

Summary
Hepatocellular carcinoma (HCC) is the second most common cause of cancer related death. Non-alcoholic fatty liver disease (NAFLD) affects a large proportion of the US population and is considered a metabolic predisposition to liver cancer 1-5. However, the role of adaptive immune responses in NAFLD-promoted HCC is largely unknown. Here, we show that dysregulation of lipid metabolism in NAFLD causes a selective loss of intrahepatic CD4+ but not CD8+ T lymphocytes leading to accelerated hepatocarcinogenesis. We also found that CD4+ T lymphocytes have greater mitochondrial mass than CD8+ T lymphocytes and generate higher levels of mitochondrially-derived reactive oxygen species (ROS). Disruption of mitochondrial function by linoleic acid, a fatty acid accumulated in NAFLD, causes more oxidative damage than other free fatty acids such as palmitic acid, and mediates selective loss of intrahepatic CD4+ T lymphocytes. In vivo blockade of ROS reversed NAFLD-induced hepatic CD4+ T lymphocyte decrease and delayed NAFLD-promoted HCC. Our results provide an unexpected link between lipid dysregulation and impaired anti-tumor surveillance.
HCC commonly arises in patients with underlying chronic liver disease, and is considered a typical inflammation-associated tumor 1. Recent epidemiology studies indicate an increase in the rate of non-alcoholic fatty liver disease (NAFLD)-induced HCC 2-5. Immune evasion mediated by numerous immune suppressor mechanisms involving different immune cell subsets have been shown to contribute to HCC initiation and progression 6, and patients with tumors containing lymphocytic infiltrates show longer survival and lower risk of recurrence 7. However, the role of adaptive immune responses in NAFLD and HCC have just begun to be understood 8.
Here we investigated how metabolic changes observed in NAFLD promoted hepatocarcinogenesis using a series of different murine NAFLD and HCC models and confirmed our results using human samples. Inducible liver-specific MYC oncogene transgenic mice 9 were fed with methionine choline-deficient diet (MCD) to induce NAFLD 10 (Fig. 1a, Extended Data Fig. 1a and b). Earlier microscopic liver tumor lesions were found in MYC-ON MCD mice (Fig. 1b upper panel). As expected, MYC-ON MCD mice showed more macroscopic liver tumors (Fig. 1b lower panel and c). Similar results were obtained in MYC mice fed choline-deficient and amino acid-defined diet (CDAA), another NAFLD diet (Extended Data Fig. 1c-e) 11. Again, more liver tumors were found in carcinogen diethylnitrosamine-challenged C57BL/6 mice 12,13 fed with CDAA or HF diet (Extended Data Fig. 1f-i). These results clearly demonstrate that diet-induced NAFLD enhances HCC in different murine hepatocarcinogenesis and NAFLD models.
Figure 1.

NAFLD induces a selective loss of intrahepatic CD4+ T lymphocytes and promotes HCC.
a, Experimental set-up. b, Upper panel: representative H&E liver sections. Black bar = 100 μm. Lower panel: representative liver images. Black bar= 10 mm. c, Liver surface tumor counts (CTR=control diet, n=10 for CTR, 17 for MCD , p=0.0067, Student’s t test,). d, e, Intrahepatic CD4+ T lymphocytes (IL CD4+) and intrahepatic CD8+ T lymphocytes (IL CD8+) were measured by flow cytometry (n=12 for ON-CTR 4wks, 15 for ON-MCD 4wks, 6 for OFF-CTR 4wks, 6 for OFF-MCD 4wks, 8 for ON-CTR 8wks, 9 for ON-MCD 8wks, 6 for OFF-CTR 8wks, 6 for OFF-MCD 8wks *p<0.05, two-way ANOVA). All data are Mean ± SEM.
Next, we studied the immune cell subsets in mice with NAFLD and HCC. Consistent with previous reports, dendritic cells, macrophages and CD11b+Gr1+ cells increased (Extended Data Fig. 2a,b) 14-16. Unexpectedly, significantly fewer CD3hiCD4+ T lymphocytes, which corresponded to conventional intrahepatic CD4+ T lymphocytes, were found in mice with NAFLD (Fig. 1d and Extended Data Fig. 2a-c, e). No significant difference of intrahepatic CD3loCD4+ cells representing NKT cells (Extended Data Fig. 2d, f) or splenic CD4+ T lymphocytes was observed (Extended Data Fig. 2g). Unlike CD4+ T lymphocytes, intrahepatic CD8+ T lymphocytes remained unchanged (Fig. 1e, Extended Data Fig. 2a, b). The liver-specific reduction of CD4+ but not CD8+ T lymphocytes was also observed in the two other dietary NAFLD models in both tumor-free and tumor-bearing settings (Extended Data Fig. 2h-t), illustrating a tumor-independent but NAFLD-dependent mechanism. In addition, fewer CD4+ but not CD8+ T lymphocytes were found in leptin-deficient (ob/ob) mice (Extended Data Fig. 2u-x).
CD4+ T lymphocytes in NAFLD mice were characterized. Higher levels of CD69 and CD44hiCD62lo subset were found in MYC-ON MCD mice (Extended Data Fig. 3a-d). Hepatic but not splenic CD4+ T lymphocytes also consistently produced more IFNγ but not IL-4 (Extended Data Fig. 3e-g). Although T-bet, GATA3 and Foxp3 frequency did not change, more RORγt was detected in MYC-ON MCD mice (Extended Data Fig.3 h). Accordingly, intrahepatic CD4+ T lymphocytes produced more IL17 in NAFLD (Extended Data Fig.3i, j). No change of regulatory T lymphocyte (Treg) frequency was found, and absolute numbers decreased (Extended Data Fig.3h, k), consistent with a previous report 17. In addition, the Treg function remained unchanged in NAFLD (Extended Data Fig.3l). Together, our results indicate that NAFLD caused activation of hepatic CD4+ T lymphocytes in mice.
CD4+ T lymphocytes have been reported to inhibit HCC initiation and mediate tumor regression 18,19. It has also been reported that a considerable fraction of non-synonymous cancer mutations is immunogenic and that the majority of the immunogenic mutanome is recognized by CD4+ T lymphocytes 20. Therefore, we studied tumor-specific CD4+ T lymphocytes. AFP-specific CD4+ T lymphocytes in MYC-ON MCD mice were detected, suggesting that MYC tumors induced anti-tumor CD4+ T lymphocyte responses (Extended Data Fig. 3m). Next, we depleted CD4+ T lymphocytes to study their relevance on tumor growth. CD4 antibody depletion (Extended Data Fig. 3n) caused more hepatic tumor lesions in MYC-ON MCD mice (Fig. 2a-c). In control diet-fed MYC mice CD4 antibody depletion also promoted tumors but at a later time point (Extended Data Fig. 3o, p). These results suggested that loss of CD4+ T lymphocytes strongly contributed to HCC development in MYC mice.
Figure 2.

Depletion of intrahepatic CD4+ T lymphocytes accelerates tumor development in MYC-ON MCD mice.
a, Experimental set-up. b, c, Representative H&E staining images and microscopic tumor counts. Black bar = 200 μm. Mean ± SEM; n=5 for IgG, 8 for anti-CD4, *p<0.05, Student’s t test.
Next, we studied CD4+ T lymphocyte survival in mice with NAFLD, and higher Annexin V+ level was found (Fig. 3a, Extended Data Fig. 4a). We hypothesized that the extensive hepatic lipid accumulation induced the CD4+ T lymphocyte loss. Hepatocytes isolated from MCD mice, which showed accumulation of lipid droplets (Extended Data Fig. 4b), or control mice were co-cultured with splenocytes. Interestingly, a significant increase in Annexin V+7AAD+ cells was seen in CD4+ but not CD8+ T lymphocytes (Extended Data Fig. 4c-e). No cell-to-cell contact was required (Fig. 3b). Higher lipid levels were detected in hepatic CD4+ T lymphocytes in MCD mice (Extended Data Fig. 4f, g). This prompted us to examine whether lipids released from lipid-laden hepatocytes were taken up by CD4+ T lymphocytes and caused cell death. To test this hypothesis, we first measured the hepatic FFA composition. Consistent with previous reports, palmitic acid (C16:0), stearic acid (C18:0), linoleic acid (C18:2), arachidonic acid (C20:4) and docosahexaenoic acid (C22:6) are the abundant FFAs (Fig. 3c, Extended Data Table 1). Although the total amount of FFAs did not change significantly (Extended Data Table 1), the levels of C16:0 and C22:6 decreased. C18:2 was the only abundant FFA, which accumulated in the liver after MCD treatment (Fig. 3c). Our data is supported by previous reports of hepatic C18:2 accumulation in HF diet-induced NAFLD mice and ob/ob mice 8,21. Next, we depleted FFAs from conditioned hepatocyte-culture medium. As expected FFA-depleted conditioned medium no longer caused CD4+ T lymphocyte death (Fig. 3d). FFAs in conditioned medium from lipid-laden hepatocytes were further analyzed, and C16:0, C18:0 and C18:2 were identified as predominant FFAs. (Extended Data Fig. 4h, i).
