

Abstract
Binding to mucin is the initial step for enteropathogens to establish pathogenesis. An open reading frame, gbpA, of Vibrio vulnificus was identified and characterized in this study. Compared with wild type, the gbpA mutant was impaired in binding to mucin-agar and the mucin-secreting HT29-methotrexate cells, and the impaired mucin binding was restored by the purified GbpA provided exogenously. The gbpA mutant had attenuated virulence and ability of intestinal colonization in a mouse model, indicating that GbpA is a mucin-binding protein and essential for pathogenesis of V. vulnificus. The gbpA transcription was growth phase-dependent, reaching a maximum during the exponential phase. The Fe-S cluster regulator (IscR) and the cyclic AMP receptor protein (CRP) coactivated, whereas SmcR, a LuxR homologue, repressed gbpA. The cellular levels of IscR, CRP, and SmcR were not significantly affected by one another, indicating that the regulator proteins function cooperatively to regulate gbpA rather than sequentially in a regulatory cascade. The regulatory proteins directly bind upstream of the gbpA promoter PgbpA. DNase I protection assays, together with the deletion analyses of PgbpA, demonstrated that IscR binds to two specific sequences centered at −164.5 and −106, and CRP and SmcR bind specifically to the sequences centered at −68 and −45, respectively. Furthermore, gbpA was induced by exposure to H2O2, and the induction appeared to be mediated by elevated intracellular levels of IscR. Consequently, the combined results indicated that IscR, CRP, and SmcR cooperate for precise regulation of gbpA during the V. vulnificus pathogenesis.
Keywords: bacterial adhesion, gene regulation, mucin, oxidative stress, quorum sensing, CRP, GbpA, IscR, LuxR homologue, Vibrio vulnificus
Introduction
Epithelial surfaces of the intestine are the most common portals by which enteropathogenic bacteria enter the deeper tissues of a mammalian host. The epithelial surfaces are covered by a mucous layer, which is produced by specialized cells found throughout the entire intestinal tract (1). The mucous layer is the first barrier that the enteropathogens encounter, and it prevents the pathogens from reaching and persisting on the intestinal epithelial surfaces and thereby is a major component of innate immunity (1). The mucous layer is composed of a variety of factors, but its characteristic physico-chemical properties are attributable to the presence of mucins, which are complex linear polymorphic glycoproteins (1, 2). Mucins are highly glycosylated large glycoproteins (with a molecular mass ranging from 5 × 103 to 4 × 106 Da), and up to 85% of their dry weight is carbohydrates (3). Although mucin contains extensively different types of carbohydrates, the residue, N-acetyl-d-glucosamine (GlcNAc), is one of the most abundant sugars in the carbohydrate side chains (4).
Adhesion to the mucosal surfaces followed by colonization on the mucosal tissue is considered to constitute the first stages of the infectious process (5). Accordingly, mutants of enteropathogenic bacteria that have difficulty in adhesion to the mucous layer were substantially defective in intestinal colonization, leading to attenuated virulence (6, 7). Although numerous factors (known as adhesins) are involved in the adhesion of enteropathogens, information on the adhesins with specificity toward mucin carbohydrates is still limited (8). The GlcNAc-binding protein A of Vibrio cholerae (VcGbpA) is a lectin-like mucus adhesin and is characterized at the molecular level (6, 9–11). VcGbpA is a common adhesin required for V. cholerae to adhere to chitinous and intestinal surfaces (6). VcGbpA plays an important role in the survival of V. cholerae by attachment to the surface of chitinous zooplankton in the aquatic ecosystem (6, 11). VcGbpA is a mucin-binding protein that binds to GlcNAc residues of mucin and contributes to intestinal colonization and virulence in a mouse model (6, 11). Structural analysis demonstrated that VcGbpA possesses a four-domain structure of which domains 1 and 4 interact with chitin and domain 1 is also crucial for mucin binding and intestinal colonization. In contrast, domains 2 and 3 anchor to the V. cholerae surfaces (11). It has been reported that VcGbpA expression was induced by mucin and negatively regulated by cyclic di-guanosine monophosphate at the post-transcription level and by quorum sensing at the post-translation level (9, 10, 12). However, neither the promoter(s) of the gbpA gene nor any trans-acting regulatory protein(s) required for the transcription of gbpA has been identified previously.
Vibrio vulnificus is a Gram-negative bacterium commonly associated with human disease caused by ingestion of undercooked oysters or contact of the organism with an open wound (13). Like many other enteropathogenic bacteria, V. vulnificus also expresses diverse adhesin molecules. The V. vulnificus adhesins include a flagellum, a type IV pilus, a lipoprotein, and OmpU that are crucial for adhesion to human epithelial cells in vitro and virulence in mice (14–18). However, there is still no information about the factors responsible for the initial adhesion of the pathogen to mucin. In this study, a V. vulnificus open reading frame (ORF) encoding a homologue of VcGbpA was identified. Construction of the gbpA mutant and evaluation of its phenotypes provided evidence that V. vulnificus GbpA (VvGbpA) is also a mucin-binding protein and plays a crucial role in the pathogenesis of the organism. Efforts to understand the regulatory mechanisms of the gbpA expression were initiated by determining the gbpA mRNA levels in cells of different growth phases. Because IscR (iron-sulfur (Fe-S) cluster regulator) (19), cyclic AMP receptor protein (CRP)2 (20), and SmcR (LuxR homologue) (21) were previously reported to affect the pathogenesis of V. vulnificus (22–26), influences of the global regulatory proteins on the expression of gbpA were also examined. Genetic and biochemical studies demonstrated that IscR and CRP coactivated and SmcR repressed gbpA in a growth phase-dependent manner. Furthermore, the three regulatory proteins regulate gbpA cooperatively rather than sequentially and exert their effects by directly binding to the gbpA promoter PgbpA. Deletion analyses of the upstream region of PgbpA and DNase I protection assays were performed to identify the binding sequences of IscR, CRP, and SmcR. Finally, the influence of hydrogen peroxide (H2O2) on the intracellular levels of IscR was examined to explain how IscR can mediate the induction of gbpA by oxidative stress.
Experimental Procedures
Strains, Plasmids, and Culture Conditions
The strains and plasmids used in this study are listed in Table 1. Unless otherwise noted, the V. vulnificus strains, wild type MO6-24/O and its mutants, were grown in Luria-Bertani (LB) medium supplemented with 2% (w/v) NaCl (LBS) at 30 °C, and their growth was monitored spectrophotometrically at 600 nm (A600). Anaerobic conditions were obtained by using an anaerobic chamber with an atmosphere of 90% N2, 5% CO2, and 5% H2 (Coy Laboratory Products, Grass Lake, MI). For anaerobic culture, the media were pre-incubated to remove dissolved O2 in the anaerobic chamber, which was verified by adding 0.00001% (w/v) resazurin salt (Sigma) to the media (27).
TABLE 1.
Plasmids and bacterial strains used in this study
| Strain or plasmid | Relevant characteristicsa | Ref. or source |
|---|---|---|
| Bacterial strains | ||
| V. vulnificus | ||
| MO6-24/O | Wild type, clinical isolate; virulent | 61 |
| MORR | MO6-24/O with spontaneous Rifr mutation, virulent | Laboratory collection |
| MORSR | MO6-24/O with spontaneous Rifr, Smr mutation, virulent | Laboratory collection |
| JK093 | MO6-24/O with ΔiscR | 26 |
| JK128 | MO6-24/O with iscR 3CA encoding the apo-locked IscR, IscR3CA | 32 |
| HS03 | MO6-24/O with smcR::nptI; Kmr | 22 |
| DI0201 | MO6-24/O with Δcrp | 41 |
| SO111 | MO6-24/O with ΔgbpA | This study |
| KK141 | MORSR ΔgbpA, Rifr, Smr | This study |
| KK142 | MO6-24/O with ΔiscRΔcrp | This study |
| E. coli | ||
| S17–1λpir | λ-pir lysogen; thi pro hsdR hsdM+ recA RP4–2 Tc::Mu-Km::Tn7;Tpr Smr; host for π-requiring plasmids; conjugal donor | 29 |
| BL21 (DE3) | F− ompT hsdSB (rB− mB−) gal dcm (DE3) | Laboratory collection |
| Plasmids | ||
| pGEM-T Easy | PCR product cloning vector; Apr | Promega |
| pKK1401 | pGEM-T Easy with 390-bp fragment of gbpA upstream region; Apr | This study |
| pDM4 | R6K γ ori sacB; suicide vector; oriT of RP4; Cmr | 28 |
| pSO1101 | pDM4 with ΔgbpA Cmr | This study |
| pBS0907 | pDM4 with Δcrp; Cmr | 31 |
| pJK1113 | pKS1101 with nptI; Apr, Kmr | 32 |
| pKK1402 | pJK1113 with gbpA; Apr, Kmr | This study |
| pKK1403 | pJK1113 with iscR; Apr, Kmr | This study |
| pKK1404 | pJK1113 with crp; Apr, Kmr | This study |
| pKK1405 | pJK1113 with smcR; Apr, Kmr | This study |
| pET22a(+) | His6 tag fusion expression vector; Apr | Novagen |
| pSO1201 | pET22a (+) with gbpA; Apr | This study |
| pJK0928 | pET28a (+) with iscR; Apr | 31 |
| pHK0201 | pRSET A with crp; Apr | 41 |
| pHS104 | pRSET C with SmcR; Apr | 22 |
| pBBR-lux | Broad host range vector with luxCDABE operon; Cmr | 44 |
| pKK1407 | pBBR_lux with 555-bp fragment of gbpA upstream region; Cmr | This study |
| pKK1408 | pBBR_lux with 451-bp fragment of gbpA upstream region; Cmr | This study |
| pKK1409 | pBBR_lux with 411-bp fragment of gbpA upstream region; Cmr | This study |
| pKK1410 | pBBR_lux with 341-bp fragment of gbpA upstream region; Cmr | This study |
a The following abbreviations are used: Rif, rifampicin-resistant; Smr, streptomycin-resistant; Kmr, kanamycin-resistant; Tpr, trimethoprim-resistant; Apr, ampicillin-resistant; Cmr, chloramphenicol-resistant.
