

Abstract
Cell-cell fusion of human villous trophoblasts, referred to as a process of syncytialization, acts as a prerequisite for the proper development and functional maintenance of the human placenta. Given the fact that the main components of the Hedgehog signaling pathway are expressed predominantly in the syncytial layer of human placental villi, in this study, we investigated the potential roles and underlying mechanisms of Hedgehog signaling in trophoblastic fusion. Activation of Hedgehog signaling by a variety of approaches robustly induced cell fusion and the expression of syncytial markers, whereas suppression of Hedgehog signaling significantly attenuated cell fusion and the expression of syncytial markers in both human primary cytotrophoblasts and trophoblast-like BeWo cells. Moreover, among glioma-associated oncogene (GLI) family transcriptional factors in Hedgehog signaling, knockdown of GLI2 but not GLI1 and GLI3 significantly attenuated Hedgehog-induced cell fusion, whereas overexpression of the GLI2 activator alone was sufficient to induce cell fusion. Finally, GLI2 not only stabilized glial cell missing-a, a pivotal transcriptional factor for trophoblastic syncytialization, but also formed a transcriptional heterodimer with glial cell missing-a to transactivate syncytin-1, a trophoblastic fusogen, and promote trophoblastic syncytialization. Taken together, this study uncovered a so far uncharacterized role of Hedgehog/GLI2 signaling in trophoblastic fusion, implicating that Hedgehog signaling, through GLI2, could be required for human placental development and pregnancy maintenance.
Keywords: complex, Hedgehog signaling pathway, placenta, protein stability, trophoblast, GCMa, GLI2, cell fusion, transactivation
Introduction
Cell-cell fusion is essential for the development of a new organism (sexual fusion) or generation of diverse giant multinucleate cells that comprise distinct organs (nonsexual cell fusion). Fused cells undergo dramatic changes in biological behavior and acquire new developmental fates (1, 2). Cell-cell fusion occurring in the human placenta to form syncytiotrophoblasts (STBs)3 is referred to as a process of syncytialization and is a critical event for the proper morphogenesis and functional maintenance of placentas (3). In human blastocysts, trophectoderm cells at the embryonic pole generate primary STBs by intercellular fusion during preimplantation (4). In human placental villi, secondary STBs, characterized by a single, multinucleated, and highly dynamic syncytium, are maintained by continuous fusion of underlying cytotrophoblasts (CTBs), which begins 1–2 weeks after implantation and lasts until delivery (5). Multinucleated STBs are critically important for not only successful implantation but also pregnancy maintenance (6). In placental villi, STBs directly in contact with maternal blood play a pivotal role in the maintenance of placental functions, including immune tolerance, controlling fetomaternal exchanges of nutrients and waste products, and producing and secreting pregnancy-associated hormones such as human chorionic gonadotropin β (βhCG), estradiol, and progesterone (7). Hormones, growth factors, cytokines, protein kinases, and transcriptional factors regulate the complex physiology of syncytialization (8). Among transcriptional factors, glial cell missing 1 (GCM1), encoded by the GCM1 gene, has been found to be involved in the syncytialization of murine labyrinthine layer (9). Loss of GCM1 in the murine placenta leads to cell cycle arrest and defects in trophoblastic fusion and placental development (10, 11). In humans, the homolog of GCM1 encoding GCMa is expressed exclusively in the STBs of human placental villi and appears to be essential for the transactivation of fusogens such as syncytin-1, cell cycle exit, and syncytiotrophoblast formation in chorionic villi (12, 13).
The hedgehog (HH) family of secreted proteins comprises Sonic hedeghog (SHH), Indian hedgehog, and Desert hedgehog and has conserved roles in the development of various organs in metazoans ranging from Drosophila to humans (14–16). In vertebrates, binding of HH to the receptor patched 1 (PTCH1) leads to the relief of smoothened (SMO) from PTCH1 inhibition, resulting in activation of the GLI transcriptional factors (GLI1 and GLI2) that induce the transcription of target genes, including cyclin D, cyclin E, myc, and PTCH1 and GLI1. In the absence of HH, PTCH1 represses SMO activity and, thereby, GLI3 undergoes proteolytical cleavage into its repressor within the primary cilium, resulting in inactivation of HH signaling (17). Our previous studies have shown that the main components of the HH signaling pathway are expressed robustly in both human and murine placentas (18, 19). In human placental villi, Desert hedgehog and Indian hedgehog are expressed predominantly in the villous core and trophoblast layers, respectively, whereas SHH is expressed in both trophoblast layers and villous cores. PTCH1 and SMO are expressed robustly in the STB layers, whereas GLI2 and GLI3, in contrast to GLI1, which is barely detectable, are distributed abundantly in both STB and CTB layers (19, 20). In addition, we have indicated that HH signaling is required for the synthesis of human placental hormones, including estradiol and progesterone (20). Given the fact that syncytialization is a prerequisite for the production of placental hormones, we hypothesize that HH signaling could have some implication in the cell-cell fusion of trophoblasts. In this study, we demonstrated that GLI2 activated by HH induces the syncytialization of human placental trophoblasts through formation of a transcriptional complex with GCMa.
Experimental Procedures
Cell Culture
All cell lines used in this study were obtained from the ATCC. The human trophoblast-like BeWo cells were maintained in Ham's F-12K (Kaighn's) medium (Gibco BRL)/DMEM supplemented with 10% FBS (Life Technologies), 100 units/ml penicillin, and 100 mg/ml streptomycin as described previously (19). 293-EcR-SHH cells (SHH-expressing cells) and HEK293T cells were used for the production of biologically active murine SHH conditional medium (SM) and control medium (CM), respectively, in the presence of ecdysone (19). 293FT packaging cells for generating lentiviruses were cultured as described previously (20). Human placentas were obtained from uncomplicated normal term (38- to 40-week) pregnancies after elective cesarean section without labor, following a protocol approved by the Ethics Committee of the School of Medicine of Zhejiang University. Primary CTBs were purified using a 5–65% Percoll (Sigma) gradient and cultured in DMEM containing 10% newborn calf serum (Life Technologies) as described previously (19, 20). All cells were maintained in 5% CO2 and 95% air at 37 °C.
RNA Isolation and Quantitative Real-time PCR
Total RNA was isolated from BeWo cells and CTBs using TRIzol reagent (Takara Biotechnology, Dalian, China) according to the instructions of the manufacturer. 5 μg of total RNA in a volume of 20 μl was reverse-transcribed using SuperScript III reagent (Life Technologies) and an oligo(deoxythymidine) primer with incubation at 42 °C for 1 h. After the termination of cDNA synthesis, the mRNA levels of target genes were determined by quantitative RT-PCR as described previously (19, 20). The relative amounts of the mRNA levels of the target genes were normalized to β-actin, GAPDH, or α-tubulin levels, and the relative difference in mRNA levels was calculated by the 2−ΔΔCt method.
