

Abstract
Membrane-bound cGMP-dependent protein kinase (PKG) II is a key regulator of bone growth, renin secretion, and memory formation. Despite its crucial physiological roles, little is known about its cyclic nucleotide selectivity mechanism due to a lack of structural information. Here, we find that the C-terminal cyclic nucleotide binding (CNB-B) domain of PKG II binds cGMP with higher affinity and selectivity when compared with its N-terminal CNB (CNB-A) domain. To understand the structural basis of cGMP selectivity, we solved co-crystal structures of the CNB domains with cyclic nucleotides. Our structures combined with mutagenesis demonstrate that the guanine-specific contacts at Asp-412 and Arg-415 of the αC-helix of CNB-B are crucial for cGMP selectivity and activation of PKG II. Structural comparison with the cGMP selective CNB domains of human PKG I and Plasmodium falciparum PKG (PfPKG) shows different contacts with the guanine moiety, revealing a unique cGMP selectivity mechanism for PKG II.
Keywords: allosteric regulation, cyclic nucleotide, ligand-binding protein, protein kinase G (PKG), receptor structure-function, serine/threonine protein kinase, X-ray crystallography, NO-cGMP signaling, cGMP-dependent protein kinase, cyclic nucleotide-binding domain (CNB), second messenger
Introduction
Regulation of protein activity in response to cyclic nucleotide (cNT)3 binding is an important mechanism of cellular signal transduction (1). This regulation is mediated in large part by cNT-dependent protein kinases (cGMP- and cAMP-dependent protein kinases, PKG and PKA, respectively) that phosphorylate specific downstream signaling proteins in a cNT-dependent manner (2). The ability of each kinase to selectively bind either cGMP or cAMP determines its activation and signaling specificity. However, even with the recent structural information on the PKG-cGMP interactions found in human PKG I and Plasmodium falciparum PKG (PfPKG), a complete mechanism for PKG activation has yet to emerge (3, 4).
Two types of PKGs, PKG I and II, exist in mammalian cells (5–8). They have distinct subcellular localization, tissue expression, substrates, and cNT analog specificities (2, 9–11). In particular, membrane-bound PKG II is a crucial regulator of intestinal secretion, bone growth, renin secretion, and circadian rhythms. It is highly expressed in the intestinal mucosa, kidney, chondrocytes, and brain. It phosphorylates several downstream substrates including Src homology 2 domain-containing tyrosine phosphatases, cystic fibrosis transmembrane conductance regulator (CFTR), and Glu1R subunit of AMPA receptors. PKG II is also known as a major regulator of CFTR and AMPA receptor trafficking (10, 12–15).
PKG I and II have the same domain organization (16), which can be roughly divided into N-terminal regulatory (R) and C-terminal catalytic (C)-domains. The R-domain can be further divided into individual functional domains including an N-terminal dimerization domain, an autoinhibitory domain, and two tandem cNT binding domains (N-terminal CNB-A and C-terminal CNB-B) that bind cGMP (see Fig. 1A). Although PKG I and II have a large degree of sequence similarity over the length of the whole protein (overall sequence identity 49–50%), their sequences share no similarity at the C-terminal region of the CNB-B domain, which bridges the R- and C-domains.
FIGURE 1.

Domain organization and overall structure of PKG II CNB-A and-B bound with cyclic nucleotides. A, domain organization of PKG II with each CNB domain highlighted. B, affinity measurements of PKG II CNB domains for cGMP and cAMP. Competition FP curves are shown in black. C, overall structures of the PKG II CNB-A bound with cyclic nucleotides. The secondary structure elements are labeled. The PBC is colored in yellow, the αB helix is in red, the N-terminal helices are in teal, and the β barrel is in light brown. cAMP is colored by atom type. The N and C termini are labeled with their corresponding residue numbers seen in the final model. Cyclic nucleotides are shown as sticks, and the atoms are colored as follows: carbons in cGMP, yellow; carbons in cAMP, gray; oxygen, red; nitrogen, blue. D, overall structure of the PKG II CNB-B-bound cGMP. A zoom in-view shows the bound cGMP with an |Fo − Fc| omit map (contoured at 1.0 σ level). All structure images were generated using PyMOL (Schrödinger, LLC).
The cNT affinity and selectivity in PKG I are well studied (4, 17). The PKG I CNB-A domain binds both cGMP and cAMP with similarly high affinities, showing little selectivity. In contrast, the lower affinity CNB-B domain provides high cGMP selectivity. Crystal structures of the PKG Iβ CNB-B domain revealed a conserved arginine at β5, which provides guanine-specific interactions, and a tyrosine, which provides cNT capping. These CNB-B residues are critical for cGMP binding and activation. Unlike PKG Iβ, the CNB-B domain of PKG II is reported to have a higher cGMP affinity than the CNB-A (18). However, little is known about which CNB domain provides high cGMP selectivity and drives cGMP-dependent activation. Here, we report the first co-crystal structures of PKG II CNB-A and -B bound with cGMP or cAMP. These structures reveal a new mode of cGMP selectivity unseen in human PKG Iβ and PfPKG and demonstrate the key role of CNB-B in PKG II activation.
Experimental Procedures
Protein Expression and Purification
CNB-A (residues 137–277) and CNB-B (residues 269–418) were cloned into pNic28-Bsa4 using ligation-independent cloning and transformed into BL21 (DE3) Escherichia coli cells (19). CNB domain mutants were cloned into the pQTEV plasmid and transformed into BL21 (DE3) E. coli cells (20). Cells carrying the CNB expression constructs were grown at 37 °C until A600 nm reached 0.6 and induced with 0.5 mm isopropyl β-d-1-thiogalactopyranoside. Cultures were grown for an additional 18 h at 18 °C and harvested by centrifugation. Cells were resuspended in 25 mm potassium phosphate (pH 7.5), 150 mm sodium chloride, and 1 mm β-mercaptoethanol and lysed using a cell disrupter (Constant Systems, Northamptonshire, UK). The lysate was cleared by centrifugation at 35,000 × g for 2 h at 4 °C. Cleared lysate was passed through a 0.22-μm filter. The proteins were purified by immobilized metal ion affinity chromatography using a Profinia system (Bio-Rad); proteins were eluted with resuspension buffer supplemented with 250 mm imidazole. The eluate was incubated with tobacco etch virus protease to cleave the His tag. Tobacco etch virus protease was removed using a second immobilized metal ion affinity chromatography step. Buffer exchange and aggregate removal were performed using FPLC size-exclusion chromatography on a Hi-Load 16/60 Superdex-75 column (GE Healthcare) in 25 mm Tris (pH 7.5), 150 mm sodium chloride, and 1 mm TCEP-HCl.