Generation and Complementation of the gbpA and iscR crp Mutants
The gbpA gene was inactivated in vitro by deletion of the ORF of gbpA (318-bp of 1458-bp) using the PCR-mediated linker-scanning mutation method as described previously (27). Briefly, pairs of primers GBPA01F and -R (for amplification of the 5′-amplicon) or GBPA01-F and -R (for amplification of the 3′-amplicon) were designed and used (Table 2). The gbpA gene with a 318-bp deletion was amplified by PCR using the mixture of both amplicons as the template and GBPA01-F and GBPA02-R as primers. The resulting ΔgbpA was ligated into SpeI-SphI-digested pDM4 (28) to generate pSO1101 (Table 1). Escherichia coli S17-1 λ pir, tra strain (29) containing pSO1101 was used as a conjugal donor to V. vulnificus MO6-24/O and MORSR (MO6-24/O with rifampicin and streptomycin resistance, see Ref. 30) to generate the gbpA mutant SO111 and KK141, respectively (Table 1). Similarly, pBS0907, which was constructed previously to carry a mutant allele of V. vulnificus crp on pDM4 (Table 1) (31), was used to generate the iscR crp double mutant of V. vulnificus. E. coli S17-1 λ pir, tra containing pBS0907 was used as a conjugal donor in conjugation with the iscR mutant JK093 as a recipient (26). The resulting iscR crp double mutant was named KK142 (Table 1). The conjugation and isolation of the transconjugants were conducted using the method described previously (31).
TABLE 2.
Oligonucleotides used in this study
| Name | Oligonucleotide Sequence (5′ → 3′)a,b | Use |
|---|---|---|
| For mutant construction | ||
| GBPA01-F | GAGATGCACATCAGCAACGCG | Deletion of the gbpA ORF |
| GBPA01-R | AAAGGATCCAGCGAACTTACACAGTGT | |
| GBPA02-F | GCTGGATCCTTTGTCTTCCCAGATG | Deletion of the gbpA ORF |
| GBPA02-R | CGCAACAACGGAATCAAACGC | |
| For mutant complementation | ||
| GBPA03-F | GGATCCGCCAAATAAAGTCAG | Amplification of the gbpA ORF |
| GBPA03-R | GGATCCTTACAGTTTGTCCCAC | |
| ISCR01-F | ATCCATGGCTATGAAACTGACATCTAAAGG | Amplification of the iscR ORF |
| ISCR01-R | ATTCTAGATTAAGAGCGGAAATTTACACCG | |
| CRP01-F | GAGATACCATGGTTCTAGGTAAACCTCA | Amplification of the crp ORF |
| CRP01-R | GTTAATTCTAGATTAACGAGTACCGTAAACAAC | |
| SMCR01-F | ATCCATGGACTCAATCGCAAAGAGAC | Amplification of the smcR ORF |
| SMCR02-R | ATTCTAGATTATTCGTGCTCGCGTTTATA | |
| For GbpA overexpression | ||
| GBPA04-F | CCATGGCTAAAAAACAACCGCAAAAAACC | Amplification of the gbpA ORF |
| GBPA04-R | CTCGAGCAGTTTGTCCCACGCCATT | |
| For promoter deletion analysis | ||
| GBPA003 | GAGCTCTAAGTGCTCAATGACATAGTAAAG | Deletion of the gbpA regulatory region |
| GBPA004 | GAGCTCTCACACTTTTTCGAGAAATTA | |
| GBPA005 | GAGCTC ACATCTATAAATAACGCTTCTAAAT | |
| GBPA006 | GAGCTCTTATGCCTGACATCACAC | |
| GBPA007 | ACTAGTCACCATTTTCCACTGCAG | |
| For primer extension analysis, EMSA, and DNase I protection assay | ||
| GBPA05-F | ATTGCCATAGCTGGTGGTTTTCA | Amplification of the gbpA upstream region |
| GBPA05-R | CCCCGCTATCTTGGGTATGGTAAAAA | Amplification of the gbpA upstream region, Extension of the gbpA transcript |
| For qRT-PCR | ||
| GBPA_qRT-F | TGAAAGCCTGGGGTGAAGCA | Quantification of the gbpA expression |
| GBPA_qRT-R | ATCGCGTAGCGTTGAGAGCG | |
a The oligonucleotides were designed using the V. vulnificus MO6-24/O genomic sequence (GenBankTM accession number CP002469 and CP002470, www.ncbi.nlm.nih.gov).
b Regions of oligonucleotides not complementary to the corresponding genes are underlined.
To complement the gbpA, iscR, crp, and smcR (constructed previously (22)) mutations, each ORF of gbpA, iscR, crp, and smcR was amplified by PCR using a pair of specific primers as listed in Table 2. The amplified gbpA, iscR, crp, and smcR ORFs were cloned into pJK1113 under an arabinose-inducible promoter PBAD (32) to create pKK1402, pKK1403, pKK1404, and pKK1405, respectively (Table 1). The plasmids were transferred into the appropriate mutants by conjugation as described above. For complementation tests, when the cultures reached an A600 of 0.3, arabinose was added to a final concentration of 0.1 mm to induce the expression of the recombinant genes on the plasmids.
Mucin Binding Assay
Pig gastric mucin powder (Sigma) was sterilized by mixing with 95% (v/v) ethanol for 1 h, dried at 70 °C for 24 h (33), and then added to the 1.5% agar (w/v) solution, which was autoclaved and cooled down to 60 °C to the final concentration of 3% (w/v). The mucin-agar solution (2 ml) was solidified in each well of 12-well culture dishes (Nunc, Roskilde, Denmark). On the mucin-agar, V. vulnificus cultures (100 μl, ∼107 colony-forming units (cfu)) were added to each well, and various amounts of purified GbpA were exogenously provided to the well when required. After incubation for 1 h at 30 °C, the nonadherent bacteria were removed by washing with 1 ml of phosphate-buffered saline (PBS) twice, and the adherent bacterial cells were recovered by treating with 200 μl of 0.1% Triton X-100 (Sigma) solution for 20 min and enumerated as cfu per well.
Development of the Mucin-secreting Cells and Adhesion Assay
The human colonic HT29 cells (ATCC®HTB-38TM) (ATCC, Manassas, VA) in McCoy's 5A media (Gibco-BRL, Gaithersburg, MD) containing 1% (v/v) fetal bovine serum (MCF) were developed into mucin-secreting cells, named HT29-methotrexate (MTX), as described previously (34). The HT29-MTX cells were fixed with paraformaldehyde and treated with 4′, 6-diamidino-2-phenylindole (DAPI). Mucin secretion of the HT29-MTX cells was detected with the anti-mucin 5AC primary antibody (Merck Millipore, Darmstadt, Germany), labeled with the fluorescein isothiocyanate (FITC)-conjugated secondary antibody (Abcam, Cambridge, UK), and visualized using a confocal laser scanning microscope (LSM710, Zeiss, Jena, Germany) (35, 36).
The 12-well culture dishes (Nunc) were seeded with the HT29-MTX cells (∼1 × 107 cells per well), infected with the V. vulnificus strains at a multiplicity of infection of 10 for 30 min. The culture dishes were washed two times with PBS to remove nonadherent bacteria and treated with 0.1% Triton X-100 for 20 min to recover adherent bacteria. The recovered bacterial cells were enumerated as cfu per well.
Mouse Lethality and Competition Assay
Mouse lethality of the wild type and gbpA mutant was compared as described previously (26). Groups of (n = 10) 7-week-old ICR female mice (specific pathogen-free; Seoul National University) were starved without food and water for 12 h until infection. Then the mice, without iron-dextran pretreatment, were intragastrically administered with 100 μl of the inoculum, representing ∼109 cells of either the wild type or the gbpA mutant. Mouse survivals were recorded for 24 h.
Previous mouse colonization assays demonstrated that V. vulnificus initially and mostly colonizes in the small intestine and disseminates to other organs (37, 38). Therefore, colonization of each strain in the mouse small intestine was determined by competition assays as described previously (30). Briefly, four ICR female mice (7 weeks old) were infected as described above for mouse mortality, except that 100 μl of the inoculum, prepared by mixing MORR (MO6-24/O with rifampicin resistance (30)) and KK141 at a 1:1 ratio, representing ∼106 cfu of each strain, was given intragastrically to the mice. The mice were sacrificed at 1–24 h postinfection, and their intestines were collected, washed, and homogenized. Equal amounts of the homogenates were spread on LBS agar containing either rifampicin (100 μg/ml) alone to enumerate the sum of the wild type and gbpA mutant cells or rifampicin and streptomycin (100 μg/ml) to specifically count the gbpA mutant cells. The ratio of cfu recovered from the intestines to the number of cfu inoculated is defined as a colonization index (39). All manipulations of mice were approved by the Animal Care and Use Committee of Seoul National University, and mice were humanely euthanized at the end point analysis.
