

Abstract
The γ-secretase protease and associated regulated intramembrane proteolysis play an important role in controlling receptor-mediated intracellular signaling events, which have a central role in Alzheimer disease, cancer progression, and immune surveillance. An increasing number of γ-secretase substrates have a role in cytokine signaling, including the IL-6 receptor, IL-1 receptor type I, and IL-1 receptor type II. In this study, we show that following TNF-converting enzyme-mediated ectodomain shedding of TNF type I receptor (TNFR1), the membrane-bound TNFR1 C-terminal fragment is subsequently cleaved by γ-secretase to generate a cytosolic TNFR1 intracellular domain. We also show that clathrin-mediated internalization of TNFR1 C-terminal fragment is a prerequisite for efficient γ-secretase cleavage of TNFR1. Furthermore, using in vitro and in vivo model systems, we show that in the absence of presenilin expression and γ-secretase activity, TNF-mediated JNK activation was prevented, assembly of the TNFR1 pro-apoptotic complex II was reduced, and TNF-induced apoptosis was inhibited. These observations demonstrate that TNFR1 is a γ-secretase substrate and suggest that γ-secretase cleavage of TNFR1 represents a new layer of regulation that links the presenilins and the γ-secretase protease to pro-inflammatory cytokine signaling.
Keywords: apoptosis, c-Jun N-terminal kinase (JNK), cell death, gamma-secretase, NF-kappaB, presenilin, receptor internalization, tumor necrosis factor (TNF)
Introduction
The biological activities of the tumor necrosis factor-α (TNF) pro-inflammatory cytokine are resolved by two distinct cell surface receptors, TNFR13 and TNFR2, which elicit a diversity of cellular responses, such as inflammation, cell proliferation, cell differentiation, and initiation of apoptosis (1–10). TNFR1 initiates either pro-inflammatory or pro-apoptotic signaling through the selective recruitment of intracellular adaptor and effector proteins (1, 3, 6, 7, 11). Ligand binding and trimerization of TNFR1 enables the recruitment of TNFR1-associated death domain protein (TRADD) (12, 13), which functions as a scaffold enabling the recruitment of receptor-interacting protein kinase 1 (RIPK1) (14–16), TNF receptor-associated factor 2 (TRAF2) or TRAF5 (12), and the cellular inhibitor of apoptosis proteins (cIAPs) cIAP1 and cIAP2 (11), which collectively form a signaling composite called complex I (17–19). The resulting lysine 63-linked polyubiquitination of RIPK1 by TRAF2 and the cIAPs (20–24) enables an interaction with the IκB kinase complex that mediates the phosphorylation and degradation of IκB-inhibitory proteins and activation of the transcription factor NF-κB to promote non-apoptotic signaling pathways (25–27). NF-κB also increases expression of anti-apoptotic genes, including cIAPs and FLICE inhibitory protein (c-FLIP), further ensuring a non-apoptotic signaling pathway.
The importance of receptor internalization as a regulatory mechanism for the segregation and divergence of intracellular signaling pathways is highlighted by studies on internalization of TNFR1 and TNFR2, the Fas receptor (FasR/CD95), IL-1 receptor I, Toll-like receptor 4, and TRAIL receptors (18, 28–33). The current favored model for TNFR1 signaling proposes that TNFR1 recruits TRADD, RIPK1, and TRAF2, forming a pro-survival complex I at the cell membrane (28, 31, 34–36), enabling activation of mitogen-activated protein kinases (MAPK) and NF-κB and propagation of anti-apoptotic signaling. Thereafter and subsequent to TNFR1 ubiquitination and internalization (36), TRADD enables the recruitment of FADD and procaspase-8 at endosomal vesicles and thereby forms the pro-apoptotic complex II, also designated the death-inducing signaling complex. Clathrin-mediated internalization of TNFR1 is a prerequisite for efficient recruitment of FADD and procaspase-8 and formation of pro-apoptotic complex II from endosomal compartments (37). Pharmacological inhibition of TNFR1 endocytosis prevents TNF-mediated JNK activation and antagonizes formation of complex II and pro-apoptotic signaling (37). Furthermore, TNFR1 contains a region (amino acids 205–214) termed the TNFR1 internalization domain, deletion or mutagenesis of which prevents TNFR1 internalization and TNF-mediated JNK activation and pro-apoptotic signaling (28). Adenoviruses have also evolved a mechanism to prevent internalization of TNFR1 and thereby selectively prevent TNF-mediated apoptosis of infected cells (35). Finally, a recent study has reported that Lys-63-linked polyubiquitination of TNFR1 is critical for the internalization and pro-apoptotic signaling of TNFR1 (36), indicating a high degree of regulation governing the spatial segregation of disparate TNF-mediated signaling events.
In addition to undergoing endocytosis, as a means of regulating TNF-mediated signaling, TNFR1 also undergoes ectodomain shedding and produces biologically active, soluble TNFR1 (sTNFR1) fragments that are shed from the membrane, which results in reduced cell surface availability of TNFR1 and reduced TNF signaling. TACE/ADAM17, a member of the A disintegrin and metalloprotease (ADAM) family of transmembrane and secreted proteases, is responsible for ectodomain shedding of TNFR1 (38, 39). Recently, it has emerged that several proteins that undergo ectodomain shedding are subsequently cleaved by the γ-secretase protease (40–42). This sequential proteolysis of specific type I membrane proteins is called regulated intramembrane proteolysis (43). The presenilin proteins (PS1 and PS2) are the catalytic subunit of the γ-secretase complex (44, 45), and genetic inactivation of PS1 results in complete loss of γ-secretase activity (46, 47). PS1 associates with nicastrin, presenilin enhancer 2 (PEN-2), and the anterior pharynx defective-1 (APH-1) proteins to form an active γ-secretase complex (48, 49). To date, over 100 different γ-secretase substrates have been identified, indicating that γ-secretase has a generic role in the proteolysis of membrane proteins and regulation of receptor-mediated signaling pathways (42, 50). Several cytokines and cytokine receptors involved in immune signaling, including IL-1 receptor I (40), IL-1 receptor II (52), IL-6 receptor (53), and CX3CL1 and CXCL16 (54), also undergo regulated intramembrane proteolysis, highlighting the importance of γ-secretase in the regulation of growth factor and cytokine signaling (50).
Given that the TNFR superfamily member p75 neurotrophin receptor (p75NTR) undergoes regulated intramembrane proteolysis (55–58) and that TNFR1 fits the profile of a γ-secretase substrate, we screened a number of TNF superfamily members to detect involvement of the γ-secretase protease in their proteolysis and signaling. Using pharmacological inhibitors and genetic knock-out approaches, in this study, we report the identification of TNFR1 as a γ-secretase substrate and demonstrate that loss of presenilin expression and γ-secretase activity antagonized TNF-mediated JNK MAPK activation, reduced pro-apoptotic complex II assembly, and inhibited TNF-induced caspase activation and apoptosis.
Experimental Procedures
Reagents and Antibodies
γ-Secretase inhibitors DAPT and L-685,458, protein synthesis inhibitor cycloheximide, TACE/ADAM17-specific metalloprotease inhibitor TAPI-1, proteasomal inhibitor epoxomicin, and phorbol 12-myristate 13-acetate (PMA) were purchased from Calbiochem. Recombinant human and murine TNFα were purchased from Peprotech. Antibodies used in this study are as follows. Human C terminus-specific TNFR1 (C2521; monoclonal) (WB, 1:1000; IP, 1:100; catalog no. 3736), RIPK1 (monoclonal) (WB, 1:1000; catalog no. 3493), cleaved PARP (monoclonal) (WB, 1:1000; catalog no. 9548), cleaved caspase-3 (polyclonal) (WB, 1:1000; catalog no. 9661), phospho-P38 (catalog no. 9211), phospho-IκBα (catalog no. 9246), and phospho-JNK (catalog no. 9255) were from Cell Signaling Technology (Danvers, MA). Full-length TNFR1 (H5; monoclonal) (WB, 1:200; IP, 1:100; sc-8436), mouse C terminus-specific TNFR1 (E11; monoclonal) (WB, 1:200; sc-374186), TRAF2 (C20; polyclonal) (WB, 1:200; sc-876), FADD (H181; polyclonal) (IP, 1:100; sc-5559), P38 (sc-7972), JNK (sc-571), and IκBα (sc-371) were from Santa Cruz Biotechnology, Inc. FADD (1F7; monoclonal) (WB, 1:1000; catalog no. ADI-AAM-212-E) was from Enzo Life Science. Monoclonal N-terminal PS1 antibody (catalog no. 614.1) (1:100) was as described previously (59, 60). Monoclonal PS1 N-terminal fragment (MAB1563) and anti-human PS1 C-terminal fragment (CTF) (MAB5232) were purchased from Chemicon; anti-Myc (catalog no. 1667149) was from Roche Applied Sciences; and anti-FLAG, anti-β-tubulin, and anti-β-actin and dynasore hydrate were purchased from Sigma-Aldrich. HRP-conjugated anti-mouse (WB, 1:6000) and anti-rabbit (WB, 1:3000) antibodies were from Dako Cytomation. Infrared secondary antibodies IRDye 680 goat anti-rabbit IgG and IRDye 800CW goat anti-mouse IgG (WB, 1:10,000) were procured from LI-COR (Dublin, Ireland).
Cell Culture
Human embryonic kidney 293T (HEK293T) cells, human breast adenocarcinoma MCF-7 cells, and wild type and presenilin-deficient murine embryonic fibroblasts (MEFs) (61, 62), were maintained in DMEM supplemented with 10% (v/v), fetal bovine serum, 2 mm glutamine, penicillin (50 units/ml), and streptomycin (50 μg/ml) at 37 °C in humidified air containing 5% CO2. MCF-7 and MEFs were further supplemented with 1% (v/v) non-essential amino acids and 1% (v/v) sodium pyruvate.
Mice
Psen1+/−Psen2−/− partial knock-out mice were generated on a C57BL/6J background as described (61, 62). Wild type C57BL6 were purchased from Halren Laboratories. Mouse colonies were maintained in pathogen-free conditions at Charles River UK or the Biological Services Unit at University College Cork. All animal experiments were done in accordance with the regulations and guidelines of the Irish Department of Health, and protocols were approved by the University College Cork Animal Experimentation Ethics Committee.