Crystallization, Data Collection, Phasing, Model Building, and Refinement
For the co-crystallization of the CNB-A·cAMP complex, cAMP powder (Sigma-Aldrich) was added to purified PKG II CNB-A, and the mixture was concentrated to reach 55 mg/ml for protein and 70 mm for cAMP using a 10-kDa cutoff Amicon Ultra. Crystals appeared in 200 mm calcium chloride, 200 mm cadmium chloride, 200 mm cobalt (II) chloride, and 20% PEG 3350 within 2 days. Diffraction experiments were performed on a Rigaku FR-E DW SuperBright, equipped with a Rigaku HTC detector and X-stream 2000. Data for all three structures were processed using either CCP4 or HKL2000, and both Rmerge and CC½ (the Pearson correlation coefficient) were utilized to determine resolution cutoffs (21–23). Experimental phasing was performed using molecular replacement (MR). An initial model was built using Autobuild (24). The final model was manually built using Coot and CheckMyMetal and refined using Phenix.Refine (25–27)
To obtain crystals of the CNB-A·cGMP complex, cGMP powder (Sigma-Aldrich) was added to the purified PKG II CNB-A and the mixture was concentrated to reach final concentrations of 55 mg/ml for protein and 33 mm for cGMP using a 10 kDa-cutoff Amicon Ultra (Millipore). Co-crystals of the CNB-A·cGMP complex were obtained using the vapor diffusion method in 200 mm sodium malonate at pH 5.0, 5–10% ethylene glycol, and 20% PEG, after 1 day incubation at 22 °C. The crystals were harvested and immersed in 20% ethylene glycol, immediately before being flash-cooled in liquid nitrogen. The crystals were then diffracted at the Advanced Light Source at Berkeley, CA. MR was performed using Phenix.Phaser (28). The structure of the CNB-A·cAMP complex (Protein Data Bank (PDB) ID: 5C6C) was used as a search model. Refinement and model building were performed as described above.
To crystallize PKG II CNB-B bound to cGMP, cGMP powder (Sigma-Aldrich) was added to purified PKG II CNB-B and the mixture was concentrated to reach final concentrations of 20 mg/ml for protein and 20 mm for cGMP using a 10 kDa-cutoff Amicon Ultra (Millipore). Crystals were obtained using the vapor diffusion method in 20 mm calcium chloride, 100 mm, sodium acetate (pH 4.6), and 30% 2-methyl-2,4-pentanediol at 4 °C. Crystals appeared after 2 days and were diffracted at the Advanced Light Source at Berkeley, CA. MR was performed using a truncated PKG Iβ CNB-B·cGMP (PDB ID: 4KU7) structure as a search model using Phenix.Phaser (28). Refinement and model building were performed as described above.
Fluorescence Polarization Assay
Fluorescence polarization (FP) was used to measure the binding affinity of 8-fluo-cNT to the CNB domain constructs. CNB domain constructs were titrated via serial dilution to measure the KD of 8-fluo-cNT in a direct binding assay. The direct binding assay was conducted in 150 mm sodium chloride, 20 mm MOPS (pH 7.0), 0.005% (v/v) Tween 20, and 1 nm 8-fluo-cNT (BioLog Life Science Institute, Bremen, Germany) in a black, flat-bottomed 384-well plate (PerkinElmer, OptiPlate). The FP signal was read for 2 s per well at an excitation wavelength of 485 nm and an emission wavelength of 535 nm, on BioTek Synergy2 equipped with a FITC optics cube. Data were analyzed and fit to a sigmoidal dose-response curve using GraphPad Prism 5.03 (GraphPad Software, San Diego, CA) to generate KD values.
Competition experiments were performed using protein concentrations slightly lower than the KD obtained from the direct binding assay, and 8-fluo-cNT was added to a final concentration of 1 nm. cNT ligands were titrated via a serial dilution. Polarization data were measured and analyzed as described above to generate half-maximal effective concentration (EC50) values.
Kinase-Glo Assay
HEK293T cells were grown to 80–90% confluency in Dulbecco's modified Eagle's medium (BioWhittaker®, Walkersville, MD) and transfected with FLAG-tagged wild-type and mutant PKG II (residues 40–762) constructs using Lipofectamine 2000. Cells were harvested 36 h after transfection and stored at −80 °C. Cell pellets were thawed on ice and then lysed by sonication in 1× TBS with protease inhibitors. Protein samples were purified by loading the lysate onto anti-FLAG-M2-agarose beads (Sigma-Aldrich), washing with TBS, and eluting with TBS containing 0.5 mg/ml 3× FLAG peptide (Sigma-Aldrich). Kinase activity was determined using the Kinase-Glo® Luminescent Kinase Assay (Promega, Madison, WI). Human PKG II was incubated for 2–3 h at 25 °C in kinase reaction buffer containing Tris buffer, pH 7.5, 0.1 mg/ml BSA, 3 mm Glasstide (GL Biochem, Shanghai, China), 0.01 mm ATP, 10 mm MgCl2, and various concentrations of cGMP (3 nm–5 mm). Kinase activity for each sample was indirectly determined by correlating ATPase activity to kinase activity. Although not equivalent, it is assumed that ATPase activity can be reasonably correlated to phosphotransferase activity of PKG II in our Kinase-Glo assay. Using concentrations of 0 and 2.5 mm cGMP in kinase reaction buffer, we determined maximal ATPase activity of each purified sample without detectable basal activity under these conditions. Depending on apparent maximal ATPase activity, purified kinase was diluted until activity equal to a decrease of 300,000–600,000 relative luminescence units was observed after 2–3 h. After incubation at room temperature, Kinase-Glo buffer was added in a 1:1 ratio with the kinase reaction. The Kinase-Glo reaction was allowed to equilibrate for 10 min prior to measuring luminescence on a BioTek Synergy2 in white, flat-bottomed, small volume 384 well microplates. ATP conversion was plotted against the logarithmic cNT concentration, and apparent activation constants (Ka:cGMP, the concentration of cGMP required for half-maximal activity) were calculated from sigmoidal dose-response curves using GraphPad Prism 5.03.