RNA Purification and Transcript Analysis
Total RNA from the V. vulnificus strains grown aerobically to various levels of A600 were isolated using an RNeasy® mini kit (Qiagen, Valencia, CA). When necessary, the strains grown anaerobically to an A600 of 0.5 were exposed to various concentrations of H2O2 for 10 min and harvested to isolate total RNA. For the primer extension analysis, a 26-base primer GBPA05-R (Table 2) complementary to the regulatory region of gbpA was end-labeled with [γ-32P]ATP and added to the RNA. The primer was then extended with SuperScript II RNase H− reverse transcriptase (Invitrogen). The cDNA product was purified and resolved on a sequencing gel alongside ladders generated from pKK1401 with the same primer. The plasmid pKK1401 was constructed by cloning the 390-bp gbpA upstream region extending from −301 to +88 and amplified by PCR using a pair of primers GBPA05-F and -R (Table 2) into pGEM-T Easy (Promega, Madison, WI). The primer extension product was visualized using a phosphorimager analyzer (BAS1500; Fuji Photo Film Co., Ltd., Tokyo, Japan).
For qRT-PCR, the concentrations of total RNA from the strains were measured by using a NanoVue Plus spectrophotometer (GE Healthcare). cDNA was synthesized from 1 μg of the total RNA by using the iScriptTM cDNA synthesis kit (Bio-Rad), and real time PCR amplification of the cDNA was performed by using the Chromo 4 real time PCR detection system (Bio-Rad) with a pair of specific primers (Table 2), as described previously (40). Relative expression levels of the gbpA mRNA in the same amounts of total RNA were calculated by using the 16S rRNA expression level as the internal reference for normalization.
Protein Purification and Western Blot Analysis
The ORF of gbpA was amplified by PCR using a pair of primer GBPA04-F and -R (Table 2) and cloned into a His6 tag expression vector, pET-22a(+) (Novagen, Madison, WI), to result in pSO1201 (Table 1). The His-tagged GbpA protein was expressed in E. coli BL21 (DE3) and purified by affinity chromatography according to the manufacturer's procedure (Qiagen). In a similar way, pJK0928, pHK0201, and pHS104, which were constructed previously (Table 1) (22, 26, 41), were used to overexpress and purify the His-tagged IscR, CRP, and SmcR, respectively. The purified His-tagged proteins were used to raise rabbit polyclonal antibodies against GbpA, IscR, CRP, and SmcR of V. vulnificus, respectively (AB Frontier, Seoul, South Korea). For Western blot analyses, total proteins were isolated from the strains grown aerobically to an A600 of 0.5 or anaerobically to an A600 of 0.5 and then exposed to various concentrations of H2O2 for 10 min. The concentrations of the total proteins were determined by using Bradford method (42). The same amounts of the total proteins (10 μg) were resolved on SDS-PAGE and immunoblotted as described previously (27).
Electrophoretic Mobility Shift Assay (EMSA)
The 390-bp gbpA upstream region extending from −301 to +88 was amplified by PCR using [γ-32P]ATP-labeled GBPA05-F and unlabeled GBPA05-R as primers (Table 2). The labeled 390-bp DNA (5 nm) probe was incubated with various concentrations of purified IscR for 30 min at 30 °C in a 20-μl reaction mixture containing 1× binding buffer (43) and 0.1 μg of poly(dI-dC) (Sigma). The protein-DNA binding reactions with purified CRP or SmcR were the same as those with IscR, except that the CRP or SmcR-binding buffer was used as a 1× binding buffer (41, 22). Electrophoretic analyses of the DNA-protein complexes were performed, as described previously (32), and visualized as described above for the transcript analysis.
Construction of a Set of gbpA-luxCDABE Transcriptional Fusions
The primer GBPA007 (Table 2), including an SpeI restriction site followed by bases corresponding to the 5′ end of the gbpA coding region, was used in conjunction with one of the following primers to amplify the DNA upstream of gbpA: GBPA003 (for pKK1407), GBPA004 (for pKK1408), GBPA005 (for pKK1409), or GBPA006 (for pKK1410) (Table 2). The primers were designed to amplify the gbpA regulatory region extending up to −220, −106, −76, and −6 bp, respectively. A SacI restriction site was added to these primers to facilitate cloning of the PCR products. The DNA fragments were inserted into the SpeI-SacI-digested pBBR-lux (44) carrying promoterless luxCDABE genes, thereby creating four gbpA-lux reporter constructs. All constructions were confirmed by DNA sequencing. The gbpA-lux reporters were then transferred into the V. vulnificus strains by conjugation. The cellular luminescence of the cultures grown to an A600 of 0.5 were measured with a luminometer (Tecan Infinite M200 reader, Männedorf, Switzerland) and expressed in arbitrary relative light units (RLU) as described previously (22).
DNase I Protection Assay
The same labeled 390-bp DNA probe developed for EMSA was used for DNase I protection assays. The binding of IscR, CRP, or SmcR to the labeled DNA (25 nm) was performed as described above for EMSA, and DNase I digestion of the DNA-protein complexes followed the procedures described previously (31). After precipitation with ethanol, the digested DNA products were resolved on a sequencing gel alongside sequencing ladders of pKK1401 generated using GBPA05-F (Table 2) as the primer. The gels were visualized as described above for the transcript analysis.
Data Analyses
Averages and standard deviations (S.D.) were calculated from at least three independent experiments. Mouse mortality was evaluated using the log rank test program. All other data were analyzed by Student's t tests with the SAS program (SAS software, SAS Institute Inc.). Significance of differences between experimental groups was accepted at a p value of <0.05.
Results
Identification and Sequence Analysis of GbpA
A search of the V. vulnificus MO6-24/O genome sequence database (GenBankTM CP002469 and CP002470) (45) for homology to the amino acid sequences deduced from VcGbpA singled out a protein, hereafter named VvGbpA. The amino acid sequence analysis predicted that pre-VvGbpA protein contains an N-terminal signal peptide for the type II secretion system, suggesting that VvGPA is a secreted protein (data not shown) (46). The deduced mature VvGbpA is composed of 485 amino acids with a theoretical mass of 52.83 kDa and a pI of 4.75. The amino acid sequence of VvGbpA was 80% identical to that of VcGbpA (data not shown) and exhibited a four domain modular structure consisting of two chitin binding domains and two bacterial surface binding domains as observed in VcGbpA (11). The predicted profile of the hydrophobicity was significantly similar to that of VcGbpA, indicating that VvGbpA is a soluble protein (data not shown) (11). All of this information suggested that VvGbpA is a secreted but cell surface-associated protein as is VcGbpA.
GbpA Is Essential for Mucin Binding and Virulence of V. vulnificus
To examine the function of VvGbpA, the mucin binding ability of wild type and the gbpA mutant was examined. The number of the gbpA mutants that adhered to the mucin-agar in a well of 12-well culture dishes was significantly lower than that of the parental wild type (Fig. 1A). This indicated that the gbpA mutant was defective in mucin binding. In addition, the extracellular provision of purified VvGbpA was able to rescue the defect of the gbpA mutant in mucin binding ability in a dose-dependent manner. The mucin binding ability of the gbpA mutant incubated in the presence of 5 μm VvGbpA was comparable with that of the parental wild type, in terms of the bacterial numbers adherent to the mucin-agar (Fig. 1A).
FIGURE 1.

Effect of GbpA on mucin binding and host cell adhesion of V. vulnificus. Upper panel, mucin binding activities of the strains. A, strains (∼107 cfu) were added to each well of 12-well culture dishes containing the mucin-agar and various amounts of GbpA provided exogenously as indicated. After 1 h of incubation, the adherent bacterial cells were enumerated as cfu per well. WT, wild type; gbpA, gbpA mutant. B, mucin-secreting HT29-MTX cells (∼107 cells) seeded in each well of 12-well culture dishes were infected at a multiplicity of infection of 10 with the strains as indicated. After a 30-min incubation, the adherent bacterial cells were enumerated as cfu per well. Error bars represent the S.D. *, p < 0.05, and **, p < 0.005 relative to the wild type. WT (pJK1113, empty vector), wild type; gbpA (pJK1113), gbpA mutant; gbpA (pKK1402), complemented strain. Lower panel, development of the mucin-secreting HT29-MTX cells. C, bright field image of HT29-MTX cells. D, nucleus of HT29-MTX cells was stained blue with DAPI. E, mucin of HT29-MTX cells was stained green with the anti-MUC5AC primary antibody and then labeled with FITC-conjugated secondary antibody. F, merged image of C–E. Images are visualized using a confocal laser scanning microscope. Scale bar, 40 μm.
To further understand the role of VvGbpA in binding to mucin, the HT29-MTX cells, monolayers of which mimic human intestinal epithelial cells that produce and secrete mucins (34), were developed. Mucin secretion of the HT29-MTX cells was confirmed using a confocal laser scanning microscope (Fig. 1, C–F). The wild type and gbpA mutant were incubated with the HT29-MTX cells, and the bacterial adherents to the cells were enumerated (Fig. 1B). The results revealed that adhesion of the gbpA mutant to the HT29-MTX cells was about 2-fold lower than that of the wild type. The impaired adhesion of the gbpA mutant was restored to the wild type level by complementation with a functional gbpA gene (pKK1402). These results indicated that VvGbpA is a mucin-binding protein and plays an important role in the adhesion to the mucin-secreting human epithelial cells.