Plasmids, Site-directed Mutagenesis, and Transfections
pcDNA3.1-PS1 was generated in-house as described previously (40). The pBABE-TNFR1 (human) expression plasmid was a gift from Dr. Martin S. Kluger (Yale University School of Medicine). For generation of the pBABE-TNFR1 W210A mutant, site-directed mutagenesis was performed using a two-primer pair method outlined by the QuikChangeTM site-directed mutagenesis kit (Stratagene) according to the manufacturer's instructions. The mutagenesis primer pairs were 5′-CGCTACCAACGGGCGAAGTCCAAGCTC-3′ (forward) and 5′-GAGCTTGGACTTGCGCCGTTGGTAGCG-3′ (reverse). A plasmid encoding pEGFPN1-human dynamin or -dynamin-K44A (dominant negative) was from Dr. Pietro De Camilli (Addgene plasmid 22163 and Addgene plasmid 22197). The pcDNA3.1-HA-RIPK1 and pcDNA3-Myc-TRADD was obtained from Tularick Inc. Transient transfections of HEK293T were performed using the calcium phosphate precipitation method as described previously (40).
Flow Cytometry Analysis
To quantify surface expression of TNFR1, cells were harvested in PBS-EDTA and washed once in wash buffer consisting of filtered 1% BSA in PBS. Cells were kept on ice. Cells were then incubated for 1 h with primary antibody (TNFR1 H5, Santa Cruz Biotechnology) in wash buffer, followed by three washes. Cells were then incubated for 30 min with secondary antibody (Alexa Fluor anti-mouse 488) diluted in wash buffer. Cells were washed twice with wash buffer and once with PBS and resuspended and fixed with 1% paraformaldehyde. Cells were analyzed using an AccuriC6 flow cytometer (BD Biosciences) running Accuri FloJo software. Corrected mean fluorescence intensities were calculated by subtracting the mean fluorescence intensity with irrelevant nonbinding antibody from the mean fluorescence intensity with the specific antibody. To quantify the percentage of annexin V-positive cells, wild type and presenilin-deficient MEFs were treated as indicated and assayed for programmed cell death using an annexin V-FITC apoptosis detection kit (eBioscience) as per the manufacturer's instructions. Briefly, cells were harvested and washed once in PBS and then in 1× binding buffer. Washed cells were resuspended in binding buffer (1–5 × 106 cells/ml), and aliquots (100 μl) of resuspended cells were incubated with Annexin V-FITC (5 μl) for 15 min at room temperature in the dark. Cells were washed once with 2 ml of 1× binding buffer and resuspended in 200 μl followed by flow cytometric analysis of annexin V-reactive cells using an Accuri C6 flow cytometer and CFlow Plus software (BD Biosciences).
Immunoprecipitation
Equal concentrations of cell lysates were precleared for 1 h at 4 °C with 25 μl of Protein G-Sepharose beads. Precleared lysates were immunoprecipitated overnight at 4 °C with 2–5 μg of the indicated antibody, followed by incubation with 25 μl of Protein G-Sepharose beads for 3 h. Immunoprecipitates were then washed two times in 500 mm NaCl lysis buffer followed by one wash in 150 mm NaCl lysis buffer. Washed Protein G beads were boiled in SDS loading buffer for 5 min on a heating block and subjected to Western blotting analysis.
Western Blotting
After the indicated treatments, total or immunoprecipitated protein extracts were obtained from cells. Cells were harvested in ice-cold PBS and lysed with lysis buffer (50 mm Tris-HCl (pH 7.4), 150 mm NaCl, 1 mm EDTA, 1% (v/v) Triton X-100, 1% sodium deoxycholate, and 0.1% SDS) freshly supplemented with 1 mm sodium orthovanadate and protease inhibitor mixture (CompleteTM, Molecular Biochemicals) on ice. Lysates were centrifuged (13,200 rpm, 20 min, 4 °C), the supernatants were collected, and protein yield was quantified using a bicinchoninic acid (BCA) assay (Pierce). Normalized samples were prepared with Laemmli sample buffer containing β-mercaptoethanol and electrophoresed on an SDS-polyacrylamide gel. Proteins were transferred to a nitrocellulose membrane (Millipore). After blocking for 1 h with 5% nonfat milk in TBS-T (Tris-buffered saline containing 0.1% Tween 20), the membrane was probed with the primary antibody and washed three times in 5% nonfat milk in TBS-T, followed by the secondary antibody. Immunoreactivity was visualized by the Odyssey Imaging System (LI-COR Biosciences) or by the ECL Western blotting detection system (GE Healthcare). Signal intensity was analyzed within a linear range using ImageJ (National Institutes of Health, Bethesda, MD).
Enzyme-linked Immunosorbent Assay
MEFs were seeded (0.1 × 106 cells/ml; 0.1 ml) in 96-well plates and were allowed to grow for 24 h. Cells were stimulated with murine TNFα (10 ng/ml) for 18 h. Supernatant was collected and analyzed for cytokines IL-10, IL-6, TNFα, IFNγ, IL-12 (p70), IL-1β, and chemokine CXCL1 by a mouse Proinflammatory 7-Plex cytokine kit (Meso-Scale Discovery). HEK293T cells (2 × 105 cells/ml; 2 ml) transiently expressing pBABE-human TNFR1 were seeded in 6-well plates and allowed to grow for 24 h. Soluble TNFR1 levels in conditioned medium were quantified using a murine-specific ELISA kit (Invitrogen).
Cell Viability Assays
Wild type and presenilin-deficient MEFs (2 × 105 cells/ml; 2 ml) were grown in 6-well plates. 24 h after culture, cells were stimulated with murine TNFα (40 ng/ml) and cycloheximide (10 μg/ml) for the indicated times. Cell lysates were probed by immunoblotting with antibodies against cleaved PARP, cleaved caspase-3, and β-actin. Images were captured using the ×40 objective on a Leica DMIL inverted microscope.
In Vivo Challenge with TNFα
Wild type and Psen1+/−/Psen2−/− mice 5–6 months old were injected with murine TNFα (100 μg/kg) intraperitoneally. Blood was collected by cardiac puncture 2 h postinjection. Serum was separated and analyzed for cytokines IL-10, IL-6, TNFα, IFNγ, IL-12 (p70), IL-1β, and chemokine CXCL1 by the ultrasensitive mouse Proinflammatory 7-Plex cytokine kit from Meso Scale Discovery. For all experiments, wild type and Psen1+/−/Psen2−/− mice were matched for age and sex.
Statistical Analysis
All data are typically presented as a pool of three experiments (mean ± S.E.) or as a single experiment representative of two or more independent experiments done in triplicates (mean ± S.E.). Treatment groups were compared using either one-way or two-way ANOVA analyses with GraphPad Prism version 5.0, followed by a Bonferroni post-test or using the unpaired Student's t test with Microsoft Excel. A p value of 0.05 was considered significant (*, p < 0.05; **, p < 0.01; ***, p < 0.001).
Results
Ectodomain Shedding Is a Prerequisite for γ-Secretase Cleavage of TNFR1
Several cell surface proteins undergo constitutive ectodomain shedding and can also be stimulated to release extracellular domains through the activation of cell signaling pathways. Phorbol esters, such as PMA, activate the PKC pathway (25) and induce TACE/ADAM17-mediated shedding of cell surface proteins (63). TNFR1 is a 455-amino acid protein with a large extracellular domain (ECD), a single transmembrane domain (TM), and a 221-amino acid intracellular domain (ICD) that contains the N-terminal death domain (Fig. 1a). TNFR1 has been shown to undergo ADAM17- and ADAM8-mediated cleavage, resulting in the release of sTNFR1 ectodomains and generation of a membrane-anchored TNFR1 CTF (38, 64). The majority of known γ-secretase substrates also undergo ectodomain shedding in the extracellular domain as a prerequisite for γ-secretase-mediated cleavage of the remaining membrane-bound CTF. The identification of members of the IL-1/Toll-like receptor superfamily, including IL-1 receptor I, IL-1 receptor II, and IL-6 receptor, as substrates for γ-secretase-dependent regulated intramembrane proteolysis (40, 52, 53) prompted us to examine whether TNFR1 undergoes ectodomain shedding and subsequent cleavage by γ-secretase.
FIGURE 1.
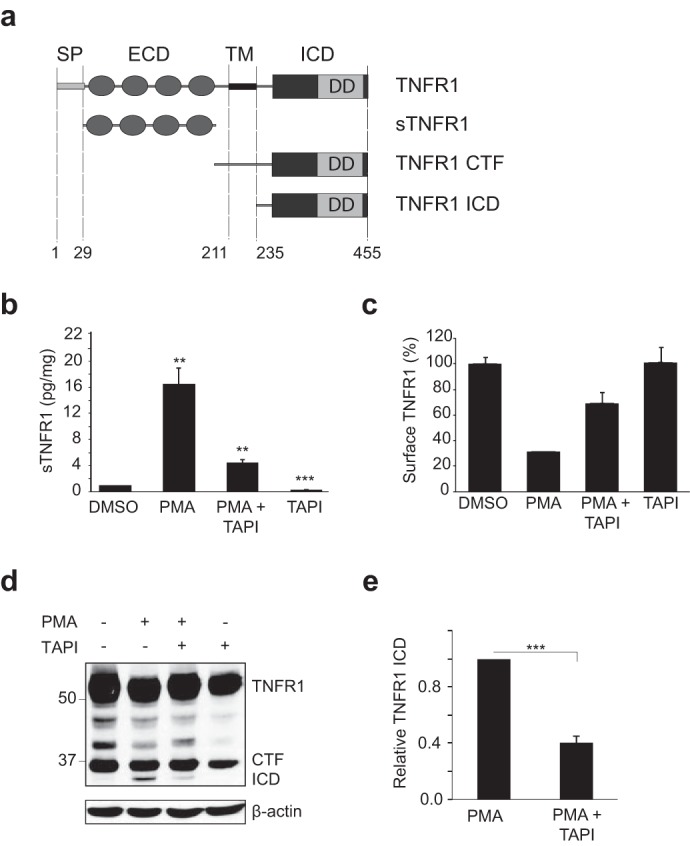
Ectodomain shedding is a prerequisite for γ-secretase cleavage of TNFR1. a, schematic representation of the domains of human TNFR1. Human TNFR1 (accession number AAA61201) is a 455-amino acid protein with a signal peptide (SP) at position 1–29; an extracellular domain (ECD) at position 29–211, containing four cysteine repeat domains; a 23-amino acid transmembrane domain (TM); and a 221-amino acid ICD at positions 235–455, containing the pro-apoptotic death domain (DD). Proteolytic cleavage by TACE/ADAM17 results in the generation of a soluble TNFR1 extracellular domain (sTNFR1) and membrane-anchored TNFR1 CTF. b, ELISA of sTNFR1 in conditioned medium from HEK293T cells transiently expressing TNFR1, left untreated or pretreated with TAPI-1 (50 μm for 2 h) and/or treated with PMA (200 ng/ml for 2 h). Data are expressed as a pictogram of sTNFR1/mg of total protein ± S.E. (error bars) versus TNFR1 control (n = 3). **, p < 0.01 (two-way ANOVA). c, FACS analysis of TNFR1 expression in HEK293T cells left untreated or pretreated with TAPI-1 (50 μm for 2 h) and/or treated with PMA (200 ng/ml for 2 h). Cells were stained with anti-TNFR1 and Alexa Fluor anti-mouse 488 antibodies. d, immunoassay of TNFR1 in corresponding whole cell lysates from HEK293T cells transiently expressing TNFR1 and left untreated or treated as in b and c. e, densitometry analysis of TNFR1 ICD normalized to total β-actin for all experimental conditions. The amount of TNFR1 ICD is expressed as TNFR1 ICD immunoreactivity ± S.E. (n = 3). ***, p < 0.001 (unpaired t test). Western blotting data are from one experiment representative of three independent experiments.