In Vivo Phosphorylation Assay
COS-7 cells were transfected with pcDNA3.1-hPKGII WT-FLAG or pcDNA3.1-hPKGIID412A/R415A-FLAG together with pFLAG-mVASP using Lipofectamine 2000 (Invitrogen; Life Technologies). The medium was replaced with fresh serum-free medium 24 h after transfection. After 6 h, cells were treated with 8-Br-cGMP for 1 h, and then they were scraped in ice-cold lysis buffer (20 mm Tris-HCl, pH 7.5, 150 mm sodium chloride, 0.5% Nonidet P-40, and 1 mm EDTA) supplemented with protease inhibitors (10 μg/ml leupeptin and 10 μg/ml aprotinin) and phosphatase inhibitor mixture (Nacalai Tesque, Kyoto, Japan). The cell lysates were centrifuged at 10,000 × g for 10 min at 4 °C to remove cellular debris, and the supernatants were subjected to SDS-PAGE and immunoblotted with anti-phospho-VASP (Ser-239) (Cell Signaling Technology) or anti-FLAG M2 (Sigma-Aldrich) antibodies. After incubation with secondary horseradish peroxidase-conjugated antibodies, blots were developed using SuperSignal West Pico Substrate (Thermo Scientific). The images were captured using an LAS-4000 image analyzer (Fujifilm, Tokyo, Japan). The intensity of bands was quantified using ImageJ software (National Institutes of Health). The phosphorylation rate of vasodilator-stimulated phosphoprotein (VASP) by PKG II WT-FLAG (100 μm 8-Br-cGMP) was taken as 100%.
Results
CNB-B Provides High cGMP Selectivity
To determine which CNB domain provides the high cGMP selectivity required for cGMP-specific activation, we measured EC50 values of each isolated CNB domain for both cNTs using a FP competition assay. Our measurements showed that the CNB-A binds cGMP and cAMP with EC50 values of 43.8 and 418 nm, respectively, showing only 10-fold preference for cGMP (Fig. 1B). In major contrast, the CNB-B binds cGMP and cAMP with EC50 values of 31.4 nm and 15.6 μm, respectively, showing an almost 500-fold preference for cGMP.
Structure Determination and Overall Structures of CNB-A and -B
To understand the structural basis of cGMP selectivity in CNB-A and -B, we solved their crystal structures in the presence of cGMP or cAMP (Fig. 1, C and D). In all, we solved three structures. For CNB-A, we determined its structure bound to cAMP or cGMP. Crystals of CNB-B were only obtained in the presence of cGMP. All of the structures showed a typical CNB fold consisting of an N-terminal helical bundle, an eight-stranded β-barrel, a conserved short helix-loop motif within the β-barrel known as the phosphate binding cassette (PBC), and a variable number of helices at the C terminus (Fig. 1, C and D).
The structure of the CNB-A·cAMP complex was solved at 2.01 Å by MR using the structure of PKG Iβ CNB-A (PDB ID: 3OCP) as a search model. The CNB-A·cAMP complex was crystallized in a P212121 space group with two molecules in the asymmetric unit. The structure of the CNB-A·cGMP complex was solved at 1.69 Å using the CNB-A·cAMP complex as a MR search model. Although the CNB-A·cGMP complex crystallized in the same space group as the cAMP containing complex, its asymmetric unit contained six molecules. All six molecules show clear electron density for residues 151–267, but lack electron density for residues at their termini. Each of the six molecules show the same conformation, with root mean square deviation values of less than 1 Å for at least 116 Cα atoms.
The structure of the CNB-B·cGMP complex was solved at 1.94 Å, using the PKG Iβ CNB-B domain (PDB ID 4KU7) as a MR search model (4). The asymmetric unit contained only one molecule showing clear density for residues 269–418. CNB-A and -B have the same overall fold, but residues corresponding to the αC-helix of CNB-A were unstructured. Statistics for crystallographic data and structural refinement for all three structures are summarized in Table 1.
TABLE 1.
Data and refinement statistics
| CNB-A·cAMP | CNB-A·cGMP | CNB-B·cGMP | |
|---|---|---|---|
| Data collection | |||
| Wavelength (Å) | 1.5416 | 0.97931 | 0.97931 |
| Space group | P212121 | P212121 | P212121 |
| Cell dimensions | |||
| a, b, c (Å) | 46.71, 63.76, 99.54 | 46.71, 103.5, 176.7 | 40.34, 50.48, 69.17 |
| α, β, δ (°) | 90, 90, 90 | 90, 90, 90 | 90, 90, 90 |
| Resolution (Å) | 42.29–2.01 | 51.73–1.80 | 50–1.94 |
| Rsym or Rmerge | 0.116 (0.751)a | 0.149 (0.503) | 0.073 (0.577) |
| I/σI | 12.1 (1.7) | 14.0 (3.3) | 37.7 (4.8) |
| CC½a,b | 0.997 (0.665) | 0.996 (0.904) | 0.998 (0.847) |
| Completeness (%) | 99.8 (97.7) | 99.6 (94.4) | 99.4 (99.4) |
| Redundancy | 9.4 (4.4) | 12.2 (7.5) | 7.0 (7.2) |
| Refinement | |||
| Resolution (Å) | 39.2–2.01 | 51.7–1.80 | 40.8–1.94 |
| No. of reflections | 16,779 | 974,540 | 10,870 |
| Rwork/Rfreec | 0.1795/0.2374 | 0.1663/0.1930 | 0.1772/0.2109 |
| No. of atoms | |||
| Proteins | 1798 | 11,313 | 1189 |
| Ligand/ion | 54 | 274 | 30 |
| Water | 150 | 746 | 105 |
| B-factors | |||
| Protein | 41.4 | 31.1 | 34.1 |
| Ligand/ion | 43.6 | 28.8 | 28.2 |
| Water | 41.3 | 36.0 | 40.0 |
| r.m.s.d deviations | |||
| Bond lengths (Å) | 0.003 | 0.009 | 0.009 |
| Bond angles (°) | 1.4 | 1.2 | 1.2 |
a Highest resolution shell is shown in parenthesis.
b CC½, Pearson correlation coefficient.
c 5.0% of the observed intensities was excluded from refinement for cross validation purposes.
d r.m.s., root mean square.
The αC-helix of CNB-B Provides Isoform-specific Contacts
Although the overall structure of the PKG II CNB-B·cGMP complex is similar to PKG Iβ CNB-B, the structures of the C-terminal region and specific amino acid contacts with guanine are different (Fig. 2) (4, 29). As in PKG Iβ CNB-B, the cGMP pocket can be divided into four sites (Fig. 2A). The interactions with cGMP at the first three sites are similar between the two isoforms. However, Site 4 shows several PKG II-specific contacts.
FIGURE 2.