Western Blotting, Immunoprecipitation, and Enzyme Immunometric Assay
Western blotting and immunoprecipitation were performed as described previously (19). For Western blotting, total protein extracts were prepared, and protein concentrations were determined by using a standard Bradford assay. 50 μg of total protein was subjected to SDS-PAGE, followed by a transfer onto PVDF membranes (Millipore, Bedford, MA). The membranes were incubated with primary antibodies against ZO-1 (catalog no. sc-10804, Santa Cruz Biotechnology Inc., Santa Cruz, CA), GLI1 (catalog no. sc-20687, Santa Cruz Biotechnology), GLI2 (catalog no. ab26056, Abcam Ltd., Cambridge, UK), GLI3 (catalog no. ab69838, Abcam), FLAG (catalog no. 14793, Cell Signaling Technology, Danvers, MA), His (catalog no. sc-803, Santa Cruz Biotechnology), GCMa (catalog no. sc-101173, Santa Cruz Biotechnology), Syncytin 1 (catalog no. sc-50369, Santa Cruz Biotechnology), PP13 (catalog no. sc-131938, Santa Cruz Biotechnology), and β-actin (catalog no. sc-69879, Santa Cruz Biotechnology), followed by incubation in secondary antibodies. For immunoprecipitation, cells were prepared in whole cell lysis buffer, and the lysates were immunoprecipitated with various antibodies followed by SDS-PAGE and immunoblotting. Immunosignals were developed by using an enhanced chemiluminescence system. National Institutes of Health Image ImageJ (http://rsb.info.nih.gov/ij/) was used to quantify the immunoreactive bands, and the normalized antigen signals were calculated from target protein-derived and β-actin-derived signals. βhCG levels in serum-free culture supernatants of BeWo cells and CTBs were determined by Enzyme Immunometric Assay kit as described previously (21).
Immunofluorescence and Syncytialization Assay
Immunofluorescence was performed on chamber slides (Nalge Nunc International, Naperville, IL). After rinsing in PBS, BeWo cells or CTBs were fixed in ice-cold methanol and permeabilized with 0.1% Triton X-100 in PBS (PBST). After incubation with blocking buffer for 30 min, cells were incubated with primary antibody against ZO-1 (catalog no. sc-10804, Santa Cruz Biotechnology), FLAG (catalog no. 14793, Cell Signaling Technology) or His (catalog no. sc-803, Santa Cruz Biotechnology) overnight at 4 °C. After washing with PBST, cells were incubated further with Alexa Fluor 488-conjugated secondary antibody (Life Technology). The nuclei were counterstained with DAPI, and immunostaining was analyzed using a laser-scanning microscope. To quantify syncytialization in BeWo cells and CTBs without bias, we performed a syncytium formation analysis as described previously (22). Briefly, three different non-overlapping fields were selected randomly for each time point under the microscope. The fusion index was calculated using the format of (N − S) / T, where N is the number of nuclei in the syncytia, S is the number of syncytia, and T is the total number of nuclei.
Generation of Lentiviruses Expressing GLI1, GLI2, or GLI3 shRNA
The generation of lentiviruses expressing shRNA has been described previously (19). The hairpin shRNA templates of complementary oligonucleotide-containing overhangs were digested and inserted into XbaI and NotI sites of a lentiviral shRNA expression vector, Pll3.7. The sequences of oligonucleotides have been described previously (19). 293FT packaging cells were transfected with each construct by Lipofectamine 2000 reagent, lentivirus-containing supernatants were harvested, and viruses with titers of more than 1 × 106 cfu/ml were used for infection in BeWo cells and CTBs in the presence of 8 μg/ml Polybrene (Sigma).
Cycloheximide Chase Analysis
CTBs were treated with cycloheximide up to the indicated time with or without pretreatment with recombinant SHH protein (SHH-N) for 24 h. At the indicated time, cells were lysed, and cell lysates were prepared for immunoblotting assays as described above.
In Situ Proximity Ligation Assay for GLI2 and GCMa Interaction
An in situ proximity ligation assay was performed according to the instructions of the manufacturer (Olink Bioscience, Uppsala, Sweden) (23). Briefly, CTBs were stained with anti-GLI2 rabbit purified polyclonal antibody at 1:200 and anti-GCMa mouse monoclonal antibody at 1:100. Signals were detected using a Duolink® 100 detection kit 613 (red), and nuclei were counterstained with DAPI (blue). Each red dot represents the protein-protein interaction complex. The images were analyzed using an optimized freeware (BlobFinder) obtained from the Centre for Image Analysis at Uppsala University.
Reporter Construction, Transient Transfection, and Dual-Luciferase Assays
Complex human syncytin-1 (ERVWE1) promoter regions were amplified from the genomic DNA of BeWo cells by using specific primers. The PCR products were cloned into the pGL3-Basic vector (Promega, Madison, WI) to generate the luciferase reporter constructs of syncytin-1. All of these constructs were verified by a DNA sequencer. Transient transfection was performed using Lipofectamine 2000 according to the instructions of the manufacturer. After the cells were harvested, the cellular lysates were prepared and used for a Dual-Luciferase assay according to the instructions of the manufacturer (Promega). Firefly luciferase levels were normalized to Renilla luciferase levels.
Chromatin Immunoprecipitation Assays
ChIP was performed as described previously using a commercial kit (catalog no. 17-295, Millipore, Billerica, MA) (19). Briefly, cells were fixed with 1% formaldehyde to cross-link transcription factors into chromatin DNA and resuspended with lysis buffer supplemented with protease inhibitor mixture. Then, cells were sonicated to shear chromatin DNA to lengths between 200 and 1000 basepairs. Immunoprecipitation was performed with GLI2 and GCMa antibodies or control IgG. After incubation with protein A-agarose/salmon sperm DNA, the antibody-protein-DNA-agarose complex was washed and harvested for subsequent reverse cross-linking. The sheared DNA fragments from reverse cross-linking were extracted for further PCR amplification using the specific primers.
Statistical Analysis
All numerous data are expressed as mean ± S.D. and analyzed by one-way analysis of variance and Tukey-Kramer multiple comparison tests (SPSS 13.0J software, SPSS, Inc., Chicago, IL). Statistical significance was assessed at p < 0.05 and p < 0.01. Experiments were performed independently in triplicate, and the results were qualitatively identical. Representative experiments are shown.