To experimentally examine the role of VvGbpA in pathogenesis, mouse mortality and colonization activity of the gbpA mutant were evaluated. As shown in Fig. 2A, the survival of mice inoculated intragastrically with the gbpA mutant was consistently and significantly prolonged (p = 0.0174, log rank test) compared with that of mice inoculated with the parental wild type. Therefore, for the mouse model infected intragastrically, the gbpA mutant appeared to be significantly less virulent than its parental wild type. Mice were also coinoculated intragastrically with MORR (wild type) and KK141 (gbpA mutant), and both strains colonized on the small intestine were recovered and enumerated (Fig. 2B). The colonization index of the KK141 ranged from 10−3 to 10−2 and was consistently and significantly (about 100-fold) lower than that of MORR, demonstrating that the MORR clearly outcompeted the KK141 in the small intestine. These results indicated that VvGbpA is a virulence factor essential for the intestinal colonization of V. vulnificus. Taken together, it is apparent that VvGbpA is a mucin-binding protein contributing to the pathogenesis of V. vulnificus.
FIGURE 2.

Mouse lethality and colonization activity of the V. vulnificus strains. A, 7-week-old specific pathogen-free female ICR mice were intragastrically infected with the wild type (WT) or the gbpA mutant (gbpA) (Table 1) at doses of ∼109 cfu as indicated. Mouse survival was monitored for 24 h. B, four mice were intragastrically infected with an inoculum prepared by mixing equal numbers of MORR and KK141 (Table 1), and then the bacterial cells colonized on the small intestine were enumerated as cfu at the indicated time intervals. Each circle corresponds to the ratio of cfu recovered from the intestines to the cfu inoculated (the colonization index) for an individual mouse. The median values are displayed as a solid line (MORR) or dashed line (KK141) on the graph. WT, wild type; gbpA, gbpA mutant; MORR, wild type with rifampicin resistance; KK141, gbpA mutant with rifampicin and streptomycin resistance.
Expression of gbpA Is Growth Phase-dependent and Regulated by IscR, CRP, and SmcR
To examine whether the production of VvGbpA is influenced by growth phase, levels of the gbpA mRNA of the wild type culture were analyzed at various growth stages (Fig. 3, A and B). The gbpA transcript reached maximum levels in the exponential phase and then decreased in the stationary phase, indicating that expression of gbpA is growth phase-dependent. To extend our understanding on the regulation of gbpA, the levels of the gbpA transcript in the wild type and mutants that lack transcription factors IscR, CRP, and SmcR (Table 1) were compared. The level of the gbpA transcript decreased in the iscR mutant and crp mutant (Fig. 3C). The result indicated that both IscR and CRP act as positive regulators for the gbpA expression. In contrast, the gbpA expression of the smcR mutant was greater than that of the wild type, indicating that SmcR negatively regulates gbpA (Fig. 3C). The cellular levels of VvGbpA determined by Western blot analyses also varied in the mutants (Fig. 3D). The magnitude of variation in the VvGbpA proteins did not significantly differ from that observed in the gbpA transcripts, indicating that the regulation of the gbpA expression occurs mostly at the transcription level. The levels of the gbpA transcript and VvGbpA protein that varied in the iscR, crp, and smcR mutant were restored to the levels comparable with those in the wild type by introducing pKK1403, pKK1404, and pKK1405 carrying recombinant iscR, crp, and smcR, respectively (Fig. 3, E and F). Taken together, these results suggested that IscR and CRP activate and SmcR represses the gbpA transcription.
FIGURE 3.

Effects of growth phases and global regulatory proteins on the gbpA expression. Upper panel, growth kinetics of V. vulnificus and growth phase-dependent expression of gbpA. A, growth of the wild type culture in LBS was monitored spectrophotometrically at 600 nm (A600), and total RNAs were isolated from the cells harvested at different growth phases (from left, at A600 of 0.5, 1.0, 1.5, 2.0, and 2.5) as indicated by arrows. B, gbpA mRNA levels were determined by qRT-PCR analyses, and the gbpA mRNA level in the cells grown to an A600 of 0.5 was set as 1. Error bars represent the S.D. *, p < 0.05, and **, p < 0.005 relative to the cells grown to an A600 of 0.5. Lower panel, expression of gbpA in V. vulnificus with different genetic backgrounds. Samples were harvested from the cultures of the wild type (WT) and isogenic mutants grown aerobically to an A600 of 0.5 and analyzed to determine the gbpA mRNA and GbpA protein levels. C and E, gbpA mRNA levels were determined by qRT-PCR analyses, and the gbpA mRNA level in the wild type was set as 1. **, p < 0.005 relative to the wild type. D and F, protein samples were resolved by SDS-PAGE, and GbpA was detected by Western blotting using the rabbit anti-V. vulnificus GbpA serum. (pJK1113), wild type; iscR (pJK1113), iscR mutant; smcR (pJK1113), smcR mutant; crp (pJK1113), crp mutant; iscR (pKK1403), crp (pKK1404), and smcR (pKK1405), complemented strains.
IscR and CRP Coactivate gbpA Additively
To further examine the roles of IscR and CRP in the gbpA expression, the iscR crp double mutant KK142 was constructed, and gbpA expression was determined. Inactivation of both crp and iscR resulted in significant reduction of the gbpA expression, and the residual gbpA mRNA level in the iscR crp double mutant corresponded to only one-tenth of that in the wild type (Fig. 4). The presence of either IscR (crp mutant) or CRP (iscR mutant) alone increased the gbpA expression, but their gbpA transcript levels were much lower than that obtained by IscR and CRP together (wild type), indicating that IscR and CRP coactivate the gbpA expression additively (Fig. 4). To determine whether an increased amount of IscR would compensate for a lack of CRP in the activation of gbpA, the iscR expression plasmid pKK1403 was introduced into the iscR crp double mutant KK142. When iscR was induced by arabinose, the cellular level of IscR in KK142 (pKK1403) was higher than that in the crp single mutant (Fig. 4). However, the level of the gbpA transcript in KK142 (pKK1403) was only about 80% that in the wild type (Fig. 4), indicating that IscR, even when overproduced, is unable to activate gbpA to the wild type level in the absence of CRP. Similarly, overproduced CRP was unable to completely compensate for the lack of IscR in the activation of gbpA (Fig. 4). The results indicated that both IscR and CRP are required simultaneously to activate gbpA to the wild type level.
FIGURE 4.

IscR and CRP coactivate gbpA additively. Samples were harvested from the cultures of the wild type (WT) and isogenic mutants grown aerobically to an A600 of 0.5 and analyzed to determine the gbpA mRNA levels and IscR and CRP protein levels. The gbpA mRNA levels were determined by qRT-PCR analyses, and the gbpA mRNA level in the wild type was set as 1. Error bars represent the S.D. The cellular levels of IscR and CRP were determined by Western blot analyses using the rabbit anti-V. vulnificus IscR and anti-V. vulnificus CRP sera, respectively. WT, wild type; iscR, iscR mutant; crp, crp mutant; iscR crp, iscR crp double mutant.
IscR, CRP, and SmcR Function Cooperatively Rather than Sequentially to Regulate gbpA
Different mechanisms are possible for this coactivation of gbpA by IscR and CRP. For example, multiple activators function sequentially in a regulatory cascade, where one activator influences the accumulation of another regulator(s), which in turn is directly responsible for the activation of gbpA. To test this possibility, the cellular levels of IscR, CRP, SmcR, and VvGbpA were determined from the same amount of total protein isolated from the wild type and its isogenic mutants (Fig. 5).
FIGURE 5.
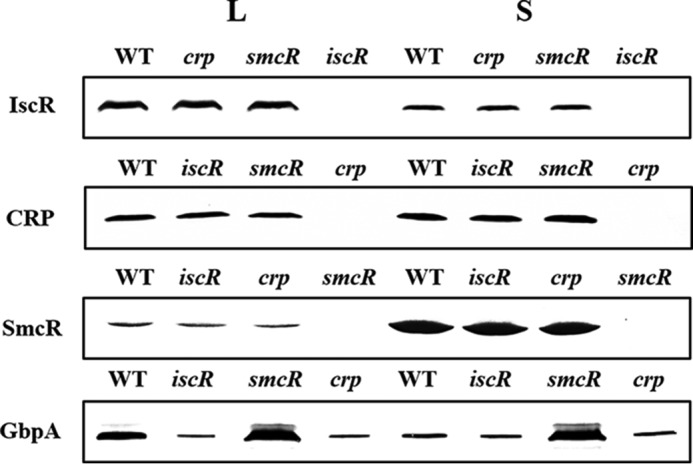
Cellular levels of IscR, CRP, and SmcR are unaffected by one another. The wild type and isogenic mutants were grown aerobically to an A600 of 0.5 (log phase, L) and 2.0 (stationary phase, S). The cells were then examined for the presence of IscR, CRP, SmcR, and GbpA proteins by Western blot analyses using the rabbit anti-V. vulnificus IscR, anti-V. vulnificus CRP, and anti-V. vulnificus SmcR, anti-V. vulnificus GbpA sera, respectively. WT, wild type; iscR, iscR mutant; crp, crp mutant; smcR, smcR mutant.