In HEK293T cells expressing TNFR1, the constitutive release of the sTNFR1 ectodomain in conditioned medium was clearly detectable by ELISA (Fig. 1b). Treatment with PMA (200 ng/ml; 2 h) induced a substantial increase in TNFR1 ectodomain shedding and generation of sTNFR1. Consistent with ADAM17 being the predominant protease involved in TNFR1 ectodomain shedding, both constitutive and PMA-induced shedding of sTNFR1 were inhibited by TAPI-1, a pharmacological inhibitor of ADAM/TACE enzymatic activity (Fig. 1b, lanes 3 and 4). To quantify the cell surface distribution and shedding of TNFR1, we used FACS analysis of intact HEK293T cells expressing TNFR1 (Fig. 1c). Stimulation with PMA induced a 72% reduction in cell surface TNFR1. In contrast, co-treatment with TAPI-1 prevented PMA-induced loss of cell surface TNFR1, where a reduction of only 33% was observed (Fig. 1c). Immunoblotting analysis of whole cell lysates from the same cell cultures with an anti-TNFR1 C terminus-specific antibody revealed the detection of a 30–34-kDa CTF, which corresponds to the remaining membrane-anchored TNFR1 following sTNFR1 shedding (Fig. 1d). Interestingly, in cells treated with PMA, the generation of a second but smaller anti-TNFR1 reactive fragment, namely the TNFR1 intracellular domain (ICD), of 26–30 kDa was detected, generation of which was also diminished on treatment with TAPI-1 (Fig. 1, d and e). TNFR1 has a molecular mass of 55–65 kDa by SDS-PAGE and has a theoretically predicted molecular mass of 50,494 Da. Following sTNFR1 shedding, the remaining membrane-anchored TNFR1 CTF and the ICD have a predicted molecular mass of 27,080 and 24,662 Da, respectively, which correspond to the detected molecular masses of 30–34 kDa and 26–30 kDa by SDS-PAGE (Fig. 1d). Therefore, we hypothesized that the TNFR1 ICD is the product of γ-secretase cleavage of TNFR1 CTF, generated following sTNFR1 shedding.
TNFR1 Is a γ-Secretase Substrate
To examine whether or not TNFR1 is indeed a γ-secretase substrate, HEK293T cells expressing TNFR1 were pretreated with a pharmacological inhibitor of γ-secretase activity, DAPT (10 μm; 8 h) and subsequently stimulated with PMA (200 ng/ml; 2 h) to induce TNFR1 ectodomain shedding (Fig. 2a). In untreated cells, constitutive generation of TNFR1 CTF and ICD was detected (Fig. 2a, long exposure, lane 1), and stimulation of cells with PMA alone resulted in increased generation of the TNFR1 ICD (Fig. 2a, lane 2). Importantly, pretreatment of cells with the γ-secretase inhibitor DAPT alone or in combination with PMA completely suppressed formation of the TNFR1 ICD fragment. Inhibition of PMA-stimulated TNFR1 ICD formation by DAPT was paralleled by an accumulation of TNFR1 CTF levels (Fig. 2, a (lane 3) and b), suggesting a precursor-product relationship and that the TNFR1 ICD is indeed the product of γ-secretase-mediated cleavage of TNFR1 CTF. The potency of the pharmacological inhibitors of γ-secretase activity used in this study (DAPT and L-685,458) was verified in independent dose-response assays against the known γ-secretase substrates Notch, IL-1R1 and TNFR1 (data not shown).
FIGURE 2.

TNFR1 is a substrate for γ-secretase-dependent regulated intramembrane proteolysis. a, immunoassay of TNFR1 in whole cell lysates from HEK293T cells transiently transfected with TNFR1 left untreated or pretreated with the γ-secretase inhibitor DAPT (10 μm for 8 h) and/or treated with PMA (200 ng/ml for 2 h), followed by Western blotting analysis with an anti-TNFR1 C terminus-specific antibody. b, densitometry analysis of TNFR1 CTF normalized to total β-actin for all experimental conditions (n = 3). **, p < 0.01 (two-way ANOVA). Error bars, S.E. c, immunoassay of cell lysates from HEK293T cells transiently expressing TNFR1 and co-expressing wild type PS1 or dominant negative PS1 (PS1D257A/D385A) mutant, left untreated or treated with PMA (200 ng/ml for 2 h) as indicated, followed by Western blotting analysis with an anti-TNFR1 (C terminus-specific), anti-PS1 (C terminus-specific), and anti-β-actin antibodies. d, immunoassay of whole cell lysates from HEK293T cells transiently expressing TNFR1, pretreated with the γ-secretase inhibitor DAPT (10 μm for 8 h), alone or in combination with TNFα (30 ng/ml for 2 h) as indicated, followed by Western blotting analysis with an anti-TNFR1 C terminus-specific antibody. Western blotting data are from one experiment representative of three independent experiments.
Next, to verify that γ-secretase complexes did indeed mediate this proteolytic cleavage of TNFR1, HEK293T cells expressing TNFR1 were co-transfected with either wild type or catalytically inactive PS1D257A/D385A mutant (Fig. 2c), which abolishes endoproteolysis of PS1 and PS1-dependent γ-secretase activity (51). PMA stimulation of cells expressing TNFR1 alone or in combination with wild type PS1 resulted in robust generation of TNFR1 ICD; however, in cells expressing the PS1D257A/D385A mutant, PMA-stimulated generation of TNFR ICD was abolished, paralleled by increased detection of TNFR1 CTF (Fig. 2c, last lane). To examine the proteolysis of TNFR1 under more physiological conditions, first HEK293T cells expressing TNFR1 were pretreated with the γ-secretase inhibitor DAPT and stimulated with recombinant human TNFα (30 ng/ml; 2 h) (Fig. 2d). Similar to stimulation with PMA, cells treated with TNFα resulted in increased generation of TNFR1 ICD, which was inhibited in cultures pretreated with DAPT (Fig. 2d, lane 3). This responsiveness to TNFα ligand, PMA, DAPT and the catalytically inactive PS1D257A/D385A mutant is consistent with the cleavage profile of other receptors that undergo regulated intramembrane proteolysis and establish the 30–34 kDa band as being the predicted TNFR1 CTF and the 26–30-kDa fragment as the predicted γ-secretase-dependent TNFR1 ICD.
Next, the cleavage of endogenous TNFR1 was assessed in MCF-7 cells (Fig. 3a). Cells were treated in the absence or presence of the proteasome inhibitor epoxomicin, a compound used to inhibit the rapid degradation of proteins often associated with γ-secretase-mediated proteolysis. Western blotting analysis showed that in addition to the 55-kDa full-length TNFR1 protein, the endogenous 30–34-kDa TNFR1 CTF was present in unstimulated MCF-7 cells (Fig. 3a, lane 1). Significantly, the constitutively generated endogenous 26–30-kDa TNFR1 ICD was stabilized and clearly detected in the presence of epoxomicin (Fig. 3a, compare lanes 1 and 6). To verify that the generation of the 26–30-kDa ICD fragment was the result of cleavage of endogenous TNFR1 by γ-secretase, MCF-7 cells were pretreated with the γ-secretase inhibitor DAPT or L-685,458, in the absence or presence of TNFα and epoxomicin, as indicated (Fig. 3a, lanes 4 and 5). Western blotting analysis of TNFR1 revealed that stimulation with TNFα in the presence of either γ-secretase inhibitor DAPT or L-685,458 inhibited generation of the 26–30-kDa ICD (Fig. 3, a and b) and promoted an accumulation of the 30–34-kDa CTF (Fig. 3, a and c), consistent with the release of the ICD by γ-secretase cleavage of TNFR1 CTF.
FIGURE 3.

Cleavage of endogenous TNFR1. a, immunoassay of endogenous TNFR1 in whole cell lysates from MCF-7 cells left untreated or pretreated with the γ-secretase inhibitor DAPT (1 μm for 16 h) or L-685,458 (1 μm for 16 h) and/or treated with TNFα (30 ng/ml for 2 h) and/or epoxomycin (Epox) (1 μm for 2 h) as indicated, followed by Western blotting analysis with an anti-TNFR1 C terminus-specific antibody. Western blotting data are from one experiment representative of three independent experiments. Cell lysates were also probed for levels of β-actin to act as loading controls. b, densitometry analysis of TNFR1 ICD for all experimental conditions. The amount of TNFR1 ICD is expressed as TNFR1 ICD immunoreactivity ± S.E. (error bars) (n = 3); **, p < 0.01; ***, p < 0.001 (two-way ANOVA). c, densitometry analysis of TNFR1 CTF for all experimental conditions. The amount of TNFR1 CTF is expressed as TNFR1 CTF immunoreactivity ± S.E. (n = 3). d, immunoassay of endogenous TNFR1 in whole cell lysates from wild type and PS DKO MEFs treated with epoxomycin (1 μm for 2 h) and/or PMA (200 ng/ml for 1 h), followed by Western blotting analysis with an anti-TNFR1 C terminus-specific antibody. Western blotting data are from one experiment representative of three independent experiments. Cell lysates were also probed for levels of β-actin to act as loading controls. e, densitometry analysis of TNFR1 ICD normalized to total β-actin for all experimental conditions. The amount of TNFR1 ICD is expressed as TNFR1 ICD immunoreactivity ± S.E. (n = 3); *, p < 0.01; ***, p < 0.001 (two-way ANOVA). f, densitometry analysis of TNFR1 CTF normalized to total β-actin for all experimental conditions. The amount of TNFR1 CTF is expressed as TNFR1 CTF immunoreactivity ± S.E. (n = 3); ***, p < 0.001 (two-way ANOVA).
