studies suggest that ascorbate directly regulates TET activity and that it may play a critical role in regulating DNA demethylation during development.
Given the importance of precise regulation of DNA methylation in various biological processes, it is not surprising that TET activity is regulated by multiple factors, such as intracellular metabolites, nutritional and developmental signals, stress, and chemical exposure. Note that both ATP and hydroquinone also stimulate TET-mediated 5mC oxidation (12, 89).
GENOME-WIDE MAPPING OF OXIDIZED 5-METHYLCYTOSINE DERIVATIVES
An important approach to understanding the function of TET proteins is to study the genomic distribution of 5mC oxidation products. Investigators have developed methods for mapping newly discovered cytosine derivatives that are proving to be valuable in revealing these proteins’ potential functions (Figure 4a) (90).
Figure 4.

Sequencing methods for 5-methylcytosine (5mC) derivatives. (a) Timeline of sequencing-method development for cytosine derivatives. (b) Affinity-based profiling methods for oxidized 5mC derivatives. Antibody-based DNA immunoprecipitation (DIP) methods are available for 5mC (5mC-DIP-Seq, MBD-Seq), 5-hydroxymethylcytosine (5hmC) (5hmC-DIP-Seq, CMS-IP, JBP1-IP), 5-formylcytosine (5fC) (5fC-DIP-Seq), and 5-carboxylcytosine (5caC) (5caC-DIP-Seq). 5hmC, 5fC, and 5caC can also be chemically and/or enzymatically labeled with biotin. These methods include 5hmC-selective chemical labeling (hMe-Seal) and GLIB (glucosylation, periodate oxidation, biotinylation) for 5hmC profiling, 5fC chemical pull-down, and 5fC-selective chemical labeling (fC-Seal) for 5fC profiling. Abbreviations: CMS, 5-methylenesulfonate; DIP-Seq, DNA immunoprecipitation sequencing; JBP, J-binding protein; MBD-Seq, methyl-binding protein sequencing.
Typically, two types of methods are employed to map the genome-wide distribution of 5mC. The first type is affinity-based profiling that utilizes a specific antibody [DNA immunoprecipitation sequencing (DIP-Seq)] or binding protein (e.g., methyl-binding protein sequencing) to enrich 5mC-containing DNA fragments (Figure 4b) (91). Subsequent high-throughput sequencing of the enriched DNA fragments provides genome-wide distribution of 5mC. Such profiling is inexpensive, but its shortcomings include poor resolution (a few hundred base pairs) and a lack of information about the relative abundance of 5mC at each modification site. The second approach is bisulfite-based base-resolution sequencing (BS-Seq) of 5mC. It relies on first treating DNA with sodium bisulfite (NaHSO3), which deaminates unmodified cytosine but not 5mC. Subsequent polymerase chain reaction (PCR) amplification converts the deaminated cytosine to thymine and 5mC to cytosine (Figure 5). By coupling this bisulfite conversion with high-throughput sequencing, BS-Seq enables quantitative assessment of the abundance of 5mC at base resolution. The only shortcoming of BS-Seq is its relatively high cost (92).
Figure 5.

Bisulfite sequencing (BS-Seq) and modified BS-Seq for base-resolution detection of oxidized 5-methylcytosine (5mC). With bisulfite treatment and polymerase chain reaction (PCR) amplification, BS-Seq reads out the sum of 5mC and 5-hydroxymethylcytosine (5hmC). Oxidative bisulfite sequencing (oxBS-Seq) oxidizes DNA with potassium perruthenate (KRuO4) before bisulfite treatment and PCR amplification. It reads out 5hmC through subtraction from BS-Seq signals. TET-assisted bisulfite sequencing (TAB-Seq) requires the protection of 5hmC with glucosylation before oxidizing DNA with TET proteins. After bisulfite treatment and PCR amplification, TAB-Seq directly reads out 5hmC. 5fC chemical modification–assisted bisulfite sequencing (fCAB-Seq) protects 5fC with EtONH2 before bisulfite treatment and PCR amplification, and after subtracting BS-Seq signals, it reads out 5fC. 5caC chemical modification–assisted bisulfite sequencing (caCAB-Seq) protects 5-carboxylcytosine (5caC) with 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC) coupling before bisulfite treatment and PCR amplification, and after subtracting BS-Seq signals, it reads out 5caC.
The discovery of 5hmC, 5fC, and 5caC requires new sequencing methods to reveal the biological functions associated with these types of modifications. The presence of these cytosine derivatives in the genome complicates base-resolution sequencing of 5mC in BS-Seq because 5hmC behaves similarly to 5mC in BS-Seq (93, 94), whereas 5fC and 5caC behave similarly to unmodified cytosine (Figure 5) (12, 95, 96). As a result, all of the 5mC signals obtained in traditional BS-Seq experiments represent the sum of 5mC and 5hmC.
Recently, researchers developed several sequencing strategies to facilitate mapping of the various cytosine derivatives and to understand the DNA demethylation process. Both affinity-based profiling and base-resolution methods are currently available for 5hmC, 5fC, and 5caC (Figure 4a) (16, 90). We describe these mapping methods and focus on recent advances in base-resolution methods, as well as the biology learned from these studies.
Affinity-Based Methods
Several profiling methods have been developed to map the genomic distribution of the oxidized derivatives of 5mC. Antibodies against all three 5mC oxidation derivatives are available, and antibody-based DIP-Seq has been used to map the genome-wide distribution of 5hmC (70, 75, 97–100), 5fC (58), and 5caC (58). These methods often suffer from high background and bias toward densely modified genomic regions (16). For 5hmC, two approaches involving modifying 5hmC before immunoprecipitation appear to have higher specificity and lower background compared with simply using anti-5hmC antibodies (101). In one approach, 5hmC is converted to 5-methylenesulfonate (CMS) with sodium bisulfite; an anti-CMS antibody has been developed and used for immunoprecipitation (101). In another strategy, 5hmC is converted to glucosylated 5hmC (g5hmC) by T4 bacteriophage β-glucosyltransferase (βGT); thereafter, JBP1, a natural g5hmC-binding protein, is used for immunoprecipitation to enrich g5hmC-containing DNA (102).
Another major type of profiling involves chemically or enzymatically adding a biotin tag to the cytosine modifications to pull them down with avidin/streptavidin beads (Figure 4b). The strong and selective biotin–avidin/streptavidin interaction makes these methods more sensitive, thereby decreasing background signal (103). βGT is a highly efficient and sequence-independent enzyme that can glucosylate 5hmC in synthetic oligos and genomic DNA with a turnover of (~1 × 108) min−1 (104). Two approaches that use βGT to add a biotin to 5hmC have been developed. The first one, 5hmC-selective chemical labeling (hMe-Seal), uses βGT to transfer an azide-modified glucose to 5hmC; thereafter, a cyclooctyne–biotin probe is used to attach a biotin group to 5hmC via azide-cyclooctyne click chemistry (105). The second method, known as GLIB (glucosylation, periodate oxidation, biotinylation), uses βGT to transfer an unmodified glucose to 5hmC, resulting in g5hmC. Next, sodium periodate (NaIO4) is used to oxidize the vicinal hydroxyl groups in the glucose of g5hmC to aldehyde groups, which then react with an aminooxy-biotin probe to attach a biotin group to 5hmC (101).
On the basis of hMe-Seal, a similar 5fC-selective chemical labeling method (fC-Seal) has been developed (59). In fC-Seal, endogenous 5fC in genomic DNA is first blocked by unmodified regular glucose via βGT. Then, 5fC is selectively reduced to 5hmC by sodium borohydride (NaBH4). hMe-Seal is then employed to label the newly generated 5hmC (from 5fC) with biotin (59). 5fC and 5caC can also be directly labeled with a biotin group by use of aminooxyaldehyde condensation (5fC chemical pull-down) (106) and 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC)-mediated amino–carboxyl coupling (107), respectively. However, both transformations have side reactions with other modified cytosine bases or backbones of the DNA, resulting in high background in the subsequent pull-down. For 5hmC, βGT-mediated glucosylation, coupled with restriction endonuclease digestion, has also been used for genome-wide profiling of 5hmC (108, 109).
With the application of these profiling methods, we have a general picture of the genome-wide distribution of the oxidized 5mC derivatives in various cell lines and tissues (16); ESCs and neuronal cells are the two most-studied systems. Note that discrepancies among published studies exist; they may be due to different methodologies and data analyses (16, 103). The potential bias of certain methods also contributes to incomplete results and sometimes inaccurate conclusions. Nevertheless, in mouse and human ESCs, 5hmC is clearly enriched at distal regulatory elements such as enhancers and transcription factor–binding sites; transcription start sites, especially bivalent promoters; and gene bodies, especially exons (70, 75, 97–99, 101, 110). In ESCs, the distributions of 5fC and 5caC generally resemble those of 5hmC but show a preference for distal regulatory elements, such as enhancers and bivalent and silent promoters (58, 59). The levels of 5fC and 5caC further increase at these regions upon TDG depletion, marking sites of TET/TDG-mediated demethylation. These recent studies indicate genome-wide active demethylation in mouse ESCs (58, 59). In mouse and human neuronal cells, studies have consistently shown that 5hmC is enriched at gene bodies and that the enrichment level positively correlates with the gene-expression level (100, 105, 111–113). 5hmC is also depleted from transcription start sites (111). Compared with studies in ESCs, these observations suggest a distinct and perhaps more active regulatory role of 5hmC in neuronal cells.
Base-Resolution Mapping of Oxidized 5-Methylcytosine Derivatives
Affinity-based profiling methods have provided valuable initial biological insights into 5hmC, 5fC, and 5caC; however, low resolution and a lack of quantitative information at each modification site are limitations. Quantitative measurements of these modifications at base resolution are highly desirable to obtain more precise biological information. For this purpose, several high-resolution sequencing methods, including single-molecule sequencing, modified BS-Seq, and restriction endonuclease–coupled sequencing, have been developed.
Third-generation sequencing technologies, involving single-molecule sequencing and amplification-free sample preparation, can directly detect DNA modifications during sequencing (114). These technologies include the well-established techniques of single-molecule, real-time (SMRT) sequencing and nanopore sequencing. SMRT sequencing monitors the fluorescent and kinetic signal of individual DNA polymerases during each cycle of DNA synthesis, enabling the detection of various base modifications, including 5hmC (115), 5fC (116), and 5caC (116). Through the integration of hMe-Seal, SMRT sequencing has been used to detect base-resolution 5hmC in mouse ESC DNA (115). Nanopore sequencing, which records the signal that arises when individual DNA molecules pass through nanoscale pores, has also been used to detect 5hmC in synthetic oligos (117–119). These single-molecule sequencing technologies promise to provide base-resolution maps of various nucleic acid modifications in the future, but further technological developments are needed for genome-wide mapping in mammalian cells.