Figure 3.

Lipid-laden hepatocytes cause CD4+ T lymphocyte death through releasing C18:2.
a, Ex vivo cell death of intrahepatic CD4+ T lymphocytes from MYC-ON NAFLD mice. (n=7 for CTR, 9 for MCD, Student’s t test). b, Lymphocyte survival after incubation with hepatocyte conditioned medium (CM) (n=4, two-way ANOVA). c, Hepatic total FFA composition analysis (n=6, *p<0.05, ANOVA). d, FFA depletion (Dep) from conditioned medium (n=3 for None, 9 for other treatments; two-way ANOVA). e, f, Lymphocyte survival after FFA treatment (n=4, one-way ANOVA). g, h, CD4+ and CD8+ T lymphocytes in high or low-C18:2 diet fed mice (n=5, Student’s t test). All data are Mean ± SEM, *p<0.05.
Then, isolated CD4+ T lymphocytes were incubated with individual FFA to study their effect on cell survival. C18:2 treatment caused a substantial higher level of 7AAD+Annexin V+ cells. Other tested FFAs did not show an effect (Fig. 3e). Unlike CD4+ T lymphocytes, cell death in CD8+ T lymphocytes was not affected at the tested concentration (Fig. 3f). Similar results were found in activated T lymphocytes (Extended Data Fig. 4j). Dose-response and time course analysis confirmed that CD4+ T lymphocytes were more susceptible to C18:2-induced cell death than CD8+ T lymphocytes (Extended Data Fig. 4k-m). The increase of caspase 3/7 activity confirmed that CD4+ T lymphocytes died via apoptosis (Extended Data Fig. 4n). Similar cell death between CD4+ and CD8+ T lymphocytes was observed in H2O2-induced cell death model showing that the effect is specific to C18:2 (Extended Data Fig. 4o). Interestingly, mice fed with a high C18:2 diet showed a reduction in CD4+ but not CD8+ T lymphocytes (Fig. 3g, h) suggesting that C18:2 is sufficient to cause CD4+ T lymphocyte death in vivo.
The mechanism of how C18:2 induced CD4+ T lymphocyte death was studied. No difference in cellular C18:2 uptake by CD4+ versus CD8+ T lymphocytes was found (Extended Data Fig. 4p). Direct assessment revealed greater mitochondrial mass in CD4+ lymphocytes (Fig. 4a). Microarray analysis revealed that oxidative phosphorylation and mitochondrial dysfunction pathways were specifically altered in CD4+ but not CD8+ T lymphocytes following C18:2 treatment (Extended Data Fig. 5). CPT1a, the rate-limiting enzyme for importing FFAs into mitochondria, increased in parallel with the decrease of a number of genes coding components for electron transport complex (Extended Data Fig. 6a). C18:2 was more potent than other FFAs in upregulating CPT1a (Fig. 4b). A similar effect was observed in Jurkat cells, a human CD4+ derived T leukemia cell line (Extended Data Fig. 6b). Knockdown of CPT1a rescued Jurkat cells from C18:2-induced cell death (Fig.4c, Extended Data Fig. 6c). All these results pointed towards mitochondria as the critical mediator for CD4+ T lymphocyte death. Inside mitochondria, FFAs are β-oxidized to fuel ATP generation via electron transport chain (ETC). Fatty acid oxidation (FAO) was measured, and C18:2 showed a greater FAO rate than C16:0 (Fig. 4d). Higher FAO favors more NADH entering the ETC to generate ATP. However, our array data also suggested that C18:2 impaired ETC function (Extended Data Fig. 6a). Indeed, mitochondrial membrane potential, which is maintained by proper ETC activity, was significantly decreased by C18:2 in CD4+ but not CD8+ T lymphocytes (Fig. 4e). Disrupted ETC can become a major site of premature electron leakage to oxygen to generate reactive oxygen species (ROS) and lead to cell death22.
Figure 4.

Mitochondrial ROS mediates C18:2-induced CD4+ T lymphocyte death.
a, Mitochondrial mass analysis. b, CPT1a mRNA levels in FFA-treated CD4+ T lymphocytes (n=6). c, CPT1a knockdown on C18:2-induced Jurkat cell death (n=6). d, Oxidation rate of C18:2 or C16:0 in lymphocytes (n=3). e, Mitochondrial membrane potential in C18:2-treated lymphocytes. f, g, Oxygen consumption rate assay of activated CD4+ and CD8+ T lymphocytes treated with FFAs (n=8). h, Ex vivo ROS levels of intrahepatic CD4+ T lymphocytes (n=6 for CTR, 8 for MCD). i, Mitochondrial ROS levels in lymphocytes. j, Effect of NAC or catalase on hepatocyte-caused lymphocyte death (n=7). Hep: hepatocytes. k, l, In vivo effect of NAC treatment on intrahepatic CD4+ T lymphocytes and tumor development (n=3 for CTR, 4 for MCD, 10 for MCD+NAC, 5 for MCD+NAC+anti-CD4). m-o, MitoTEMPO treatment, mitochondrial ROS and survival in CD4+ T lymphocytes in vitro and in vivo (n=4 for CTR, 4 for MCD+PBS, 5 for MCD+MitoTEMPO). p, Human lymphocyte survival after FFA treatment (n=6). q, CD4/CD8 ratio of intrahepatic T lymphocytes in patient biopsies (n=6 for Normal, 16 for NASH, 8 for ASH, 15 for HBV/HCV). All data are Mean ± SEM, *p<0.05, one-way or two-way ANOVA analysis was used.
To assess mitochondrial respiration, oxygen consumption analysis was performed. Normalized oxygen consumption rates (OCR) were significantly higher in CD4+ T lymphocytes compared to CD8+ T lymphocytes, consistent with previous reports 23. Treatment with oligomycin, an inhibitor of mitochondrial ATP synthase, revealed substantial levels of ATP synthase-dependent oxygen consumption in both CD4+ and CD8+ T lymphocytes (Figure 4f, Extended Figure 6d). C18:2 abrogated the oligomycin-sensitive fraction of OCR in CD4+ and CD8+ T lymphocytes without reducing total oxygen consumption levels (Figure 4f, Extended Figure 6d). These data are consistent with a shift in oxygen consumption from ATP synthase dependent to ATP synthase independent ROS production. In contrast, C16:0 failed to eliminate ATP synthase dependent oxygen consumption in CD4+ T lymphocytes (Figure 4g, Extended 6e). Consistently, increased total ROS production was found in CD4+ T lymphocytes when co-cultured with C18:2 vs C16:0 (Extended Data Fig. 6f). Moreover, elevated ROS levels were detected in hepatic CD4+ T lymphocytes ex vivo under NAFLD condition (Fig. 4h) Lastly, mitochondrial superoxide was confirmed to selectively increase in CD4+ T lymphocytes after C18:2 treatment (Fig. 4i), and CPT1a knockdown blocked C18:2-induced mitochondrial ROS production (Extended Data Fig. 6g). Taken together, these data suggest that greater levels of mitochondrial-derived ROS accumulate in CD4+ T lymphocytes following C18:2 treatment leading to their depletion.
Therefore, we tested the role of ROS in NAFLD-associated CD4+ T lymphocyte death and HCC development in vivo. Blocking ROS with catalase or N-acetylcysteine (NAC) abrogated cell death in vitro in CD4+ T lymphocytes when incubated with hepatocytes from MCD-MYC mice (Fig. 4j). Similarly, catalase and NAC prevented C18:2-induced CD4+ T lymphocyte death in vitro (Extended Data Fig. 6h). Oxidative stress is an important factor in NAFLD progression 24. To test whether ROS mediate hepatic CD4+ T lymphocyte loss in vivo, we treated MCD diet-fed mice with NAC. Although NAC treatment did not influence steatosis (Extended Data Fig. 6i, j), it effectively reversed the loss of hepatic CD4+ T lymphocytes (Fig. 4k). More importantly, NAC treatment significantly delayed NASH-promoted tumor development (Fig. 4l and Extended Data Fig. 6j). Tumor lesions occurred despite NAC treatment when CD4+ T lymphocytes were removed, suggesting prevention of CD4+ T lymphocyte death mediates at least partially the anti-tumor effect of NAC. Similar results were obtained using mitoTEMPO 25, a specific mitochondrial antioxidant in both in vitro and in vivo settings (Fig. 4m-o).