To further validate this proposal, we used immortalized MEFs derived from wild type (PS WT) and Psen1−/− and Psen2−/− double-knock-out (PS DKO) mice and assessed their responsiveness to PMA-induced cleavage of TNFR1 (Fig. 3, d–f). MEF cells were pretreated with PMA (200 ng/ml for 1 h) and the proteasome inhibitor epoxomicin (1 μm for 2 h) before harvesting, because constitutively generated endogenous TNFR1 ICD was not detectable (data not shown). Western blotting analysis revealed that constitutive generation of both TNFR1 CTF and TNFR1 ICD was observed in wild type MEFs, and PMA stimulation increased generation of the ICD fragment (Fig. 3, d (lanes 1 and 2) and e). Both constitutive and PMA-stimulated TNFR1 CTF were only weakly detected in wild type MEFs, suggesting rapid proteolysis by γ-secretase and conversion to TNFR1 ICD. In contrast, in PS DKO MEFs, although constitutive and PMA-induced production of TNFR1 CTF was observed (Fig. 3, d and f), no generation of TNFR1 ICD was evident (Fig. 3, d (lanes 3 and 4) and e), consistent with TNFR1 being a γ-secretase substrate. Our results collectively demonstrate that TNFR1 is a substrate for γ-secretase-mediated regulated intramembrane proteolysis in multiple cell types.
TNFR1 Internalization Is Required for γ-Secretase-mediated Cleavage
Endocytosis is an important regulatory mechanism controlling TNFR1 signaling (28, 35–37). After identifying TNFR1 as a new γ-secretase substrate, we next studied the subcellular occurrence of this cleavage event. Dynasore is a pharmacological inhibitor of dynamin (65, 66), a large GTPase that is a required component for clathrin-mediated endocytosis and of certain mechanisms of clathrin-independent endocytosis (67–69). To determine whether γ-secretase cleavage of TNFR1 occurred before or after receptor internalization, HEK293T cells expressing TNFR1 were untreated or pretreated with dynasore (50 μm for 2 h) and subsequently stimulated with PMA (200 ng/ml for 2 h) to induce ectodomain shedding and γ-secretase cleavage of TNFR1. In cells stimulated with PMA alone, generation of TNFR1 ICD (Fig. 4, a and b) and sTNFR1 (Fig. 4c) was clearly detected. In contrast, pretreatment with dynasore reduced PMA-induced generation of TNFR1 ICD (Fig. 4, a and b) and, as expected due to increased surface localization of TNFR1, increased release of constitutive and PMA-stimulated sTNFR1 (Fig. 4c). Dynamin-1 acts by facilitating release of newly formed endocytic vesicles from the plasma membrane and thereby plays a critical role in clathrin-mediated and non-clathrin-mediated endocytosis (69). To further examine the subcellular γ-secretase cleavage of TNFR1, we tested the ability of a dominant-negative K44A mutant of dynamin-1 (Dyn K44A) to inhibit γ-secretase cleavage of TNFR1. HEK293T cells were cotransfected with TNFR1 and wild type dynamin-1 (Dyn WT) or Dyn K44A mutant. In cells expressing Dyn WT, both constitutive and PMA-induced generation of TNFR1 ICD were observed (Fig. 4, d and e), and release of sTNFR1 was detected in conditioned medium (Fig. 4f). In contrast, in cells expressing the Dyn K44A mutant, a reduction in PMA-stimulated TNFR1 ICD formation was evident (Fig. 4, d and e), whereas constitutive and PMA-stimulated generation of TNFR1 CTF (Fig. 4d) and release of sTNFR1 (Fig. 4f) were comparable with Dyn WT-expressing cells.
FIGURE 4.

TNFR1 Internalization is required for γ-secretase-mediated cleavage. a, immunoassay of TNFR1 in whole cell lysates from HEK293T cells transiently transfected with TNFR1 left untreated or pretreated with clathrin-mediated endocytosis inhibitor dynasore (50 μm for 2 h) and/or treated with PMA (200 ng/ml for 2 h) followed by Western blotting analysis with an anti-TNFR1 C terminus-specific antibody. b, densitometry analysis of TNFR1 ICD normalized to total β-actin for all experimental conditions. The amount of TNFR1 ICD is expressed as TNFR1 ICD immunoreactivity ± S.E. (error bars) (n = 3). *, p < 0.05 (unpaired t test). c, ELISA of soluble TNFR1 (sTNFR1) in conditioned medium from HEK293T cells treated as in A. Data are expressed as a pictogram of sTNFR1 per milligram of total protein ± S.E. versus TNFR1 control (n = 3). d, immunoassay of HEK293T cell lysates transiently co-transfected with TNFR1 and Dyn WT or Dyn K44A and/or treated with PMA (200 ng/ml for 2 h) as indicated, followed by immunoblotting analysis with an anti-TNFR1 C terminus-specific antibody and anti-dynamin and anti-β-actin antibodies. e, densitometry analysis of TNFR1 ICD normalized to total β-actin for all experimental conditions. The amount of TNFR1 ICD is expressed as TNFR1 ICD immunoreactivity ± S.E. (n = 3). ***, p < 0.001 (unpaired t test). *, p < 0.05 (two-way ANOVA). f, ELISA of soluble TNFR1 (sTNFR1) in conditioned medium from HEK293T cells treated as in d. Data are expressed as a pictogram of sTNFR1/mg of total protein ± S.E. versus TNFR1 control (n = 3). g, immunoblotting analysis of HEK293T cells transiently expressing TNFR1 and TNFR1 W210A and/or treated with PMA (200 ng/ml for 2 h) as indicated, followed by immunoblotting analysis with an anti-TNFR1 C terminus-specific antibody. h, densitometry analysis of TNFR1 ICD normalized to total β-actin for all experimental conditions. The amount of TNFR1 ICD is expressed as TNFR1 ICD immunoreactivity ± S.E. (n = 3). ***, p < 0.001 (unpaired t test). i, ELISA of soluble TNFR1 (sTNFR1) in conditioned medium from HEK293T cells as in g. Data are expressed as pictogram of sTNFR1/mg of total protein ± S.E. versus TNFR1 control (n = 3).
Prompted by these results, we further examined the requirement of receptor internalization for γ-secretase cleavage of TNFR1. Schneider-Brachert et al. (28) identified a highly conserved internalization motif (YXXW) within the TNFR1 internalization domain and demonstrated that the single point mutation TNFR1 W210A is sufficient to inhibit TNFR1 internalization. HEK293T cells expressing wild type TNFR1 or TNFR1 W210A mutant were stimulated with PMA and lysed to assess the requirement of TNFR1 internalization for γ-secretase cleavage (Fig. 4, g and h). Again in cells expressing TNFR1, PMA stimulation increased γ-secretase-mediated generation of TNFR1 ICD, generation of which was completely inhibited in cells expressing the internalization-defective TNFR1 W210A mutant (Fig. 4, g and h). Conditioned medium from cells expressing wild type TNFR1 or TNFR1 W210A mutant was also collected, and sTNFR1 levels were measured (Fig. 4i). Increased levels of sTNFR1 were detected in cells expressing TNFR1 W210A, which we attributed to increased localization of the TNFR1 W210A receptor at the plasma membrane. Inhibition of TNFR1 internalization when bearing the TNFR1 W210A mutation or when TNFR1 was co-expressed with the Dyn K44A mutant was confirmed by FACS analysis of cell surface TNFR1, accumulation of TNFR1 CTF in cytosolic fractions, and inhibition of TNF-induced JNK activation (data not shown). Collectively, these findings indicate that receptor internalization is required for γ-secretase cleavage of TNFR1.
Presenilin Deficiency Is Associated with Enhanced Formation of TNFR1 Complex I and Reduced Assembly of Complex II
Activation of TNFR1 can lead to several distinct and opposing biological outcomes necessitating the accurate regulation of TNFR1 signaling (1, 26, 27). One regulatory mechanism ensuring TNFR1 signaling output is the spatial segregation of contrasting TNFR1 signaling complexes (71). TNFR1 complex I is located on the plasma membrane and controls expression of pro-inflammatory responses involving activation of the MAPK pathways (p38 and ERK) as well as the canonical NF-κB transcription factor that prevents induction of cell death, whereas complex II is formed after TNFR1 internalization and activates cell death processes (30).
Given the ability of γ-secretase to cleave TNFR1, we next investigated the importance of TNFR1 ectodomain shedding and γ-secretase cleavage in the recruitment and assembly of TNFR1 signaling complexes. First, HEK293T cells co-expressing TNFR1 and RIPK1 were either untreated or pretreated with the γ-secretase inhibitor DAPT and subsequently stimulated with PMA to induce TNFR1 ectodomain shedding and generation of TNFR1 CTF, as indicated (Fig. 5a, lane 3). TNFR1 was immunoprecipitated and assessed for co-precipitated levels of RIPK1. Under unstimulated conditions, modest association between TNFR1 and RIPK1 was evident, consistent with previous reports that recruitment of RIPK1 to TNFR1 requires TRADD (14). Interestingly, increased interaction between RIPK1 and TNFR1 was evident in cells stimulated to undergo PMA-mediated ectodomain shedding (Fig. 5a, lane 4), whereas inhibition of γ-secretase activity modestly reduced both basal and PMA-induced recruitment of RIPK1 to TNFR1 (Fig. 5a). In similar experiments, HEK293T cells co-expressing TNFR1 and TRADD were untreated or pretreated with DAPT alone or in combination with PMA (Fig. 5b). TNFR1 was immunoprecipitated and probed for the presence of co-precipitated TRADD. TNFR1 recruited TRADD under unstimulated conditions, and increased amounts of TRADD coprecipitated with TNFR1 following PMA-induced ectodomain shedding. Inhibition of γ-secretase activity did not significantly affect PMA-induced recruitment of TRADD to TNFR1 (Fig. 5b, lane 6). These studies suggest that TNFR1 ectodomain shedding affects recruitment of TRADD and RIPK1 to TNFR1, but the interaction was not critically regulated by γ-secretase activity, further implying that γ-secretase cleavage of TNFR1 may function downstream of receptor ectodomain shedding, internalization, and complex I formation.
FIGURE 5.