cGMP binding pocket of CNB-B and its comparison with the PKG I CNB-B. A, detailed interactions between CNB-B and cGMP. Zoomed-in views for each cGMP binding site are shown on the top (Sites 1–3) and either side (Site 4). Residues that interact with cGMP are shown as sticks and colored using the following color scheme: side chain carbon atoms, black; oxygen, red; nitrogen, blue. Residues that provide van der Waals interactions are shown with gray surface, and cGMP is shown with red surface. Hydrogen bonds are shown as dotted lines with their distances marked in Å units. The backbone amide of 359 and carbonyl of Val-365 are marked with blue and red dots. B, cGMP binding pockets of PKG II and PKG I with sequence alignment. The cGMP pocket of PKG II in red is shown on the left panel, and that of PKG I (PDB code: 4KU7) in gray is shown on the right. The disordered β4-β5 loop is indicated with a dotted line. Top, sequence alignment of PKG II; bottom, sequence alignment of PKG Iβ. Conserved residues are shaded in yellow. Key cGMP contact residues in PKG II (Leu-408, Asp-412, and Arg-415) and PKG I (Arg-297 and Tyr-351) are shaded in red. C, a side-by-side comparison of the cGMP interactions at the O6 and N7 positions of cGMP by PKG I and II. Left, Lys-347 and Asp-411 of PKG II and their coordination of a water molecule, in blue. Right, Arg 297 of PKG I and the hydrogen bond interactions with cGMP are shown.
The first site is shared with other CNB domains (3, 4, 17, 29). It includes Glu-357 and Arg-366, which capture the sugar phosphate moiety through hydrogen bonds. At the second site, a highly conserved serine, Ser-367, substitutes for a threonine (Thr-307) of PKG Iβ and provides cGMP-specific interactions by linking the axial oxygen of the cyclic phosphate to the N2 of guanine through hydrogen bonds. The backbone carbonyl of Val-365 additionally interacts with the axial oxygen of cGMP via an ordered water molecule. A water molecule is not seen in Site 2 of the PKG Iβ CNB-B·cGMP complex, because the methyl group of Thr-307 occupies this position (Fig. 2, A and B). At Site 3, Lys-347 is analogous to Arg-297 of PKG Iβ, which functions as a “cAMP filter” by promoting cGMP binding (Fig. 2C). Similarly to Arg-297 of PKG Iβ, Lys-347 hydrogen-bonds with cGMP at N7, and in concert with Asp-411, a water molecule is coordinated to hydrogen-bond with the O6 position of cGMP. The side chain of Lys-347 also interacts with the guanine moiety through van der Waals contact (Fig. 2A).
Due to the large structural differences at the αC-helix of PKG II, cGMP interactions at Site 4 are distinct from those observed in PKG I and can be described in two parts: 1) charged contacts that recognize guanine through hydrogen bonds, and 2) hydrophobic contacts that shield cGMP from solvent (Fig. 2A). The charged contacts at Site 4 are located at the last turn of the αC-helix and include Asp-412 and Arg-415, which make direct contacts with guanine. Specifically, the carboxylate group of Asp-412 forms hydrogen bonds with both the N2 and the unprotonated N1 amine, whereas the guanidinium group of Arg-415 interacts with the C-6 carbonyl group of guanine through a hydrogen bond. Additionally, Asp-412 and Arg-415 interact with each other and with Gln-335 at the β4-β5 loop through hydrogen bonds. Thus, together these residues form an extended hydrogen-bonding network, which positions the side chains of Asp-412 and Arg-415 for binding cGMP.
The amino acids that form hydrophobic contacts at Site 4 are located mainly at the PBC and the αC-helix. They include Lys-358, Leu-408, and Asn-409, and shield one side of cGMP from solvent (Fig. 2A). In particular, Lys-358 at the PBC forms a hydrogen bond with Asn-409 at the αC-helix. This positions the side chain of Lys-358 close to cGMP, so that its hydrophobic arm shields the sugar phosphate. Additionally, the side chain of Leu-408 forms a capping interaction with the guanine ring, sandwiching it against two hydrophobic residues (Val-333 and Ile-346) at the base of the cGMP pocket (Fig. 2A). Despite little primary sequence similarity at this region, PKG II Leu-408 structurally aligns with PKG Iβ Tyr-351 and serves as a capping residue (Fig. 3). In addition, due to the unique set of αC-helix contacts in PKG II CNB-B, the cGMP binding pocket is more shielded from solvent when compared with PKG Iβ CNB-B (Fig. 3).
FIGURE 3.
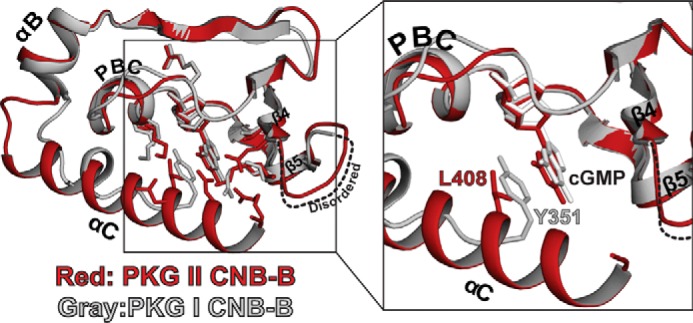
Capping interactions in the CNB-B domains of PKG I and II. Two structures (PKG I in gray and PKG II in red) are aligned at the β-barrel, and key cGMP interacting residues are shown. A zoomed-in view on the right shows the cGMP pockets only with the capping residues.
CNB-A Shows an Open Pocket with Few Guanine-specific Contacts
Both of our PKG II CNB-A structures show “open” cyclic nucleotide binding pockets, which lack the C-terminal helix and Site 4 contacts seen in CNB-B (Fig. 4A). The C-terminal helix of CNB-A lacks the sequence/structural requirements to form a lid portion of the “hinge and lid,” structural features often found in CNBs (see Rehman et al. (1)). Indeed, the αC-helix of PKG II CNB-A is not ordered in either cGMP-bound or cAMP-bound structures as observed in type I PKG (17). Therefore, CNB-A lacks a shielded pocket or the large number of cGMP-specific contacts that we see in CNB-B.
FIGURE 4.

Structural comparison between the CNB-A domain bound with cGMP and cAMP. A, the cGMP-bound (black) and the cAMP-bound (gray) structures aligned at the β barrel region. An enhanced view of the binding pocket highlighting key contacts with cyclic nucleotides is shown on the right. B, cyclic nucleotide binding pocket of CNB-A. Only β4 through PBC of CNB-A are shown with cyclic nucleotides. Cyclic nucleotides and interacting residues are shown as sticks. Cyclic nucleotides and van der Waals contact residues are shown with transparent surfaces. Hydrogen bonds are shown as dotted lines.