C18:2 has also been identified as an important fatty acid in the context of NAFLD in humans 26,27. We tested whether C18:2 also affects human CD4+ T lymphocyte survival. Consistent with our mouse data, C18:2, but no other tested FFAs, caused selective CD4+ but not CD8+ T lymphocyte death (Fig. 4p, Extended Data Fig. 7a). Similarly, C18:2 but not C16:0 increased ROS level in human CD4+ T lymphocytes (Extended Data Fig. 7b). Finally, intrahepatic CD4+ T lymphocytes in liver biopsies from patients with NASH, alcoholic steatohepatitis (ASH) and viral hepatitis were determined (Extended Data Table 2). While ALT and AST levels did not differ among patients with different liver diseases (Extended Data Fig.7c, d), fewer CD4+ T lymphocytes were found in NASH and ASH patients than viral hepatitis patients (Extended Data Fig. 7e and Extended Data Fig. 8), and the CD4/CD8 ratio was significantly lower in NASH patients supporting the selective CD4+ T lymphocyte loss (Fig. 4q). Interestingly, lower CD4 counts were also found in ASH patients, which has very similar histological features compared with NASH.
Dysregulation of lipid metabolism and accumulation of lipids in the liver is part of the etiology of NAFLD. So far NASH has been described as causing NFκB dysregulation, activation of the inflammasome, TLR activation and affecting innate immune responses through multiple pathways or directly affecting hepatocytes 28-30. Our results extend these findings by describing a novel link between obesity-induced lipid accumulation and selective CD4+ T lymphocyte loss, suggest a critical role for CD4+ T lymphocytes in the disease progression from NAFLD to HCC.
Methods
Murine studies
LAP-tTA and TRE-MYC mice were previously described and MYC expression in the liver was activated by removing doxycycline treatment (100 μg/ml) from the drinking water of 4-weeks old double transgenic mice for both TRE-MYC and LAP-tTA as previously described 9,13. C57BL/6 mice were obtained from NCI/Frederick (Frederick, USA). Chemically induced HCC was established by intraperitoneal injection of diethylnitrosoamine (DEN) (Sigma) into two-week old male pups at a dose of 20 μg/g bodyweight 13. Twelve-week old male B6.Cg-Lepob/J (ob/ob) mice or wild type control mice were obtained from Charles River. Foxp3-GFP mice were previously described 31. NAFLD was induced by feeding mice with methionine choline-deficient (MCD) diet (Cat# 960439, MP biomedical, USA), choline-deficient and amino acid-defined (CDAA) diet (Cat# 518753, Dyets, USA) or high fat diet (Cat# F3282, Bio Serv, USA) for the indicated time 10,11,32. The MCD diet was supplied with corn oil (10%, w/w), and no fish oil was added. Control diet was purchased from MP Biomedical (Cat#960441). Custom-made high or low linoleic acid mouse diets were purchased from Research Diets, Inc. The modified diets were based on AIN-76A standard mouse diet, and are isocaloric (4.45kcal/g) and contained same high-fat content (23%, w/w). Linoleic acid-rich safflower oil and saturated fatty acid-containing coconut oil were supplied at different ratio to yield 2% (w/w) for low linoleic acid diet or 12% (w/w) for high linoleic acid diet. C57BL/6 mice were fed with high or low linoleic acid diet for 4 weeks. MYC mice were injected i.p. with 50μg CD4 antibody (clone GK1.5, BioXcell) every week for the indicated time period to deplete CD4+ T cells 33. N-acetylcysteine (NAC) was given in drinking water (10 mg/ml) 34 for the indicated time period to prevent excess ROS production. Mitochondrial specific antioxidant mitoTEMPO was purchased from Sigma. Mice received mitoTEMPO at a dose of 0.7 mg/kg per day 25 by osmotic minipumps (ALZET). At the experimental end points, mice were sacrificed. For flow cytometry analysis, single cell suspensions were prepared from spleen, liver and blood as described previously. Red blood cells were lysed by ACK Lysis Buffer (Quality Biologicals). Parts of live tissue were fixed by 10% formaldehyde and subjected to H&E staining. Free fatty acids were purchased from Sigma.
Oil red O staining
Lipid accumulation was detected by Oil Red O staining in frozen liver sections using custom service of Histo Serv (Germantown, Maryland).
Flow cytometry
Cells were surface-labeled with indicated antibodies for 15 minutes at 4°C. Flow cytometry was performed on BD FACSCalibur or BD LSRII platforms and results were analyzed using FlowJo software version 9.3.1.2 (TreeStar Inc). The following antibodies were used for flow cytometry analysis: anti-CD3-FITC (clone 17A2, BD Pharmingen), anti-CD4-PE (clone RM4-4, Biolegend), anti-CD4-APC (clone RM4-5, eBioscience), anti-CD8-Alexa Fluor 700 (clone 53-6.7 Biolegend), anti-CD45, anti-CD44-PE (clone IM7, eBioscience), anti-CD62L-PerCP/Cy5.5 (MEL-14, Biolegend), anti-CD69-pacific blue (clone H1.2F3, Biolegend), PBS57/CD1d-tetramer-APC (NIH core facility). To determine cytokine production, cells were stimulated with PMA and ionomycine for 30 minutes, and then were fixed and permeabilized using cytofix/cytoperm kit (BD Pharmingen) followed by anti-IFNγ-PE (clone XMG1.2, BD Pharmingen), anti-IL17-PerCP/Cy5.5 (clone TC11-18H10.1, Biolegend) staining. Cell death and apoptosis were detected with Annexin V-PE (BD Pharmingen) and 7-AAD (BD Pharmingen) staining according to manufacturer’s instructions. Intrahepatic CD4+ lymphocytes were gated on CD3hiCD4+ population from total live hepatic infiltrating mononuclear cells. Absolute numbers were calculated by multiplying frequencies obtained from flow by total live mononuclear cell count, then divided by liver weight. The antibodies used for human PBMC staining are following: anti-CD3-PE (clone SK7, BD Pharmingen), anti-CD4-FITC(clone RPA-T4, BD Pharmingen), anti-CD8-APC (clone RPA-T8, BD Pharmingen).
Treg suppressive function assay
Murine Treg assays were performed as described 31. Briefly, liver Treg cells were isolated as CD4+GFP+ by flow cytometry assisted cell sorting from Foxp3-GFP mice kept on MCD or control diet for 4 weeks. CD4+GFP− Teff cells (5 × 104) were stimulated for 72 h in the presence of irradiated T depleted splenocytes (5 × 104) plus CD3ε mAb (1 µg/mL), with or without Treg added at different ratios. 3H-Thymidine was added to the culture for the last 6 h and incorporated radioactivity was measured.
AFP-specific T cell response
Freshly isolated splenocytes from MYC-ON MCD mice were incubated with 5 μg/ml of mouse alpha-fetoprotein protein (MyBioSource Inc) for 24 hours. Golgiplug was added the last 6 hours. Then, cells were fixed and permeabilized using cytofix/cytoperm kit (BD Pharmingen) followed by anti-IFNγ-PE (clone XMG1.2, BD Pharmingen) staining.
Hepatocyte isolation
Primary mouse hepatocytes were isolated from MYC mice and cultured according to previous report 35. Briefly, mice were anesthetized and the portal vein was cannulated under aseptic conditions. The livers were perfused with EGTA solution (5.4 mM KCl, 0.44 mM KH2PO4, 140 mM NaCl, 0.34 mM Na2HPO4, 0.5 mM EGTA, 25 mM Tricine, pH 7.2) and Gey’s balanced salt solution (Sigma), and digested with 0.075% collagenase solution. The isolated mouse hepatocytes were then cultured with complete RPMI media in collagen I coated plates.
Hepatic fatty acid profiling
Hepatic fatty acid composition was measured at LIPID MAPS lipidomics core at the University of California (San Diego) using esterified and non-esterified (total) fatty acid panel. Briefly, liver tissues were homogenized and lipid fraction was extracted using modified Bligh Dyer liquid/liquid extraction method. The lipids were saponified and the hydrolyzed fatty acids were extracted using a liquid/liquid method. The extracted fatty acids were derivatized using Pentaflourylbenzylbromine (PFBB) and analyzed by gas chromatography (GC) using an Agilent GC/MS ChemStation. Individual analytes were monitored using selective ion monitoring (SIM). Analytes were monitored by peak area and quantified using the isotope dilution method using a deuterated internal standard and a standard curve.
Free fatty acid identification
Isolated primary hepatocytes from MYC mice fed with MCD or control diet were cultured in complete RPMI for 24 hrs. Supernatant were harvested and FFAs were identified by gas chromatography-mass spectrometry.