Presenilin deficiency is associated with enhanced formation of TNF-induced TNFR1 complex I and reduced assembly of complex II. a, HEK293T cells were co-transfected with the indicated constructs encoding TNFR1 and RIPK1, left untreated or pretreated with the γ-secretase inhibitor DAPT (10 μm; 8 h) and/or treated with PMA (200 ng/ml; 2 h), as indicated. Cell lysates were immunoprecipitated with an anti-TNFR1 antibody, and immunoprecipitates were probed with anti-TNFR1 or anti-RIPK1 antibody. Cell lysates, before immunoprecipitation (Input), were also analyzed by immunoblotting with anti-RIPK1, anti-TNFR1, and anti-β-actin antibodies. b, HEK293T cells were co-transfected with the indicated constructs encoding TNFR1 and TRADD, left untreated or pretreated with the γ-secretase inhibitor DAPT (10 μm for 8 h) and/or treated with PMA (200 ng/ml for 2 h), as indicated. Cell lysates were immunoprecipitated with an anti-TNFR1 antibody, and immunoprecipitates were probed with anti-TNFR1 or anti-TRADD antibody. Cell lysates, before immunoprecipitation (Input), were also analyzed by immunoblotting with anti-TRADD and anti-TNFR1 antibodies. Western blotting data are from one experiment representative of three independent experiments. c, wild type and PS DKO MEFs were treated with TNFα (40 ng/ml) for increasing times (0, 10, 20, 30, 45, and 60 min). Cell lysates were immunoprecipitated with an anti-TNFR1 antibody. Immunoprecipitates and cell lysates (Input) were probed for levels of endogenous RIPK1, TRAF2, and TNFR1 by immunoblotting. Cell lysates were also probed for levels of β-actin to act as loading controls. d, wild type and PS DKO MEFs were treated with TNFα (50 ng/ml) for the indicated times (0, 6, and 10 h). Cell lysates were immunoprecipitated with an anti-FADD antibody. Immunoprecipitates and cell lysates (Input) were probed for levels of endogenous RIPK1 and FADD by immunoblotting. Cell lysates were also probed for levels of β-actin to act as loading controls. *, light chain. Western blotting data are from one experiment representative of three independent experiments.
In order to define the relevance of γ-secretase cleavage of TNFR1 in TNF signaling, we next assessed TNF-induced formation of complex I and II in PS DKO MEFs. Wild type and PS DKO MEFs were stimulated with TNF for increasing times (0–30 min), and immunoprecipitated TNFR1 was examined for composition of endogenous TNFR1 complex I components (Fig. 5c). In wild type MEFs, we confirmed previously established observations that TNF stimulates a time-dependent recruitment and ubiquitination of RIPK1 and TRAF2 to TNFR1, forming complex I (Fig. 5c) (34–36). However, in PS DKO MEFs, increased levels of RIPK1 and TRAF2 co-precipitated with TNFR1, indicating increased formation of complex I in the absence of presenilins and γ-secretase activity. These data suggest that the loss of presenilins and/or γ-secretase activity increased the formation of TNF-induced complex I.
To determine the effect of presenilin and γ-secretase deficiency on assembly of TNFR1 complex II, wild type and PS DKO MEFs were stimulated with TNF for increased time periods (0–10 h), and formation of complex II was examined by immunoprecipitation of FADD and probing for RIPK1. In wild type cells, TNF induced ubiquitination of RIPK1, and binding of RIPK1 and FADD was clearly detected (Fig. 5d). In contrast, such formation of complex II was not observed in PS DKO MEFs, where only modest association between FADD and RIPK1 was observed (Fig. 5d). These data suggest that the loss of presenilins and/or γ-secretase activity disrupts assembly of TNF-induced complex II.
Presenilin Deficiency Is Associated with Reduced TNFα-mediated JNK Activation and Inhibition of CXCL1 Production
We next evaluated TNFα-mediated TNFR1 signal transduction in wild type and PS DKO MEFs. First to investigate the dependence of complex I-mediated activation of MAPK and NFκB signaling pathways upon presenilins and/or γ-secretase activity, we stimulated wild type and PS DKO MEFs with TNFα (10 ng/ml) for increasing time periods (10, 20, 30, 45, and 60 min) (Fig. 6a). In wild type MEFs, TNFα promoted a time-dependent activation of NF-κB, as indicated by TNFα-induced phosphorylation and degradation of IκB. PS DKO MEFs did not show any detectable alteration in TNFα-induced activation of NF-κB transcription factor when compared with wild type cells (Fig. 6a). Likewise, PS DKO MEFs showed no detectable defect in p38 MAPK activation compared with wild type cells, as detected by phospho-p38 antibody. However, a very significant reduction in TNFα-stimulated activation of JNK MAPK was observed in PS DKO MEFs when compared with wild type cells (Fig. 6a), demonstrating that loss of presenilins and γ-secretase activity prevented TNFα-induced JNK MAPK activation.
FIGURE 6.

Presenilin deficiency is associated with reduced JNK activity and inhibition of CXCL1 chemokine production. a, immunoblotting analysis of phosphorylated (P-) and total IκBα, p38, and JNK in lysates of WT and PS DKO MEFs stimulated with TNFα (10 ng/ml) for increasing times (0–60 min) (indicated above the lanes). Data are from one experiment representative of three independent experiments. b, ELISA of IL-6, IFNγ, IL-10, IL-12 (p70), IL-1β, and CXCL1 in medium from WT and PS DKO MEFs treated with TNFα (10 ng/ml) for 18 h. ***, p < 0.001 (two-way ANOVA analyses followed by Bonferroni post-test). Data are presented as an average of three independent experiments performed in duplicate (mean ± S.E. (error bars)). c, ELISA of IL-6, IFNγ, IL-10, IL-12 (p70), IL-1β, and CXCL1 in serum from WT and Psen1+/−/Psen2−/− mice, 2 h after intraperitoneal injection with endotoxin-free PBS or TNFα (100 μg/kg). Values are represented as mean ± S.E. of duplicate determinations per sample (n = 7–8 mice/group). Each symbol represents an individual mouse; small horizontal lines indicate the mean and S.E. (error bars). **, p < 0.01 (unpaired two-tailed Student's t test).
To further examine the loss of TNFα-mediated activation of JNK MAPK in PS DKO MEFs, we next examined whether presenilin deficiency altered TNFα-mediated pro-inflammatory cytokine production. Wild type and PS DKO MEFs were stimulated with TNFα (10 ng/ml) for 18 h, and production of cytokines IL-10, IL-6, IL-1β, IL-12 (p70), IFNγ, and chemokine CXCL1 (chemotactic cytokine) was analyzed by multiplex ELISA (Fig. 6b). In TNFα-stimulated wild type and PS DKO MEFs a comparable increase in the production of cytokines IL-10, IL-6, IL-1β, IL-12 (p70), and IFNγ was observed. However, production of the chemokine CXCL1 was significantly reduced in PS DKO MEFs (Fig. 6b), suggesting that lack of presenilins affects TNFα-mediated CXCL1 chemokine production. Interestingly, and consistent with our reported defect in JNK MAPK activation in PS DKO MEFs, it has been reported that CXCL1 production is dependent upon TNFα-stimulated JNK MAPK activation (72–74). To further validate this observation, we used wild type and the partial knock-out Psen1+/−/Psen2−/− murine model, which have the maximum reduction in presenilin expression that is compatible with viability. Age-matched animals were injected with TNFα (100 μg/kg) intraperitoneally, and production of cytokines IL-10, IL-6, IL-1β, IL-12 (p70), IFNγ, and CXCL1 in serum was analyzed 2 h postinjection. Consistent with the data obtained in vitro from MEFs, presenilin deficiency reduced chemokine CXCL1 production compared with the wild type counterpart, whereas production of all other examined cytokines and IFNγ was unaffected (Fig. 6c). These results are consistent with a model in which loss of presenilins negatively impacts TNFα-mediated JNK MAPK signaling.
Presenilin-deficient MEFs Have Increased Resistance to Apoptosis in Response to TNFα/Cycloheximide Co-stimulation
Given that presenilins and/or γ-secretase activity are important in the assembly/formation of complex II, we further investigated the effect of presenilin deficiency in a model of TNFα-induced apoptosis. Co-stimulation of cells with TNFα and the protein synthesis inhibitor cycloheximide is well known to induce apoptotic cell death, wherein cycloheximide prevents the induction of anti-apoptotic proteins. Co-treatment of wild type and PS DKO MEFs with TNFα and cycloheximide prompted the cells to develop apoptotic morphology, where they became rounded and non-adherent (Fig. 7a). Consistent with our data demonstrating reduced formation of pro-apoptotic complex II, PS DKO MEF cultures exhibited a dramatically reduced number of cells with apoptotic morphologies and annexin V positivity in response to the co-treatment (Fig. 7, a and b). Furthermore, whereas wild type MEFs displayed increased caspase-3 activation (Fig. 7c) and PARP cleavage (Fig. 7d) in response to the co-treatment, significantly reduced activation (cleavage) of caspase-3 and cleavage of PARP were evident in PS DKO MEFs. These molecular apoptotic signatures add further support to our hypothesis that γ-secretase cleavage of TNFR1 has a role in regulating the formation of complex II and induction of apoptosis in response to TNFα.
FIGURE 7.

Presenilin-deficient MEFs show increased resistance to TNF-induced apoptosis. a, wild type and PS DKO MEFs were treated in the absence (Control) or presence of murine TNFα (50 ng/ml) and cycloheximide (CHX) (10 μg/ml) for 6 h. Cells were photographed using a phase-contrast microscope at ×40 magnification. b, WT and PS DKO MEFs were treated in the absence (Control) or presence of murine TNFα (50 ng/ml) and cycloheximide (10 μg/ml) for 4 h. Percentage of apoptotic cells showing annexin V positivity. The columns show the means ± S.E. (error bars) of the mean of three independent experiments; **, p < 0.01 (two-way ANOVA). c and d, wild type and PS DKO MEFs were treated in the absence (0 h) or presence of murine TNFα (40 ng/ml) and cycloheximide (10 μg/ml) for 2, 4, 6, or 8 h, as indicated. Cell lysates were probed by immunoblotting for levels of cleaved caspase 3 c and cleaved PARP d. Cell lysates were also probed for levels of β-actin to act as loading controls. Data are from one experiment representative of at least three independent experiments.
Discussion
Our findings are summarized in the model depicted in Fig. 8, where, upon TNF ligand binding and receptor trimerization, TNFR1 undergoes TACE/ADAM17-mediated ectodomain shedding, releases sTNFR1, and generates membrane-anchored TNFR1 CTF, which is subsequently cleaved by the γ-secretase protease to generate a cytosolic TNFR1 ICD. Based on our data presented in this study, TNF-activated TNFR1 undergoes TACE/ADAM17-induced ectodomain shedding, and following receptor internalization, TNFR1 CTF undergoes γ-secretase cleavage. Furthermore, presenilins are required for TNFα-mediated JNK MAPK activation, assembly of complex II, and induction of apoptosis.
FIGURE 8.