The CNB-A·cGMP complex shows similar contacts with cGMP as the CNB-B·cGMP complex at the first three interaction sites (Fig. 4B). Charged residues Glu-233 and Arg-242, at the PBC, bind the sugar phosphate of cGMP, and Thr-243 at the PBC and Leu-222/Ser-223 at the β5 stand interact with the guanine moiety. In particular, Thr-243 plays an analogous role to Ser-367 of CNB-B and similarly interacts with cGMP in the syn conformation (Fig. 5). The side chains of Leu-222/Ser-223 of CNB-A, analogous to Ile-346/Lys-347 of CNB-B, provide similar van der Waals contacts.
FIGURE 5.

Structural comparison between CNB-A and -B bound with cGMP. A, the structures of CNB-A (black) and -B (red) are aligned at the β barrel region. An enhanced view of the binding pocket highlighting key contacts with cGMP is shown on the right. B, cyclic nucleotide binding pockets of CNB-A and -B. Only β4 through PBC are shown with cyclic nucleotides. Cyclic cGMP and interacting residues are shown as sticks. Cyclic cGMP and van der Waals contact residues are shown with transparent surfaces.
The structure of the PKG II CNB-A·cAMP complex shows the same open pocket with cAMP bound in an anti conformation (Figs. 1C and 4). The pocket interacts with cAMP using the same residues in a manner similar to the CNB-A·cGMP complex, except for the interaction with Thr-242. Due to the missing N2 group in cAMP, Thr-242 only interacts with the cyclic phosphate (Fig. 4B).
Mutating Key cGMP Contacts Reduces cGMP Selectivity and Kinase Activation
Next, we tested the roles of the newly identified CNB-B contacts in cGMP binding. Because they appeared to provide the majority of cGMP-specific interactions, we individually mutated Gln-335, Asp-411, Asp-412, and Arg-415 to alanine (Fig. 2A). Wild-type and mutant CNB-B domains were purified, and their affinities for cGMP and cAMP were measured using an FP competition assay (Table 2). We found that mutating any of these residues increases the EC50 value for cGMP, while leaving EC50 values for cAMP relatively unchanged. Notably, mutating Asp-412 or Arg-415 increases the EC50 for cGMP from 31 nm to as much as 1.1 μm without significantly changing it values for cAMP (from 15 to 25 μm). However, mutating Gln-335 and Asp-411 showed modest increases in its EC50 value for cGMP (2.9- and 8.5-fold, respectively), while slightly increasing the EC50 for cAMP (1.7- and 1.6-fold, respectively).
TABLE 2.
Affinity measurements for CNB-A, CNB-B, and mutants
KD is the equilibrium constant for association. EC50 is the half-maximal effective concentration.
| PKGII(CNB-B) | 8-Fluo-cGMPa | cGMPb | cAMPb |
|---|---|---|---|
| Wild type | 23.5 ± 0.03 nm | 31 ± 1 nm | 15 ± 1 μm |
| Q335A | 31 ± 1 nm | 90 ± 4 nm | 25 ± 3 μm |
| D411A | 1.8 ± 0.2 nm | 262 ± 5 nm | 23.4 ± 4 μm |
| D412A | 17 ± 0.4 nm | 1065 ± 39 nm | 13.2 ± 1 μm |
| R415A | 47 ± 1 nm | 964 ± 101 nm | 25 ± 1 μm |
| PKGII(CNB-A) | |||
| Wild type | 44 ± 1 nmc | 43.8 ± 1 nm | 418 ± 19 nm |
a KD ± S.E. direct measurements. FP measurements were at least in triplicate (n = 3).
b EC50 ± S.E.a competition experiments. FP measurements were at least in triplicate (n = 3).
c This value is for 8-Fluo-cAMP rather than for 8-Fluo-cGMP.
Because D412A and R415A dramatically reduced the CNB-B domain's affinity for cGMP, we generated these mutations in full-length PKG II and measured the resulting effects on kinase activation (Fig. 6, A and B). Consistent with the dramatic increases seen in their EC50 values for cGMP, the D412A and R415A mutants showed a large increase in their activation constants (1.6 and 3.0 μm, respectively).
FIGURE 6.

CNB-B recognition of cGMP is critical to kinase activation. A, key cGMP contact residues, whose role in kinase activation was tested. B, activation curves are shown. Ka:cGMP values were measured using the Kinase-Glo assay, performed at least in triplicate with S.E. shown. C, in vivo kinase assay using VASP as a substrate. COS-7 cells were transfected with PKG II WT-FLAG or PKG II D412A/R415A-FLAG together with FLAG-VASP. After 6 h, cells were treated with the indicated concentrations of 8-Br-cGMP for 1 h. D, Western blots (IB) with anti-phospho-VASP (Ser-239) or anti-FLAG M2 antibodies illustrate the effect of the mutations on VASP phosphorylation. Statistical significances were determined by Student's t test. *, p < 0.05; **, p < 0.01.
Finally, we investigated the role of Asp-412 and Arg-415 in PKG II activation by performing an in vivo kinase assay using VASP as a substrate (Fig. 6, C and D). We co-expressed human WT or D412A/R415A mutant PKG II with VASP and monitored the phosphorylation in response to increasing levels of 8-bromo-cGMP (8-Br-cGMP). Although WT PKG II maximally phosphorylated VASP Ser-239 at 30 μm 8-Br-cGMP, the D412A/R415A mutant showed less than 40% of maximal VASP phosphorylation even at 100 μm 8-Br-cGMP. These results are consistent with our in vitro cGMP binding and kinase activation measurements, demonstrating the crucial role of these contacts for PKG II activation in mammalian cells.
Discussion
Although understanding the cNT selectivity of PKG II is crucial for understanding its activation mechanism, little information has been available about its binding affinities for either cGMP or cAMP. We resolved this by measuring cNT binding affinity in isolated CNB domains. We found CNB-B to be the higher affinity cGMP binding site and to have almost 500-fold selectivity for cGMP over cAMP. CNB-A was found to have a slightly lower affinity for cGMP than CNB-B and showed significantly less cGMP selectivity with a 10-fold preference.