Western blot analysis revealed that neither activator affected the cellular level of the other, i.e. compared with the wild type the iscR mutant strain did not exhibit any significant changes in the cellular level of CRP and vice versa (Fig. 5). From these results, it is unlikely that IscR (or CRP) indirectly activates gbpA by increasing the cellular level of CRP (or IscR), which directly activates gbpA. Similarly, neither IscR nor CRP influences the cellular levels of SmcR, indicating that the activation of gbpA by IscR and CRP is not the result of decreasing cellular levels of SmcR, which directly represses gbpA. Consequently, it appears that IscR, CRP, and SmcR function cooperatively to regulate gbpA rather than sequentially in a regulatory cascade. It is noteworthy that the level of IscR, which activates gbpA, was higher in the log phase cells than in the stationary phase cells. Although levels of CRP did not vary significantly in cells of different growth phases, SmcR, which represses gbpA, accumulated more in cells of stationary phase (Fig. 5). Consistent with this, the level of VvGbpA was higher in the log phase cells than the stationary phase cells of the wild type (Fig. 5). Therefore, it was postulated that the growth phase-dependent expression of gbpA (Fig. 3, A and B) is attributed to this variation in the levels of IscR and SmcR in cells of different growth phase.
Mapping the Regulatory Region of gbpA
To map the promoter of gbpA, a transcription start site of gbpA was determined by a primer extension analysis. A single reverse transcript was produced from primer extension of RNA isolated from the wild type grown aerobically to an A600 of 0.5 (Fig. 6A). Several attempts to identify other transcription start sites using different sets of primers were not successful (data not shown). The 5′ end of the gbpA transcript was located 235-bp upstream of the translational initiation codon of gbpA and was subsequently designated +1 (Fig. 6B). The putative promoter constituting this transcription start site was named PgbpA, and the sequences for −10 and −35 regions of PgbpA were assigned on the basis of similarity to consensus sequences of the E. coli σ70 promoter (Fig. 6B).
FIGURE 6.

Transcription start site and sequences of the gbpA regulatory region. A, transcription start site of gbpA was determined by primer extension of the RNA isolated from the wild type grown aerobically to an A600 of 0.5. Lanes C, T, A, and G represent the nucleotide sequencing ladders of pKK1401. The asterisk indicates the transcription start site of gbpA. B, transcription start site of gbpA is indicated by a bent arrow, and the positions of the putative −10 and −35 regions are underlined. The sequences for binding of IscR (ISCRB1 and ISCRB2, white boxes), CRP (CRPB, shaded boxes), and SmcR (SMCRB, gray boxes) were determined later in this study (Fig. 10). The nucleotides showing enhanced cleavage are indicated by black boxes. The consensus sequences for binding of IscR (type 2), CRP, and SmcR are, respectively, indicated above the V. vulnificus DNA sequence. R, A or G; Y, C or T; W, A or T; x, any nucleotide.
The pKK reporters carrying the upstream regulatory region of PgbpA, which was deleted up to different 5′ ends and fused transcriptionally to luxCDABE, were constructed (Fig. 7A). The reporters were transferred into the wild type and isogenic mutants, and culture luminescence was used to quantify the PgbpA activity (Fig. 7B). The luminescence produced by pKK1407 carrying PgbpA deleted up to −220 was ∼6.0 × 103 RLU in the wild type but significantly reduced in the iscR and crp mutants, supporting our previous observation that IscR and CRP activate PgbpA. The RLU of the smcR mutant containing pKK1407 increased, confirming the SmcR repression of PgbpA (Fig. 7B). Deletion up to −106 significantly decreased the PgbpA activity as determined based on the reduced luminescence of the strains containing pKK1408. Interestingly, the RLU of pKK1408 in the iscR mutant was indistinguishable from that in the wild type, indicating the absence of the region(s) necessary for IscR to activate PgbpA in pKK1408. Similarly, the comparable RLU produced by pKK1409 in the crp mutant and wild type indicated that the region upstream from −76 is probably required for CRP activation of PgbpA. In contrast, the smcR mutant containing pKK1409 (and pKK1408) produced still greater luminescence compared with the wild type, indicating that the region downstream from −76 is essential for SmcR repression of PgbpA. The undetectable luminescence produced by pKK1410 indicates that deletion up to −6 completely impaired the PgbpA activity. Taken together, the results suggested that the regulatory region extending from −220 to −6 consecutively harbors the cis-elements necessary for IscR, CRP, and SmcR regulation of PgbpA.
FIGURE 7.

Deletion analysis of the PgbpA regulatory region. A, construction of gbpA-lux fusion pKK reporters. PCR fragments carrying the gbpA regulatory region with 5′ end deletions were subcloned into pBBR-lux (44) to create each pKK reporter. Solid lines, the upstream region of gbpA; black blocks, the gbpA coding region; open blocks, luxCDABE. The wild type gbpA regulatory region is shown on top with the proposed −10 and −35 regions, and the binding sites for IscR (ISCRB1 and ISCRB2, white boxes), CRP (CRPB, shaded box), and SmcR (SMCRB, gray box) were determined later in this study (Fig. 10). B, cellular luminescence determined from the wild type (black bars), iscR mutant (dark gray bars), crp mutant (gray bars), and smcR mutant (open bars) containing each pKK reporter as indicated. Cultures grown aerobically to an A600 of 0.5 were used to measure the cellular luminescence. Error bars represent the S.D. WT, wild type; iscR, iscR mutant; crp, crp mutant; smcR, smcR mutant.
IscR, CRP, and SmcR Regulate gbpA by Directly Binding to PgbpA
There are still several possible ways for IscR, CRP, and SmcR to affect PgbpA activity. One is by binding directly to the PgbpA regulatory region to regulate the promoter, whereas another is by modulating the cellular level of unidentified trans-acting factor(s), which in turn binds directly to the PgbpA regulatory region. To distinguish these two possibilities, the 390-bp labeled DNA probe encompassing the PgbpA regulatory region (from −301 to +88) was incubated with increasing amounts of IscR and then subjected to electrophoresis. Because IscR was isolated, purified, and used under aerobic conditions, most of the purified IscR would be in the Fe-S clusterless apo-form (32, 47). The addition of IscR resulted in two retarded bands in a concentration-dependent manner, indicating that at least two binding sites for IscR are present in the PgbpA regulatory region (Fig. 8A). The binding of IscR was also specific, because assays were performed in the presence of 0.1 μg of poly(dI-dC) as a nonspecific competitor. In a second EMSA, the same but unlabeled 390-bp DNA fragment was used as a self-competitor to confirm the specific binding of IscR. The unlabeled 390-bp DNA competed for the binding of IscR in a dose-dependent manner (Fig. 8A), confirming that IscR binds specifically to the PgbpA regulatory region. In similar DNA binding assays, CRP and SmcR each displayed specific binding to the PgbpA regulatory region (Fig. 8, B and C). These results suggested that IscR, CRP, and SmcR regulate gbpA by specifically binding to PgbpA.
FIGURE 8.
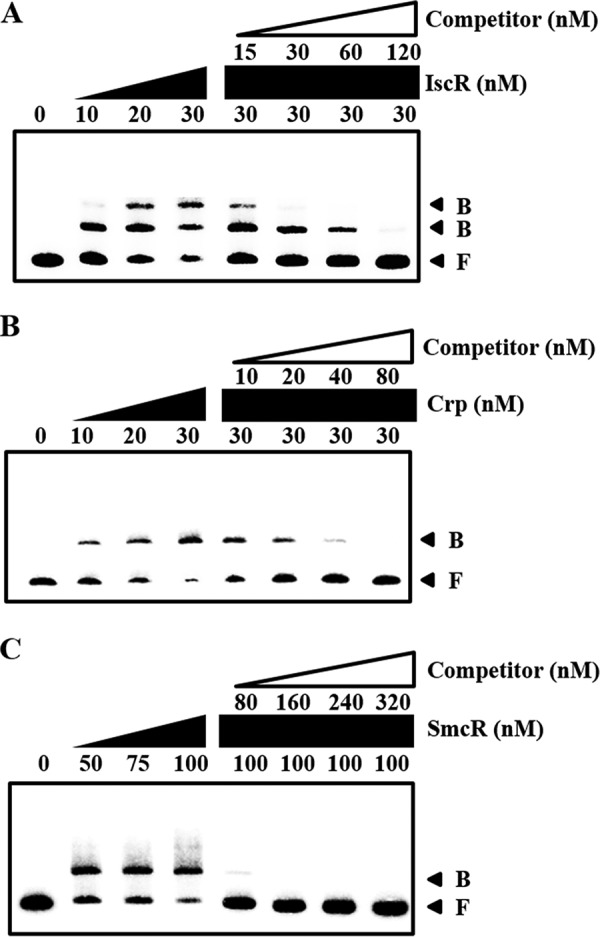
Specific bindings of IscR, CRP, and SmcR to PgbpA. A 390-bp DNA fragment of the gbpA regulatory region was radioactively labeled and then used as a probe DNA. The radiolabeled probe DNA (5 nm) was mixed with increasing amounts IscR (A), CRP (B), and SmcR (C) as indicated. For competition analysis, the same but unlabeled 390-bp DNA fragment was used as a self-competitor DNA. Various amounts of the self-competitor DNA were added to a reaction mixture containing the 5 nm labeled DNA prior to the addition of 30 nm IscR (A), 30 nm CRP (B), and 100 nm SmcR (C). B, bound DNA; F, free DNA.