Currently, a widely used strategy for base-resolution mapping of oxidized 5mC bases is to modify BS-Seq by exploring different properties of cytosine derivatives in bisulfite treatment (Figure 5). As a reminder, cytosine, 5fC, and 5caC are deaminated and converted to thymine after bisulfite treatment and PCR amplification, whereas 5mC and 5hmC remain as cytosine. Two methods have recently been developed for genome-wide base-resolution 5hmC mapping. The first method, oxidative bisulfite (oxBS) sequencing (oxBS-Seq), uses potassium perruthenate (KRuO4) to chemically oxidize 5hmC to 5fC; therefore, only 5mC is retained as cytosine in oxBS. Base-resolution information about 5hmC can be obtained by subtracting oxBS-Seq signals (5mC only) from BS-Seq (5mC and 5hmC) (95). In the second method, TET-assisted bisulfite sequencing (TAB-Seq), 5hmC is protected from TET oxidation through conversion to g5hmC using βGT; then, 5mC is oxidized to 5caC by use of mouse TET1 protein. Thus, TAB-Seq directly reads out 5hmC as the only cytosine signal in the subsequent BS-Seq data (96). Subtraction of TAB-Seq signals from BS-Seq signals yields base-resolution information about 5mC. Both oxBS-Seq and TAB-Seq can deliver quantitative-abundance information about 5hmC and 5mC. Due to the extensive DNA degradation caused by KRuO4 oxidation, so far oxBS-Seq has been applied to only a fraction of the mammalian genome in reduced representation BS-Seq (95), whereas TAB-Seq has been used to map both genome-wide and loci-specific 5hmC (32, 96, 113, 120). However, note that, at normal sequencing depth, TAB-Seq can detect only relatively abundant 5hmC sites (>20%) (96).
Very recently, modified BS-Seq for base-resolution 5fC and 5caC mapping became available (Figure 5). 5fC chemical modification–assisted bisulfite sequencing (fCAB-Seq) utilizes O-ethylhydroxylamine (EtONH2) to chemically block 5fC from deamination in BS-Seq (59). Similarly, 5caC chemical modification–assisted bisulfite sequencing (caCAB-Seq) uses EDC-based coupling to chemically block 5caC from bisulfite deamination (107). Together with conventional BS-Seq, fCAB-Seq and caCAB-Seq reveal quantitative base-resolution information about 5fC and 5caC, respectively. Due to the low abundance of 5fC and 5caC, a maximum sequencing depth is needed to confidently detect them at base resolution. For instance, fCAB-Seq requires ~1,000× or higher coverage to map 5fC in a single locus (59). Thus, currently it is still impractical to use fCAB-Seq or caCAB-Seq in whole-genome sequencing. However, fCAB-Seq can be combined with chromatin immunoprecipitation to map a fraction of the genome enriched with 5fC (59). Coupling enrichment with CAB-Seq could be a practical approach to obtaining genome-wide, base-resolution information about 5fC and 5caC.
A unique technique known as AbaSI coupled with sequencing (Aba-Seq) has also been developed for high-resolution genome-wide mapping of 5hmC (121). AbaSI is a member of a family of 5hmC-dependent restriction endonucleases that recognize and cut g5hmC; the recognition sequence is 5′-g5hmCN11–13↓N9–16G-3′/3′-GN9–10↓N11–13X-5′ (where N represents any base, ↓ indicates a cutting site, and the activity is as follows: X = g5hmC > 5hmC > 5mC > cytosine). After βGT glucosylation and AbaSI digestion, Aba-Seq sequences the enriched cleaved ends to determine the 5hmC sites. This approach is not exactly base resolution. A preference for symmetrical 5hmC, as well as residual activity toward other forms of cytosine, may complicate data analysis; however, Aba-Seq has the advantage of detecting low-abundance 5hmC sites with a relatively low sequencing depth (121).
The application of base-resolution methods has revealed new aspects of 5mC oxidation that were difficult to determine through affinity-based profiling. TAB-Seq has been used to map 5hmC sites in both mouse and human ESCs (96) and frontal cortex (120). In contrast to the prevalence of non-CpG methylation (CpH, where H = A, C, or T) in ESCs and mature neurons, nearly all the 5hmC in these cells is found in a CpG context (96, 120), consistent with the finding that the active cavity of TET2 specifically recognizes CpG di-nucleotides (28). In addition, TAB-Seq results revealed that most 5hmC sites exist in distal regulatory elements, such as enhancers, and near (but not on) transcription factor–binding sites with reciprocal low levels of 5mC; this finding implicates dynamic DNA methylation or demethylation processes at these elements (96). Recently, fCAB-Seq was used to confirm the presence of 5fC at selective loci and the preference of 5fC for poised enhancers compared with active enhancers (59).
BIOLOGICAL FUNCTIONS OF TET-MEDIATED OXIDATION OF 5-METHYLCYTOSINE
With increasing biochemical knowledge about the TET-mediated DNA demethylation, as well as emerging technologies for mapping 5mC oxidation products, we are beginning to understand the functions of TET-mediated 5mC oxidation in the biological processes in which DNA demethylation takes place. Two major waves of global DNA demethylation occur in mammalian development (4, 122). The first wave occurs at the beginning of the mammalian life cycle upon fertilization of eggs by sperm. The second wave takes place in the developing PGCs. Given their roles in DNA demethylation, TET proteins have been extensively studied in these systems.
Role in Preimplantation Development
Approximately 30 years ago, studies using methylation-sensitive restriction enzyme digestion demonstrated that the sperm genome is relatively hypermethylated compared with the egg genome and that a global demethylation event takes place during preimplantation development (123). These observations were later confirmed by immunostaining and locus-specific BS-Seq (124, 125). Interestingly, the paternal and maternal pronuclei undergo DNA demethylation with distinct kinetics (Figure 6). Loss of 5mC signal in the sperm-derived paternal genome takes place immediately after fertilization, before the first round of DNA replication commences, suggesting active erasure of 5mC. By contrast, egg-derived maternal genome is demethylated during the subsequent cleavage divisions, indicating passive demethylation. These observations raised two important questions regarding the identity of the involved enzymes and the molecular mechanism underlying the asymmetric DNA methylation reprogramming. Follow-up studies demonstrated that PGC7 (also known as DPPA3 or Stella), a maternal factor that is essential for early development, protects the maternal genome from active demethylation (126). However, the enzyme responsible for active demethylation itself had been elusive until recently.
Figure 6.

Dynamics of 5-methylcytosine (5mC) and its oxidation derivatives during preimplantation development. After fertilization, 5mC in the paternal genome is quickly converted to 5-hydroxymethylcytosine/5-formylcytosine/5-carboxylcytosine (5hmC/5fC/5caC), which is then diluted through a replication-dependent process, whereas the maternal genome simply goes through replication-dependent passive DNA demethylation. At the blastocyst stage, DNA methylation patterns in the inner cell mass are quickly reestablished. The expression levels of Tet1, Tet2, Tet3, and Tdg are indicated (the dashed line represents cytoplasmic localization).
The first indication that TET proteins might be responsible for the rapid loss of 5mC in paternal genome of zygotes came from immunostaining studies that used antibodies specific for 5hmC. These studies demonstrated that loss of 5mC coincides with the appearance of 5hmC in the paternal genome (66, 67). Consistent with the idea that TET3 is responsible for this process, small interfering RNA–mediated depletion or targeted knockout of Tet3 prevented 5mC-to-5hmC conversion in the paternal genome (66, 127). Interestingly, although TET3 actively converts 5mC to 5hmC, high-resolution chromosome immunostaining revealed that the 5hmC generated in zygotes is relatively stable and is gradually lost in a replication-dependent manner during preimplantation development (128). Similar studies also revealed that the generation and disappearance of 5fC and 5caC follow the same pattern as those of 5hmC (129), implicating a dilution mechanism of the oxidized 5mC derivatives during preimplantation development (Figure 6).
TET3-mediated oxidation of 5mC in the paternal genome appears to be a well-controlled process. TET3 is a maternal protein that is uniformly distributed in the egg cytoplasm. Upon fertilization, TET3 is specifically enriched in the paternal pronucleus but not the maternal pronucleus (127). The paternal pronucleus–specific localization of TET3 might be linked to the maternal pronucleus–specific localization of the PGC7 protein (130). A recent study indicated that the maternal pronucleus–specific histone H3K9 dimethylation (H3K9me2) is responsible for attracting PGC7 to the maternal pronucleus (130), but how PGC7 prevents TET3 from entering maternal pronucleus remains to be determined. Notably, PGC7 may also contribute to the protection of imprinted genes from demethylation during preimplantation development. For example, methylation of H19 and Rasgrf1 differentially methylated regions (DMRs) is lost in embryos derived from PGC7-null oocytes (126). However, not all paternally imprinted genes are affected by the loss of PGC7 [e.g., the DMR of the Dlk1–Gtl2/Meg3 domain is unaffected (126)], indicating that PGC7 is not the only factor involved in protecting methylated DNA from demethylation.
Role in Primordial Germ Cell Reprogramming
During early embryonic development, global DNA methylation reaches its lowest point at E3.5 in the ICM; then, remethylation begins and reaches completion by E6.5 (131). Because PGCs originate from proximal epiblasts at ~E6.5, they largely inherit the newly established DNA methylation pattern of the epiblast cells. Erasure of this DNA methylation pattern, including the parent-of-origin-specific DNA methylation imprints, is essential in resetting the PGC epigenome to prepare these cells for germ cell development (132). In line with the requirement for demethylation in this process, both TET1 and TET2 are expressed in developing PGCs (Figure 7), and several recent studies have demonstrated an important role of TET-mediated 5mC oxidation during this process (65, 68, 69, 133, 134).
Figure 7.