Results
Induction of Cell-Cell Fusion of Trophoblasts by HH
Human primary CTBs and trophoblast-like BeWo cells capable of mimicking the biological behavior of placental villous trophoblasts are valuable models for the study of syncytialization (24). To investigate the effects of HH on syncytialization, we treated CTBs with recombinant human SHH protein ranging from 0–100 ng/ml or SM and performed immunofluorescent staining and Western blotting for ZO-1 to define the intercellular contact between aggregated cells and quantify the protein levels, respectively. SHH-N dose-dependently induced the cell-cell fusion of CTBs, and SHH-N at 50 and 100 ng/ml increased the fusion index 1.9- and 3.0-fold, respectively (Fig. 1, A and C). Consistently, SM treatment for 24 and 48 h led to decreases in ZO-1 protein levels by 50% and 67%, respectively, compared with CM treatment (Fig. 1B). To confirm the universality of SHH in inducing syncytialization, we treated trophoblast-like BeWo cells with either CM or SM for 24 and 48 h. Immunofluorescent staining for ZO-1 demonstrated that SM similarly stimulated the formation of large syncytia in BeWo cells and increased the fusion index ∼2.5- and 3.7-fold, respectively (Fig. 1, D and E). Likewise, SM treatment decreased ZO-1 protein levels by 70% and 78% at 24 and 48 h, respectively, over the CM treatment (Fig. 1F). However, pretreatment with 5E1 SHH-neutralizing antibody almost completely abolished not only the SM-induced protein levels of GLI1, a target of the HH pathway, but also the SM-negated protein levels of ZO-1 (Fig. 1G). Finally, to investigate the expression of syncytialization markers, including βhCG, syncytin-1, and placental protein 13 (PP13) (25), we performed Enzyme Immunometric Assay (EIA), quantitative RT-PCR, and Western blotting assays. In CTBs, SM treatment for 24 and 48 h increased syncytin-1 protein levels 1.5- and 2.3-fold and increased PP13 protein levels 1.2- and 1.9-fold, respectively (Fig. 1H). In both CTBs and BeWo cells, treatment with SM for 48 h resulted in 0.6- and 1.8-fold increases in βhCG levels, respectively, compared with CM treatment (Fig. 1I). Similarly, SM treatment for 24 and 48 h increased syncytin-1 mRNA levels 2.7- and 3.0-fold in BeWo cells and 2.5- and 3.0-fold in CTBs, respectively. However, SM treatment for 24 and 48 h increased PP13 mRNA levels up to 1.8- and 2.0-fold in BeWo cells and up to 2.0- and 3.6-fold in CTBs, respectively (Fig. 1J). Because syncytin-2 has been regarded to play an important role in trophoblast fusion, we also detected syncytin-2 mRNA levels and found that, similar to syncytin-1, syncytin-2 mRNA levels were up-regulated by either SHH-N or SM stimulation in both CTBs and BeWo cells (data not shown). Taken together, HH induces syncytialization not only in human primary CTBs but also in human trophoblast-like BeWo cells.
FIGURE 1.

Induction of syncytialization by SHH. A and C, immunofluorescence localization of ZO-1 in CTBs after 48 h of culture with SHH-N. The fusion index was analyzed. B, ZO-1 protein levels in CTBs after 24 and 48 h of culture in either SM or CM. D and E, immunofluorescence localization of ZO-1 in BeWo cells after 24 and 48 h of culture in SM or CM. The fusion index was analyzed. F, ZO-1 protein levels in BeWo cells after 24 and 48 h of culture in SM or CM. G, the 5E1 anti-SHH neutralizing antibody (Nab) abrogated SM-induced decreases in ZO-1 protein and increases in GLI1 protein. H, protein levels of syncytin-1 and PP13 in CTBs after 24 and 48 h of culture in either SM or CM. I and J, βhCG production and mRNA levels of syncytin-1 and PP13 in SM or CM after 24- and 48-h treatment in BeWo cells and CTBs. mRNA and protein abundance were normalized to β-actin. *, p < 0.05; **, p < 0.01 versus CM or vehicle; n = 6. Error bars show mean ± S.D. Scale bars = 20 μm.
Involvement of SMO in HH-induced Syncytialization
To determine whether HH-induced syncytialization is SMO-dependent, we activated HH signaling by either treatment with a SMO agonist, purmorphamine (Pur), or overexpression of a constitutively active form of SMO (SMO*) and suppressed HH signaling by a SMO inhibitor, cyclopamine (Cyc). Immunofluorescent staining indicated that treatment with purmorphamine led to the apparent disappearance of ZO-1 at intercellular contact and appearance of a larger number of multinuclear cells than vehicle treatment (Fig. 2A). Purmorphamine at 1 and 2 μm increased the fusion index 1.4- and 3.2-fold, respectively (Fig. 2B), and increased βhCG production up to 1.5- and 2.0-fold, respectively (Fig. 2E). Moreover, activation of SMO by purmorphamine decreased ZO-1 mRNA and protein levels and increased GLI1 protein levels and syncytialization marker mRNA and protein levels in dose-dependent manners. Purmorphamine at 2 μm negated ZO-1 mRNA and protein levels by 0.6- and 0.8-fold, respectively, whereas it induced the protein levels of GLI1 by 2.4-fold, the mRNA levels of syncytin-1 and PP13 by 2.8- and 1.5-fold, respectively, and it induced the protein levels of syncytin-1 and PP13 by 2.8- and 2.0-fold, respectively (Fig. 2, C and D). Likewise, overexpression of SMO* decreased the mRNA levels of ZO-1 0.7-fold and increased the mRNA levels of syncytin-1 and PP13 2.2- and 1.9-fold, respectively, and increased the protein levels of syncytin-1 and PP13 1.8- and 1.5-fold, respectively, and βhCG production 1.2-fold (Fig. 2, F–H). Conversely, pretreatment of cells with cyclopamine significantly reversed the effects caused by SM. Immunofluorescent staining showed that cyclopamine significantly increased ZO-1 protein levels at intercellular contact and dose-dependently attenuated SM-induced decreases in the fusion index (Fig. 2, I and J). Moreover, cyclopamine dose-dependently reversed not only SM-negated mRNA and protein levels of ZO-1 but also SM-induced mRNA and protein levels of syncytin-1 and PP13 and protein levels of GLI1 (Fig. 2L). Cyclopamine at 5 μm decreased the SM-induced fusion index by 52%, mRNA levels of syncytin-1 and PP13 by 63% and 50%, protein levels of syncytin-1 and PP13 by 69% and 63%, GLI1 protein levels by 60%, and βhCG production by 49%, whereas it increased SM-negated ZO-1 mRNA and protein levels 1.7- and 0.6-fold, respectively (Fig. 2, K and L). Therefore, HH-induced trophoblastic syncytialization is SMO-dependent, and activation of SMO alone is sufficient to induce trophoblastic syncytialization.