Model of TNFR1-regulated intramembrane proteolysis and TNF-mediated pro-survival and apoptosis signaling pathways. TNF binding and trimerization of TNFR1 enables the recruitment of TRADD, RIPK1, TRAF2 or TRAF5, and the cIAPs, which collectively form a signaling composite called complex I (A). The resulting lysine 63-linked polyubiquitination of RIPK1 by TRAF2 and the cIAPs enables an interaction with the IκB kinase complex that mediates the phosphorylation and degradation of IκB inhibitory proteins and activation of the transcription factor NF-κB to promote cell survival and pro-inflammatory signaling pathways. Following TNFR1 ectodomain shedding (B) and internalization of TNFR1 (C), γ-secretase cleavage of TNFR1 (D) enables interactions between cytosolic TRADD and FADD, which enable the recruitment of procaspase-8, establishing the pro-apoptotic complex II (E), leading to the induction of apoptosis.
It had been commonly assumed that signaling events that are initiated by cell surface receptors, including G-protein-coupled receptors, Toll-like receptors, and the very prominent death receptors (TNFR1, FasR, and TRAIL-R1/2), are exclusively started and terminated at the cell surface. Recent studies reveal that many of these receptor-mediated signaling events do not always follow this established paradigm (7, 19, 24, 30, 71, 75, 76). In the new model, following ligand binding and cell surface receptor activation, receptors initiate signaling events from the plasma membrane and, subsequent to receptor internalization, can also propagate distinct signaling events from endosomal membranes. A high degree of regulation surrounds receptor-mediated signaling pathways, including post-translational modification of receptors, involving ubiquitination, phosphorylation, and proteolysis. In this study, we have added to the regulatory complexity of TNFα-mediated signaling through the identification of TNFR1 as a novel substrate for γ-secretase protease. TNFR1 undergoes TACE/ADAM17-mediated ectodomain shedding and produces biologically active sTNFR1 fragments, which results in reduced cell surface availability of TNFR1 and reduced TNF signaling. In this study, we have shown that, following ectodomain shedding, γ-secretase is capable of catalyzing the proteolytic cleavage of membrane-anchored TNFR1 CTF to generate the TNFR1 ICD. Consistent with regulated intramembrane proteolysis being a sequential proteolysis event (42, 43, 50), we show that inhibition of TACE/ADAM17-mediated TNFR1 ectodomain shedding prevented the cleavage of TNFR1 CTF by γ-secretase and generation of TNFR1 ICD. Using pharmacological inhibitors of γ-secretase activity, we demonstrated that TNF induced both TACE/ADAM17 and γ-secretase cleavage of TNFR1. Through genetic knock-out of the presenilins, we have further demonstrated the essential role of the presenilins in γ-secretase cleavage of TNFR1. Although it has been shown that γ-secretase cleavage of the insulin-like growth factor 1 receptor, Notch, IL-1 receptor I, p75NTR, or ErbB-4 receptor occurs in the presence of their corresponding ligands (40, 50, 58, 77–79), ligand-induced cleavage has not been shown for other receptors, such as the growth hormone receptor. Here we demonstrate that TNFα stimulation can induce ectodomain shedding, generation of TNFR1 CTF, and subsequent γ-secretase cleavage of TNFR1, perhaps pointing to a general mechanism of ligand-mediated regulated intramembrane proteolysis. Collectively, these molecular signatures of other γ-secretase substrates support our conclusion that TNFR1 is a genuine γ-secretase substrate.
TNFα-induced activation of pro-survival (MAPK and NF-κB) and apoptotic signaling pathways has been extensively studied and well elucidated (1, 6, 7, 16, 70). TNFR1 internalization and the formation of distinct TNFR1 signaling complexes or receptosomes provide the structural basis for spatial compartmentalization of TNFR1-mediated pro- and anti-apoptotic signaling pathways. A model was initially proposed (18) and subsequently refined wherein TNFR1-mediated signaling arises from two sequential signaling complexes: a plasma membrane-located pro-survival signaling complex I consisting of TNFR1, TRADD, RIPK1, TRAF2, and the cIAPs, which transmit a pro-inflammatory and cell survival signal via the activation of MAPK and NFκB, and, subsequent to TNFR1 internalization, an intracellular pro-apoptotic signaling complex II, which contains TRADD, RIPK1, TRAF2, FADD, and procaspase-8 (28, 30, 31, 34–37). In various cell types, TNFR1 internalization is mediated by clathrin-coated pit formation. We demonstrated the critical role of TNFR1 internalization in γ-secretase cleavage of TNFR1 by using a pharmacological inhibitor (dynasore) of dynamin (65), a large GTPase that is a required component for clathrin-mediated endocytosis, or expression of catalytically inactive dynamin (dynamin K44A) or by mutagenesis of the TNFR1 internalization domain (TNFR1 W210A). Indeed, expression of the TNFR1 W210A mutant that completely abolished TNFR1 internalization promoted the cell surface accumulation of TNFR1, increased generation of sTNFR1, prevented cytosolic localization of TNFR1 CTF, and inhibited formation of the γ-secretase-generated TNFR1 ICD.
Inhibition of TNFR1 internalization also has a role in mediating TNF cytotoxicity and promotion of pro-apoptosis signaling pathways (34, 35). It has been shown that inhibition of clathrin-coated pit formation by monodansyl cadaverine inhibited activation of the JNK MAPK and endolysosomal acid sphingomyelinase signaling pathways and blocked TNF-stimulated apoptosis (37). By contrast, inhibition of TNFR1 internalization had no effect on TNF-mediated activation of the plasma membrane-associated neutral sphingomyelinase or activation of the NF-κB signaling pathway. It was subsequently shown that inhibition of TNFR1 internalization prevented the recruitment of the pro-apoptotic proteins FADD and procaspase-8, inhibited the formation of complex II, and reduced TNF-induced apoptotic cell death (34). Consistent with these reports, in this study, we found that deficiency of the presenilins and γ-secretase activity inhibited activation of the JNK MAPK, antagonized JNK-dependent CXCL1 chemokine production, reduced formation of TNFR1 complex II, and blocked TNF-stimulated apoptosis. In contrast, loss of γ-secretase activity coincided with increased TNFR1 recruitment of TRAF2 and RIPK1 adaptor proteins and formation of complex I, and loss of γ-secretase activity had no measurable effect on TNF-mediated cell surface signaling events, activation of the p38 MAPK and NF-κB signaling pathways, and cytokine production.
From these data and other published works, we propose that mechanistically γ-secretase acts as an intracellular regulator of TNFR1-mediated pro-survival and pro-apoptotic signaling pathways. We propose that, following internalization of TNFR1, the proteolytic activity of γ-secretase regulates the release of cytosolic TNFR1 ICD. In addition, the presenilins are required for the recruitment and assembly of TNFR1 complex II, thereby facilitating TNFR1-mediated pro-apoptotic signaling pathways. Further research will no doubt elucidate the full regulatory mechanisms behind the pro-apoptotic switch in TNFR1 signaling.
In conclusion, our findings have added to the regulatory intricacy of TNF-mediated signaling through the identification of TNFR1 as a γ-secretase substrate. The presenilins are also required for TNF-mediated JNK MAPK activation, assembly of complex II, caspase activation, and induction of apoptosis. These regulatory steps may serve as a novel therapeutic target in the modulation of cellular responses to TNF, by inhibition of γ-secretase-mediated cleavage of TNFR1 to antagonize TNF-induced cell death and promote cell survival and pro-inflammatory signaling or stimulation of γ-secretase to promote TNFα-mediated cell death.
Author Contributions
J. C.-G., C. C.-V., V. A., N. S., and J. V. M. conceived and designed the experiments; J. C.-G., C. C.-V., V. A., N. S., and E. H. performed the experiments; J. C.-G., C. C.-V., V. A., N. S., J. C. P., and J. V. M. analyzed the data; and J. C.-G., C. C.-V., and J. V. M. wrote the paper. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgments
We thank B. de Strooper (KU Leuven, Belgium) and P. Saftig (CAU, Kiel, Germany) for the PS deficient MEFs. We thank members of the Signal Transduction Laboratory for experimental assistance and helpful discussion.
This work was supported by Science Foundation Ireland Grants 02/IN1/B218 and 09/IN.1/B2624 and Irish Research Council for Science, Engineering, and Technology Grant RS/2012/407. The authors declare that they have no conflicts of interest with the contents of this article.
- TNFR1 and 2
- TNF type I and II receptor, respectively
- TRADD
- TNFR1-associated death domain protein
- RIPK1
- receptor-interacting protein kinase 1
- TRAF
- TNF receptor-associated factor
- cIAP
- cellular inhibitor of apoptosis protein
- sTNFR1
- soluble TNFR1
- ADAM
- A disintegrin and metalloprotease
- PMA
- phorbol 12-myristate 13-acetate
- WB
- Western blotting
- IP
- immunoprecipitation
- PARP
- poly(ADP-ribose) polymerase
- MEF
- murine embryonic fibroblast
- CTF
- C-terminal fragment
- ICD
- intracellular domain
- DAPT
- N-[N-(3,5-difluorophenacetyl)-l-alanyl]-S-phenylglycine t-butyl ester
- DKO
- double knock-out
- Dyn
- dynamin
- PS
- presenilin
- FADD
- Fas-associated death domain protein
- TRAIL
- TNF-related apoptosis-inducing ligand
- ANOVA
- analysis of variance.