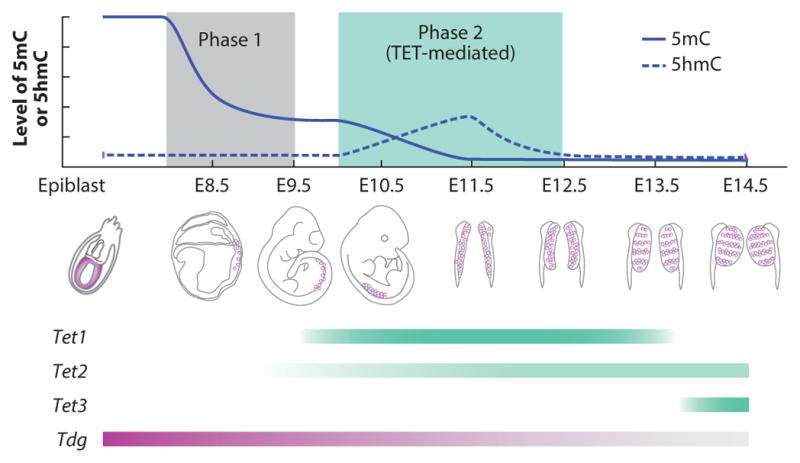
DNA methylation dynamics during primordial germ cell (PGC) reprogramming. Genome-wide DNA demethylation in developing PGCs takes place in two steps. First, during embryonic day (E)7.25 to E9.5, the bulk of the genome is demethylated in a replication-dependent but TET-independent manner. Second, at E9.5, TET1 and possibly TET2 convert the remaining 5-methylcytosine (5mC) to 5-hydroxymethylcytosine (5hmC), which is then diluted through a replication-dependent process to complete the demethylation in PGC reprogramming. The expression levels of Tet1, Tet2, Tet3, and Tdg are indicated.
Previous studies have demonstrated that global erasure of DNA demethylation takes place at E8.5–E12.5 PGCs, leading to almost complete erasure of DNA methylation by E13.5 (53, 135, 136). Several recent studies have revealed that PGC demethylation occurs in two distinct phases and employs both replication-dependent and -independent mechanisms (68, 69, 137, 138). At E9.5, ~70% of the DNA methylation in PGCs is lost (Figure 7) (138). Passive demethylation is probably responsible for this phase of DNA demethylation because Uhrf1, the gene that encodes for the cofactor essential for DNA methylation maintenance, is not expressed (Figure 7) (139). However, the methylation pattern of some regions, such as imprinting control regions, is maintained during this period, and these regions are not demethylated until the second phase (E9.5–E12.5), after PGCs enter the genital ridge. The second phase coincides with the upregulation of TET1 expression and the generation of 5hmC (Figure 7), indicating that TET1 functions in the second phase. Oxidation of 5mC by TET1 (and possibly TET2) in this phase is a well-controlled active process, but erasure of the resulting 5hmC is probably a replication-dependent process (68, 69, 137). Several sequence classes that escape the first wave of DNA demethylation are demethylated in this phase. These sequence classes, including DMRs of imprinted genes, promoters of gametogenesis-related genes, and CpG islands of the X chromosome (68, 137, 138), suggest that TET proteins are functionally important in the removal of locus-specific 5mC in the second stage of PGC reprogramming.
In support of the locus-specific effect of TET proteins in PGC reprogramming, depletion of Tet1 alone or of both Tet1 and Tet2 does not lead to a global increase of 5mC levels in E13.5 PGCs or sperm (65, 140). Hypermethylation of the DMRs of some imprinted genes has been observed in some offspring of Tet1 and Tet2 double-knockout mice (140), implicating incomplete demethylation of DMRs during PGC reprogramming. Definitive evidence for the involvement of TET1 in imprinting erasure came from an analysis of the offspring of homozygous Tet1-null male mice crossed with wild-type female mice (141). Although all these mice were heterozygous, they exhibited various imprinting abnormality–related defects, including early embryonic lethality, placental and embryonic growth defects, and postnatal growth retardation. RNA-Seq and BS-Seq of embryonic tissues revealed defective demethylation at the DMRs of various imprinted genes, including Peg10 and Peg3. Similar defects were observed in the placentas and were traced back to the sperm of the mutant mice. Finally, reduced representation BS-Seq analysis of E13.5 PGCs of mutant male mice showed hypermethylation in the DMRs of imprinted genes and gametogenesis-related gene promoters (141); both groups of genes belong to the late-demethylation gene groups during PGC reprogramming (138), a finding consistent with TET1 playing a role in the second phase of PGC reprogramming. Collectively, these studies have established a critical function of TET1 in imprinting erasure during PGC reprogramming.
Role in Stem Cell and Somatic Cell Reprogramming
DNA methylation dynamics has also been observed during stem cell differentiation and somatic cell reprogramming. Mouse ESCs are derived from the ICM of E3.5 blastocysts. At that time, global DNA methylation has reached its lowest point and de novo establishment of DNA methylation pattern is about to begin. In ESCs cultured under the standard condition (in a medium containing serum and leukemia inhibitory factor), the machinery for both de novo DNA methylation (DNMT3A and DNMT3B) and DNA demethylation (TET1, TET2, and TDG) is present, making ESCs a unique system for study of the regulation of these cytosine-modifying enzymes. The biological functions of TET proteins in ESCs have been reviewed elsewhere (16, 142, 143), but whether the biological functions of TET proteins are related to their catalytic activities is not very well understood. TET proteins may upregulate gene expression through their oxidative demethylation function (144) or by recruiting OGT to gene promoters (74, 76–78), and they may also downregulate gene expression by facilitating the recruitment of Polycomb repressive complex 2 and/or SIN3A corepressor complex (75, 144).
The function of TET-mediated 5mC oxidation has been more clearly demonstrated in ESCs cultured under the 2i condition. Mass spectrometry and genome-wide BS-Seq results showed that changing ESC culture conditions from serum to 2i induces genome-wide demethylation, resulting in a hypomethylated genome that resembles the cells in the ICM of E3.5 blastocysts (145–147). Detailed analyses of DNA methylation showed that, after switching to the 2i condition, DNA methylation decreases in a stepwise manner concomitantly with an increase in the 5hmC level, and that the demethylated regions significantly overlap with TET1-binding sites in ESCs (146, 147). Furthermore, a combination of Tet1 knockout and Tet2 knockdown substantially reduced 5hmC and delayed demethylation upon 2i treatment (146), suggesting that TET-mediated 5mC oxidation is involved in this 2i-induced demethylation process. Because 2i-induced demethylation is a slow process that takes ~12 days (147), the underlying mechanism is probably passive dilution of 5hmC. More detailed genome-wide studies of both 5mC and 5hmC that compare wild-type and Tet1 and Tet2 double-deficient ESCs are required to elucidate how and to what extent TET1 and TET2 are involved in this demethylation process.
Previous mass spectrometry analyses showed that 5fC and 5caC are relatively abundant in ESCs compared with other cells and tissues (11), suggesting that TET/TDG-mediated active DNA demethylation activity is present in ESCs. Indeed, a recent genome-wide mapping study of 5fC/5caC in mouse ESCs revealed the accumulation of 5fC/5caC at poised/active distal regulatory elements and poised/inactive promoters of lineage-specific genes, especially upon Tdg depletion (58, 59). These findings suggest that dynamic DNA methylation/demethylation is present at those loci. Given that the level of de novo DNA methylation activity in ESCs is high, the presence of TET/TDG-mediated active DNA demethylation activity may help precisely and efficiently correct methylation errors accidentally generated by the de novo DNMTs and ensure a proper DNA methylation pattern in ESCs and during differentiation.
In addition to contributing to ESC maintenance, TET proteins also involve reprogramming of somatic cells to generate induced pluripotent stem cells. For example, at the early stage of the transduction of the Yamanaka factors (OCT4, SOX2, KLF4, and c-MYC), TET2 is recruited to the Nanog and Esrrb loci to activate their expression (148). In addition, both TET1 and TET2 can associate with NANOG and facilitate induced pluripo-tent stem cell generation in an enzymatic activity–dependent manner (149). Remarkably, TET1 can not only enhance the reprogramming efficiency, but also replace OCT4 in the reprogramming cocktail (150). Furthermore, beyond transcription factor–mediated reprogramming, TET proteins also contribute to cell fusion–mediated somatic cell reprogramming (151).
Role in Cancer
Aberrant DNA methylation is the most common molecular lesion in cancer cells; it usually causes global DNA hypomethylation and locus-specific promoter hypermethylation (6, 152). The first indication that TET proteins may have a role in cancer was the finding that TET1 is a fusion partner of MLL in rare cases of AML (24, 25). TET1 has also been implicated in suppressing breast cancer growth and metastasis through demethylation of specific genes (153). In addition, TET2 mutations have frequently been found in myeloid malignancies (~15%) (154–156). Indeed, genetic studies demonstrated that TET2 is critical for hematopoietic stem cell self-renewal and differentiation in mouse models, and confirmed that Tet2 inactivation leads to the development of myeloid malignancies (157–160). A global loss of 5hmC levels has been observed in various types of cancers, such as human skin, myeloid, breast, liver, lung, pancreatic, and prostate cancers (36, 161–163), suggesting that impairment of TET-mediated 5mC oxidation may be common in cancer development. Indeed, TET proteins have been reported as targets of oncogenic miRNAs (72, 73), and downregulation of TET expression is frequently observed in cancer cells (162). Therefore, TET proteins generally function as tumor suppressors. However, a recent study also indicated that TET1 plays an oncogenic role in MLL-rearranged leukemia (164), although whether this role is relevant to its 5mC oxidation activity has yet to be determined.
Not only are mutations in TET genes themselves associated with tumorigenesis; mutations in the pathways affecting the generation of cofactors of TET-mediated reactions also contribute to tumor formation. As discussed above, tumor-associated mutations in IDH1 and IDH2 or inactivating mutations in FH and SDH can cause the accumulation of αKG analogs (2HG, fumarate, and succinate) that can inhibit TET-mediated 5mC oxidation. Interestingly, although both are frequently found in AML, IDH1 and IDH2 mutations and TET2 mutations are mutually exclusive (81), suggesting that neomorphic mutations in IDH1 and IDH2 contribute to tumorigenesis by inactivating TET2 in AML. Thus, the inhibition of TET-mediated 5mC oxidation and the consequent global loss of 5hmC appear to be hallmarks of a subset of tumors. Further studies are needed to understand how and to what extent the dysregulation of TET activity contributes to aberrant DNA methylation and tumorigenesis.
RNA METHYLATION AND DEMETHYLATION
Methylation modifications are widely spread in mammalian RNA in the forms of N7-methylguanosine, N6-methyl-2′-O-methyladenosine, 2′-O-methylated nucleosides, 5mC, and m6A (17). The reversibility of RNA methylation was first demonstrated in 2011, when investigators found that FTO exhibits robust m6A RNA demethylase activity (2), indicating that reversal of this most prevalent mRNA modification in mammalian cells is possible (165). To date, two AlkB family dioxygenases, FTO and ALKBH5 (15), are known to efficiently convert m6A to adenosine (Figure 1c). FTO and ALKBH5 are efficient mammalian m6A demethylases with turnover numbers ranging from 0.1 to 0.3 min−1 (2, 15), approximately 10-fold lower than that of the E. coli prototype AlkB (166). FTO affects human obesity and energy homeostasis (167–169) and may reverse m6A methylation in specific mRNAs that are important for neuronal signaling in midbrain (170). ALKBH5, however, affects mRNA export and RNA metabolism. Alkbh5 deficiency leads to increased levels of m6A in mRNA, and male Alkbh5-null mice exhibit impaired fertility due to their deficiency in spermatogenesis (15). Potential m6A-specific binding proteins have also been suggested (171, 172).