FIGURE 2.

SMO is involved in HH-induced syncytialization. A and B, immunofluorescence localization of ZO-1 after 48 h of culture with Pur. The fusion index was analyzed. C–E, mRNA and protein levels of ZO-1, syncytin-1, and PP13; protein levels of ZO-1 and GLI1; and βhCG production after treatment with Pur for 48 h in BeWo cells. F–H, mRNA and protein levels of ZO-1, syncytin-1, and PP13 and βhCG production after overexpression of SMO* in BeWo cells. I and J, immunofluorescence localization of ZO-1 after 48 h of culture with cyclopamine (Cyc). The fusion index was analyzed. K and L, mRNA levels of ZO-1, syncytin-1, and PP13; protein levels of GLI1, ZO-1, syncytin-1, and PP13; and βhCG production after treatment with cyclopamine for 48 h in BeWo cells. RNA and protein abundance were normalized to β-actin. * and †, p < 0.05; **and ‡, p < 0.01 versus vehicle, empty vector, CM, or SM with vehicle; n = 6. Error bars show mean ± S.D. Scale bars = 20 μm.
Requirement of GLI2 but Not GLI1 and GLI3 in HH-induced Syncytialization
To assess the potential involvement of GLI transcriptional factors in HH-induced syncytialization, we generated lentiviruses expressing GLI1, GLI2, or GLI3 shRNA that knocked down the expression of GLI1, GLI2, and GLI3 by as much as 60–70% (Fig. 3, A–C). Immunofluorescence showed that knockdown of GLI2, but not GLI1 and GLI3, apparently reversed the SM-induced disappearance of ZO-1 staining between the mononucleated trophoblast cell membranes and the appearance of multinuclear cells (Fig. 3D). Quantification of cell fusion further indicated that SM increased the fusion index by 80%, and that knockdown of GLI2, but not GLI1 and GLI3, attenuated SM-induced cell fusion by 78% (Fig. 3E). Likewise, knockdown of GLI2 increased the SM-negated protein levels of ZO-1 2.0-fold and reduced the SM-induced mRNA levels of syncytin-1 and PP13 0.68- and 0.55-fold, respectively, whereas knockdown of GLI1 or GLI3 affected neither the protein levels of ZO-1 nor the mRNA levels of syncytin-1 and PP13 (Fig. 3F). The pivotal role of GLI2 in syncytialization was verified further in human primary CTBs. Knockdown of GLI2, but not GLI1 and GLI3, attenuated the SM-induced mRNA levels of syncytin-1 and PP13 by 70% and 55%, the protein levels of syncytin-1 and PP13 by 70% and 62%, and βhCG production by 55%, respectively (Fig. 3, G–I). On the other hand, overexpression of GLI2 suppressed the mRNA levels of ZO-1 by 0.75-fold, but induced the mRNA levels of syncytin-1 and PP13 by 5.6- and 3.5-fold, respectively. However, overexpression of GLI1 or GLI3 did not significantly affect these mRNA levels (Fig. 3J). In the rescue experiments, overexpression of ΔN-GLI2 (N-terminally truncated GLI2), a constitutively active form of GLI2, was able to reverse GLI2-shRNA-induced ZO-1 mRNA levels and GLI2-shRNA-negated syncytin-1 and PP13 mRNA levels (data not shown). Taken together, these data demonstrate that GLI2, but not GLI1 and GLI3, is required for HH-induced syncytialization both in trophoblast-like BeWo cells and CTBs, and that GLI2 alone is sufficient to induce trophoblastic syncytialization.
FIGURE 3.

GLI2 is essential for HH-induced syncytialization. A–C, knockdown efficiencies of GLI1, GLI2, or GLI3 shRNA-expressing lentiviruses. D and E, immunofluorescence localization of ZO-1 after GLIs were knocked down in BeWo cells in the presence of CM or SM. The fusion index was analyzed. Ctrl, control. F, protein level of ZO-1 and mRNA levels of ZO-1, syncytin-1, and PP13 after GLIs were knocked down in BeWo cells in the presence of CM or SM. G–I, mRNA and protein levels of syncytin-1 and PP13 and βhCG production after GLIs were knocked down in CTBs in the presence of CM or SM. J, mRNA levels of ZO-1, syncytin-1, and PP13 in BeWo cells transfected with the indicated plasmids. RNA and protein abundance were normalized to β-actin. * and †, p < 0.05; ** and ‡, p < 0.01 versus CM with control shRNA, SM with control shRNA, or empty vector; n = 6. Error bars show mean ± S.D. Scale bars = 20 μm.
Formation and Requirement of the Transcriptional Complex of GLI2 and GCMa in Syncytialization
To understand the mechanism(s) underlying GLI2 regulating syncytialization, we focused on GCMa, a critical transcriptional factor governing human placental trophoblast syncytialization (26). We performed GCMa binding sites (GBS)-luciferase report assays by generating 8 × GBS-driven luciferase reporter construct and its matched control mutant with mutation of GCMa binding sites. Either treatment with SM or overexpression of SMO* induced GBS luciferase activity by 6.5- and 6.3-fold compared with CM treatment or overexpression of control YFP vector, respectively, whereas the variant harboring a mutation at GCMa binding sites failed to respond to these treatments (Fig. 4, A and B). In contrast, cyclopamine dose-dependently attenuated SM-induced GBS-luciferase activity, and cyclopamine at 5 μm decreased luciferase activity by 70% (Fig. 4A). Likewise, overexpression of GLI2 induced GBS-luciferase activity by 5.2-fold, and overexpression of ΔN-GLI2 induced GBS-luciferase activity by 11.2-fold, respectively (Fig. 4C). However, overexpression of GLI1 or GLI3 did not affect luciferase activities (data not shown). Conversely, knockdown of GLI2 by lentiviral shRNA attenuated SM-induced GBS-luciferase activity by 70%, whereas knockdown of GLI1 or GLI3 did not affect SM-induced luciferase activities (Fig. 4D). To determine whether the GCMa expression level was regulated