Identification of Binding Sites for IscR, CRP, and SmcR
To determine the precise location of the IscR-, CRP-, and SmcR-binding sites in the PgbpA regulatory region, DNase I protection assays were performed using the same 390-bp DNA probe used for DNA binding assays. As shown in Fig. 9A, two regions extending from −184 to −145 (ISCRB1, centered at −164.5) and −124 to −88 (ISCRB2, centered at −106) were clearly protected by IscR (Fig. 9A). Both sequences were equally protected by the same level of IscR, indicating that IscR bound to the two sites with a comparable affinity. The sequences of ISCRB1 and ISCRB2 revealed a 29-bp imperfect palindrome and scored about 83 and 79% identity to a consensus sequence of the type 2 IscR-binding site, respectively, to which the apo-form of IscR most likely binds in E. coli (Fig. 6B) (48). The CRP footprint extended from −82 to −54 (CRPB, centered at −68) (Fig. 9B), and the sequences of CRPB scored 86% identity to a consensus sequence for CRP binding (Fig. 6B) (49). The positioning of ISCRB and CRPB suggested that both IscR and CRP may act as class I activators interacting with the C-terminal domains of RNA polymerase α subunits (50). The sequences protected by SmcR extended from −58 to −32 (SMCRB, centered at −45) and revealed 73% identity to a consensus sequence for SmcR binding (51). In contrast to ISCRB and CRPB, SMCRB overlaps with the sequences of the −35 region of PgbpA (Fig. 6B), and thus the bound SmcR could prevent RNA polymerase binding, supporting the SmcR repression of PgbpA. Several nucleotides also showed enhanced cleavages, indicating that binding of the regulators altered the configuration of the DNA of PgbpA (Fig. 9, A–C). These results confirmed that IscR, CRP, and SmcR regulate gbpA by binding to specific sequences of PgbpA.
FIGURE 9.

Sequences for binding of IscR, CRP, and SmcR to PgbpA. A 390-bp DNA fragment of the gbpA regulatory region was radioactively labeled and then used as a DNA probe. The radiolabeled DNA probe (25 nm) was incubated with increasing amounts of IscR (A), CRP (B), and SmcR (C) as indicated. The regions protected by IscR, CRP, and SmcR are indicated by white boxes, shaded boxes, and gray boxes, respectively. The nucleotides showing enhanced cleavage are indicated by black boxes. Lanes C, T, A, and G represent the nucleotide sequencing ladders of pKK1401.
IscR Activates PgbpA by Sensing Reactive Oxygen Species
Recently, it has been discovered that IscR senses reactive oxygen species and activates the expression of numerous virulence genes (26, 32). This prompted us to examine the effect of oxidative stress on the gbpA expression by measuring the levels of gbpA transcript and VvGbpA protein in the strains grown anaerobically and exposed to a range of H2O2. The levels of the gbpA transcript and VvGbpA protein in the wild type were gradually elevated along with increasing concentrations of H2O2 (Fig. 10, A and B). In contrast, the H2O2-dependent increase of the gbpA expression was not evident in the iscR mutant (Fig. 10, A and B), indicating that the activation of PgbpA in response to oxidative stress is mediated by IscR. Because the cellular level of IscR also increased by exposure to H2O2 (Fig. 10B), the increased activity of PgbpA by oxidative stress was possibly attributed to the increased level of IscR. The levels of CRP and SmcR were not significantly changed in the wild type and iscR mutant exposed to H2O2 (Fig. 10B).
FIGURE 10.

Effects of oxidative stress and apo-IscR on the activity of PgbpA. Total RNAs and proteins were isolated either from the cultures grown anaerobically to an A600 of 0.5 and then exposed to various concentrations of H2O2 for 10 min as indicated (A and B) or from the cultures grown aerobically to an A600 of 0.5 (C and D). A and C, gbpA mRNA levels were determined by qRT-PCR analyses, and the gbpA mRNA level in the wild type unexposed to H2O2 (A) or the wild type (C) was set to 1. Error bars represent the S.D. **, p < 0.005 relative to the wild type unexposed to H2O2 (A) or to the wild type (C). B and D, protein samples were resolved by SDS-PAGE, and IscR (or IscR3CA), CRP, SmcR, and GbpA were detected by Western blotting using the rabbit anti-V. vulnificus IscR, anti-V. vulnificus CRP, anti-V. vulnificus SmcR, and anti-V. vulnificus GbpA sera, respectively. WT, wild type; iscR, iscR mutant; iscR3CA, a strain expressing apo-locked IscR3CA.
Previous reports that the [2Fe-2S] cluster in IscR is disrupted by oxidative stress to result in the clusterless apo-form (19, 32, 48, 52) led us to examine whether apo-IscR indeed activates PgbpA in vivo. The level of gbpA transcript in the iscR3CA mutant, of which the iscR coding region on the chromosome was replaced with iscR3CA encoding an apo-locked IscR3CA (Table 1) (32), was compared with those of the wild type and iscR mutant. When determined by qRT-PCR (Fig. 10C) and Western blot analyses (Fig. 10D), the PgbpA activity of the iscR3CA mutant was almost 3- and 13-fold greater than that of the wild type and iscR mutant, respectively, indicating that apo-IscR is able to activate PgbpA in vivo. Furthermore, it was noted that the IscR3CA level of the iscR3CA mutant was significantly greater than the IscR level of the wild type, in which both holo- and apo-IscR coexist (Fig. 10D) (52). In contrast, the levels of CRP and SmcR did not significantly differ among the strains (Fig. 10D). These results indicated that the increased activity of PgbpA in the iscR3CA mutant was possibly attributed to the elevated cellular level of IscR3CA. This elevated IscR3CA level in the iscR3CA mutant was perhaps not surprising because apo-IscR de-represses its own expression (19, 52). Taken together, the results led us to propose a model in which IscR senses reactive oxygen species and shifts to the apo-form, leading to the de-repression of the isc operon, elevating apo-IscR protein levels, and accordingly, activating PgbpA.
Discussion
There are several lines of evidence that V. vulnificus cells embed themselves in oyster tissues and form biofilms to persist in the oyster as the primary route of infection (13, 53). Furthermore, biofilms are likely a form of pathogenic V. vulnificus cells and an important source for new outbreaks as they provide a means to reach a concentrated infective dose consumed by humans (25). Therefore, it is a reasonable hypothesis that cells of the V. vulnificus biofilms entering the host intestine might be detached at first and become free-living planktonic cells that disperse to epithelial surfaces for adhesion (25). Adhesion to the intestinal epithelia is a prerequisite step for the establishment of a successful infection, and thus interference with the adhesion is an efficient way to prevent or treat infections of V. vulnificus. Efforts to develop the anti-adhesion therapies were initiated by identification and characterization of the adhesins of V. vulnificus. Previous studies demonstrated that V. vulnificus expresses different types of adhesin molecules essential for adhesion to human cell lines (14–18). Additionally, this study identified GbpA required for adhesion to mucin and the mucin-secreting HT29-MTX cells (Fig. 1, A and B). These observations suggested that V. vulnificus adheres to epithelial surfaces through multiple adhesive interactions as observed in other pathogens (5).
Nevertheless, little is known about the regulatory mechanisms adopted by V. vulnificus to modulate the expression of the adhesins. No information on the expression pattern or level of the adhesins during infection of the pathogen has been reported in previous studies. As a result of this study, the expression of gbpA is growth phase-dependent and decreases in the stationary phase cells (Fig. 3). SmcR, a master regulator of the V. vulnificus quorum sensing (21, 22, 51, 54), represses gbpA at the transcriptional level (Fig. 3). This result, along with elevated levels of SmcR in the stationary phase cells (Fig. 5), indicated that the decrease of gbpA expression in the stationary phase cells attributes most likely to SmcR repression. It is noteworthy that most individual cells in biofilms are close to stationary phase physiology with reduced growth rates and increased resistance to stress (55–57). Considering the V. vulnificus infection in the form of biofilms, the repression of gbpA by the elevated SmcR in the biofilm (stationary phase) cells could save the limited nutrients that can alternatively be used for expression of the genes responsible for the detachment of the biofilms. Consistent with this, SmcR appears to enhance the detachment of V. vulnificus biofilms entering the host intestine and thereby promote the dispersal of the pathogen to establish a new infectious cycle on the intestinal surfaces (25). Furthermore, GbpA is probably surplus upon establishing colonization and may be even detrimental to the detachment of individual cells from the established colony. SmcR, a cell density-dependent regulator, is believed to sense cell densities higher than critical levels in the colony and then render V. vulnificus to leave the congested colony for new colonization loci by repressing gbpA, which is crucial for pathogenesis.
Upon arrival onto new loci, CRP and IscR activate the gbpA expression to facilitate adhesion that is generally accompanied by the onset of accelerated (perhaps exponential) growth to colonize (Fig. 3). CRP, which is a central regulator of energy (catabolic) metabolism (20), may recognize host environments by sensing the starvation of specific nutrients imposed by the host cells and endogenous bacterial flora. IscR increases at the exponential phase (Fig. 5), indicating that the maximum expression of gbpA at the exponential phase attributes possibly to increased IscR (Fig. 3). The [2Fe-2S] cluster in IscR is disrupted by oxidative stress, and the resulting clusterless IscR (apo-IscR) increases the cellular level of IscR, most likely in its apo-form (19, 52, 58). There are two IscR-binding sequences, type 1 and type 2, and the type 2 sequence is recognized by apo-IscR, whereas the type 1 sequence is recognized exclusively by holo-IscR (47, 59, 60). The IscR binding sequences on PgbpA scored an 80% homology to the type 2 sequences of E. coli (Fig. 6B) (48, 60). Furthermore, the H2O2 induction of gbpA was mediated by apo-IscR (Fig. 10). The combined results suggested that IscR senses the oxidative stress imposed by the host defense system, turns into the apo-form, and activates the gbpA expression, leading to improved adhesion to the host intestinal surfaces.