Increasing evidence supports the idea that reversible RNA m6A modification plays broad and critical roles in fundamental biological processes in mammals (5, 17, 173, 174). In addition to chromatin-based epigenetic regulations, dynamic RNA modifications can add another layer of complexity to biological regulation (173, 174). Interestingly, biochemical and cellular studies have shown that FTO can oxidize m6A to short-lived, previously unknown intermediates, namely N6-hydroxymethyladenosine (hm6A) and N6-formyladenosine (f 6A), in a stepwise manner (Figure 1c) (172); this process is similar to the oxidation of 5mC to 5hmC and then 5fC by the TET proteins. hm6A and f 6A are relatively stable under physiological conditions, with a half-life of ~3 h, and can be detected in mammalian cells (172). These new RNA modifications, introduced by the demethylase, may add to the dynamics of reversible RNA methylation–dependent regulation.
Transcriptome-wide m6A distribution in mice and humans has been determined using antibody-based profiling approaches (170, 171, 175). The sequencing results revealed that m6A is preferentially enriched around stop codons, in 3′UTRs, and within long internal exons and that its distribution is dynamically modulated throughout development. m6A in mRNA may affect mRNA splicing, trafficking, translation, and stability (5, 17). Future studies will focus on the fundamental roles of m6A and its dynamic regulation in specific biological contexts.
PERSPECTIVES
DNA and RNA molecules carry sequence information to guide downstream events, including transcription and translation. Such information determines not only what products (RNAs or proteins) to make, but also (in part) how much of the products to make. To increase the diversity of regulation so that organisms can cope with environmental and developmental challenges, cells have developed other regulatory mechanisms, including methylation on both DNA and RNA. Although DNA and RNA methylation had been regarded as a relatively stable modification, the finding that both DNA and RNA methylation can be reversed by oxidation reactions has opened the door to an understanding of DNA and RNA methylation dynamics. Studies performed during the past 5 years have not only revealed enzymes involved in DNA and RNA demethylation, but also provided many new technologies to perform high-resolution mapping of the various modified bases. With these tools in hand, future studies will reveal many more exciting insights into molecular mechanisms and biological functions of the dynamic methylation of DNA and RNA.
SUMMARY POINTS.
5mC in DNA and m6A in mRNA carry important regulatory information. Active removal of these modifications is initiated by the AlkB-like Fe(II)/αKG-dependent dioxygenases. Specifically, TET proteins can oxidize 5mC to generate 5hmC, 5fC, and 5caC; FTO and ALKBH5 can oxidatively reverse m6A methylation in mRNA.
hm6A and f 6A are short-lived intermediates formed from FTO-mediated m6A oxidation and can spontaneously resolve within hours to achieve demethylation. However, 5hmC, 5fC, and 5caC are quite stable under physiological conditions. These bases can be gradually removed by passive dilution. Alternatively, 5fC and 5caC can be actively reverted to unmodified cytosine through TDG-initiated BER.
Various technologies have been developed for genome-wide mapping of 5mC oxidation derivatives.
FUTURE ISSUES.
Both AM-PD and AM-AR take place in different biological contexts. Although AM-PD is involved mainly in global DNA demethylation during preimplantation development and PGC reprogramming, AM-AR is probably involved in locus-specific demethylation in response to environmental stimuli, such as nuclear hormone and growth factors (176–178). Future studies are needed to reveal the mechanistic details of these two types of TET-mediated demethylation pathways.
In addition to the TET/TDG/BER pathway, is there another enzymatic pathway for AM-AR? Does a 5caC decarboxylase exist? Can DNMTs directly remove the oxidized 5-substituents of the 5mC derivatives?
How is the processivity of TET-mediated 5mC oxidation regulated? Why does the oxidation reaction mostly stall at 5hmC? Detailed kinetics analyses of TET proteins may help address these issues.
In preimplantation development and PGC reprogramming, some parts of the genome are resistant to demethylation. What is the underlying mechanism, and how are the resistant regions selected?
What are the roles of 5hmC, 5fC, and 5caC as epigenetic modifications, and are there specific proteins that bind and interpret them?
Although TET proteins are present in all metazoans with 5mC in their genomes, oxidation of 5mC is not detected in zebrafish during early embryogenesis. What is the role of TET proteins in zebrafish? Understanding the functions of TET proteins in nonmammalian organisms may provide evolutionary insight into the role of dynamic regulation of DNA methylation.
What is the biological function of m6A in mRNA, and how is it regulated?
Acknowledgments
We thank Dr. Xianchi Dong for his help in preparing Figure 2b and Drs. Hao Wu and Damian Sendler for critical reading of this manuscript. We apologize to colleagues whose work cannot be cited owing to space constraints. Our research was supported by National Institutes of Health grants GM68804 and U01DK089565 (to Y.Z.) and GM071440 and HG006827 (to C.H.). Y.Z. is an investigator of the Howard Hughes Medical Institute.
Glossary
- 5mC
5-methylcytosine
- m6A
N6-methyladenosine
- SAM
S-adenosylmethionine
- αKG
α-ketoglutarate
- 5hmC
5-hydroxymethylcytosine
- 5fC
5-formylcytosine
- 5caC
5-carboxylcytosine
- TDG
thymine-DNA glycosylase
- BER
base excision repair
- DSBH
double-stranded β-helix
- AML
acute myeloid leukemia
- MLL
mixed-lineage leukemia
- 5hmU
5-hydroxymethyluracil
- JBP
J-binding protein
- ESCs
embryonic stem cells
- AM-AR
active modification (AM) of 5mC followed by active restoration (AR) of unmodified cytosine
- AM-PD
active modification (AM) of 5mC followed by replication-dependent passive dilution (PD)
- DNMT
DNA methyltransferase
- PGC
primordial germ cell
- UDG
uracil-DNA glycosylase
- UTR
untranslated region
- OGT
O-GlcNAc transferase
- 2HG
2-hydroxyglutarate
- IDH
isocitrate dehydrogenase
- FH
fumarate hydratase
- SDH
succinate dehydrogenase
- DIP-Seq
DNA immunoprecipitation sequencing
- BS-Seq
bisulfite sequencing
- g5hmC
glucosylated 5hmC
- βGT
β-glucosyltransferase
- DMR
differentially methylated region
- 2i condition
serum-free medium supplemented with two small-molecule kinase inhibitors, ERK1/2 inhibitor PD0325901 and GSK3βinhibitor CHIR99021
Footnotes
DISCLOSURE STATEMENT
The authors are not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
Contributor Information
Chuan He, Email: chuanhe@uchicago.edu.
Yi Zhang, Email: yzhang@genetics.med.harvard.edu.
LITERATURE CITED
- 1.Goll MG, Bestor TH. Eukaryotic cytosine methyltransferases. Annu Rev Biochem. 2005;74:481–514. doi: 10.1146/annurev.biochem.74.010904.153721. [DOI] [PubMed] [Google Scholar]
- 2.Jia G, Fu Y, Zhao X, Dai Q, Zheng G, et al. N6-Methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat Chem Biol. 2011;7:885–87. doi: 10.1038/nchembio.687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Klose RJ, Bird AP. Genomic DNA methylation: the mark and its mediators. Trends Biochem Sci. 2006;31:89–97. doi: 10.1016/j.tibs.2005.12.008. [DOI] [PubMed] [Google Scholar]
- 4.Smith ZD, Meissner A. DNA methylation: roles in mammalian development. Nat Rev Genet. 2013;14:204–20. doi: 10.1038/nrg3354. [DOI] [PubMed] [Google Scholar]
- 5.Fu Y, He C. Nucleic acid modifications with epigenetic significance. Curr Opin Chem Biol. 2012;16:516–24. doi: 10.1016/j.cbpa.2012.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Baylin SB, Jones PA. A decade of exploring the cancer epigenome—biological and translational implications. Nat Rev Cancer. 2011;11:726–34. doi: 10.1038/nrc3130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Walport LJ, Hopkinson RJ, Schofield CJ. Mechanisms of human histone and nucleic acid demethylases. Curr Opin Chem Biol. 2012;16:525–34. doi: 10.1016/j.cbpa.2012.09.015. [DOI] [PubMed] [Google Scholar]
- 8.Yi C, He C. DNA repair by reversal of DNA damage. Cold Spring Harb Perspect Biol. 2013;5:a012575. doi: 10.1101/cshperspect.a012575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science. 2009;324:930–35. doi: 10.1126/science.1170116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ito S, D’Alessio AC, Taranova OV, Hong K, Sowers LC, Zhang Y. Role of Tet proteins in 5mC to 5hmC conversion, ES-cell self-renewal and inner cell mass specification. Nature. 2010;466:1129–33. doi: 10.1038/nature09303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ito S, Shen L, Dai Q, Wu SC, Collins LB, et al. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science. 2011;333:1300–3. doi: 10.1126/science.1210597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.He YF, Li BZ, Li Z, Liu P, Wang Y, et al. Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science. 2011;333:1303–7. doi: 10.1126/science.1210944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Maiti A, Drohat AC. Thymine DNA glycosylase can rapidly excise 5-formylcytosine and 5-carboxylcytosine: potential implications for active demethylation of CpG sites. J Biol Chem. 2011;286:35334–38. doi: 10.1074/jbc.C111.284620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang L, Lu X, Lu J, Liang H, Dai Q, et al. Thymine DNA glycosylase specifically recognizes 5-carboxylcytosine-modified DNA. Nat Chem Biol. 2012;8:328–30. doi: 10.1038/nchembio.914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zheng G, Dahl JA, Niu Y, Fedorcsak P, Huang CM, et al. ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol Cell. 2013;49:18–29. doi: 10.1016/j.molcel.2012.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pastor WA, Aravind L, Rao A. TETonic shift: biological roles of TET proteins in DNA demethylation and transcription. Nat Rev Mol Cell Biol. 2013;14:341–56. doi: 10.1038/nrm3589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jia G, Fu Y, He C. Reversible RNA adenosine methylation in biological regulation. Trends Genet. 2013;29:108–15. doi: 10.1016/j.tig.2012.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shen L, Zhang Y. 5-Hydroxymethylcytosine: generation, fate, and genomic distribution. Curr Opin Cell Biol. 2013;25:289–96. doi: 10.1016/j.ceb.2013.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Koh KP, Rao A. DNA methylation and methylcytosine oxidation in cell fate decisions. Curr Opin Cell Biol. 2013;25:152–61. doi: 10.1016/j.ceb.2013.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Loenarz C, Schofield CJ. Physiological and biochemical aspects of hydroxylations and demethylations catalyzed by human 2-oxoglutarate oxygenases. Trends Biochem Sci. 2011;36:7–18. doi: 10.1016/j.tibs.2010.07.002. [DOI] [PubMed] [Google Scholar]