by GLI2, we further examined both the mRNA and protein levels of GCMa in BeWo cells cultured in CM or SM for 48 h. GCMa protein levels were induced significantly by 1.3-fold, in response to SM (Fig. 4E). However, quantitative RT-PCR assays, in which different housekeeping gene mRNA levels were used to normalize the GCM1 mRNA levels, indicated that SM did not affect the mRNA levels of the Gcm1 gene or housekeeping genes in the process of syncytialization (Fig. 4F), suggesting that GCM1 is not a direct downstream target of GLI2. In CTBs, SHH-N at 100 ng/ml consistently increased GCMa protein levels by 2.2-fold, and purmorphamine at 5 μm induced GCMa protein levels by 4.0-fold (Fig. 4, G and H). Finally, knockdown of GLI2, but not GLI1 and GLI3, significantly attenuated SM-induced GCMa protein levels by 80% (Fig. 4I). These results prompted us to speculate that GLI2 enhanced the stability of GCMa protein. To this end, we blocked de novo protein synthesis with cycloheximide in CTBs cultured with SHH-N or vehicle and analyzed the protein levels by Western blotting. Activation of the HH pathway by SHH-N extended the half-life of endogenous GCMa protein from ∼60 to ∼180 min (Fig. 4, J and K). Therefore, HH up-regulates the transcriptional activity and stability of GCMa through GLI2. Activation of HH signaling results in proteolytical cleavage of GLI2 into its activator, which enters the nucleus and transactivates target genes (27). Immunostaining results showed that full-length GLI2 was expressed in both the cytoplasm and nucleus, whereas the active form of GLI2 was mainly localized in the nucleus (data not shown). To determine the possible interaction between GLI2 and GCMa, we employed immunoprecipitation analysis. In cells expressing FLAG-tagged GCMa and His-tagged ΔN-GLI2, protein complexes precipitated with a FLAG antibody contained His-ΔN-GLI2 in addition to FLAG-tagged GCMa, as expected. However, in cells expressing FLAG-tagged GCMa alone, the protein complexes contained no His-ΔN-GLI2 (Fig. 5A). Similarly, protein complexes precipitated with IgG contained no GCMa and GLI2, whereas protein complexes precipitated with a GCMa antibody contained abundant endogenous GLI2 (Fig. 5B), consistent with immunostaining that showed co-localization of ΔN-GLI2 and GCMa in HEK293T cells (Fig. 5C). To further determine the interaction between GLI2 and GCMa, we conducted an in situ proximity ligation assay in CTBs and performed quantitative measurements as well. Treatment of SM led to a robust increase in endogenous GLI2-GCMa interaction and the GLI2-GCMa complex was apparently co-localized in the cellular nucleus (Fig. 5, D and E). To confirm the involvement of the GLI2-GCMa complex in syncytialization, we knocked down GLI2 or GCMa in combination with or without overexpression of GCMa or ΔN-GLI2 and performed syncytialization assays. Overexpression of either GCMa or ΔN-GLI2 led to significant increases in trophoblastic fusion and βhCG production. However, knockdown of GCMa by GCMa shRNA-expressing lentiviruses robustly attenuated ΔN-GLI2-induced trophoblastic fusion and βhCG production, whereas knockdown of GLI2 by GLI2 shRNA-expressing lentiviruses robustly attenuated GCMa-induced trophoblastic fusion and βhCG production (Fig. 5, F and G). Taken together, these results demonstrate that trophoblastic syncytialization is dependent on GLI2-GCMa complex formation.
FIGURE 4.

GLI2 potentiates the transcriptional activity and stability of GCMa. A–D, Dual-Luciferase assays of GBS (GCMa binding site) in BeWo cells after treatment under the indicated conditions. Cyc, cyclopamine; Ctrl, control. E and F, protein levels of GLI1 and GCMa and mRNA levels of GCM1 in BeWo cells treated with either CM or SM for 48 h. G and H, GCMa protein levels in CTBs treated with SHH-N or Pur for 48 h. I, GCMa protein levels after BeWo cells were infected with lentiviruses expressing GLIs shRNA in the presence of CM or SM for 48 h. J, endogenous GCMa protein levels in CTBs exposed to SHH-N and treated for different times with cycloheximide (CHX). K, quantification via arbitrary and statistical analysis of the bands in J. Protein abundance was normalized to β-actin. The RNA abundance in F was normalized to β-actin, GAPDH, and α-tublin. * and †, p < 0.05; ** and ‡, p < 0.01 versus CM, CM with control shRNA, SM with control shRNA, cycloheximide without SHH, or empty vector; n = 6. Error bars show mean ± S.D. Scale bars = 20 μm.
FIGURE 5.

Complex formation of GLI2 and GCMa. A, co-immunoprecipitation of FLAG-tagged GCMa and His-tagged ΔN-GLI2 in HEK293T cells. Immunoprecipitation (IP), α-FLAG; Western blotting, α-His. B, co-immunoprecipitation of endogenous GCMa and GLI2 in BeWo cells. Immunoprecipitation, GCMa; Western blotting, GLI2. C, immunofluorescent staining of His-tagged ΔN-GLI2 and FLAG-tagged GCMa in HEK293T cells. D, in situ proximity ligation assay in CTBs after treatment with SM or CM for 48 h. Red spots indicate the GLI2-GCMa interaction. Nuclei were stained with DAPI (blue). E, quantitative analysis of the proximity ligation (PL) assay spots (n = 20). F and G, fusion index and βhCG production after BeWo cells were transient transfected and infected with the indicated plasmids and lentiviruses. RNA and protein abundance were normalized to β-actin. ** and ‡, p < 0.01 versus CM, empty vector with control shRNA, ΔN-GLI2 with control shRNA, or GCMa with control shRNA; n = 6. Error bars show mean ± S.D. Scale bars = 20 μm (C) and 80 μm (D).