Microarray analysis
Splenocytes from MYC mice were cultured with or without 50 μM linoleic acid for 24 hrs. CD4+ and CD8+ T lymphocytes were sorted and total RNA was extracted using miRNeasy mini kit (Qiagen). Array analysis was performed in Department of Transfusion Medicine, clinical center at NIH. Mouse gene 2.0 ST array (Affymetrix) was used and performed according to manufacturer’s instruction. Data were log-transformed (base 2) for subsequent statistical analysis. Partek Genomic Suite 6.4 (Partek Inc., St. Louis, MO, USA) was used for the identification of differentially expressed transcripts. The Ingenuity Pathway Analysis tool (http://www.ingenuity.com, Ingenuity System Inc., Redwood City, CA, USA) was used for analysis of functional pathways. Microarray data were uploaded to GEO (CEO# GSE67918).
RNA isolation and Real-Time PCR
RNA was extracted from frozen tissues with RNeasyMini Kit (Qiagen). Complementary DNA was synthesized by iScript™cDNA synthesis kit (BioRad). Sequence of primers used for quantitative RT-PCR can be obtained from authors. The reactions were run in triplicates using iQSYBR green supermix kit (BioRad). The results were normalized to endogenous GAPDH expression levels.
CD4+ T cell isolation and co-culture with fatty acids
CD4+ T lymphocytes were isolated from spleen of MYC mice by negative autoMACS selection using CD4+ T lymphocytes isolation kit (Miltenyi Biotec) or flow cytometry cell sorting. Human CD4+ T lymphocytes were prepared from PBMC by autoMACS using CD4+ T lymphocytes isolation kit (Miltenyi Biotec). The purity of CD4+ T lymphocytes was above 90% after autoMACS separation and above 95% after flow cytometry cell sorting. Palmitic acid, stearic acid, oleic acid, linoleic acid and docosahexaenoic acid were purchased from sigma. Fatty acids were dissolved in DMEM with 2% fatty acid free BSA (Sigma, Cat#A8806) after solvent was evaporated, then followed by two rounds of vortexing and 30 seconds of sonication. Isolated CD4+ T lymphocytes or splenocytes were incubated with different fatty acids or conditioned medium from hepatocyte culture for 3 days. Unless specifically described, fatty acids were used at 50 μM concentration. For fatty acid depletion, active charcoal (Cat#C-170, Fisher) was used as described before 36. Briefly, 0.5 gram of active charcoal was added into every 10 ml of conditioned medium. Then pH was lowered to 3.0 by addition of 0.2N HCl. The solution was rotated at 4°C for 2 hours. Charcoal was then removed by centrifugation, and clarified solution was then brought back to pH7.0 by addition of 0.2 N NaOH. NAC (10 mM), catalase (1000 U/ml) or mitoTEMPO (10 μM) was used to inhibit ROS production, mitochondrial ROS levels were determined by mitoSOX staining 24 hrs after treatment, cell death and apoptosis were measured by Annexin V and 7-AAD staining 3 days after treatment.
Caspase activity assay
Caspase activity assay was measured by caspase-Glo 3/7 assay kit (Promega) according to manufacturer’s protocol.
BODIPY staining
Fresh prepared liver infiltrating mononuclear cells were washed and resuspended in 500 μl of BODIPY 493/503 at 0.5 μg/ml in PBS. Cells were stained for 15 min at room temperature. Then cells were subjected to flow cytometry analysis.
RNA interference assay
Two pZIP lentiviral shRNA vectors targeting human CPT1a and a control vector (NT#4) were purchased from TransOMIC Technologies. Lentivirus were packed in 293T cells. Jurkat cells were purchased from DSMZ-German Collection of Microorganisms and Cell Cultures, and no authenticated test has been performed by us. Cells were cultured in complete RPMI medium and have been tested to be mycoplasma free. Jurkat cells were infected with shRNA lentivirus. Puromycin was added to eliminate non-transduced cells. Doxycycline (100 ng/ml) was added to induce shRNA and GFP expression for 3 days. Efficiency of shRNAs was confirmed by Western. Jurkat cells with CPT1a knockdown were treated with 200 µM linoleic acid for 24 hrs. Mitochondrial ROS production and cell survival were measured in GFP+-transduced cells.
Fatty acid oxidation assay
Fatty acid oxidation was measured according to previous publication 37. 1-14C-linoleic acid and 1-14C-palmitic acid were purchased from PerkinElmer. Briefly, isolated CD4+ or CD8+ T lymphocytes were pretreated with linoleic acid or kept in regular media. After 24 hrs. cell media was changed to media containing 50 μM cold linoleic acid plus 1μCi 1-14C-linoleic acid/ml or 50 μM cold palmitic acid plus 1μCi 1-14C-palmitic acid /ml. After 2 hours medium was removed and mixed with concentrated perchloric acid (final concentration 0.3M) plus BSA (final concentration 2%) to precipitate the radiolabeled fatty acids. Samples were vortexed and centrifuged (10,000x g for 10 min). Radioactivity was determined in the supernatant to measure water-soluble β-oxidation products.
Mitochondrial membrane potential and ROS staining
Mitochondrial membrane potential was measured by TMRM (immunochemistry technologies) staining according to manufacturer’s protocol. Briefly, cells were kept in culture medium with 100 nM of TMRM for 20 minutes in CO2 incubator at 37°C. After washing twice, cells were processed to flow cytometry analysis. Mitochondria associated superoxide was detected by mitoSOX (life technologies) staining according to manufacturer’s protocol. Briefly cells were first subjected to surface marker staining. Then cells were stained with 2.5 μM mitoSOX for 30 minutes in CO2 incubator at 37°C. After washing twice, cells were processed for flow cytometry analysis.
Oxygen consumption assay
OCR was measured using an XFe96 Extracellular Flux Analyzer (Seahorse Bioscience) as previously published 38. AutoMACS sorted mouse CD4+ and CD8+ T lymphocytes were attached to XFe96 cell culture plates using Cell-Tak (BD Bioscience) in RPMI media with 11 mM glucose. Cells were activated with 1:1 CD3:CD28 beads (Miltenyi BioTech) and vehicle vs 50 µM linoleic acid was added. Twenty-four hours after activation, cells were incubated in serum-free XF Base Media (Seahorse Bioscience) supplemented with 10 mM glucose, 2 mM pyruvate, and 2 µM glutamine, pH 7.4, along with 50 µM linoleic acid if previously present, for 30 minutes at 37°C in a CO2-free cell culture incubator before beginning the assay. Five consecutive measurements, each representing the mean of 8 wells, were obtained at baseline and following sequential addition of 1.25 µM oligomycin, 0.25 µM trifluorocarbonylcyanide phenylhydrazone (FCCP), and 1 µM each of rotenone and antimycin A (all drugs from Seahorse Bioscience). OCR values were normalized to cell number as measured by the CyQUANT Cell Proliferation Assay Kit (Life Technologies).
Human studies
Human liver samples were stained as previously described 8. For immuno staining, formalin-fixed, paraffin-embedded human liver tissue samples were retrieved from the archives of the Institute of Surgical Pathology, University Hospital Zurich. The study was approved by the local ethics committee (“Kantonale Ethikkommission Zürich”, application number KEK-ZH-Nr. 2013-0382). Human PBMC from healthy donors were obtained on an NIH approved protocol and prepared as described previously 39. Informed consent was obtained from all subjects.
Statistic analysis
Sample size for animal studies were guided by previous study in our laboratory in which the same MYC transgenic mouse stain was used. No animals were excluded. Neither randomization nor blinding was done during the in vivo study. However, mice from same littermates were evenly distributed into control or treatment groups whenever possible. Sample size for the patient studies was guided by a recent publication also studying NASH induced HCC, but focused on different aspects 8. Statistical analysis was performed with GraphPad Prism 6 (GraphPad Software). Significance of the difference between groups was calculated by Student’s unpaired t-test, one-way or two-way ANOVA (Tukey’s and Bonferroni’s multiple comparison test). Welch’s corrections were used when variances between groups were unequal. p<0.05 was considered as statistically significant.
Extended Data
Extended Data Figure 1.