Our affinity data are semi-consistent with a previous study examining PKG II cNT affinity (18). In that study, key cGMP interacting residues in CNB-A and -B were predicted based on the structures of other CNB domains. These residues were then mutated to disable cNT binding to each pocket in full-length PKG II, and cGMP dissociation rates were measured. Using these mutants, they found that CNB-B has a slower dissociation rate (t½ = 4.0 s) when compared with CNB-A (t½ = 0.18 s), which suggested that CNB-B is the higher affinity site. Although our finding, that CNB-B has a higher affinity than CNB-A, is consistent with these results, we only observed a marginal difference between the EC50 values for cGMP. This finding does not fit with the large difference in the dissociation rates seen in the previous study. The discrepancy can be explained by differences in cGMP association rates of the CNBs, as affinity is a function of association and disassociation. In addition, the previous study proposed a model that binding to one CNB would affect cGMP binding to the other, suggesting allosteric communication between the two binding pockets (18). This effect would have been lost in our isolated domains.
Our PKG II CNB structures reveal specific amino acid contacts that explain their differences in cGMP selectivity. The overall structures of CNB-A and -B are similar, but only in CNB-B does the αC-helix make contact with and shield the cGMP pocket. Although the CNB-A protein used for crystallization contained residues that can form the αC helix, these residues were disordered in the crystal, suggesting that they do not provide stable contacts for cGMP. In contrast, residues in the CNB-B αC-helix specifically interact with the guanine moiety through hydrogen bonds (Asp-412 or Arg-415 at Site 4). Mutating these residues to alanine reduces the affinity of CNB-B for cGMP, without significantly affecting its cAMP affinity. These results demonstrate the critical role of these residues in cGMP binding and selectivity. In addition, kinase assays using full-length PKG II show that these mutations dramatically increased activation constant for cGMP (Ka:cGMP), demonstrating their critical role in kinase activation.
Structural comparison between cGMP selective human PKG Iβ and PfPKG CNB domains suggests a distinct cGMP selectivity mechanism for PKG II CNB-B (3, 4). Crystal structures combined with mutational analysis of the carboxyl CNB domains of PKG Iβ (CNB-B) and PfPKG (PfCNB-D) demonstrate that two structural features are required for achieving high cGMP selectivity. First, they both have a conserved arginine at β5 that forms hydrogen bonds with the guanine moiety and has been shown to reduce cAMP binding in PKG Iβ. The second feature is a hydrophobic cap that interacts with one side of the bound cGMP and shields it from solvent. In PKG Iβ CNB-B, a single aromatic residue at a C-terminal loop provides this capping interaction. PfCNB-D shows a set of charged residues from the PBC and the C-terminal helix interacting through hydrogen bonds, which together provide the capping interaction (Fig. 7). In PKG II, the αC helix provides two charged residues, which recognize the guanine moiety through hydrogen bonds. Due to the C-terminal helix that shields the entire cGMP pocket, the structure of PKG II CNB-B is more similar to that of PfPKG. PKG II CNB-B lacks the cGMP interacting arginine at β5, but it possesses Lys-347 at this position. Another distinct feature in PKG II's CNB-B is its capping interaction with cGMP, which shows characteristics of both PKG Iβ and PfPKG. Leu-408 at the αC-helix structurally aligns with Tyr-351 in PKG Iβ and provides a similar hydrophobic capping interaction with cGMP. PKG II Lys-358 (at the PBC) and Asn-409 (at the αC helix) structurally align with PfPKG Arg-484 (at the PBC) and Asp-533 (at the αC helix). They form similar hydrogen bonds, which shield the sugar phosphate of cGMP (Fig. 7).
FIGURE 7.
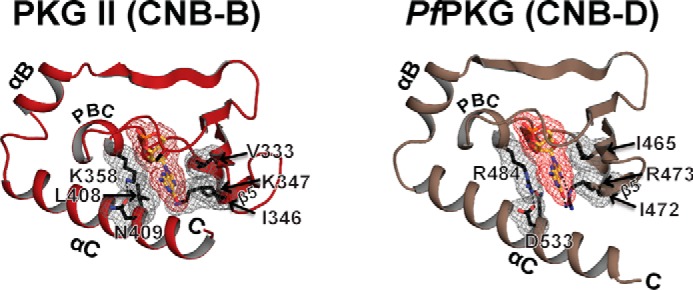
cGMP binding pockets of PKG II and PfPKG CNB-D. The cGMP pocket of PKG II in red is shown on the left panel, and that of PKG I (PDB code: 4KU7) in tan is shown on the right. Key cGMP contact residues in PKG II and PfPKG are shown with gray mesh.
Our structures may explain previously reported cGMP analog specificity for the PKG isoforms and suggest that the CNB-B domains provide this specificity (30). Studies on the activation of PKG I and II isoforms by cGMP analogs showed that 8-substituted cGMP analogs differentially activate PKG isoforms. PKG II is significantly more sensitive to 8-pCPT-cGMP than cGMP (activation constants (Ka) showing 3.5–80 nm versus 800 nm) and is poorly activated by PET-cGMP (Ka of 4.7 μm). This trend is reversed in PKG Iα, where PET-cGMP is more potent than cGMP (16–26 nm versus 110 nm) and 8-pCPT-cGMP activates PKG I at a similar concentration as cGMP (50 nm). Structural comparison of all four CNB domains of PKG I and II suggests that the CNB-B domains are responsible for the observed analog specificity because of the structural differences at the C-terminal region. In particular, PKG II CNB-B has an additional hydrophobic pocket near the C8 position of cGMP (consisting of Lys-347, Tyr-354, Leu-349, Tyr-404, Glu-357, Phe-355, and Leu-408), which can provide van der Waals contacts and accommodate modifications at the C8 position, such as 8-pCPT. In contrast, the more open pocket of PKG I CNB-B lacks these interactions, which may explain PKG I's lower affinity for this and other 8-position analogs (31). Lastly, PET-cGMP is a less potent activator of PKG II because of the derivatization of the purine ring of cGMP into 1,N2-ethenoguanosine. This prevents the favorable hydrogen bonding between Asp-412 and the N1 and C2 positions of cGMP and may reduce the binding affinity (32).
The work presented here, combined with previous work by our lab, offers a path to structure-based design of isoform-specific PKG agonists (3, 4, 17). These agonists have the potential to be new chemical tools that may contribute to generating new insights into cyclic nucleotide research. Furthermore, due to its high affinity and selectivity, PKG II CNB-B is a good candidate as a scaffold for the generation of FRET-based protein biosensors, which could be used to measure the spatiotemporal dynamics of NO-cGMP signaling in cells. Historically, cGMP-responsive FRET biosensors have been constructed using the PKG I CNBs (33–36). Based on our previous work examining PKG I CNBs (4, 17), we can explain different cGMP selectivity profiles seen in these sensors (37).