- 21.Yi C, Yang CG, He C. A non-heme iron–mediated chemical demethylation in DNA and RNA. Acc Chem Res. 2009;42:519–29. doi: 10.1021/ar800178j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Krebs C, Galonić Fujimori D, Walsh CT, Bollinger JM., Jr Non-heme Fe(IV)-oxo intermediates. Acc Chem Res. 2007;40:484–92. doi: 10.1021/ar700066p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fromme JC, Verdine GL. Base excision repair. Adv Protein Chem. 2004;69:1–41. doi: 10.1016/S0065-3233(04)69001-2. [DOI] [PubMed] [Google Scholar]
- 24.Ono R, Taki T, Taketani T, Taniwaki M, Kobayashi H, Hayashi Y. LCX, leukemia-associated protein with a CXXC domain, is fused to MLL in acute myeloid leukemia with trilineage dysplasia having t(10;11)(q22;q23) Cancer Res. 2002;62:4075–80. [PubMed] [Google Scholar]
- 25.Lorsbach RB, Moore J, Mathew S, Raimondi SC, Mukatira ST, Downing JR. TET1, a member of a novel protein family, is fused to MLL in acute myeloid leukemia containing the t(10;11)(q22;q23) Leukemia. 2003;17:637–41. doi: 10.1038/sj.leu.2402834. [DOI] [PubMed] [Google Scholar]
- 26.Iyer LM, Abhiman S, Aravind L. Natural history of eukaryotic DNA methylation systems. Prog Mol Biol Transl Sci. 2011;101:25–104. doi: 10.1016/B978-0-12-387685-0.00002-0. [DOI] [PubMed] [Google Scholar]
- 27.Iyer LM, Tahiliani M, Rao A, Aravind L. Prediction of novel families of enzymes involved in oxidative and other complex modifications of bases in nucleic acids. Cell Cycle. 2009;8:1698–710. doi: 10.4161/cc.8.11.8580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hu L, Li Z, Cheng J, Rao Q, Gong W, et al. Crystal structure of TET2-DNA complex: insight into TET-mediated 5mC oxidation. Cell. 2013;155:1545–55. doi: 10.1016/j.cell.2013.11.020. [DOI] [PubMed] [Google Scholar]
- 29.Hashimoto H, Pais JE, Zhang X, Saleh L, Fu ZQ, et al. Structure of a Naegleria Tet-like dioxygenase in complex with 5-methylcytosine DNA. Nature. 2013;506:391–95. doi: 10.1038/nature12905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McDonough MA, Loenarz C, Chowdhury R, Clifton IJ, Schofield CJ. Structural studies on human 2-oxoglutarate dependent oxygenases. Curr Opin Struct Biol. 2010;20:659–72. doi: 10.1016/j.sbi.2010.08.006. [DOI] [PubMed] [Google Scholar]
- 31.Upadhyay AK, Horton JR, Zhang X, Cheng X. Coordinated methyllysine erasure: structural and functional linkage of a Jumonji demethylase domain and a reader domain. Curr Opin Struct Biol. 2011;21:750–60. doi: 10.1016/j.sbi.2011.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xu Y, Xu C, Kato A, Tempel W, Abreu JG, et al. Tet3 CXXC domain and dioxygenase activity cooperatively regulate key genes for Xenopus eye and neural development. Cell. 2012;151:1200–13. doi: 10.1016/j.cell.2012.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu N, Wang M, Deng W, Schmidt CS, Qin W, et al. Intrinsic and extrinsic connections of Tet3 dioxygenase with CXXC zinc finger modules. PLoS ONE. 2013;8:e62755. doi: 10.1371/journal.pone.0062755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ko M, An J, Bandukwala HS, Chavez L, Aijo T, et al. Modulation of TET2 expression and 5-methylcytosine oxidation by the CXXC domain protein IDAX. Nature. 2013;497:122–26. doi: 10.1038/nature12052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Borst P, Sabatini R. Base J: discovery, biosynthesis, and possible functions. Annu Rev Microbiol. 2008;62:235–51. doi: 10.1146/annurev.micro.62.081307.162750. [DOI] [PubMed] [Google Scholar]
- 36.Ko M, Huang Y, Jankowska AM, Pape UJ, Tahiliani M, et al. Impaired hydroxylation of 5-methylcytosine in myeloid cancers with mutant TET2. Nature. 2010;468:839–43. doi: 10.1038/nature09586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kriaucionis S, Heintz N. The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain. Science. 2009;324:929–30. doi: 10.1126/science.1169786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wu SC, Zhang Y. Active DNA demethylation: Many roads lead to Rome. Nat Rev Mol Cell Biol. 2010;11:607–20. doi: 10.1038/nrm2950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu CK, Hsu CA, Abbott MT. Catalysis of three sequential dioxygenase reactions by thymine 7-hydroxylase. Arch Biochem Biophys. 1973;159:180–87. doi: 10.1016/0003-9861(73)90443-8. [DOI] [PubMed] [Google Scholar]
- 40.Pfaffeneder T, Hackner B, Truß M, Münzel M, Müller M, et al. The discovery of 5-formylcytosine in embryonic stem cell DNA. Angew Chem Int Ed. 2011;50:7008–12. doi: 10.1002/anie.201103899. [DOI] [PubMed] [Google Scholar]
- 41.Kohli RM, Zhang Y. TET enzymes, TDG and the dynamics of DNA demethylation. Nature. 2013;502:472–79. doi: 10.1038/nature12750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Smiley JA, Kundracik M, Landfried DA, Barnes VR, Sr, Axhemi AA. Genes of the thymidine salvage pathway: thymine-7-hydroxylase from a Rhodotorula glutinis cDNA library and iso-orotate decarboxylase from Neurospora crassa. Biochim Biophys Acta. 2005;1723:256–64. doi: 10.1016/j.bbagen.2005.02.001. [DOI] [PubMed] [Google Scholar]
- 43.Schiesser S, Hackner B, Pfaffeneder T, Müller M, Hagemeier C, et al. Mechanism and stem-cell activity of 5-carboxycytosine decarboxylation determined by isotope tracing. Angew Chem Int Ed. 2012;51:6516–20. doi: 10.1002/anie.201202583. [DOI] [PubMed] [Google Scholar]
- 44.Chen CC, Wang KY, Shen CK. The mammalian de novo DNA methyltransferases DNMT3A and DNMT3B are also DNA 5-hydroxymethylcytosine dehydroxymethylases. J Biol Chem. 2012;287:33116–21. doi: 10.1074/jbc.C112.406975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liutkevičiutė Z, Lukinavičius G, Masevičius V, Daujotytė D, Klimašauskas S. Cytosine-5-methyltransferases add aldehydes to DNA. Nat Chem Biol. 2009;5:400–2. doi: 10.1038/nchembio.172. [DOI] [PubMed] [Google Scholar]
- 46.Hashimoto H, Liu Y, Upadhyay AK, Chang Y, Howerton SB, et al. Recognition and potential mechanisms for replication and erasure of cytosine hydroxymethylation. Nucleic Acids Res. 2012;40:4841–49. doi: 10.1093/nar/gks155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Valinluck V, Sowers LC. Endogenous cytosine damage products alter the site selectivity of human DNA maintenance methyltransferase DNMT1. Cancer Res. 2007;67:946–50. doi: 10.1158/0008-5472.CAN-06-3123. [DOI] [PubMed] [Google Scholar]
- 48.Guo JU, Su Y, Zhong C, Ming GL, Song H. Hydroxylation of 5-methylcytosine by TET1 promotes active DNA demethylation in the adult brain. Cell. 2011;145:423–34. doi: 10.1016/j.cell.2011.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bransteitter R, Pham P, Scharff MD, Goodman MF. Activation-induced cytidine deaminase deaminates deoxycytidine on single-stranded DNA but requires the action of RNase. Proc Natl Acad Sci USA. 2003;100:4102–7. doi: 10.1073/pnas.0730835100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nabel CS, Jia H, Ye Y, Shen L, Goldschmidt HL, et al. AID/APOBEC deaminases disfavor modified cytosines implicated in DNA demethylation. Nat Chem Biol. 2012;8:751–58. doi: 10.1038/nchembio.1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhu JK. Active DNA demethylation mediated by DNA glycosylases. Annu Rev Genet. 2009;43:143–66. doi: 10.1146/annurev-genet-102108-134205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rai K, Huggins IJ, James SR, Karpf AR, Jones DA, Cairns BR. DNA demethylation in zebrafish involves the coupling of a deaminase, a glycosylase, and Gadd45. Cell. 2008;135:1201–12. doi: 10.1016/j.cell.2008.11.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Popp C, Dean W, Feng S, Cokus SJ, Andrews S, et al. Genome-wide erasure of DNA methylation in mouse primordial germ cells is affected by AID deficiency. Nature. 2010;463:1101–5. doi: 10.1038/nature08829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bhutani N, Brady JJ, Damian M, Sacco A, Corbel SY, Blau HM. Reprogramming towards pluripotency requires AID-dependent DNA demethylation. Nature. 2010;463:1042–47. doi: 10.1038/nature08752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Stivers JT, Jiang YL. A mechanistic perspective on the chemistry of DNA repair glycosylases. Chem Rev. 2003;103:2729–59. doi: 10.1021/cr010219b. [DOI] [PubMed] [Google Scholar]