MCD, CDAA and HF diet induce NASH and promote HCC.
a, Representative imagines of Oil Red O staining of MYC-ON mice fed MCD or CTR. Black bar = 100 μm. b, Serum ALT levels analysis. Mean ± SEM; n=4, *p<0.05, one-way ANOVA. c-e, The effect of CDAA diet on tumor development in MYC transgenic mice. Experimental set-up, representative liver images and liver surface tumor counts are shown. Black bar=10 mm. Mean ± SEM; n= 6 for CDAA and n=5 mice for CTR, p=0.0345, Student’s t test. f-i, The effect of CDAA and HF diet on liver carcinogenesis in DEN-injected C57BL/6 mice. Experimental set-up, representative tumor-free H&E stainings, macroscopic liver images and surface tumor counts are shown. Black bar=100 μm. Mean ± SEM; n=13 for CTR, n=9 for HF, n=10 for CDAA, *p<0.05, one-way ANOVA.
Extended Data Figure 2.
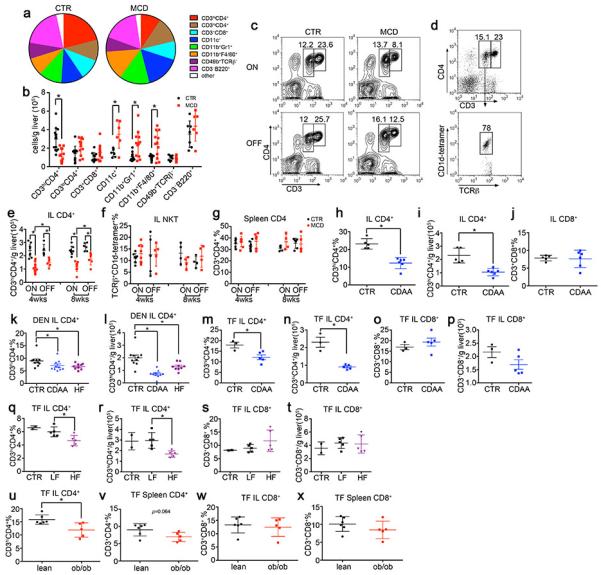
Immune cell monitoring in NAFLD-HCC.
MYC mice were fed with MCD diet or CTR diet. Intrahepatic immune cells were determined by flow cytometry, a, composition and b, absolute numbers of different intrahepatic immune cell subsets in MYC-ON mice, which were kept for 4 weeks on MCD diet or CTR diet. Mean ± SEM; n≥6, *p<0.05, one-way ANOVA. c, Representative contour plots of intrahepatic CD4+ T lymphocytes. d, Representative dot plots of CD1d-tetramer staining in CD3loCD4+ population. e-g, Absolute number of intrahepatic CD4+ T lymphocytes, frequencies of NKT cells and splenic CD4+ T lymphocytes were measured by flow cytometry. Mean ± SEM; n=4, *p<0.05, two-way ANOVA. h-j, Intrahepatic CD4+ and CD8+ T lymphocyte levels in MYC-ON mice fed with CDAA diet for 16 weeks. Mean ± SEM; n=6 for CDAA and n=5 for CTR, *p<0.05, Student’s t test. k, l, Intrahepatic CD4+ T lymphocyte levels in DEN-injected BL/6 male mice treated with CDAA diet, HF diet or CTR for 7 months. Mean ± SEM; n=13 for CTR, n=9 for HF, n=10 for CDAA, *p<0.05, one-way ANOVA. m-p, Intrahepatic CD4+ and CD8+ T lymphocytes in tumor free C57BL/6 mice treated with CDAA diet for 16 weeks. TF: tumor free; Mean ± SEM; n=3 for CTR, n=5 for CDAA, *p<0.05, Student’s t test. q-t, Intrahepatic CD4+ and CD8+ T lymphocytes in tumor free C57BL/6 mice treated with HF or LF diet for 6 months. Mean ± SEM; n=2 for CTR, n=5 for LF, n=5 for HF, *p<0.05, one-way ANOVA. u-x, CD4+ and CD8+ T lymphocytes in 12-week old male ob/ob or wild type lean mice. Mean ± SEM, n=5, *p<0.05, Student’s t test w, x, MYC mice were fed with MCD or CTR. Macrophage and CD11b+Gr1+ populations were measured. Mean ± SEM; n≥4, *p<0.05, two-way ANOVA.
Extended Data Figure 3.
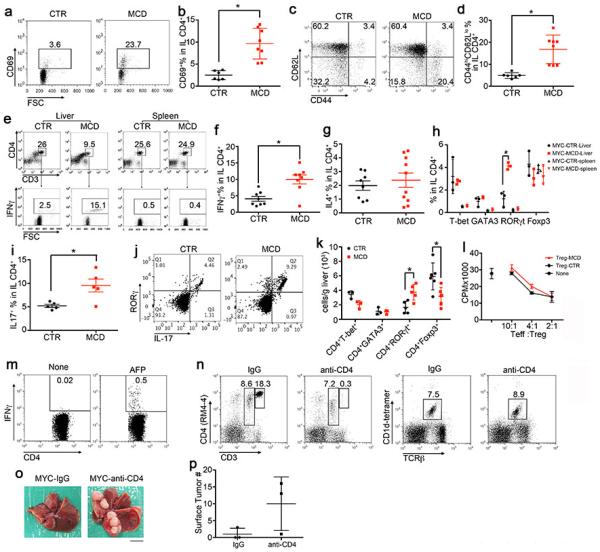
Intrahepatic CD4+ lymphocytes are activated in NAFLD, and CD4 depletion enhances HCC.
MYC-ON mice were fed with MCD or CTR for 4 weeks. a-d, CD69 and CD44hiCD62Llo subsets in intrahepatic CD4+ T lymphocytes were measured. Mean ± SEM; n=8 for MCD and n=6 for CTR, *p<0.05, Student’s t test. e-g. Ex vivo IFNγ, IL4 production in intrahepatic CD4+ T lymphocytes were determined. Mean ± SEM; n=8, *p<0.05, Student’s t test. h, Ex vivo staining of T-bet, GATA3, RORγt and Foxp3 levels in intrahepatic and splenic CD4+ T lymphocytes. Mean ± SEM; n=3, *p<0.05, two-way ANOVA. i, Ex vivo IL17 production by intrahepatic CD4+ T lymphocytes. Mean ± SEM; n=5, *p<0.05, Student’s t test. j, Representative dot plots of RORγt/IL17 staining in intrahepatic CD4+ T lymphocytes. k, Absolute number of intrahepatic CD4+ lymphocyte subsets. Mean ± SEM; n=3, *p<0.05, two-way ANOVA. l, Suppressive function assay of isolated hepatic Tregs from Foxp3-GFP mice kept on MCD or CTR for 4 weeks. m, Detection of AFP-specific CD4+ T lymphocytes in spleen from MYC-MCD mice. n, Selective depletion of intrahepatic CD4+ T lymphocytes but not NKT by i.p. injection of 50 μg GK1.5. o, p, MYC-on mice on CTR received 50 μg of GK1.5 antibody or isotype control i.p. once per week for 8 weeks. Representative liver imagines and surface tumor counts are shown. Black bar=10 mm. Mean ± SEM, n=3.
Extended Data Figure 4.

Lipid-laden hepatocytes release linoleic acid and induce CD4+ T lymphocyte death via apoptosis.
a, Representative contour plots of ex vivo 7AAD/Annexin V staining of intrahepatic CD4+ T lymphocytes from MYC-ON mice fed with MCD or CTR. b, Representative phase-contrast images of primary hepatocytes from MYC-ON mice after MCD or CTR treatment. c-e, Isolated primary hepatocytes from MYC-ON mice on MCD or CTR were cocultured with isolated CD4+ T lymphocytes or splenocytes. Cell death levels were measured by flow cytometry. Mean ± SEM; n=4, one-way or two-way ANOVA. f, g, BODIPY 493/503 staining of CD4+ T lymphocytes in liver, spleen or blood from MYC-ON mice with MCD or CTR. Mean ± SEM; n=4, *p<0.05, two-way ANOVA. h,i, Identification of free fatty acids in hepatocyte conditioned medium by GC/MS. Mean ± SEM; n=3, *p<0.05, two-way ANOVA. j, Anti-CD3/28 beads-activated splenocytes were treated with different free fatty acids, and cell death level in CD4+ or CD8+ T lymphocytes were determined. Mean ± SEM; n=4, *p<0.05, two-way ANOVA. k-m, Dose-response curve and time course of linoleic acid-induced cell death in CD4+ or CD8+ T lymphocytes. n, caspase3/7 activity in CD4+ lymphocytes after linoleic acid treatment. Mean ± SEM; n=9, *p<0.05, Student’s t test. o, Dose-response curve of H2O2-induced cell death in CD4+ or CD8+ T lymphocytes. p, Uptake of linoleic acid by CD4+ and CD8+ T lymphocytes after incubation with 50 μM linoleic acid for 2 hrs. Mean ± SEM; n=6, *p<0.05. two-way ANOVA.
Extended Data Figure 5.