Author Contributions
J. C. C. and C. K. designed the experiments required for addressing the questions in this study. T. M. L, D. E. C., J. E. L., and J. J. K. designed the constructs. G. Y. H, J. J. K., and J. C. C. identified and optimized crystallization conditions. A. S. R., J. C. C., and B. S. collected diffraction images. J. C. C., and J. J. K. performed mutagenesis with single domain and full-length PKG II. J. C. C. and A. S. R. solved the structures and refined the crystallographic models. J. C. C. and K. Y. L. purified single domain mutants and performed binding assays. J. C. C purified full-length PKG II and mutants and performed in vitro kinase activation assay. S. M. and K. Y. performed the in vivo kinase activation assays. J. C. C. and C. K. wrote the manuscript. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgments
We thank A. Fields and H. Rehmann for critically reading the manuscript. We also thank M. Young and C. Zhao for their assistance with crystallization. The Berkeley Center for Structural Biology is supported in part by the National Institutes of Health, NIGMS, and the Howard Hughes Medical Institute. The Advanced Light Source is supported by the Director, Office of Science, Office of Basic Energy Sciences, of the United States Department of Energy under Contract Number DE-AC02-05CH11231.
This work was supported by National Institutes of Health Grant 2R01GM090161 (to C. K.), the Training Program in Pharmacological Sciences fellowship, NIGMS Grant T32GM089657 (to J. C. C.), and the Houston Area Molecular Biophysics Program, NIGMS Grant T32GM008280 (to G. Y. H.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the author and does not necessarily represent the official views of the National Institutes of Health.
The atomic coordinates and structure factors (codes 5C8W, 5C6C, and 5BV6) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- cNT
- cyclic nucleotide
- PKG
- cGMP-dependent protein kinase
- CNB
- cyclic nucleotide binding domain
- CNB-A
- N-terminal CNB
- CNB-B
- C-terminal CNB
- PfPKG
- Plasmodium falciparum PKG
- MR
- molecular replacement
- FP
- fluorescence polarization
- VASP
- vasodilator-stimulated phosphoprotein
- TCEP
- tris(2-carboxyethyl)phosphine
- 8-Br-cGMP
- 8-bromoguanosine 3′,5′-cyclic monophosphate sodium salt
- PET-cGMP
- β-phenyl-1,N2-ethenoguanosine 3′,5′-monophosphate
- PBC
- phosphate binding cassette
- 8-pCPT-cGMP
- 8-para-chlorophenylthio-cGMP
- 8-Fluo-cGMP
- 8-(2-[fluoresceinyl]aminoethylthio)guanosine-3′,5′-cyclic monophosphate
- 8-Fluo-cAMP
- 8-(2-[fluoresceinyl]aminoethylthio)adenosine-3′,5′-cyclic monophosphate.
References
- 1. Rehmann H., Wittinghofer A., and Bos J. L. (2007) Capturing cyclic nucleotides in action: snapshots from crystallographic studies. Nat. Rev. Mol. Cell Biol. 8, 63–73 [DOI] [PubMed] [Google Scholar]
- 2. Francis S. H., and Corbin J. D. (1999) Cyclic nucleotide-dependent protein kinases: intracellular receptors for cAMP and cGMP action. Crit. Rev. Clin. Lab. Sci. 36, 275–328 [DOI] [PubMed] [Google Scholar]
- 3. Kim J. J., Flueck C., Franz E., Sanabria-Figueroa E., Thompson E., Lorenz R., Bertinetti D., Baker D. A., Herberg F. W., and Kim C. (2015) Crystal structures of the carboxyl cGMP binding domain of the Plasmodium falciparum cGMP-dependent protein kinase reveal a novel capping triad crucial for merozoite egress. PLoS Pathog. 11, e1004639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Huang G. Y., Kim J. J., Reger A. S., Lorenz R., Moon E.-W., Zhao C., Casteel D. E., Bertinetti D., Vanschouwen B., Selvaratnam R., Pflugrath J. W., Sankaran B., Melacini G., Herberg F. W., and Kim C. (2014) Structural basis for cyclic-nucleotide selectivity and cGMP-selective activation of PKG I. Structure 22, 116–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Butt E., Geiger J., Jarchau T., Lohmann S. M., and Walter U. (1993) The cGMP-dependent protein kinase: gene, protein, and function. Neurochem Res. 18, 27–42 [DOI] [PubMed] [Google Scholar]
- 6. Wolfe L., Corbin J. D., and Francis S. H. (1989) Characterization of a novel isozyme of cGMP-dependent protein kinase from bovine aorta. J. Biol. Chem. 264, 7734–7741 [PubMed] [Google Scholar]
- 7. Jarchau T., Häusler C., Markert T., Pöhler D., Vanderkerckhove J., De Jonge H. R., Lohmann S. M., and Walter U. (1994) Cloning, expression, and in situ localization of rat intestinal cGMP-dependent protein kinase II. Proc. Natl. Acad. Sci. U.S.A. 91, 9426–9430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Uhler M. D. (1993) Cloning and expression of a novel cyclic GMP-dependent protein kinase from mouse brain. J. Biol. Chem. 268, 13586–13591 [PubMed] [Google Scholar]
- 9. Schlossmann J., and Desch M. (2009) cGK substrates. Handb. Exp. Pharmacol. 163–193 [DOI] [PubMed] [Google Scholar]
- 10. Vaandrager A. B., Smolenski A., Tilly B. C., Houtsmuller A. B., Ehlert E. M., Bot A. G., Edixhoven M., Boomaars W. E., Lohmann S. M., and de Jonge H. R. (1998) Membrane targeting of cGMP-dependent protein kinase is required for cystic fibrosis transmembrane conductance regulator Cl− channel activation. Proc. Natl. Acad. Sci. U.S.A. 95, 1466–1471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hofmann F., Feil R., Kleppisch T., and Schlossmann J. (2006) Function of cGMP-dependent protein kinases as revealed by gene deletion. Physiol. Rev. 86, 1–23 [DOI] [PubMed] [Google Scholar]
- 12. Pfeifer A., Aszódi A., Seidler U., Ruth P., Hofmann F., and Fässler R. (1996) Intestinal secretory defects and dwarfism in mice lacking cGMP-dependent protein kinase II. Science 274, 2082–2086 [DOI] [PubMed] [Google Scholar]