- 56.Bennett MT, Rodgers MT, Hebert AS, Ruslander LE, Eisele L, Drohat AC. Specificity of human thymine DNA glycosylase depends on N-glycosidic bond stability. J Am Chem Soc. 2006;128:12510–19. doi: 10.1021/ja0634829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Williams RT, Wang Y. A density functional theory study on the kinetics and thermodynamics of N-glycosidic bond cleavage in 5-substituted 2′-deoxycytidines. Biochemistry. 2012;51:6458–62. doi: 10.1021/bi300797q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shen L, Wu H, Diep D, Yamaguchi S, D’Alessio AC, et al. Genome-wide analysis reveals TET-and TDG-dependent 5-methylcytosine oxidation dynamics. Cell. 2013;153:692–706. doi: 10.1016/j.cell.2013.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Song CX, Szulwach KE, Dai Q, Fu Y, Mao SQ, et al. Genome-wide profiling of 5-formylcytosine reveals its roles in epigenetic priming. Cell. 2013;153:678–91. doi: 10.1016/j.cell.2013.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cortellino S, Xu J, Sannai M, Moore R, Caretti E, et al. Thymine DNA glycosylase is essential for active DNA demethylation by linked deamination-base excision repair. Cell. 2011;146:67–79. doi: 10.1016/j.cell.2011.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cortázar D, Kunz C, Selfridge J, Lettieri T, Saito Y, et al. Embryonic lethal phenotype reveals a function of TDG in maintaining epigenetic stability. Nature. 2011;470:419–23. doi: 10.1038/nature09672. [DOI] [PubMed] [Google Scholar]
- 62.Millar CB, Guy J, Sansom OJ, Selfridge J, MacDougall E, et al. Enhanced CpG mutability and tumorigenesis in MBD4-deficient mice. Science. 2002;297:403–5. doi: 10.1126/science.1073354. [DOI] [PubMed] [Google Scholar]
- 63.Kemmerich K, Dingler FA, Rada C, Neuberger MS. Germline ablation of SMUG1 DNA glycosylase causes loss of 5-hydroxymethyluracil- and UNG-backup uracil-excision activities and increases cancer predisposition of Ung−/−Msh2−/−mice. Nucleic Acids Res. 2012;40:6016–25. doi: 10.1093/nar/gks259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Law JA, Jacobsen SE. Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat Rev Genet. 2010;11:204–20. doi: 10.1038/nrg2719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yamaguchi S, Hong K, Liu R, Shen L, Inoue A, et al. Tet1 controls meiosis by regulating meiotic gene expression. Nature. 2012;492:443–47. doi: 10.1038/nature11709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wossidlo M, Nakamura T, Lepikhov K, Marques CJ, Zakhartchenko V, et al. 5-Hydroxymethylcytosine in the mammalian zygote is linked with epigenetic reprogramming. Nat Commun. 2011;2:241. doi: 10.1038/ncomms1240. [DOI] [PubMed] [Google Scholar]
- 67.Iqbal K, Jin SG, Pfeifer GP, Szabó PE. Reprogramming of the paternal genome upon fertilization involves genome-wide oxidation of 5-methylcytosine. Proc Natl Acad Sci USA. 2011;108:3642–47. doi: 10.1073/pnas.1014033108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hackett JA, Sengupta R, Zylicz JJ, Murakami K, Lee C, et al. Germline DNA demethylation dynamics and imprint erasure through 5-hydroxymethylcytosine. Science. 2013;339:448–52. doi: 10.1126/science.1229277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yamaguchi S, Hong K, Liu R, Inoue A, Shen L, et al. Dynamics of 5-methylcytosine and 5-hydroxy-methylcytosine during germ cell reprogramming. Cell Res. 2013;23:329–39. doi: 10.1038/cr.2013.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ficz G, Branco MR, Seisenberger S, Santos F, Krueger F, et al. Dynamic regulation of 5-hydroxy-methylcytosine in mouse ES cells and during differentiation. Nature. 2011;473:398–402. doi: 10.1038/nature10008. [DOI] [PubMed] [Google Scholar]
- 71.Koh KP, Yabuuchi A, Rao S, Huang Y, Cunniff K, et al. Tet1 and Tet2 regulate 5-hydroxy-methylcytosine production and cell lineage specification in mouse embryonic stem cells. Cell Stem Cell. 2011;8:200–13. doi: 10.1016/j.stem.2011.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Song SJ, Ito K, Ala U, Kats L, Webster K, et al. The oncogenic microRNA miR-22 targets the TET2 tumor suppressor to promote hematopoietic stem cell self-renewal and transformation. Cell Stem Cell. 2013;13:87–101. doi: 10.1016/j.stem.2013.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Song SJ, Poliseno L, Song MS, Ala U, Webster K, et al. MicroRNA antagonism regulates breast cancer stemness and metastasis via TET-family-dependent chromatin remodeling. Cell. 2013;154:311–24. doi: 10.1016/j.cell.2013.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Shi FT, Kim H, Lu W, He Q, Liu D, et al. Ten-eleven translocation 1 (Tet1) is regulated by O-linked N-acetylglucosamine transferase (OGT) for target gene repression in mouse embryonic stem cells. J Biol Chem. 2013;288:20776–84. doi: 10.1074/jbc.M113.460386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Williams K, Christensen J, Pedersen MT, Johansen JV, Cloos PA, et al. TET1 and hydroxymethyl-cytosine in transcription and DNA methylation fidelity. Nature. 2011;473:343–48. doi: 10.1038/nature10066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Deplus R, Delatte B, Schwinn MK, Defrance M, Méndez J, et al. TET2 and TET3 regulate GlcNAcylation and H3K4 methylation through OGT and SET1/COMPASS. EMBO J. 2013;32:645–55. doi: 10.1038/emboj.2012.357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Chen Q, Chen Y, Bian C, Fujiki R, Yu X. TET2 promotes histone O-GlcNAcylation during gene transcription. Nature. 2013;493:561–64. doi: 10.1038/nature11742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Vella P, Scelfo A, Jammula S, Chiacchiera F, Williams K, et al. Tet proteins connect the O-linked N-acetylglucosamine transferase OGT to chromatin in embryonic stem cells. Mol Cell. 2013;49:645–56. doi: 10.1016/j.molcel.2012.12.019. [DOI] [PubMed] [Google Scholar]
- 79.Yildirim O, Li R, Hung JH, Chen PB, Dong X, et al. Mbd3/NURD complex regulates expression of 5-hydroxymethylcytosine marked genes in embryonic stem cells. Cell. 2011;147:1498–510. doi: 10.1016/j.cell.2011.11.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Spruijt CG, Gnerlich F, Smits AH, Pfaffeneder T, Jansen PWTC, et al. Dynamic readers for 5-(hydroxy)methylcytosine and its oxidized derivatives. Cell. 2013;152:1146–59. doi: 10.1016/j.cell.2013.02.004. [DOI] [PubMed] [Google Scholar]
- 81.Figueroa ME, Abdel-Wahab O, Lu C, Ward PS, Patel J, et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell. 2010;18:553–67. doi: 10.1016/j.ccr.2010.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Xu W, Yang H, Liu Y, Yang Y, Wang P, et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of α-ketoglutarate-dependent dioxygenases. Cancer Cell. 2011;19:17–30. doi: 10.1016/j.ccr.2010.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ward PS, Patel J, Wise DR, Abdel-Wahab O, Bennett BD, et al. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting α-ketoglutarate to 2-hydroxyglutarate. Cancer Cell. 2010;17:225–34. doi: 10.1016/j.ccr.2010.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Dang L, White DW, Gross S, Bennett BD, Bittinger MA, et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature. 2009;462:739–44. doi: 10.1038/nature08617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Xiao M, Yang H, Xu W, Ma S, Lin H, et al. Inhibition of α-KG-dependent histone and DNA demethylases by fumarate and succinate that are accumulated in mutations of FH and SDH tumor suppressors. Genes Dev. 2012;26:1326–38. doi: 10.1101/gad.191056.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Yin R, Mao S-Q, Zhao B, Chong Z, Yang Y, et al. Ascorbic acid enhances Tet-mediated 5-methyl-cytosine oxidation and promotes DNA demethylation in mammals. J Am Chem Soc. 2013;135:10396–403. doi: 10.1021/ja4028346. [DOI] [PubMed] [Google Scholar]
- 87.Minor EA, Court BL, Young JI, Wang G. Ascorbate induces ten-eleven translocation (Tet) methyl-cytosine dioxygenase–mediated generation of 5-hydroxymethylcytosine. J Biol Chem. 2013;288:13669–74. doi: 10.1074/jbc.C113.464800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Blaschke K, Ebata KT, Karimi MM, Zepeda-Martínez JA, Goyal P, et al. Vitamin C induces Tet-dependent DNA demethylation and a blastocyst-like state in ES cells. Nature. 2013;500:222–26. doi: 10.1038/nature12362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Coulter JB, O’Driscoll CM, Bressler JP. Hydroquinone increases 5-hydroxymethylcytosine formation through ten eleven translocation 1 (Tet1) 5-methylcytosine dioxygenase. J Biol Chem. 2013;288:28792–800. doi: 10.1074/jbc.M113.491365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Song CX, Yi C, He C. Mapping recently identified nucleotide variants in the genome and transcriptome. Nat Biotechnol. 2012;30:1107–16. doi: 10.1038/nbt.2398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Beck S, Rakyan VK. The methylome: approaches for global DNA methylation profiling. Trends Genet. 2008;24:231–37. doi: 10.1016/j.tig.2008.01.006. [DOI] [PubMed] [Google Scholar]
- 92.Lister R, Pelizzola M, Dowen RH, Hawkins RD, Hon G, et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature. 2009;462:315–22. doi: 10.1038/nature08514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Huang Y, Pastor WA, Shen Y, Tahiliani M, Liu DR, Rao A. The behaviour of 5-hydroxymethylcytosine in bisulfite sequencing. PLoS ONE. 2010;5:e8888. doi: 10.1371/journal.pone.0008888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Jin SG, Kadam S, Pfeifer GP. Examination of the specificity of DNA methylation profiling techniques towards 5-methylcytosine and 5-hydroxymethylcytosine. Nucleic Acids Res. 2010;38:e125. doi: 10.1093/nar/gkq223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Booth MJ, Branco MR, Ficz G, Oxley D, Krueger F, et al. Quantitative sequencing of 5-methylcytosine and 5-hydroxymethylcytosine at single-base resolution. Science. 2012;336:934–37. doi: 10.1126/science.1220671. [DOI] [PubMed] [Google Scholar]