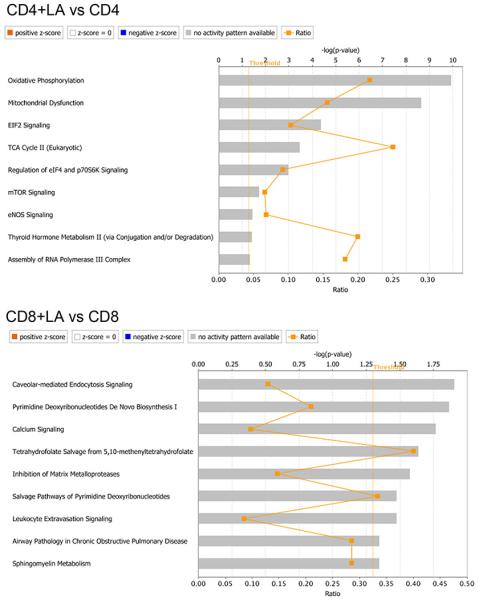
Ingenuity pathway analysis of microarray data.
CD4+ and CD8+ T lymphocytes sorted from linoleic acid-treated splenocytes were subjected to microarray analysis. Pathway analysis was done by IPA. n=3. Ratio= number of changed genes divided by total genes in the pathway.
Extended Data Figure 6.

Mitochondrial ROS mediates linoleic acid-induced CD4+ T lymphocyte death in vitro and in vivo.
a, Real-time PCR confirmed the gene changes from microarray. Mean ± SEM; n=3, *p<0.05, two-way ANOVA. b, CPT1a mRNA level in Jurkat cells after fatty acid treatment. Mean ± SEM; n=6, *p<0.05, one-way ANOVA. c, Expression of CPT1a in wild type and two knockdown Jurkat cells. NT: none-targeting control. d, e, OCR analysis of activated CD4+ and CD8+ T lymphocytes upon linoleic acid or palmitic acid incubation. f, ROS levels of CD4+ or CD8+ T lymphocytes in splenocytes treated with linoleic acid or palmitic acid. Mean ± SEM; n=8, *p<0.05, two-way ANOVA. g, Mitochondrial ROS in wild type and two CPT1 knockdown Jurkat cells. i, Cell death of CD4+ or CD8+ T lymphocytes in splenocytes treated with linoleic acid at the presence of NAC or catalase. Mean ± SEM; n=4, *p<0.05, two-way ANOVA. i, j, In vivo blocking ROS with NAC in MYC-ON mice treated with MCD. Some mice also received CD4 antibody depletion. Experimental setup and representative H&E liver sections are shown. Black bar= 200 μm.
Extended Data Figure 7.
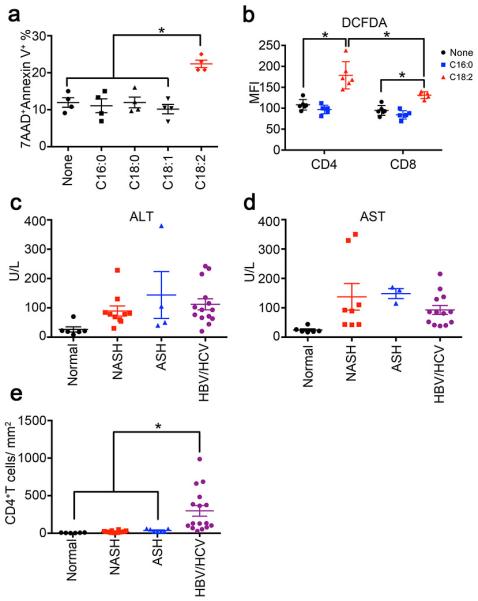
Linoleic acid induces cell death in human CD4+ T lymphocytes, and NASH patients have lower intrahepatic CD4+ T lymphocytes.
a, Cell death levels of sorted human CD4+ T lymphocytes treated with different free fatty acids. Mean ± SEM; n=4 *p<0.05, one-way ANOVA. b, ROS level of CD4+ or CD8+ T lymphocyte in PBMC treated with linoleic acid or palmitic acid. Mean ± SEM; n=6 *p<0.05, two-way ANOVA. c,d, serum ALT and AST concentration in different patients. e, Intrahepatic CD4+ T lymphocyte count in biopsies. CD4+ T lymphocytes were identified by immunohistochemistry. Mean ± SEM; normal=6, NASH=16, ASH=8, HBV/HCV=16, *p<0.05, one-way ANOVA.
Extended Data Figure 8.

Immunohistochemistry staining of intrahepatic CD4+ or CD8+ T lymphocytes in patient biopsies.
Representative CD4 or CD8 immunohistochemistry images of liver biopsies from healthy individuals, NASH, ASH patients or patients with HBV or HCV. For each condition, two different magnifications are shown. Black bar =100 μm.
Extended Data Table 1.
Fatty Acid Composition of Hepatic Lipids.
| Fatty Acid | ON-CTR | ON-MCD | OFF-CTR | OFF-MCD |
|---|---|---|---|---|
| C12:0 | 17.2 ± 4.2 | 16.7 ± 2.1 | 10.3 ± 2.2 | 18.6 ± 2.6 |
| C14:0 | 143.9 ± 8.1 | 204.8 ± 21.1 | 195.2 ± 34.4 | 237.6 ± 25.7 |
| C15:0 | 86.9 ± 6.1 | 62.2 ± 6.1 | 77.9 ± 4.2 | 53.1 ± 4.0 |
| C16:0 | 10986.0 ± 703.0 | 7576.4 ± 486.5a | 11325.9 ± 517.8 | 8274.1 ± 543.9b |
| C16:1 | 467.2 ± 49.0 | 88.5 ± 7.7 | 430.9 ± 52.6 | 169.2 ± 17.7 |
| C17:0 | 216.1 ± 18.4 | 199.0 ± 17.2 | 249.8 ± 17.0 | 171.7 ± 12.9 |
| C17:1 | 0.5 ± 0.2 | 0.0 ± 0.0 | 0.0 ± 0.0 | 0.0 ± 0.0 |
| C18:0 | 6861.1 ± 520.0 | 7228.8 ± 463.1 | 8335.6 ± 287.9 | 7700.4 ± 557.0 |
| C18:1 | 1498.2 ± 126.0 | 1432.8 ± 108.1 | 1545.8 ± 102.3 | 1773.6 ± 111.2 |
| C18:2 | 4664.8 ± 464.5 | 6492.8 ± 356.1a | 4516.3 ± 582.9 | 7584.1 ± 462.3b |
| C18:3N3 | 30.0 ± 3.8 | 183.5 ± 23.3 | 28.3 ± 4.0 | 187.7 ± 20.1 |
| C18:3N6 | 71.0 ± 8.0 | 37.8 ± 2.4 | 65.1 ± 8.0 | 48.5 ± 2.8 |
| C18:4 | 2.3 ± 0.4 | 3.7 ± 0.5 | 1.4 ± 0.3 | 4.1 ± 0.4 |
| C20:0 | 80.8 ± 6.8 | 29.0 ± 2.4 | 16.4 ± 1.1 | 13.2 ± 1.3 |
| C20:1 | 2.5 ± 0.3 | 3.1 ± 0.4 | 1.1 ± 0.2 | 1.8 ± 0.2 |
| C20:2 | 119.7 ± 8.7 | 183.0 ± 14.3 | 108.3 ± 4.3 | 158.3 ± 16.7 |
| C20:3N3 | 53.0 ± 4.0 | 38.0 ± 5.6 | 53.8 ± 5.7 | 24.1 ± 3.8 |
| C20:3N6 | 973.8 ± 28.6 | 592.2 ± 75.3 | 709.6 ± 33.2 | 529.5 ± 58.0 |
| C20:3N9 | 13.5 ± 1.2 | 3.4 ± 0.3 | 10.0 ± 0.8 | 2.2 ± 0.3 |
| C20:4 | 10503.7 ± 751.6 | 10949.5 ± 799.5 | 14287.7 ± 619.5 | 10801.1 ± 1218.5b |
| C20:5 | 118.6 ± 9.7 | 25.0 ± 2.1 | 103.3 ± 10.3 | 28.5 ± 2.0 |
| C22:0 | 19.8 ± 1.6 | 6.5 ± 0.7 | 7.8 ± 1.0 | 3.2 ± 0.2 |
| C22:1 | 20.3 ± 1.6 | 5.5 ± 0.5 | 6.1 ± 0.3 | 2.2 ± 0.3 |
| C22:2 | 7.8 ± 0.9 | 2.5 ± 0.3 | 0.2 ± 0.1 | 0.0 ± 0.0 |
| C22:3 | 2.4 ± 0.4 | 0.0 ± 0.0 | 0.0 ± 0.0 | 0.0 ± 0.0 |
| C22:4 | 140.1 ± 8.3 | 419.9 ± 42.2 | 136.7 ± 4.3 | 296.4 ± 37.5 |
| C22:5N3 | 454.8 ± 29.2 | 997.8 ± 76.6 | 523.1 ± 23.4 | 706.5 ± 114.3 |
| C22:5N6 | 336.1 ± 29.8 | 80.0 ± 6.8 | 206.1 ± 7.6 | 87.7 ± 11.4 |
| C22:6 | 9237.6 ± 687.1 | 5207.0 ± 388.0a | 10704.6 ± 426.9 | 6414.9 ± 638.9b |
| C23:0 | 1.2 ± 0.2 | 0.2 ± 0.1 | 0.2 ± 0.1 | 0.0 ± 0.0 |
| C24:0 | 6.5 ± 0.4 | 2.9 ± 0.5 | 3.3 ± 0.4 | 1.8 ± 0.1 |
| C24:1 | 1.1 ± 0.1 | 0.4 ± 0.1 | 0.2 ± 0.0 | 0.0 ± 0.0 |
| Total | 47313.0 ± 3047.1 | 41742.3 ± 2471.9 | 53664.5 ± 1953.3 | 45295.3 ± 3554.2 |
Mouse livers from MYC-on or MYC-OFF mice receiving 4 weeks of MCD or control diet treatment were subjected to total fatty acid profiling by GC/MS as described in Material and Methods. Results (in pmol/mg tissue) are expressed as mean± SEM (n=6/group,
p<0.05 for ON-CTR vs ON-MCD,
p<0.05 for OFF-CTR vs OFF-MCD, two-way ANOVA)
Extended Data Table 2.