Essential Role of the GLI2-GCMa Transcriptional Complex in Syncytialization
To further confirm the importance of the GLI2-GCMa complex in syncytialization, we cloned the syncytin-1 promoter region (ERVWE1, nt −2600 to +181) containing two potential GLI response elements (nt −1238 to −1229 and nt −1075 to −1067) and one established GCMa binding site (nt −2537 to −2530) (26) and generated a luciferase reporter construct and its deletion mutants lacking one or more responsive elements (Fig. 6A). In BeWo cells, after treatment with SM or CM for 48 h, the full-length construct (nt −2600 to +181) exhibited a robust response to SM over CM (15.4-fold), whereas all deletion mutants led to significantly diminished luciferase expression (Fig. 6B). Likewise, inhibition of HH signaling by cyclopamine ranging from 0–5 μm attenuated SM-induced luciferase activity in a dose-dependent manner, and knockdown of GLI2 attenuated SM-induced luciferase activity by 71%, whereas overexpression of ΔN-GLI2 almost completely reversed GLI2 shRNA-negated luciferase activities (Fig. 6, C–E). Conversely, activation of HH signaling, by either purmorphamine at 2 μm or overexpression of SMO*, significantly stimulated luciferase activities 14.7- and 7.2-fold, respectively, and overexpression of ΔN-GLI2 alone induced luciferase activity ∼25-fold (Fig. 6, F and G, and data not shown). To examine the physical interaction between GLI2 and the promoter region of the ERVWE1 gene, we performed ChIP-PCR in CTBs by using an antibody against GLI2. The PCR primers were designed to amplify specific genomic DNA fragments of nt −1300/-1000 and nt −1500/1300, and the ChIP assays were normalized to their corresponding input. As expected, GLI2 robustly bound to the fragment of nt −1300/−1000 but not nt −1500/−1300 (Fig. 6I). To gain further insights into the role of the GLI2-GCMa transcriptional complex, we performed luciferase report assays (nt −2600 to +181) in cells after various treatments. Concomitant overexpression of ΔN-GLI2 and GCMa increased luciferase activity by 98% compared with overexpression of ΔN-GLI2 alone and by 70% compared with overexpression of GCMa alone (Fig. 6J). Knockdown of GCMa significantly attenuated ΔN-GLI2-induced luciferase activity and syncytin-1 mRNA levels, whereas knockdown of GLI2 robustly attenuated GCMa-induced luciferase activity and syncytin-1 mRNA levels(Fig. 6, H and K). Finally, to further confirm the physical interaction between GLI2 and GCMa, we performed ChIP-PCR by using an antibody against GCMa in CTBs infected with GLI2 shRNA-expressing lentiviruses. Primers for amplifying a specific genomic DNA fragment (nt −2600 to −2400) containing a GCMa-responsive element (nt −2537 to −2530) were designed, and the PCR products were normalized to their corresponding input. GCMa bound robustly to the DNA fragment (nt −2600 to −2400) from scribbled shRNA-expressing CTBs, whereas the binding amount was reduced greatly in CTBs expressing GLI2 shRNA (Fig. 6L). Taken together, these results demonstrate that the GLI2-GCMa heterodimer is essential for transcriptional activation of syncytin-1, a key gene for trophoblastic syncytialization.
FIGURE 6.

Essential role of the transcriptional complex GLI2-GCMa in syncytialization. A, schematic of promoter regions with potential GLI consensus and non-consensus binding sites of the ERVWE1 gene encoding syncytin-1. The GCMa binding region is also shown. B–G, Dual-Luciferase assays in BeWo cells treated with the indicated conditions. Cyc, cyclopamine; Ctrl, control. H, syncytin-1 mRNA levels after BeWo cells were transiently transfected and infected with the indicated plasmids or lentiviruses. I, ChIP-PCR in CTBs using antibody against GLI2. J and K, Dual-Luciferase assays in BeWo cells treated with the indicated conditions. L, ChIP-PCR in CTBs using antibody against GCMa. The abundance was normalized to the corresponding input. ** and ‡, p < 0.01 versus empty vector with control shRNA, ΔN-GLI2 with control shRNA, CM, SM, SM with vehicle, empty vector, CM with control shRNA, SM with control shRNA, or SM with GLI2 shRNA; n = 6. Scrib., scribbled. Error bars show mean ± S.D.
Discussion
This study, for the first time, revealed that GLI2 binds to and stabilizes GCMa to form a heterodimer in transactivating fusogen and, consequently, promoting human trophoblastic syncytialization in response to HH. Therefore, our study uncovered a hitherto uncharacterized role of HH/GLI2 signaling in cell-cell fusion.
HH ligands secreted by HH-producing cells act on HH-responding cells to fulfill signaling transduction and regulate cell behavior (28, 29). Given the fact that PTCH1 and SMO are expressed robustly and predominantly in the syncytial layers of human placental villi (20), we assume that HH proteins may signal through a paracrine mechanism to regulate the syncytialization of CTBs. This hypothesis has been confirmed by the following solid evidence in this study: in both BeWo cells and primary human placental CTB culture models, activation of HH signaling induces trophoblastic fusion; inhibition of HH signaling by multiple approaches results in attenuation of trophoblastic fusion; knockdown of GLI2 or overexpression of active form of GLI2 significantly affects trophoblastic fusion; and GLI2 forms a transcriptional complex with GCMa to induce fusogen expression and trophoblastic fusion. Therefore, our findings not only uncover a so far uncharacterized role of HH signaling but also expand the list of tissues and cells influenced by HH signaling. These findings correspond to our previous results showing that knockout of SHH (SHH−/−) in murine placentas leads to defects in the expression of syncytial markers and development of the placental labyrinth and that placenta-specific knockdown of GLI2 by lentiviral GLI2-shRNA in SHH−/+ murine placentas phenocopies the labyrinthine defects of SHH−/− placentas (18). However, to determine whether GLI2 indeed promotes trophoblastic fusion, genetic evidence from trophoblast-specific knockout of GLI2 in murine placenta is worthy of further investigation.
Most importantly, this study identified that GLI2 not only stabilizes GCMa but also interacts with GCMa to control the transcription of fusogen and promote syncytialization. The interaction between GLI2 and GCMa is attested to by the following evidence: immunoprecipitation experiments indicate that both the GLI2 activator and GCMa exist in one immunocomplex, and immunofluorescent staining indicates that both the GLI2 activator and GCMa are co-localized in the nuclei of trophoblasts; proximity ligation assay results directly implicate the interaction between GLI2 and GCMa; ChIP-PCR experiments demonstrate that GLI2 interacts physically with GCMa in the promoter region of syncytin-1; and, functionally, GLI2 is essential for GCMa-mediated transactivation of syncytin-1 and trophoblastic fusion, whereas GCMa is required for GLI2-mediated transactivation of syncytin-1 and trophoblastic fusion to the same extent. Although the exact mechanism underlying GLI2 stabilizing GCMa is still unknown, to date, ours is the first report that reveals the existence of a transcriptional partner for either GCMa or GLI2. Given the fact that activation of HH signaling does not increase the mRNA levels of GCM1 but the protein levels of GCMa, we suggest that posttranslational regulation is involved in HH stabilizing GCMa. Therefore, we speculate that GLI2 possibly binds to the proteolytic domain of GCMa to prevent the degradation of GCMa in response to HH. Further experiments are needed to address this issue. Similarly, whether the interaction between GLI2 and GCMa is direct or not, and which exact domains link GLI2 and GCMa, needs to be determined.
HH family proteins have been recognized as one of the major intercellular signals that play vital roles in cell growth, cell survival, and cell fate decision (30–32). Cell-cell fusion is not only unique to placental tissues but also occurs in several different cell and tissue types, including fusion between the egg and the sperm during fertilization, formation of multinucleated chondroclasts, fusion of skeletal muscle satellite cells with skeletal muscle fibers, and so on (1, 33). Therefore, this study provides a paradigm for studying cell-cell fusion regulated by HH signaling. On the other hand, cell-cell fusion of placental trophoblasts plays critically important roles not only in implantation but also in successful pregnancy, especially in the proper development and functional maintenance of the human placenta (6, 34). Given the fact that malformation of the placenta is an underlying feature of a great variety of human developmental abnormalities and pregnancy-associated diseases, the identification of HH signaling critical for syncytialization in the human placenta may help to pinpoint the underlying mechanisms involved in the etiology of these conditions.