In summary, gbpA encoding a mucin-binding protein essential for pathogenesis of V. vulnificus was identified in this study. The gbpA expression was in a growth phase-dependent manner and was regulated positively by IscR and CRP but negatively by SmcR. The regulatory proteins regulate the gbpA expression cooperatively rather than sequentially and exerted their effects by directly binding to the regulatory region of PgbpA. Two distinct IscR-binding sequences centered at −164.5 and −106, a CRP-binding sequence centered at −68, and an SmcR-binding sequence centered at −45 were identified. The gbpA expression was induced by exposure to oxidative stress, and the induction was mediated by the elevated intracellular levels of, most probably, apo-IscR. It is still difficult to define the implications of the collaboration between IscR, CRP, and SmcR in terms of pathogenesis of V. vulnificus. However, it is likely that the collaboration allows more precise tuning of the gbpA expression by integrating the signals presumably encountered in the host intestine, such as oxidative stress, starvation of specific nutrients, and increased cell density, and thereby it enhances the overall success of V. vulnificus during pathogenesis.
Author Contributions
K. K. J., S. Y. G., J. G. L., and S. H. C. designed the research; K. K. J. and S. Y. G. performed the research; K. K. J., S. Y. G., J. G. L., and S. H. C. analyzed the data; and K. K. J. and S. H. C. wrote the paper. All authors reviewed the results and approved the final version of the manuscript.
This work was supported by Mid-career Researcher Program Grant 2015R1A2A1A13001654 through the National Research Foundation funded by Ministry of Science, ICT, and Future Planning; the R&D Convergence Center Support Program of the Ministry of Agriculture, Food and Rural Affairs; and Ministry of Food and Drug Safety, Grant 14162MFDS972 (to S. H. C.). The authors declare that they have no conflicts of interest with the contents of this article.
- CRP
- cyclic AMP receptor protein
- MTX
- methotrexate
- qRT-PCR
- quantitative real time PCR
- EMSA
- electrophoretic mobility shift assay
- RLU
- relative light unit
- F
- forward
- R
- reverse.
References
- 1. McGuckin M. A., Lindén S. K., Sutton P., and Florin T. H. (2011) Mucin dynamics and enteric pathogens. Nat. Rev. Microbiol. 9, 265–278 [DOI] [PubMed] [Google Scholar]
- 2. Neutra M. R., and Forstner J. F. (1987) in Physiology of the Gastrointestinal Tract (Johnson L. R., ed) Vol. 2, 2nd Ed., pp. 975–1009, Raven Press, Ltd., New York [Google Scholar]
- 3. Wiggins R., Hicks S. J., Soothill P. W., Millar M. R., and Corfield A. P. (2001) Mucinases and sialidases: their role in the pathogenesis of sexually transmitted infections in the female genital tract. Sex Transm. Infect. 77, 402–408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Thornton D. J., and Sheehan J. K. (2004) From mucins to mucus: toward a more coherent understanding of this essential barrier. Proc. Am. Thorac. Soc. 1, 54–61 [DOI] [PubMed] [Google Scholar]
- 5. Ofek I., Bayer E. A., and Abraham S. N. (2013) in The Prokaryotes (Rosenberg E., Delong E. F., Lory S., Stackebrandt E., and Thompson F., eds) pp. 107–123, 10.1007/978-3-642-30144-5_50, Springer-Verlag, Berlin, Heidelberg [DOI] [Google Scholar]
- 6. Kirn T. J., Jude B. A., and Taylor R. K. (2005) A colonization factor links Vibrio cholerae environmental survival and human infection. Nature 438, 863–866 [DOI] [PubMed] [Google Scholar]
- 7. Weening E. H., Barker J. D., Laarakker M. C., Humphries A. D., Tsolis R. M., and Bäumler A. J. (2005) The Salmonella enterica serotype Typhimurium lpf, bcf, stb, stc, std, and sth fimbrial operons are required for intestinal persistence in mice. Infect. Immun. 73, 3358–3366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Juge N. (2012) Microbial adhesins to gastrointestinal mucus. Trends Microbiol. 20, 30–39 [DOI] [PubMed] [Google Scholar]
- 9. Bhowmick R., Ghosal A., Das B., Koley H., Saha D. R., Ganguly S., Nandy R. K., Bhadra R. K., and Chatterjee N. S. (2008) Intestinal adherence of Vibrio cholerae involves a coordinated interaction between colonization factor GbpA and mucin. Infect. Immun. 76, 4968–4977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Jude B. A., Martinez R. M., Skorupski K., and Taylor R. K. (2009) Levels of the secreted Vibrio cholerae attachment factor GbpA are modulated by quorum-sensing-induced proteolysis. J. Bacteriol. 191, 6911–6917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wong E., Vaaje-Kolstad G., Ghosh A., Hurtado-Guerrero R., Konarev P. V., Ibrahim A. F., Svergun D. I., Eijsink V. G., Chatterjee N. S., and van Aalten D. M. (2012) The Vibrio cholerae colonization factor GbpA possesses a modular structure that governs binding to different host surfaces. PLoS Pathog. 8, e1002373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sudarsan N., Lee E. R., Weinberg Z., Moy R. H., Kim J. N., Link K. H., and Breaker R. R. (2008) Riboswitches in eubacteria sense the second messenger cyclic di-GMP. Science 321, 411–413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Froelich B., and Oliver J. (2013) Increases in the amount of Vibrio spp. in oysters upon addition of exogenous bacteria. Appl. Environ. Microbiol. 79, 5208–5213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ran Kim Y., and Haeng Rhee J. (2003) Flagellar basal body flg operon as a virulence determinant of Vibrio vulnificus. Biochem. Biophys. Res. Commun. 304, 405–410 [DOI] [PubMed] [Google Scholar]
- 15. Lee J. H., Rho J. B., Park K. J., Kim C. B., Han Y. S., Choi S. H., Lee K. H., and Park S. J. (2004) Role of flagellum and motility in pathogenesis of Vibrio vulnificus. Infect. Immun. 72, 4905–4910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Paranjpye R. N., and Strom M. S. (2005) A Vibrio vulnificus type IV pilin contributes to biofilm formation, adherence to epithelial cells, and virulence. Infect. Immun. 73, 1411–1422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Goo S. Y., Lee H. J., Kim W. H., Han K. L., Park D. K., Lee H. J., Kim S. M., Kim K. S., Lee K. H., and Park S. J. (2006) Identification of OmpU of Vibrio vulnificus as a fibronectin-binding protein and its role in bacterial pathogenesis. Infect. Immun. 74, 5586–5594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lee K. J., Lee N. Y., Han Y. S., Kim J., Lee K. H., and Park S. J. (2010) Functional characterization of the IlpA protein of Vibrio vulnificus as an adhesion and its role in bacterial pathogenesis. Infect. Immun. 78, 2408–2417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Schwartz C. J., Giel J. L., Patschkowski T., Luther C., Ruzicka F. J., Beinert H., and Kiley P. J. (2001) IscR, an Fe-S cluster-containing transcription factor, represses expression of Escherichia coli genes encoding Fe-S cluster assembly proteins. Proc. Natl. Acad. Sci. U.S.A. 98, 14895–14900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Green J., Stapleton M. R., Smith L. J., Artymiuk P. J., Kahramanoglou C., Hunt D. M., and Buxton R. S. (2014) Cyclic-AMP and bacterial cyclic-AMP receptor proteins revisited: adaptation for different ecological niches. Curr. Opin. Microbiol. 18, 1–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Shao C. P., and Hor L. I. (2001) Regulation of metalloprotease gene expression in Vibrio vulnificus by a Vibrio harveyi LuxR homologue. J. Bacteriol. 183, 1369–1375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Jeong H. S., Lee M. H., Lee K. H., Park S. J., and Choi S. H. (2003) SmcR and cyclic AMP receptor protein coactivate Vibrio vulnificus vvpE encoding elastase through the RpoS-dependent promoter in a synergistic manner. J. Biol. Chem. 278, 45072–45081 [DOI] [PubMed] [Google Scholar]
- 23. Oh M. H., Lee S. M., Lee D. H., and Choi S. H. (2009) Regulation of the Vibrio vulnificus hupA gene by temperature alteration and cyclic AMP receptor protein and evaluation of its role in virulence. Infect. Immun. 77, 1208–1215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Shao C. P., Lo H. R., Lin J. H., and Hor L. I. (2011) Regulation of cytotoxicity by quorum-sensing signaling in Vibrio vulnificus is mediated by SmcR, a repressor of hlyU. J. Bacteriol. 193, 2557–2565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kim S. M., Park J. H., Lee H. S., Kim W. B., Ryu J. M., Han H. J., and Choi S. H. (2013) LuxR homologue SmcR is essential for Vibrio vulnificus pathogenesis and biofilm detachment, and its expression is induced by host cells. Infect. Immun. 81, 3721–3730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lim J. G., and Choi S. H. (2014) IscR is a global regulator essential for pathogenesis of Vibrio vulnificus and induced by host cells. Infect. Immun. 82, 569–578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kim S., Bang Y. J., Kim D., Lim J. G., Oh M. H., and Choi S. H. (2014) Distinct characteristics of OxyR2, a new OxyR-type regulator, ensuring expression of Peroxiredoxin 2 detoxifying low levels of hydrogen peroxide in Vibrio vulnificus. Mol. Microbiol. 93, 992–1009 [DOI] [PubMed] [Google Scholar]