Overview of patient cohort for IHC analysis.
| No | Disease | Age | Gender | ALT(UlL) | AST(UlL) | Fibrosis | Cirrhosis | Activity |
|---|---|---|---|---|---|---|---|---|
| 1 | Normal | 39 | M | 70 | 44 | 0 | N/A | N/A |
| 2 | Normal | 29 | F | 18 | 26 | 0 | N/A | N/A |
| 3 | Normal | 50 | F | 8 | 19 | 0 | N/A | N/A |
| 4 | Normal | 44 | M | 21 | 20 | 0 | N/A | N/A |
| 5 | Normal | 26 | M | 19 | 17 | 0 | N/A | N/A |
| 6 | Normal | 56 | F | 22 | 21 | 0 | N/A | N/A |
| 7 | NASH | 55 | M | 78 | 43 | 4 | 1a | N/A |
| 8 | NASH | 45 | F | 30 | 92 | 5 | 3 | N/A |
| 9 | NASH | 61 | F | 54 | 89 | 4 | 2 | N/A |
| 10 | NASH | 41 | M | 81 | x | 4 | x | N/A |
| 11 | NASH | 43 | M | x | x | 4 | 1a | N/A |
| 12 | NASH | 35 | M | x | x | 4 | 1b | N/A |
| 13 | NASH | 27 | M | 78 | x | 4 | 2 | N/A |
| 14 | NASH | 69 | M | x | x | 4 | 3 | N/A |
| 15 | NASH | 39 | F | 69 | 350 | 5 | 1b | N/A |
| 16 | NASH | 54 | F | 130 | 110 | 4 | 2 | N/A |
| 17 | NASH | 59 | M | 228 | 329 | 6 | 4 | N/A |
| 18 | NASH | 49 | M | x | x | 5 | 2 | N/A |
| 19 | NASH | 27 | F | x | x | 5 | 2 | N/A |
| 20 | NASH | 44 | M | 84 | 42 | 5 | 1b | N/A |
| 21 | NASH | 64 | F | 60 | 43 | 4 | 2 | N/A |
| 22 | NASH | 50 | F | x | x | 4 | 1a | N/A |
| 23 | ASH | 63 | F | 40 | 115 | 4 | N/A | N/A |
| 24 | ASH | 51 | M | x | x | 2 | N/A | N/A |
| 25 | ASH | 75 | F | x | x | 3 | N/A | N/A |
| 26 | ASH | 46 | M | 50 | 160 | 4 | N/A | N/A |
| 27 | ASH | 58 | M | 105 | x | 4 | N/A | N/A |
| 28 | ASH | 70 | M | x | x | 1 | N/A | N/A |
| 29 | ASH | 43 | M | 380 | 170 | 0 | N/A | N/A |
| 30 | ASH | 64 | M | x | x | 3 | N/A | N/A |
| 31 | HBV | 19 | M | 234 | 86 | 1 | N/A | 2 |
| 32 | HBV | 60 | M | 115 | 86 | 3 | N/A | 1 |
| 33 | HBV | 41 | F | 76 | 52 | 1 | N/A | 2 |
| 34 | HBV | 27 | F | 71 | 41 | 1 | N/A | 2 |
| 35 | HBV | 48 | F | x | x | 1 | N/A | 1 |
| 36 | HBV | 77 | M | 63 | 137 | 1 | N/A | 1 |
| 37 | HCV | 54 | F | 44 | 38 | 3 | N/A | 2 |
| 38 | HCV | 50 | M | 102 | 85 | 1 | N/A | 1 |
| 39 | HCV | 58 | M | 66 | x | 2 | N/A | 1 |
| 40 | HCV | 57 | F | 215 | 164 | 3 | N/A | 1 |
| 41 | HCV | 55 | M | 135 | 129 | 2 | N/A | 2 |
| 42 | HCV | 73 | F | 93 | 81 | 3 | N/A | 1 |
| 43 | HCV | 71 | F | x | x | 4 | N/A | 2 |
| 44 | HCV | 60 | F | 20 | 40 | 4 | N/A | 2 |
| 45 | HCV | 70 | F | 242 | 215 | 4 | N/A | 2 |
| 46 | HCV | 57 | F | 93 | 51 | 2 | N/A | 2 |
Fibrosis grade was analyzed for NASH according to NAS (NAFLD activity score) and for others according to METAVIR score. Disease activity of viral hepatitis was analyzed according to METAVIR score. x: not available, N/A: not applicable.
Acknowledgements
We would like to thank Jay Berzofsky, Wilhelm Stoffel, Ethan M. Shevach and Snorri Thorgeirsson for helpful discussion. A.M.T. was supported by the Intramural Research Program of the NIH, NIAID. J.L. was supported by the Intramural Program Grant ZIABC011303, NIH, NCI. M.H. was supported by an ERC starting grant (LiverCancer mechanism), the Stiftung Experimentelle Biomedizin (Hofschneider Stiftung), the Pre-clinical Comprehensive Center (PCCC) and the Helmholtz foundation. A.W. was supported by grants from the Krebsliga Schweiz (Oncosuisse) and the Promedica Stiftung, Switzerland. A.H.K.,D.W.M. and T.F.G. were supported by the Intramural Research Program of the NIH, NCI.
Footnotes
Author contributions:
C.M. and A.H.K. performed experiments. C.M., A.H.K., D.W.M., and T.F. G. analyzed data. T.E., J.M.-E., D.K., P.J., D.F.S., M.T., V.K., M.E., M.H., A.M.T., H.Z., J.L. and D.F.W. assisted with experiments and analysis or provided valuable reagents. H.K and D.W.M. performed and analyzed seahorse studies. M.E. A.W. and M.H. collected/provided human specimens and performed/analyzed immunohistochemistries of human samples. C.M. and T.F.G. conceived of and designed the project. C.M. and T.F.G wrote the manuscript and all authors contributted to writing and provided feedback.
Microarray data were uploaded to GEO (CEO# GSE67918).
The authors declare no competing financial interests.