Overall, using human primary CTBs and trophoblast-like BeWo cell culture models, this study suggests that HH/GLI2 signaling is essential for trophoblastic syncytialization and that GLI2 stabilizes GCMa and forms a transcriptional heterodimer with GCMa to transactivate fusogen in the regulation of trophoblastic syncytialization (Fig. 7). Therefore, HH, through GLI2, acts as a critical signal in supporting the development and physiological function of the placenta.
FIGURE 7.
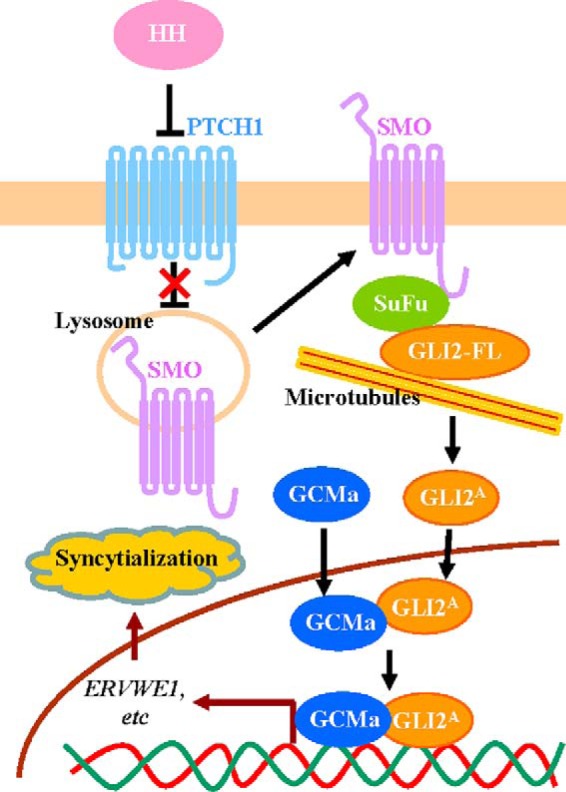
Schematic showing the proposed model for HH/GLI2-induced syncytialization in human trophoblasts. In the presence of HH ligand, binding of HH to PTCH1 relieves the inhibition of SMO, resulting in activation of GLI2A (active form of GLI2). GLI2A enters the nucleus to form a heterodimer transcriptional complex with GCMa, which increases the stability and transcriptional activity of GCMa. The GLI2-GCMa complex transactivates downstream target fusogens such as syncytin-1, which consequently promotes trophoblastic syncytialization.
Author Contributions
C. T. designed the study and performed all experiments. L. T. performed the Western blotting for syncytin-1 and PP13. X. W. and W. X. performed the quantitative RT-PCR for syncytin-2. H. R. and M. H. performed the Western blotting for GLI1, GLI2, and GLI3. J. W. performed the quantitative RT-PCR for GLI1, GLI2, and GLI3. C. Z. and X. W. wrote the manuscript. All authors read and approved the final manuscript.
Acknowledgment
We thank Dr. Sasaki for plasmids.
This work was supported by 973 Program Grant 2011CB944403; National Natural Science Foundation of China Grants 31271561, 81170016, 81170787, 81370713, and 31571493; and Natural Science Foundation of Zhejiang Province Grant LY13H150002. The authors declare that they have no conflicts of interest with the contents of this article.
- STB
- syncytiotrophoblast
- CTB
- cytotrophoblast
- hCG
- human chorionic gonadotropin
- HH
- hedgehog
- SHH
- Sonic hedgehog
- SMO
- Smoothened
- GCMa
- glial cell missing-a
- SM
- SHH conditional medium
- CM
- control medium
- Pur
- purmorphamine
- GBS
- GCMa-binding site.
References
- 1. Aguilar P. S., Baylies M. K., Fleissner A., Helming L., Inoue N., Podbilewicz B., Wang H., and Wong M. (2013) Genetic basis of cell-cell fusion mechanisms. Trends Genet. 29, 427–437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Pidoux G., Gerbaud P., Cocquebert M., Segond N., Badet J., Fournier T., Guibourdenche J., and Evain-Brion D. (2012) Review: human trophoblast fusion and differentiation: lessons from trisomy 21 placenta. Placenta 33, S81–86 [DOI] [PubMed] [Google Scholar]
- 3. Guttmacher A. E., Maddox Y. T., and Spong C. Y. (2014) The Human Placenta Project: placental structure, development, and function in real time. Placenta 35, 303–304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Huppertz B., and Borges M. (2008) Placenta trophoblast fusion. Methods Mol. Biol. 475, 135–147 [DOI] [PubMed] [Google Scholar]
- 5. Gauster M., and Huppertz B. (2010) The paradox of caspase 8 in human villous trophoblast fusion. Placenta 31, 82–88 [DOI] [PubMed] [Google Scholar]
- 6. Huppertz B., and Gauster M. (2011) Trophoblast fusion. Adv. Exp. Med. Biol. 713, 81–95 [DOI] [PubMed] [Google Scholar]
- 7. Morrish D. W., Dakour J., and Li H. (1998) Functional regulation of human trophoblast differentiation. J. Reprod. Immunol. 39, 179–195 [DOI] [PubMed] [Google Scholar]
- 8. Al-Nasiry S., Spitz B., Hanssens M., Luyten C., and Pijnenborg R. (2006) Differential effects of inducers of syncytialization and apoptosis on BeWo and JEG-3 choriocarcinoma cells. Hum. Reprod. 21, 193–201 [DOI] [PubMed] [Google Scholar]
- 9. Basyuk E., Cross J. C., Corbin J., Nakayama H., Hunter P., Nait-Oumesmar B., and Lazzarini R. A. (1999) Murine Gcm1 gene is expressed in a subset of placental trophoblast cells. Dev. Dyn. 214, 303–311 [DOI] [PubMed] [Google Scholar]
- 10. Hughes M., Dobric N., Scott I. C., Su L., Starovic M., St-Pierre B., Egan S. E., Kingdom J. C., and Cross J. C. (2004) The Hand1, Stra13 and Gcm1 transcription factors override FGF signaling to promote terminal differentiation of trophoblast stem cells. Dev. Biol. 271, 26–37 [DOI] [PubMed] [Google Scholar]
- 11. Baczyk D., Drewlo S., Proctor L., Dunk C., Lye S., and Kingdom J. (2009) Glial cell missing-1 transcription factor is required for the differentiation of the human trophoblast. Cell Death Differ. 16, 719–727 [DOI] [PubMed] [Google Scholar]
- 12. Wich C., Kausler S., Dotsch J., Rascher W., and Knerr I. (2009) Syncytin-1 and glial cells missing a: hypoxia-induced deregulated gene expression along with disordered cell fusion in primary term human trophoblasts. Gynecol. Obstet. Invest. 68, 9–18 [DOI] [PubMed] [Google Scholar]
- 13. Zhuang X. W., Li J., Brost B. C., Xia X. Y., Chen H. B., Wang C. X., and Jiang S. W. (2014) Decreased expression and altered methylation of syncytin-1 gene in human placentas associated with preeclampsia. Curr. Pharm. Des. 20, 1796–1802 [DOI] [PubMed] [Google Scholar]