- 28. Milton D. L., O'Toole R., Horstedt P., and Wolf-Watz H. (1996) Flagellin A is essential for the virulence of Vibrio anguillarum. J. Bacteriol. 178, 1310–1319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Simon R., Priefer U., and Pühler A. (1983) A broad host range mobilization system for in vivo genetic engineering: transposon mutagenesis in Gram negative bacteria. Nat. Biotechnol. 1, 784–791 [Google Scholar]
- 30. Hwang J., Kim B. S., Jang S. Y., Lim J. G., You D. J., Jung H. S., Oh T. K., Lee J. O., Choi S. H., and Kim M. H. (2013) Structural insights into the regulation of sialic acid catabolism by the Vibrio vulnificus transcriptional repressor NanR. Proc. Natl. Acad. Sci. U.S.A. 110, E2829–2837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kim B. S., Hwang J., Kim M. H., and Choi S. H. (2011) Cooperative regulation of the Vibrio vulnificus nan gene cluster by NanR protein, cAMP receptor protein, and N-acetylmannosamine 6-phosphate. J. Biol. Chem. 286, 40889–40899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lim J. G., Bang Y. J., and Choi S. H. (2014) Characterization of the Vibrio vulnificus 1-Cys peroxiredoxin Prx3 and regulation of its expression by the Fe-S cluster regulator IscR in response to oxidative stress and iron starvation. J. Biol. Chem. 289, 36263–36274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Yeung A. T., Parayno A., and Hancock R. E. (2012) Mucin promotes rapid surface motility in Pseudomonas aeruginosa. mBio 3, e00073–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lesuffleur T., Barbat A., Dussaulx E., and Zweibaum A. (1990) Growth adaptation to methotrexate of HT-29 human colon carcinoma cells is associated with their ability to differentiate into columnar absorptive and mucus-secreting cells. Cancer Res. 50, 6334–6343 [PubMed] [Google Scholar]
- 35. Kapuscinski J. (1995) DAPI: a DNA-specific fluorescent probe. Biotech. Histochem. 70, 220–233 [DOI] [PubMed] [Google Scholar]
- 36. Vieira M. A., Gomes T. A., Ferreira A. J., Knöbl T., Servin A. L., and Liévin-Le Moal V. (2010) Two atypical enteropathogenic Escherichia coli strains induce the production of secreted and membrane-bound mucins to benefit their own growth at the apical surface of human mucin-secreting intestinal HT29-MTX cells. Infect. Immun. 78, 927–938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Jeong H. G., and Satchell K. J. (2012) Additive function of Vibrio vulnificus MARTXVv and VvhA cytolysins promotes rapid growth and epithelial tissue necrosis during intestinal infection. PLoS Pathog. 8, e1002581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lee S. J., Jung Y. H., Ryu J. M., Jang K. K., Choi S. H., and Han H. J. (2016) VvpE mediates the intestinal colonization of Vibrio vulnificus by the disruption of tight junctions. Int. J. Med. Microbiol. 306, 10–19 [DOI] [PubMed] [Google Scholar]
- 39. Jeong H. G., Oh M. H., Kim B. S., Lee M. Y., Han H. J., and Choi S. H. (2009) The capability of catabolic utilization of N-acetylneuraminic acid, a sialic acid, is essential for Vibrio vulnificus pathogenesis. Infect. Immun. 77, 3209–3217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kim S. M., Lee D. H., and Choi S. H. (2012) Evidence that the Vibrio vulnificus flagellar regulator FlhF is regulated by a quorum sensing master regulator SmcR. Microbiology 158, 2017–2025 [DOI] [PubMed] [Google Scholar]
- 41. Choi H. K., Park N. Y., Kim D. I., Chung H. J., Ryu S., and Choi S. H. (2002) Promoter analysis and regulatory characteristics of vvhBA encoding cytolytic hemolysin of Vibrio vulnificus. J. Biol. Chem. 277, 47292–47299 [DOI] [PubMed] [Google Scholar]
- 42. Bradford M. M. (1976) A rapid and sensitive method for the quantification of microgram quantities of protein utilizing the principles of protein-dye binding. Anal. Biochem. 7, 248–254 [DOI] [PubMed] [Google Scholar]
- 43. Giel J. L., Rodionov D., Liu M., Blattner F. R., and Kiley P. J. (2006). IscR-dependent gene expression links iron-sulphur cluster assembly to the control of O2-regulated genes in Escherichia coli. Mol. Microbiol. 60, 1058–1075 [DOI] [PubMed] [Google Scholar]
- 44. Lenz D. H., Mok K. C., Lilley B. N., Kulkarni R. V., Wingreen N. S., and Bassler B. L. (2004) The small RNA chaperone Hfq and multiple small RNAs control quorum sensing in Vibrio harveyi and Vibrio cholerae. Cell 118, 69–82 [DOI] [PubMed] [Google Scholar]
- 45. Park J. H., Cho Y. J., Chun J., Seok Y. J., Lee J. K., Kim K. S., Lee K. H., Park S. J., and Choi S. H. (2011) Complete genome sequence of Vibrio vulnificus MO6-24/O. J. Bacteriol. 193, 2062–2063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Korotkov K. V., Sandkvist M., and Hol W. G. (2012) The type II secretion system: biogenesis, molecular architecture and mechanism. Nat. Rev. Microbiol. 10, 336–351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Yeo W. S., Lee J. H., Lee K. C., and Roe J. H. (2006) IscR acts as an activator in response to oxidative stress for the suf operon encoding Fe-S assembly proteins. Mol. Microbiol. 61, 206–218 [DOI] [PubMed] [Google Scholar]
- 48. Nesbit A. D., Giel J. L., Rose J. C., and Kiley P. J. (2009) Sequence-specific binding to a subset of IscR-regulated promoters does not require IscR Fe-S cluster ligation. J. Mol. Biol. 387, 28–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Cameron A. D., and Redfield R. J. (2006) Non-canonical CRP sites control competence regulons in Escherichia coli and many other γ-proteobacteria. Nucleic Acids Res. 34, 6001–6014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Browning D. F., and Busby S. J. (2004) The regulation of bacterial transcription initiation. Nat. Rev. Microbiol. 2, 57–65 [DOI] [PubMed] [Google Scholar]
- 51. Lee D. H., Jeong H. S., Jeong H. G., Kim K. M., Kim H., and Choi S. H. (2008) A consensus sequence for binding of SmcR, a Vibrio vulnificus LuxR homologue, and genome wide identification of the SmcR regulon. J. Biol. Chem. 283, 23610–23618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Giel J. L., Nesbit A. D., Mettert E. L., Fleischhacker A. S., Wanta B. T., and Kiley P. J. (2013) Regulation of iron-sulphur cluster homeostasis through transcriptional control of the Isc pathway by [2Fe-2S]-IscR in Escherichia coli. Mol. Microbiol. 87, 478–492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Paranjpye R. N., Johnson A. B., Baxter A. E., and Strom M. S. (2007) Role of type IV pilins in persistence of Vibrio vulnificus in Crassostera virginica oysters. Appl. Environ. Microbiol. 73, 5041–5044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Jeong H. S., Kim S. M., Lim M. S., Kim K. S., and Choi S. H. (2010) Direct interaction between quorum-sensing regulator SmcR and DNA polymerase is mediated by integration host factor to activate vvpE encoding elastase in Vibrio vulnificus. J. Biol. Chem. 285, 9357–9366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Hall-Stoodley L., Costerton J. W., and Stoodley P. (2004) Bacterial biofilms: from the natural environment to infectious diseases. Nat. Rev. Microbiol. 2, 95–108 [DOI] [PubMed] [Google Scholar]
- 56. Fux C. A., Costerton J. W., Stewart P. S., and Stoodley P. (2005) Survival strategies of infectious biofilms. Trends Microbiol. 13, 34–40 [DOI] [PubMed] [Google Scholar]
- 57. Høiby N., Ciofu O., and Bjarnsholt T. (2010) Pseudomonas aeruginosa biofilms in cystic fibrosis. Future Microbiol. 5, 1663–1674 [DOI] [PubMed] [Google Scholar]
- 58. Zheng M., Wang X., Templeton L. J., Smulski D. R., LaRossa R. A., and Storz G. (2001) DNA microarray-mediated transcriptional profiling of the Escherichia coli response to hydrogen peroxide. J. Bacteriol. 183, 4562–4570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Wu Y., and Outten F. W. (2009) IscR controls iron-dependent biofilm formation in Escherichia coli by regulating type I fimbria expression. J. Bacteriol. 191, 1248–1257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Rajagopalan S., Teter S. J., Zwart P. H., Brennan R. G., Phillips K. J., and Kiley P. J. (2013) Studies of IscR reveal a unique mechanism for metal-dependent regulation of DNA binding specificity. Nat. Struct. Mol. Biol. 20, 740–747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Wright A. C., Simpson L. M., Oliver J. D., and Morris J. G. (1990) Phenotypic evaluation of acapsular transposon mutants of Vibrio vulnificus. Infect. Immun. 58, 1769–1773 [DOI] [PMC free article] [PubMed] [Google Scholar]