References
- 1. Hayden M. S., and Ghosh S. (2014) Regulation of NF-κB by TNF family cytokines. Semin. Immunol. 26, 253–266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Aggarwal B. B. (2003) Signalling pathways of the TNF superfamily: a double-edged sword. Nat. Rev. Immunol. 3, 745–756 [DOI] [PubMed] [Google Scholar]
- 3. Ashkenazi A., and Dixit V. M. (1998) Death receptors: signaling and modulation. Science 281, 1305–1308 [DOI] [PubMed] [Google Scholar]
- 4. Chen G., and Goeddel D. V. (2002) TNF-R1 signaling: a beautiful pathway. Science 296, 1634–1635 [DOI] [PubMed] [Google Scholar]
- 5. Tartaglia L. A., Rothe M., Hu Y. F., and Goeddel D. V. (1993) Tumor necrosis factor's cytotoxic activity is signaled by the p55 TNF receptor. Cell 73, 213–216 [DOI] [PubMed] [Google Scholar]
- 6. Silke J. (2011) The regulation of TNF signalling: what a tangled web we weave. Curr. Opin. Immunol. 23, 620–626 [DOI] [PubMed] [Google Scholar]
- 7. Cabal-Hierro L., and Lazo P. S. (2012) Signal transduction by tumor necrosis factor receptors. Cell. Signal. 24, 1297–1305 [DOI] [PubMed] [Google Scholar]
- 8. Tartaglia L. A., Ayres T. M., Wong G. H., and Goeddel D. V. (1993) A novel domain within the 55 kd TNF receptor signals cell death. Cell 74, 845–853 [DOI] [PubMed] [Google Scholar]
- 9. Tartaglia L. A., and Goeddel D. V. (1992) Two TNF receptors. Immunol. Today 13, 151–153 [DOI] [PubMed] [Google Scholar]
- 10. Tartaglia L. A., Pennica D., and Goeddel D. V. (1993) Ligand passing: the 75-kDa tumor necrosis factor (TNF) receptor recruits TNF for signaling by the 55-kDa TNF receptor. J. Biol. Chem. 268, 18542–18548 [PubMed] [Google Scholar]
- 11. Shu H. B., Takeuchi M., and Goeddel D. V. (1996) The tumor necrosis factor receptor 2 signal transducers TRAF2 and c-IAP1 are components of the tumor necrosis factor receptor 1 signaling complex. Proc. Natl. Acad. Sci. U.S.A. 93, 13973–13978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hsu H., Shu H. B., Pan M. G., and Goeddel D. V. (1996) TRADD-TRAF2 and TRADD-FADD interactions define two distinct TNF receptor 1 signal transduction pathways. Cell 84, 299–308 [DOI] [PubMed] [Google Scholar]
- 13. Hsu H., Xiong J., and Goeddel D. V. (1995) The TNF receptor 1-associated protein TRADD signals cell death and NF-κB activation. Cell 81, 495–504 [DOI] [PubMed] [Google Scholar]
- 14. Hsu H., Huang J., Shu H. B., Baichwal V., and Goeddel D. V. (1996) TNF-dependent recruitment of the protein kinase RIP to the TNF receptor-1 signaling complex. Immunity 4, 387–396 [DOI] [PubMed] [Google Scholar]
- 15. Meylan E., and Tschopp J. (2005) The RIP kinases: crucial integrators of cellular stress. Trends Biochem. Sci. 30, 151–159 [DOI] [PubMed] [Google Scholar]
- 16. Humphries F., Yang S., Wang B., and Moynagh P. N. (2015) RIP kinases: key decision makers in cell death and innate immunity. Cell Death Differ. 22, 225–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Legler D. F., Micheau O., Doucey M. A., Tschopp J., and Bron C. (2003) Recruitment of TNF receptor 1 to lipid rafts is essential for TNFα-mediated NF-κB activation. Immunity 18, 655–664 [DOI] [PubMed] [Google Scholar]
- 18. Micheau O., and Tschopp J. (2003) Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell 114, 181–190 [DOI] [PubMed] [Google Scholar]
- 19. Muppidi J. R., Tschopp J., and Siegel R. M. (2004) Life and death decisions: secondary complexes and lipid rafts in TNF receptor family signal transduction. Immunity 21, 461–465 [DOI] [PubMed] [Google Scholar]
- 20. Yamamoto M., Okamoto T., Takeda K., Sato S., Sanjo H., Uematsu S., Saitoh T., Yamamoto N., Sakurai H., Ishii K. J., Yamaoka S., Kawai T., Matsuura Y., Takeuchi O., and Akira S. (2006) Key function for the Ubc13 E2 ubiquitin-conjugating enzyme in immune receptor signaling. Nat. Immunol. 7, 962–970 [DOI] [PubMed] [Google Scholar]
- 21. Lee T. H., Shank J., Cusson N., and Kelliher M. A. (2004) The kinase activity of Rip1 is not required for tumor necrosis factor-α-induced IκB kinase or p38 MAP kinase activation or for the ubiquitination of Rip1 by Traf2. J. Biol. Chem. 279, 33185–33191 [DOI] [PubMed] [Google Scholar]
- 22. Bertrand M. J., Milutinovic S., Dickson K. M., Ho W. C., Boudreault A., Durkin J., Gillard J. W., Jaquith J. B., Morris S. J., and Barker P. A. (2008) cIAP1 and cIAP2 facilitate cancer cell survival by functioning as E3 ligases that promote RIP1 ubiquitination. Mol. Cell. 30, 689–700 [DOI] [PubMed] [Google Scholar]
- 23. Varfolomeev E., Goncharov T., Fedorova A. V., Dynek J. N., Zobel K., Deshayes K., Fairbrother W. J., and Vucic D. (2008) c-IAP1 and c-IAP2 are critical mediators of tumor necrosis factor α (TNFα)-induced NF-κB activation. J. Biol. Chem. 283, 24295–24299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Varfolomeev E., Goncharov T., and Vucic D. (2015) Roles of c-IAP proteins in TNF receptor family activation of NF-κB signaling. Methods Mol. Biol. 1280, 269–282 [DOI] [PubMed] [Google Scholar]
- 25. Castagna M., Takai Y., Kaibuchi K., Sano K., Kikkawa U., and Nishizuka Y. (1982) Direct activation of calcium-activated, phospholipid-dependent protein kinase by tumor-promoting phorbol esters. J. Biol. Chem. 257, 7847–7851 [PubMed] [Google Scholar]
- 26. Wertz I. E., and Dixit V. M. (2008) Ubiquitin-mediated regulation of TNFR1 signaling. Cytokine Growth Factor Rev. 19, 313–324 [DOI] [PubMed] [Google Scholar]
- 27. Wertz I. E., and Dixit V. M. (2010) Regulation of death receptor signaling by the ubiquitin system. Cell Death Differ. 17, 14–24 [DOI] [PubMed] [Google Scholar]
- 28. Schneider-Brachert W., Tchikov V., Neumeyer J., Jakob M., Winoto-Morbach S., Held-Feindt J., Heinrich M., Merkel O., Ehrenschwender M., Adam D., Mentlein R., Kabelitz D., and Schütze S. (2004) Compartmentalization of TNF receptor 1 signaling: internalized TNF receptosomes as death signaling vesicles. Immunity 21, 415–428 [DOI] [PubMed] [Google Scholar]
- 29. Fischer R., Maier O., Naumer M., Krippner-Heidenreich A., Scheurich P., and Pfizenmaier K. (2011) Ligand-induced internalization of TNF receptor 2 mediated by a di-leucin motif is dispensable for activation of the NFκB pathway. Cell. Signal. 23, 161–170 [DOI] [PubMed] [Google Scholar]
- 30. Tchikov V., Bertsch U., Fritsch J., Edelmann B., and Schütze S. (2011) Subcellular compartmentalization of TNF receptor-1 and CD95 signaling pathways. Eur. J. Cell Biol. 90, 467–475 [DOI] [PubMed] [Google Scholar]
- 31. Schütze S., and Schneider-Brachert W. (2009) Impact of TNF-R1 and CD95 internalization on apoptotic and antiapoptotic signaling. Results Probl. Cell Differ. 49, 63–85 [DOI] [PubMed] [Google Scholar]
- 32. Brissoni B., Agostini L., Kropf M., Martinon F., Swoboda V., Lippens S., Everett H., Aebi N., Janssens S., Meylan E., Felberbaum-Corti M., Hirling H., Gruenberg J., Tschopp J., and Burns K. (2006) Intracellular trafficking of interleukin-1 receptor I requires Tollip. Curr. Biol. 16, 2265–2270 [DOI] [PubMed] [Google Scholar]
- 33. McGettrick A. F., and O'Neill L. A. (2010) Localisation and trafficking of Toll-like receptors: an important mode of regulation. Curr. Opin. Immunol. 22, 20–27 [DOI] [PubMed] [Google Scholar]
- 34. Neumeyer J., Hallas C., Merkel O., Winoto-Morbach S., Jakob M., Thon L., Adam D., Schneider-Brachert W., and Schütze S. (2006) TNF-receptor I defective in internalization allows for cell death through activation of neutral sphingomyelinase. Exp. Cell Res. 312, 2142–2153 [DOI] [PubMed] [Google Scholar]
- 35. Schneider-Brachert W., Tchikov V., Merkel O., Jakob M., Hallas C., Kruse M. L., Groitl P., Lehn A., Hildt E., Held-Feindt J., Dobner T., Kabelitz D., Krönke M., and Schütze S. (2006) Inhibition of TNF receptor 1 internalization by adenovirus 14.7K as a novel immune escape mechanism. J. Clin. Invest. 116, 2901–2913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Fritsch J., Stephan M., Tchikov V., Winoto-Morbach S., Gubkina S., Kabelitz D., and Schütze S. (2014) Cell fate decisions regulated by K63 ubiquitination of tumor necrosis factor receptor 1. Mol. Cell. Biol. 34, 3214–3228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Schütze S., Machleidt T., Adam D., Schwandner R., Wiegmann K., Kruse M. L., Heinrich M., Wickel M., and Krönke M. (1999) Inhibition of receptor internalization by monodansylcadaverine selectively blocks p55 tumor necrosis factor receptor death domain signaling. J. Biol. Chem. 274, 10203–10212 [DOI] [PubMed] [Google Scholar]
- 38. Bartsch J. W., Wildeboer D., Koller G., Naus S., Rittger A., Moss M. L., Minai Y., and Jockusch H. (2010) Tumor necrosis factor-α (TNF-α) regulates shedding of TNF-α receptor 1 by the metalloprotease-disintegrin ADAM8: evidence for a protease-regulated feedback loop in neuroprotection. J. Neurosci. 30, 12210–12218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Le Gall S. M., Bobé P., Reiss K., Horiuchi K., Niu X. D., Lundell D., Gibb D. R., Conrad D., Saftig P., and Blobel C. P. (2009) ADAMs 10 and 17 represent differentially regulated components of a general shedding machinery for membrane proteins such as transforming growth factor α, l-selectin, and tumor necrosis factor α. Mol. Biol. Cell 20, 1785–1794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Elzinga B. M., Twomey C., Powell J. C., Harte F., and McCarthy J. V. (2009) Interleukin-1 receptor type 1 is a substrate for γ-secretase-dependent regulated intramembrane proteolysis. J. Biol. Chem. 284, 1394–1409 [DOI] [PubMed] [Google Scholar]