- 14. Honselmann K. C., Pross M., Jung C. M., Wellner U. F., Deichmann S., Keck T., and Bausch D. (2015) Regulation mechanisms of the hedgehog pathway in pancreatic cancer: a review. JOP 16, 25–32 [DOI] [PubMed] [Google Scholar]
- 15. Ingham P. W. (1995) Signalling by hedgehog family proteins in Drosophila and vertebrate development. Curr. Opin. Genet. Dev. 5, 492–498 [DOI] [PubMed] [Google Scholar]
- 16. Wicking C., Smyth I., and Bale A. (1999) The hedgehog signalling pathway in tumorigenesis and development. Oncogene 18, 7844–7851 [DOI] [PubMed] [Google Scholar]
- 17. Lee J., Platt K. A., Censullo P., and Ruiz i Altaba A. (1997) Gli1 is a target of Sonic hedgehog that induces ventral neural tube development. Development 124, 2537–2552 [DOI] [PubMed] [Google Scholar]
- 18. Pan Y. B., Gong Y., Ruan H. F., Pan L. Y., Wu X. K., Tang C., Wang C. J., Zhu H. B., Zhang Z. M., Tang L. F., Zou C. C., Wang H. B., and Wu X. M. (2015) Sonic hedgehog through Gli2 and Gli3 is required for the proper development of placental labyrinth. Cell Death Dis. 6, e1653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tang C., Pan Y., Luo H., Xiong W., Zhu H., Ruan H., Wang J., Zou C., Tang L., Iguchi T., Long F., and Wu X. (2015) Hedgehog signaling stimulates the conversion of cholesterol to steroids. Cell. Signal. 27, 487–497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tang C., Mei L., Pan L., Xiong W., Zhu H., Ruan H., Zou C., Tang L., Iguchi T., and Wu X. (2015) Hedgehog signaling through GLI1 and GLI2 is required for epithelial-mesenchymal transition in human trophoblasts. Biochim. Biophys. Acta 1850, 1438–1448 [DOI] [PubMed] [Google Scholar]
- 21. Cocquebert M., Berndt S., Segond N., Guibourdenche J., Murthi P., Aldaz-Carroll L., Evain-Brion D., and Fournier T. (2012) Comparative expression of hCG β-genes in human trophoblast from early and late first-trimester placentas. Am. J. Physiol. Endocrinol. Metab. 303, E950–958 [DOI] [PubMed] [Google Scholar]
- 22. Pidoux G., Gerbaud P., Gnidehou S., Grynberg M., Geneau G., Guibourdenche J., Carette D., Cronier L., Evain-Brion D., Malassiné A., and Frendo J. L. (2010) ZO-1 is involved in trophoblastic cell differentiation in human placenta. Am. J. Physiol. Cell Physiol. 298, C1517–1526 [DOI] [PubMed] [Google Scholar]
- 23. Leuchowius K. J., Weibrecht I., and Soderberg O. (2011) In situ proximity ligation assay for microscopy and flow cytometry. Curr. Protoc. Cytom. Chapter 9, Unit 9 36 [DOI] [PubMed] [Google Scholar]
- 24. Drewlo S., Baczyk D., Dunk C., and Kingdom J. (2008) Fusion assays and models for the trophoblast. Methods Mol. Biol. 475, 363–382 [DOI] [PubMed] [Google Scholar]
- 25. Butler T. M., Elustondo P. A., Hannigan G. E., and MacPhee D. J. (2009) Integrin-linked kinase can facilitate syncytialization and hormonal differentiation of the human trophoblast-derived BeWo cell line. Reprod. Biol. Endocrinol. 7, 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Yu C., Shen K., Lin M., Chen P., Lin C., Chang G. D., and Chen H. (2002) GCMa regulates the syncytin-mediated trophoblastic fusion. J. Biol. Chem. 277, 50062–50068 [DOI] [PubMed] [Google Scholar]
- 27. Joeng K. S., and Long F. (2009) The Gli2 transcriptional activator is a crucial effector for Ihh signaling in osteoblast development and cartilage vascularization. Development 136, 4177–4185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Briscoe J., and Thérond P. P. (2013) The mechanisms of Hedgehog signalling and its roles in development and disease. Nat. Rev. Mol. Cell Biol. 14, 416–429 [DOI] [PubMed] [Google Scholar]
- 29. Choudhry Z., Rikani A. A., Choudhry A. M., Tariq S., Zakaria F., Asghar M. W., Sarfraz M. K., Haider K., Shafiq A. A., and Mobassarah N. J. (2014) Sonic hedgehog signalling pathway: a complex network. Ann. Neurosci. 21, 28–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ali S. A., Al-Jazrawe M., Ma H., Whetstone H., Poon R., Farr S., Naples M., Adeli K., and Alman B. (2016) Regulation of cholesterol homeostasis by Hedgehog signaling in osteoarthritic cartilage. Arthritis Rheum. 68, 127–137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Cobourne M. T., and Green J. B. (2012) Hedgehog signalling in development of the secondary palate. Front. Oral Biol. 16, 52–59 [DOI] [PubMed] [Google Scholar]
- 32. Cobourne M. T., Miletich I., and Sharpe P. T. (2004) Restriction of sonic hedgehog signalling during early tooth development. Development 131, 2875–2885 [DOI] [PubMed] [Google Scholar]
- 33. Shrimali R. K., and Reddy K. V. (2000) Integrins and disintegrins: the candidate molecular players in sperm-egg interaction. Indian J. Exp. Biol. 38, 415–424 [PubMed] [Google Scholar]
- 34. Gerbaud P., and Pidoux G. (2015) Review: an overview of molecular events occurring in human trophoblast fusion. Placenta 36, S35–42 [DOI] [PubMed] [Google Scholar]