- 96.Yu M, Hon G, Szulwach KE, Song CX, Zhang L, et al. Base-resolution analysis of 5-hydroxy-methylcytosine in the mammalian genome. Cell. 2012;149:1368–80. doi: 10.1016/j.cell.2012.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Wu H, D’Alessio AC, Ito S, Wang Z, Cui K, et al. Genome-wide analysis of 5-hydroxymethylcytosine distribution reveals its dual function in transcriptional regulation in mouse embryonic stem cells. Genes Dev. 2011;25:679–84. doi: 10.1101/gad.2036011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Xu Y, Wu F, Tan L, Kong L, Xiong L, et al. Genome-wide regulation of 5hmC, 5mC, and gene expression by Tet1 hydroxylase in mouse embryonic stem cells. Mol Cell. 2011;42:451–64. doi: 10.1016/j.molcel.2011.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Stroud H, Feng S, Morey Kinney S, Pradhan S, Jacobsen S. 5-Hydroxymethylcytosine is associated with enhancers and gene bodies in human embryonic stem cells. Genome Biol. 2011;12:R54. doi: 10.1186/gb-2011-12-6-r54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Jin SG, Wu X, Li AX, Pfeifer GP. Genomic mapping of 5-hydroxymethylcytosine in the human brain. Nucleic Acids Res. 2011;39:5015–24. doi: 10.1093/nar/gkr120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Pastor WA, Pape UJ, Huang Y, Henderson HR, Lister R, et al. Genome-wide mapping of 5-hydroxymethylcytosine in embryonic stem cells. Nature. 2011;473:394–97. doi: 10.1038/nature10102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Robertson AB, Dahl JA, Vågbø CB, Tripathi P, Krokan HE, Klungland A. A novel method for the efficient and selective identification of 5-hydroxymethylcytosine in genomic DNA. Nucleic Acids Res. 2011;39:e55. doi: 10.1093/nar/gkr051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Matarese F, Carrillo–de Santa Pau E, Stunnenberg HG. 5-Hydroxymethylcytosine: a new kid on the epigenetic block? Mol Syst Biol. 2011;7:562. doi: 10.1038/msb.2011.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Terragni J, Bitinaite J, Zheng Y, Pradhan S. Biochemical characterization of recombinant β-glucosyltransferase and analysis of global 5-hydroxymethylcytosine in unique genomes. Biochemistry. 2012;51:1009–19. doi: 10.1021/bi2014739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Song CX, Szulwach KE, Fu Y, Dai Q, Yi C, et al. Selective chemical labeling reveals the genome-wide distribution of 5-hydroxymethylcytosine. Nat Biotechnol. 2011;29:68–72. doi: 10.1038/nbt.1732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Raiber EA, Beraldi D, Ficz G, Burgess H, Branco MR, et al. Genome-wide distribution of 5-formylcytosine in ES cells is associated with transcription and depends on thymine DNA glycosylase. Genome Biol. 2012;13:R69. doi: 10.1186/gb-2012-13-8-r69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Lu X, Song CX, Szulwach K, Wang Z, Weidenbacher P, et al. Chemical modification–assisted bisulfite sequencing (CAB-Seq) for 5-carboxylcytosine detection in DNA. J Am Chem Soc. 2013;135:9315–17. doi: 10.1021/ja4044856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Khare T, Pai S, Koncevicius K, Pal M, Kriukiene E, et al. 5-hmC in the brain is abundant in synaptic genes and shows differences at the exon–intron boundary. Nat Struct Mol Biol. 2012;19:1037–43. doi: 10.1038/nsmb.2372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Gao F, Xia Y, Wang J, Luo H, Gao Z, et al. Integrated detection of both 5-mC and 5-hmC by high-throughput tag sequencing technology highlights methylation reprogramming of bivalent genes during cellular differentiation. Epigenetics. 2013;8:421–30. doi: 10.4161/epi.24280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Szulwach KE, Li X, Li Y, Song CX, Han JW, et al. Integrating 5-hydroxymethylcytosine into the epigenomic landscape of human embryonic stem cells. PLoS Genet. 2011;7:e1002154. doi: 10.1371/journal.pgen.1002154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Szulwach KE, Li X, Li Y, Song CX, Wu H, et al. 5-hmC-mediated epigenetic dynamics during postnatal neurodevelopment and aging. Nat Neurosci. 2011;14:1607–16. doi: 10.1038/nn.2959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Mellén M, Ayata P, Dewell S, Kriaucionis S, Heintz N. MeCP2 binds to 5hmC enriched within active genes and accessible chromatin in the nervous system. Cell. 2012;151:1417–30. doi: 10.1016/j.cell.2012.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Hahn MA, Qiu R, Wu X, Li AX, Zhang H, et al. Dynamics of 5-hydroxymethylcytosine and chromatin marks in mammalian neurogenesis. Cell Rep. 2013;3:291–300. doi: 10.1016/j.celrep.2013.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Korlach J, Turner SW. Going beyond five bases in DNA sequencing. Curr Opin Struct Biol. 2012;22:251–61. doi: 10.1016/j.sbi.2012.04.002. [DOI] [PubMed] [Google Scholar]
- 115.Song CX, Clark TA, Lu XY, Kislyuk A, Dai Q, et al. Sensitive and specific single-molecule sequencing of 5-hydroxymethylcytosine. Nat Methods. 2012;9:75–77. doi: 10.1038/nmeth.1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Clark TA, Lu X, Luong K, Dai Q, Boitano M, et al. Enhanced 5-methylcytosine detection in single-molecule, real-time sequencing via Tet1 oxidation. BMC Biol. 2013;11:4. doi: 10.1186/1741-7007-11-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Wanunu M, Cohen-Karni D, Johnson RR, Fields L, Benner J, et al. Discrimination of methylcytosine from hydroxymethylcytosine in DNA molecules. J Am Chem Soc. 2010;133:486–92. doi: 10.1021/ja107836t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Wallace EV, Stoddart D, Heron AJ, Mikhailova E, Maglia G, et al. Identification of epigenetic DNA modifications with a protein nanopore. Chem Commun. 2010;46:8195–97. doi: 10.1039/c0cc02864a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Li WW, Gong L, Bayley H. Single-molecule detection of 5-hydroxymethylcytosine in DNA through chemical modification and nanopore analysis. Angew Chem Int Ed. 2013;52:4350–55. doi: 10.1002/anie.201300413. [DOI] [PubMed] [Google Scholar]
- 120.Lister R, Mukamel EA, Nery JR, Urich M, Puddifoot CA, et al. Global epigenomic reconfiguration during mammalian brain development. Science. 2013;341:6146. doi: 10.1126/science.1237905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Sun Z, Terragni J, Borgaro JG, Liu Y, Yu L, et al. High-resolution enzymatic mapping of genomic 5-hydroxymethylcytosine in mouse embryonic stem cells. Cell Rep. 2013;3:567–76. doi: 10.1016/j.celrep.2013.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Seisenberger S, Peat JR, Reik W. Conceptual links between DNA methylation reprogramming in the early embryo and primordial germ cells. Curr Opin Cell Biol. 2013;25:281–88. doi: 10.1016/j.ceb.2013.02.013. [DOI] [PubMed] [Google Scholar]
- 123.Monk M, Boubelik M, Lehnert S. Temporal and regional changes in DNA methylation in the embryonic, extraembryonic and germ cell lineages during mouse embryo development. Development. 1987;99:371–82. doi: 10.1242/dev.99.3.371. [DOI] [PubMed] [Google Scholar]
- 124.Mayer W, Niveleau A, Walter J, Fundele R, Haaf T. Demethylation of the zygotic paternal genome. Nature. 2000;403:501–2. doi: 10.1038/35000656. [DOI] [PubMed] [Google Scholar]
- 125.Oswald J, Engemann S, Lane N, Mayer W, Olek A, et al. Active demethylation of the paternal genome in the mouse zygote. Curr Biol. 2000;10:475–78. doi: 10.1016/s0960-9822(00)00448-6. [DOI] [PubMed] [Google Scholar]
- 126.Nakamura T, Arai Y, Umehara H, Masuhara M, Kimura T, et al. PGC7/Stella protects against DNA demethylation in early embryogenesis. Nat Cell Biol. 2007;9:64–71. doi: 10.1038/ncb1519. [DOI] [PubMed] [Google Scholar]
- 127.Gu TP, Guo F, Yang H, Wu HP, Xu GF, et al. The role of Tet3 DNA dioxygenase in epigenetic reprogramming by oocytes. Nature. 2011;477:606–10. doi: 10.1038/nature10443. [DOI] [PubMed] [Google Scholar]
- 128.Inoue A, Zhang Y. Replication-dependent loss of 5-hydroxymethylcytosine in mouse preimplantation embryos. Science. 2011;334:194. doi: 10.1126/science.1212483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Inoue A, Shen L, Dai Q, He C, Zhang Y. Generation and replication-dependent dilution of 5fC and 5caC during mouse preimplantation development. Cell Res. 2011;21:1670–76. doi: 10.1038/cr.2011.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Nakamura T, Liu YJ, Nakashima H, Umehara H, Inoue K, et al. PGC7 binds histone H3K9me2 to protect against conversion of 5mC to 5hmC in early embryos. Nature. 2012;486:415–19. doi: 10.1038/nature11093. [DOI] [PubMed] [Google Scholar]
- 131.Smith ZD, Chan MM, Mikkelsen TS, Gu H, Gnirke A, et al. A unique regulatory phase of DNA methylation in the early mammalian embryo. Nature. 2012;484:339–44. doi: 10.1038/nature10960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Saitou M, Kagiwada S, Kurimoto K. Epigenetic reprogramming in mouse pre-implantation development and primordial germ cells. Development. 2012;139:15–31. doi: 10.1242/dev.050849. [DOI] [PubMed] [Google Scholar]
- 133.Hajkova P, Jeffries SJ, Lee C, Miller N, Jackson SP, Surani MA. Genome-wide reprogramming in the mouse germ line entails the base excision repair pathway. Science. 2010;329:78–82. doi: 10.1126/science.1187945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Vincent JJ, Huang Y, Chen PY, Feng S, Calvopina JH, et al. Stage-specific roles for Tet1 and Tet2 in DNA demethylation in primordial germ cells. Cell Stem Cell. 2013;12:470–78. doi: 10.1016/j.stem.2013.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Seki Y, Hayashi K, Itoh K, Mizugaki M, Saitou M, Matsui Y. Extensive and orderly reprogramming of genome-wide chromatin modifications associated with specification and early development of germ cells in mice. Dev Biol. 2005;278:440–58. doi: 10.1016/j.ydbio.2004.11.025. [DOI] [PubMed] [Google Scholar]
- 136.Hajkova P, Erhardt S, Lane N, Haaf T, El-Maarri O, et al. Epigenetic reprogramming in mouse primordial germ cells. Mech Dev. 2002;117:15–23. doi: 10.1016/s0925-4773(02)00181-8. [DOI] [PubMed] [Google Scholar]