- 13. Rangaswami H., Marathe N., Zhuang S., Chen Y., Yeh J. C., Frangos J. A., Boss G. R., and Pilz R. B. (2009) Type II cGMP-dependent protein kinase mediates osteoblast mechanotransduction. J. Biol. Chem. 284, 14796–14808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Serulle Y., Zhang S., Ninan I., Puzzo D., McCarthy M., Khatri L., Arancio O., and Ziff E. B. (2007) A GluR1-cGKII interaction regulates AMPA receptor trafficking. Neuron 56, 670–688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Golin-Bisello F., Bradbury N., and Ameen N. (2005) STa and cGMP stimulate CFTR translocation to the surface of villus enterocytes in rat jejunum and is regulated by protein kinase G. Am. J. Physiol. Cell Physiol. 289, C708–C716 [DOI] [PubMed] [Google Scholar]
- 16. Vaandrager A. B., Hogema B. M., and de Jonge H. R. (2005) Molecular properties and biological functions of cGMP-dependent protein kinase II. Front. Biosci. 10, 2150–2164 [DOI] [PubMed] [Google Scholar]
- 17. Kim J. J., Casteel D. E., Huang G., Kwon T. H., Ren R. K., Zwart P., Headd J. J., Brown N. G., Chow D.-C., Palzkill T., and Kim C. (2011) Co-Crystal structures of PKG Iβ (92–227) with cGMP and cAMP reveal the molecular details of cyclic-nucleotide binding. PLoS ONE 6, e18413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Taylor M. K., and Uhler M. D. (2000) The amino-terminal cyclic nucleotide binding site of the type II cGMP-dependent protein kinase is essential for full cyclic nucleotide-dependent activation. J. Biol. Chem. 275, 28053–28062 [DOI] [PubMed] [Google Scholar]
- 19. Savitsky P., Bray J., Cooper C. D. O., Marsden B. D., Mahajan P., Burgess-Brown N. A., and Gileadi O. (2010) High-throughput production of human proteins for crystallization: the SGC experience. J. Struct. Biol. 172, 3–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Scheich C., Niesen F. H., Seckler R., and Büssow K. (2004) An automated in vitro protein folding screen applied to a human dynactin subunit. Protein Sci. 13, 370–380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Diederichs K., and Karplus P. A. (2013) Better models by discarding data? Acta Crystallogr. D Biol. Crystallogr. 69, 1215–1222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Winn M. D., Ballard C. C., Cowtan K. D., Dodson E. J., Emsley P., Evans P. R., Keegan R. M., Krissinel E. B., Leslie A. G. W., McCoy A., McNicholas S. J., Murshudov G. N., Pannu N. S., Potterton E. A., Powell H. R., Read R. J., Vagin A., and Wilson K. S. (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 67, 235–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Minor W., Cymborowski M., Otwinowski Z., and Chruszcz M. (2006) HKL-3000: the integration of data reduction and structure solution-from diffraction images to an initial model in minutes. Acta Crystallogr. D. Biol. Crystallogr. 62, 859–866 [DOI] [PubMed] [Google Scholar]
- 24. Terwilliger T. C., Grosse-Kunstleve R. W., Afonine P. V., Moriarty N. W., Zwart P. H., Hung L.-W., Read R. J., and Adams P. D. (2008) Iterative model building, structure refinement and density modification with the PHENIX AutoBuild wizard. Acta Crystallogr. D 64, 61–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Emsley P., and Cowtan K. (2004) COOT:model building tools for molecular graphics. Acta Crystallogr. D. Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 26. Afonine P. V., Grosse-Kunstleve R. W., Echols N., Headd J. J., Moriarty N. W., Mustyakimov M., Terwilliger T. C., Urzhumtsev A., Zwart P. H., and Adams P. D. (2012) Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. D. Biol. Crystallogr. 68, 352–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zheng H., Chordia M. D., Cooper D. R., Chruszcz M., Müller P., Sheldrick G. M., and Minor W. (2014) Validating metal binding sites in macromolecule structures using the CheckMyMetal web server. Nat. Protoc. 9, 156–170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., and Zwart P. H. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D. Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Huang G. Y., Gerlits O. O., Blakeley M. P., Sankaran B., Kovalevsky A. Y., and Kim C. (2014) Neutron diffraction reveals hydrogen bonds critical for cGMP-selective activation: insights for cGMP-dependent protein kinase agonist design. Biochemistry 53, 6725–6727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Schlossmann J., and Hofmann F. (2005) cGMP-dependent protein kinases in drug discovery. Drug Discov. Today 10, 627–634 [DOI] [PubMed] [Google Scholar]
- 31. Corbin J. D., Ogreid D., Miller J. P., Suva R. H., Jastorff B., and Døskeland S. O. (1986) Studies of cGMP analog specificity and function of the two intrasubunit binding sites of cGMP-dependent protein kinase. J. Biol. Chem. 261, 1208–1214 [PubMed] [Google Scholar]
- 32. Vaandrager A. B., Edixhoven M., Bot A. G. M., Kroos M. A., Jarchau T., Lohmann S., Genieser H.-G., and de Jonge H. R. (1997) Endogenous type II cGMP-dependent protein kinase exists as a dimer in membranes and can be functionally distinguished from the type I isoforms. J. Biol. Chem. 272, 11816–11823 [DOI] [PubMed] [Google Scholar]
- 33. Kaupp U. B., and Seifert R. (2002) Cyclic nucleotide-gated ion channels. Physiol. Rev. 82, 769–824 [DOI] [PubMed] [Google Scholar]
- 34. Thunemann M., Fomin N., Krawutschke C., Russwurm M., and Feil R. (2013) Visualization of cGMP with cGi Biosensors. in Guanylate Cyclase and Cyclic GMP (Krieg T., and Lukowski R. eds), pp, 89–120, Humana Press, New York: [DOI] [PubMed] [Google Scholar]
- 35. Poppe H., Rybalkin S. D., Rehmann H., Hinds T. R., Tang X.-B., Christensen A. E., Schwede F., Genieser H.-G., Bos J. L., Doskeland S. O., Beavo J. A., and Butt E. (2008) Cyclic nucleotide analogs as probes of signaling pathways. Nat. Methods 5, 277–278 [DOI] [PubMed] [Google Scholar]
- 36. Russwurm M., Mullershausen F., Friebe A., Jäger R., Russwurm C., and Koesling D. (2007) Design of fluorescence resonance energy transfer (FRET)-based cGMP indicators: a systematic approach. Biochem. J. 407, 69–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sprenger J. U., and Nikolaev V. O. (2013) Biophysical techniques for detection of cAMP and cGMP in living cells. Int. J. Mol. Sci. 14, 8025–8046 [DOI] [PMC free article] [PubMed] [Google Scholar]