- 41. Urra S., Escudero C. A., Ramos P., Lisbona F., Allende E., Covarrubias P., Parraguez J. I., Zampieri N., Chao M. V., Annaert W., and Bronfman F. C. (2007) TrkA receptor activation by nerve growth factor induces shedding of the p75 neurotrophin receptor followed by endosomal gamma-secretase-mediated release of the p75 intracellular domain. J. Biol. Chem. 282, 7606–7615 [DOI] [PubMed] [Google Scholar]
- 42. Jurisch-Yaksi N., Sannerud R., and Annaert W. (2013) A fast growing spectrum of biological functions of γ-secretase in development and disease. Biochim. Biophys. Acta 1828, 2815–2827 [DOI] [PubMed] [Google Scholar]
- 43. Brown M. S., Ye J., Rawson R. B., and Goldstein J. L. (2000) Regulated intramembrane proteolysis: a control mechanism conserved from bacteria to humans. Cell 100, 391–398 [DOI] [PubMed] [Google Scholar]
- 44. Kimberly W. T., Xia W., Rahmati T., Wolfe M. S., and Selkoe D. J. (2000) The transmembrane aspartates in presenilin 1 and 2 are obligatory for γ-secretase activity and amyloid β-protein generation. J. Biol. Chem. 275, 3173–3178 [DOI] [PubMed] [Google Scholar]
- 45. Wolfe M. S., Xia W., Ostaszewski B. L., Diehl T. S., Kimberly W. T., and Selkoe D. J. (1999) Two transmembrane aspartates in presenilin-1 required for presenilin endoproteolysis and γ-secretase activity. Nature 398, 513–517 [DOI] [PubMed] [Google Scholar]
- 46. Wolfe M. S. (2008) γ-Secretase: structure, function, and modulation for Alzheimer's disease. Curr. Top. Med. Chem. 8, 2–8 [DOI] [PubMed] [Google Scholar]
- 47. De Strooper B., Saftig P., Craessaerts K., Vanderstichele H., Guhde G., Annaert W., Von Figura K., and Van Leuven F. (1998) Deficiency of presenilin-1 inhibits the normal cleavage of amyloid precursor protein. Nature 391, 387–390 [DOI] [PubMed] [Google Scholar]
- 48. Wakabayashi T., and De Strooper B. (2008) Presenilins: members of the γ-secretase quartets, but part-time soloists too. Physiology 23, 194–204 [DOI] [PubMed] [Google Scholar]
- 49. De Strooper B., Iwatsubo T., and Wolfe M. S. (2012) Presenilins and γ-secretase: structure, function, and role in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2, a006304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. McCarthy J. V., Twomey C., and Wujek P. (2009) Presenilin-dependent regulated intramembrane proteolysis and γ-secretase activity. Cell. Mol. Life Sci. 66, 1534–1555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Fluhrer R., Grammer G., Israel L., Condron M. M., Haffner C., Friedmann E., Böhland C., Imhof A., Martoglio B., Teplow D. B., and Haass C. (2006) A γ-secretase-like intramembrane cleavage of TNFα by the GxGD aspartyl protease SPPL2b. Nat. Cell Biol. 8, 894–896 [DOI] [PubMed] [Google Scholar]
- 52. Kuhn P. H., Marjaux E., Imhof A., De Strooper B., Haass C., and Lichtenthaler S. F. (2007) Regulated intramembrane proteolysis of the interleukin-1 receptor II by α-, β-, and γ-secretase. J. Biol. Chem. 282, 11982–11995 [DOI] [PubMed] [Google Scholar]
- 53. Chalaris A., Gewiese J., Paliga K., Fleig L., Schneede A., Krieger K., Rose-John S., and Scheller J. (2010) ADAM17-mediated shedding of the IL6R induces cleavage of the membrane stub by γ-secretase. Biochim. Biophys. Acta 1803, 234–245 [DOI] [PubMed] [Google Scholar]
- 54. Schulte A., Schulz B., Andrzejewski M. G., Hundhausen C., Mletzko S., Achilles J., Reiss K., Paliga K., Weber C., John S. R., and Ludwig A. (2007) Sequential processing of the transmembrane chemokines CX3CL1 and CXCL16 by α- and γ-secretases. Biochem. Biophys. Res. Commun. 358, 233–240 [DOI] [PubMed] [Google Scholar]
- 55. Jung K. M., Tan S., Landman N., Petrova K., Murray S., Lewis R., Kim P. K., Kim D. S., Ryu S. H., Chao M. V., and Kim T. W. (2003) Regulated intramembrane proteolysis of the p75 neurotrophin receptor modulates its association with the TrkA receptor. J. Biol. Chem. 278, 42161–42169 [DOI] [PubMed] [Google Scholar]
- 56. Kenchappa R. S., Zampieri N., Chao M. V., Barker P. A., Teng H. K., Hempstead B. L., and Carter B. D. (2006) Ligand-dependent cleavage of the P75 neurotrophin receptor is necessary for NRIF nuclear translocation and apoptosis in sympathetic neurons. Neuron 50, 219–232 [DOI] [PubMed] [Google Scholar]
- 57. Parkhurst C. N., Zampieri N., and Chao M. V. (2010) Nuclear localization of the p75 neurotrophin receptor intracellular domain. J. Biol. Chem. 285, 5361–5368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Powell J. C., Twomey C., Jain R., and McCarthy J. V. (2009) Association between Presenilin-1 and TRAF6 modulates regulated intramembrane proteolysis of the p75NTR neurotrophin receptor. J. Neurochem. 108, 216–230 [DOI] [PubMed] [Google Scholar]
- 59. Kirschenbaum F., Hsu S. C., Cordell B., and McCarthy J. V. (2001) Glycogen synthase kinase-3β regulates presenilin 1 C-terminal fragment levels. J. Biol. Chem. 276, 30701–30707 [DOI] [PubMed] [Google Scholar]
- 60. Kirschenbaum F., Hsu S. C., Cordell B., and McCarthy J. V. (2001) Substitution of a glycogen synthase kinase-3β phosphorylation site in presenilin 1 separates presenilin function from β-catenin signaling. J. Biol. Chem. 276, 7366–7375 [DOI] [PubMed] [Google Scholar]
- 61. Herreman A., Hartmann D., Annaert W., Saftig P., Craessaerts K., Serneels L., Umans L., Schrijvers V., Checler F., Vanderstichele H., Baekelandt V., Dressel R., Cupers P., Huylebroeck D., Zwijsen A., Van Leuven F., and De Strooper B. (1999) Presenilin 2 deficiency causes a mild pulmonary phenotype and no changes in amyloid precursor protein processing but enhances the embryonic lethal phenotype of presenilin 1 deficiency. Proc. Natl. Acad. Sci. U.S.A. 96, 11872–11877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Tournoy J., Bossuyt X., Snellinx A., Regent M., Garmyn M., Serneels L., Saftig P., Craessaerts K., De Strooper B., and Hartmann D. (2004) Partial loss of presenilins causes seborrheic keratosis and autoimmune disease in mice. Hum. Mol. Genet. 13, 1321–1331 [DOI] [PubMed] [Google Scholar]
- 63. Arribas J., and Borroto A. (2002) Protein ectodomain shedding. Chem. Rev. 102, 4627–4638 [DOI] [PubMed] [Google Scholar]
- 64. Peschon J. J., Slack J. L., Reddy P., Stocking K. L., Sunnarborg S. W., Lee D. C., Russell W. E., Castner B. J., Johnson R. S., Fitzner J. N., Boyce R. W., Nelson N., Kozlosky C. J., Wolfson M. F., Rauch C. T., Cerretti D. P., Paxton R. J., March C. J., and Black R. A. (1998) An essential role for ectodomain shedding in mammalian development. Science 282, 1281–1284 [DOI] [PubMed] [Google Scholar]
- 65. Macia E., Ehrlich M., Massol R., Boucrot E., Brunner C., and Kirchhausen T. (2006) Dynasore, a cell-permeable inhibitor of dynamin. Dev. Cell 10, 839–850 [DOI] [PubMed] [Google Scholar]
- 66. Stenmark H. (2009) Rab GTPases as coordinators of vesicle traffic. Nat. Rev. Mol. Cell Biol. 10, 513–525 [DOI] [PubMed] [Google Scholar]
- 67. Grant B. D., and Donaldson J. G. (2009) Pathways and mechanisms of endocytic recycling. Nat. Rev. Mol. Cell Biol. 10, 597–608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Traub L. M. (2009) Tickets to ride: selecting cargo for clathrin-regulated internalization. Nat. Rev. Mol. Cell Biol. 10, 583–596 [DOI] [PubMed] [Google Scholar]
- 69. Doherty G. J., and McMahon H. T. (2009) Mechanisms of endocytosis. Annu. Rev. Biochem. 78, 857–902 [DOI] [PubMed] [Google Scholar]
- 70. Wajant H., and Scheurich P. (2011) TNFR1-induced activation of the classical NF-κB pathway. FEBS J. 278, 862–876 [DOI] [PubMed] [Google Scholar]
- 71. Schneider-Brachert W., Heigl U., and Ehrenschwender M. (2013) Membrane trafficking of death receptors: implications on signalling. Int. J. Mol. Sci. 14, 14475–14503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Gao Y. J., Zhang L., Samad O. A., Suter M. R., Yasuhiko K., Xu Z. Z., Park J. Y., Lind A. L., Ma Q., and Ji R. R. (2009) JNK-induced MCP-1 production in spinal cord astrocytes contributes to central sensitization and neuropathic pain. J. Neurosci. 29, 4096–4108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Lo H. M., Lai T. H., Li C. H., and Wu W. B. (2014) TNF-α induces CXCL1 chemokine expression and release in human vascular endothelial cells in vitro via two distinct signaling pathways. Acta Pharmacol. Sin. 35, 339–350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Lo H. M., Shieh J. M., Chen C. L., Tsou C. J., and Wu W. B. (2013) Vascular endothelial growth factor induces CXCL1 chemokine release via JNK and PI-3K-dependent pathways in human lung carcinoma epithelial cells. Int. J. Mol. Sci. 14, 10090–10106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Irannejad R., and von Zastrow M. (2014) GPCR signaling along the endocytic pathway. Curr. Opin. Cell Biol. 27, 109–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Tsvetanova N. G., Irannejad R., and von Zastrow M. (2015) G protein-coupled receptor (GPCR) signaling via heterotrimeric G proteins from endosomes. J. Biol. Chem. 290, 6689–6696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. McElroy B., Powell J. C., and McCarthy J. V. (2007) The insulin-like growth factor 1 (IGF-1) receptor is a substrate for γ-secretase-mediated intramembrane proteolysis. Biochem. Biophys. Res. Commun. 358, 1136–1141 [DOI] [PubMed] [Google Scholar]
- 78. Berezovska O., Jack C., McLean P., Aster J. C., Hicks C., Xia W., Wolfe M. S., Weinmaster G., Selkoe D. J., and Hyman B. T. (2000) Rapid Notch1 nuclear translocation after ligand binding depends on presenilin-associated γ-secretase activity. Ann. N.Y. Acad. Sci. 920, 223–226 [DOI] [PubMed] [Google Scholar]
- 79. Schroeter E. H., Kisslinger J. A., and Kopan R. (1998) Notch-1 signalling requires ligand-induced proteolytic release of intracellular domain. Nature 393, 382–386 [DOI] [PubMed] [Google Scholar]