References
- 1.European Association For The Study Of The, L. European Organisation For, R. & Treatment Of, C. EASL-EORTC clinical practice guidelines: management of hepatocellular carcinoma. J Hepatol. 2012;56:908–943. doi: 10.1016/j.jhep.2011.12.001. [DOI] [PubMed] [Google Scholar]
- 2.Sun B, Karin M. Obesity, inflammation, and liver cancer. J Hepatol. 2012;56:704–713. doi: 10.1016/j.jhep.2011.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Michelotti GA, Machado MV, Diehl AM. NAFLD, NASH and liver cancer. Nature reviews. Gastroenterology & hepatology. 2013;10:656–665. doi: 10.1038/nrgastro.2013.183. [DOI] [PubMed] [Google Scholar]
- 4.Schuppan D, Schattenberg JM. Non-alcoholic steatohepatitis: pathogenesis and novel therapeutic approaches. J Gastroenterol Hepatol. 2013;28(Suppl 1):68–76. doi: 10.1111/jgh.12212. [DOI] [PubMed] [Google Scholar]
- 5.Wree A, Broderick L, Canbay A, Hoffman HM, Feldstein AE. From NAFLD to NASH to cirrhosis-new insights into disease mechanisms. Nature reviews. Gastroenterology & hepatology. 2013;10:627–636. doi: 10.1038/nrgastro.2013.149. [DOI] [PubMed] [Google Scholar]
- 6.Greten TF, Wang XW, Korangy F. Current concepts of immune based treatments for patients with HCC: from basic science to novel treatment approaches. Gut. 2015 doi: 10.1136/gutjnl-2014-307990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Greten TF, Duffy AG, Korangy F. Hepatocellular carcinoma from an immunologic perspective. Clin Cancer Res. 2013;19:6678–6685. doi: 10.1158/1078-0432.CCR-13-1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wolf MJ, et al. Metabolic activation of intrahepatic CD8+ T cells and NKT cells causes nonalcoholic steatohepatitis and liver cancer via cross-talk with hepatocytes. Cancer Cell. 2014;26:549–564. doi: 10.1016/j.ccell.2014.09.003. [DOI] [PubMed] [Google Scholar]
- 9.Shachaf CM, et al. MYC inactivation uncovers pluripotent differentiation and tumour dormancy in hepatocellular cancer. Nature. 2004;431:1112–1117. doi: 10.1038/nature03043. [DOI] [PubMed] [Google Scholar]
- 10.Rinella ME, et al. Mechanisms of hepatic steatosis in mice fed a lipogenic methionine choline-deficient diet. J Lipid Res. 2008;49:1068–1076. doi: 10.1194/jlr.M800042-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yang L, et al. Transforming growth factor beta signaling in hepatocytes participates in steatohepatitis through regulation of cell death and lipid metabolism in mice. Hepatology. 2014;59:483–495. doi: 10.1002/hep.26698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Park EJ, et al. Dietary and genetic obesity promote liver inflammation and tumorigenesis by enhancing IL-6 and TNF expression. Cell. 2010;140:197–208. doi: 10.1016/j.cell.2009.12.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kapanadze T, et al. Regulation of accumulation and function of myeloid derived suppressor cells in different murine models of hepatocellular carcinoma. J Hepatol. 2013;59:1007–1013. doi: 10.1016/j.jhep.2013.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xia S, et al. Gr-1+ CD11b+ myeloid-derived suppressor cells suppress inflammation and promote insulin sensitivity in obesity. J Biol Chem. 2011;286:23591–23599. doi: 10.1074/jbc.M111.237123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Baeck C, et al. Pharmacological inhibition of the chemokine CCL2 (MCP-1) diminishes liver macrophage infiltration and steatohepatitis in chronic hepatic injury. Gut. 2012;61:416–426. doi: 10.1136/gutjnl-2011-300304. [DOI] [PubMed] [Google Scholar]
- 16.Henning JR, et al. Dendritic cells limit fibroinflammatory injury in nonalcoholic steatohepatitis in mice. Hepatology. 2013;58:589–602. doi: 10.1002/hep.26267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ma X, et al. A high-fat diet and regulatory T cells influence susceptibility to endotoxin-induced liver injury. Hepatology. 2007;46:1519–1529. doi: 10.1002/hep.21823. [DOI] [PubMed] [Google Scholar]
- 18.Rakhra K, et al. CD4(+) T cells contribute to the remodeling of the microenvironment required for sustained tumor regression upon oncogene inactivation. Cancer Cell. 2010;18:485–498. doi: 10.1016/j.ccr.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kang TW, et al. Senescence surveillance of pre-malignant hepatocytes limits liver cancer development. Nature. 2011;479:547–551. doi: 10.1038/nature10599. [DOI] [PubMed] [Google Scholar]
- 20.Kreiter S, et al. Mutant MHC class II epitopes drive therapeutic immune responses to cancer. Nature. 2015;520:692–696. doi: 10.1038/nature14426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Giesbertz P, et al. Metabolite profiling in plasma and tissues of ob/ob and db/db mice identifies novel markers of obesity and type 2 diabetes. Diabetologia. 2015;58:2133–2143. doi: 10.1007/s00125-015-3656-y. [DOI] [PubMed] [Google Scholar]
- 22.Schonfeld P, Wojtczak L. Fatty acids as modulators of the cellular production of reactive oxygen species. Free Radic Biol Med. 2008;45:231–241. doi: 10.1016/j.freeradbiomed.2008.04.029. [DOI] [PubMed] [Google Scholar]
- 23.Cao Y, Rathmell JC, Macintyre AN. Metabolic reprogramming towards aerobic glycolysis correlates with greater proliferative ability and resistance to metabolic inhibition in CD8 versus CD4 T cells. PLoS One. 2014;9:e104104. doi: 10.1371/journal.pone.0104104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sumida Y, Niki E, Naito Y, Yoshikawa T. Involvement of free radicals and oxidative stress in NAFLD/NASH. Free radical research. 2013;47:869–880. doi: 10.3109/10715762.2013.837577. [DOI] [PubMed] [Google Scholar]
- 25.Porporato PE, et al. A mitochondrial switch promotes tumor metastasis. Cell reports. 2014;8:754–766. doi: 10.1016/j.celrep.2014.06.043. [DOI] [PubMed] [Google Scholar]
- 26.Feldstein AE, et al. Mass spectrometric profiling of oxidized lipid products in human nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. J Lipid Res. 2010;51:3046–3054. doi: 10.1194/jlr.M007096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Muir K, et al. Proteomic and lipidomic signatures of lipid metabolism in NASH-associated hepatocellular carcinoma. Cancer Res. 2013;73:4722–4731. doi: 10.1158/0008-5472.CAN-12-3797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Beraza N, et al. Pharmacological IKK2 inhibition blocks liver steatosis and initiation of non-alcoholic steatohepatitis. Gut. 2008;57:655–663. doi: 10.1136/gut.2007.134288. [DOI] [PubMed] [Google Scholar]
- 29.Henao-Mejia J, et al. Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature. 2012;482:179–185. doi: 10.1038/nature10809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Miura K, et al. Toll-like receptor 2 and palmitic acid cooperatively contribute to the development of nonalcoholic steatohepatitis through inflammasome activation in mice. Hepatology. 2013;57:577–589. doi: 10.1002/hep.26081. [DOI] [PMC free article] [PubMed] [Google Scholar]
Additonal References for Methods
- 31.Andersson J, et al. CD4+ FoxP3+ regulatory T cells confer infectious tolerance in a TGF-beta-dependent manner. J Exp Med. 2008;205:1975–1981. doi: 10.1084/jem.20080308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nakagawa H, et al. ER stress cooperates with hypernutrition to trigger TNF-dependent spontaneous HCC development. Cancer Cell. 2014;26:331–343. doi: 10.1016/j.ccr.2014.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ikehara Y, et al. CD4(+) Valpha14 natural killer T cells are essential for acceptance of rat islet xenografts in mice. J Clin Invest. 2000;105:1761–1767. doi: 10.1172/JCI8922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hanczko R, et al. Prevention of hepatocarcinogenesis and increased susceptibility to acetaminophen-induced liver failure in transaldolase-deficient mice by N-acetylcysteine. J Clin Invest. 2009;119:1546–1557. doi: 10.1172/JCI35722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Radaeva S, et al. Interferon-alpha activates multiple STAT signals and down-regulates c-Met in primary human hepatocytes. Gastroenterology. 2002;122:1020–1034. doi: 10.1053/gast.2002.32388. [DOI] [PubMed] [Google Scholar]
- 36.Chen RF. Removal of fatty acids from serum albumin by charcoal treatment. J Biol Chem. 1967;242:173–181. [PubMed] [Google Scholar]
- 37.Huynh FK, Green MF, Koves TR, Hirschey MD. Measurement of fatty acid oxidation rates in animal tissues and cell lines. Methods in enzymology. 2014;542:391–405. doi: 10.1016/B978-0-12-416618-9.00020-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pelletier M, Billingham LK, Ramaswamy M, Siegel RM. Extracellular flux analysis to monitor glycolytic rates and mitochondrial oxygen consumption. Methods in enzymology. 2014;542:125–149. doi: 10.1016/B978-0-12-416618-9.00007-8. [DOI] [PubMed] [Google Scholar]
- 39.Hoechst B, et al. A new population of myeloid-derived suppressor cells in hepatocellular carcinoma patients induces CD4(+)CD25(+)Foxp3(+) T cells. Gastroenterology. 2008;135:234–243. doi: 10.1053/j.gastro.2008.03.020. [DOI] [PubMed] [Google Scholar]