- 137.Kagiwada S, Kurimoto K, Hirota T, Yamaji M, Saitou M. Replication-coupled passive DNA demethylation for the erasure of genome imprints in mice. EMBO J. 2013;32:340–53. doi: 10.1038/emboj.2012.331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Seisenberger S, Andrews S, Krueger F, Arand J, Walter J, et al. The dynamics of genome-wide DNA methylation reprogramming in mouse primordial germ cells. Mol Cell. 2012;48:849–62. doi: 10.1016/j.molcel.2012.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Kurimoto K, Yabuta Y, Ohinata Y, Shigeta M, Yamanaka K, Saitou M. Complex genome-wide transcription dynamics orchestrated by Blimp1 for the specification of the germ cell lineage in mice. Genes Dev. 2008;22:1617–35. doi: 10.1101/gad.1649908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Dawlaty MM, Breiling A, Le T, Raddatz G, Barrasa MI, et al. Combined deficiency of Tet1 and Tet2 causes epigenetic abnormalities but is compatible with postnatal development. Dev Cell. 2013;24:310–23. doi: 10.1016/j.devcel.2012.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Yamaguchi S, Shen L, Liu Y, Sendler D, Zhang Y. Role of Tet1 in genomic imprinting erasure. Nature. 2013;504:460–64. doi: 10.1038/nature12805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Wu H, Zhang Y. Mechanisms and functions of Tet protein–mediated 5-methylcytosine oxidation. Genes Dev. 2011;25:2436–52. doi: 10.1101/gad.179184.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Williams K, Christensen J, Helin K. DNA methylation: TET proteins—guardians of CpG islands? EMBO Rep. 2012;13:28–35. doi: 10.1038/embor.2011.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Wu H, D’Alessio AC, Ito S, Xia K, Wang Z, et al. Dual functions of Tet1 in transcriptional regulation in mouse embryonic stem cells. Nature. 2011;473:389–93. doi: 10.1038/nature09934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Leitch HG, McEwen KR, Turp A, Encheva V, Carroll T, et al. Naive pluripotency is associated with global DNA hypomethylation. Nat Struct Mol Biol. 2013;20:311–16. doi: 10.1038/nsmb.2510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Ficz G, Hore TA, Santos F, Lee HJ, Dean W, et al. FGF signaling inhibition in ESCs drives rapid genome-wide demethylation to the epigenetic ground state of pluripotency. Cell Stem Cell. 2013;13:351–59. doi: 10.1016/j.stem.2013.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Habibi E, Brinkman AB, Arand J, Kroeze LI, Kerstens HH, et al. Whole-genome bisulfite sequencing of two distinct interconvertible DNA methylomes of mouse embryonic stem cells. Cell Stem Cell. 2013;13:360–69. doi: 10.1016/j.stem.2013.06.002. [DOI] [PubMed] [Google Scholar]
- 148.Doege CA, Inoue K, Yamashita T, Rhee DB, Travis S, et al. Early-stage epigenetic modification during somatic cell reprogramming by Parp1 and Tet2. Nature. 2012;488:652–55. doi: 10.1038/nature11333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Costa Y, Ding J, Theunissen TW, Faiola F, Hore TA, et al. NANOG-dependent function of TET1 and TET2 in establishment of pluripotency. Nature. 2013;495:370–74. doi: 10.1038/nature11925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Gao Y, Chen J, Li K, Wu T, Huang B, et al. Replacement of Oct4 by Tet1 during iPSC induction reveals an important role of DNA methylation and hydroxymethylation in reprogramming. Cell Stem Cell. 2013;12:453–69. doi: 10.1016/j.stem.2013.02.005. [DOI] [PubMed] [Google Scholar]
- 151.Piccolo FM, Bagci H, Brown KE, Landeira D, Soza-Ried J, et al. Different roles for Tet1 and Tet2 proteins in reprogramming-mediated erasure of imprints induced by EGC fusion. Mol Cell. 2013;49:1023–33. doi: 10.1016/j.molcel.2013.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Esteller M. Aberrant DNA methylation as a cancer-inducing mechanism. Annu Rev Pharmacol Toxicol. 2005;45:629–56. doi: 10.1146/annurev.pharmtox.45.120403.095832. [DOI] [PubMed] [Google Scholar]
- 153.Sun M, Song CX, Huang H, Frankenberger CA, Sankarasharma D, et al. HMGA2/TET1/HOXA9 signaling pathway regulates breast cancer growth and metastasis. Proc Natl Acad Sci USA. 2013;110:9920–25. doi: 10.1073/pnas.1305172110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Delhommeau F, Dupont S, Della Valle V, James C, Trannoy S, et al. Mutation in TET2 in myeloid cancers. N Engl J Med. 2009;360:2289–301. doi: 10.1056/NEJMoa0810069. [DOI] [PubMed] [Google Scholar]
- 155.Pronier E, Delhommeau F. Role of TET2 mutations in myeloproliferative neoplasms. Curr Hematol Malig Rep. 2012;7:57–64. doi: 10.1007/s11899-011-0108-8. [DOI] [PubMed] [Google Scholar]
- 156.Langemeijer SM, Kuiper RP, Berends M, Knops R, Aslanyan MG, et al. Acquired mutations in TET2 are common in myelodysplastic syndromes. Nat Genet. 2009;41:838–42. doi: 10.1038/ng.391. [DOI] [PubMed] [Google Scholar]
- 157.Ko M, Bandukwala HS, An J, Lamperti ED, Thompson EC, et al. Ten-eleven-translocation 2 (TET2) negatively regulates homeostasis and differentiation of hematopoietic stem cells in mice. Proc Natl Acad Sci USA. 2011;108:14566–71. doi: 10.1073/pnas.1112317108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Quivoron C, Couronne L, Della Valle V, Lopez CK, Plo I, et al. TET2 inactivation results in pleiotropic hematopoietic abnormalities in mouse and is a recurrent event during human lymphomagenesis. Cancer Cell. 2011;20:25–38. doi: 10.1016/j.ccr.2011.06.003. [DOI] [PubMed] [Google Scholar]
- 159.Li Z, Cai X, Cai CL, Wang J, Zhang W, et al. Deletion of Tet2 in mice leads to dysregulated hematopoietic stem cells and subsequent development of myeloid malignancies. Blood. 2011;118:4509–18. doi: 10.1182/blood-2010-12-325241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Moran-Crusio K, Reavie L, Shih A, Abdel-Wahab O, Ndiaye-Lobry D, et al. Tet2 loss leads to increased hematopoietic stem cell self-renewal and myeloid transformation. Cancer Cell. 2011;20:11–24. doi: 10.1016/j.ccr.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Lian CG, Xu Y, Ceol C, Wu F, Larson A, et al. Loss of 5-hydroxymethylcytosine is an epigenetic hallmark of melanoma. Cell. 2012;150:1135–46. doi: 10.1016/j.cell.2012.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Yang H, Liu Y, Bai F, Zhang JY, Ma SH, et al. Tumor development is associated with decrease of TET gene expression and 5-methylcytosine hydroxylation. Oncogene. 2013;32:663–69. doi: 10.1038/onc.2012.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Hsu CH, Peng KL, Kang ML, Chen YR, Yang YC, et al. TET1 suppresses cancer invasion by activating the tissue inhibitors of metalloproteinases. Cell Rep. 2012;2:568–79. doi: 10.1016/j.celrep.2012.08.030. [DOI] [PubMed] [Google Scholar]
- 164.Huang H, Jiang X, Li Z, Li Y, Song CX, et al. TET1 plays an essential oncogenic role in MLL-rearranged leukemia. Proc Natl Acad Sci USA. 2013;110:11994–99. doi: 10.1073/pnas.1310656110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Perry RP, Kelley DE, Friderici K, Rottman F. The methylated constituents of L cell messenger RNA: evidence for an unusual cluster at the 5′terminus. Cell. 1975;4:387–94. doi: 10.1016/0092-8674(75)90159-2. [DOI] [PubMed] [Google Scholar]
- 166.Roy TW, Bhagwat AS. Kinetic studies of Escherichia coli AlkB using a new fluorescence-based assay for DNA demethylation. Nucleic Acids Res. 2007;35:e147. doi: 10.1093/nar/gkm1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Dina C, Meyre D, Gallina S, Durand E, Körner A, et al. Variation in FTO contributes to childhood obesity and severe adult obesity. Nat Genet. 2007;39:724–26. doi: 10.1038/ng2048. [DOI] [PubMed] [Google Scholar]
- 168.Frayling TM, Timpson NJ, Weedon MN, Zeggini E, Freathy RM, et al. A common variant in the FTO gene is associated with body mass index and predisposes to childhood and adult obesity. Science. 2007;316:889–94. doi: 10.1126/science.1141634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Scuteri A, Sanna S, Chen WM, Uda M, Albai G, et al. Genome-wide association scan shows genetic variants in the FTO gene are associated with obesity-related traits. PLoS Genet. 2007;3:e115. doi: 10.1371/journal.pgen.0030115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Hess ME, Hess S, Meyer KD, Verhagen LAW, Koch L, et al. The fat mass and obesity associated gene (Fto) regulates activity of the dopaminergic midbrain circuitry. Nat Neurosci. 2013;16:1042–48. doi: 10.1038/nn.3449. [DOI] [PubMed] [Google Scholar]
- 171.Dominissini D, Moshitch-Moshkovitz S, Schwartz S, Salmon-Divon M, Ungar L, et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-Seq. Nature. 2012;485:201–6. doi: 10.1038/nature11112. [DOI] [PubMed] [Google Scholar]
- 172.Fu Y, Jia G, Pang X, Wang RN, Wang X, et al. FTO-mediated formation of N6-hydroxymethyladenosine and N6-formyladenosine in mammalian RNA. Nat Commun. 2013;4:1798. doi: 10.1038/ncomms2822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.He C. Grand challenge commentary: RNA epigenetics? Nat Chem Biol. 2010;6:863–65. doi: 10.1038/nchembio.482. [DOI] [PubMed] [Google Scholar]
- 174.Yi C, Pan T. Cellular dynamics of RNA modification. Acc Chem Res. 2011;44:1380–88. doi: 10.1021/ar200057m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Meyer KD, Saletore Y, Zumbo P, Elemento O, Mason CE, Jaffrey SR. Comprehensive analysis of mRNA methylation reveals enrichment in 3′UTRs and near stop codons. Cell. 2012;149:1635–46. doi: 10.1016/j.cell.2012.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Kangaspeska S, Stride B, Metivier R, Polycarpou-Schwarz M, Ibberson D, et al. Transient cyclical methylation of promoter DNA. Nature. 2008;452:112–15. doi: 10.1038/nature06640. [DOI] [PubMed] [Google Scholar]
- 177.Metivier R, Gallais R, Tiffoche C, Le Peron C, Jurkowska RZ, et al. Cyclical DNA methylation of a transcriptionally active promoter. Nature. 2008;452:45–50. doi: 10.1038/nature06544. [DOI] [PubMed] [Google Scholar]
- 178.Thillainadesan G, Chitilian JM, Isovic M, Ablack JN, Mymryk JS, et al. TGF-β-dependent active demethylation and expression of the p15ink4b tumor suppressor are impaired by the ZNF217/CoREST complex. Mol Cell. 2012;46:636–49. doi: 10.1016/j.molcel.2012.03.027. [DOI] [PubMed] [Google Scholar]