

Abstract
SecYEG protein of bacteria or Sec61αβγ of eukaryotes is a universally conserved heterotrimeric protein channel complex that accommodates the partitioning of membrane proteins into the lipid bilayer as well as the secretion of proteins to the trans side of the plasma or endoplasmic reticular membrane, respectively. SecYEG function is facilitated by cytosolic partners, mainly a nascent chain-ribosome complex or the SecA ATPase motor protein. Extensive efforts utilizing both biochemical and biophysical approaches have been made to determine whether SecYEG functions as a monomer or a dimer, but such approaches have often generated conflicting results. Here we have employed site-specific in vivo photo-cross-linking or cysteine cross-linking, along with co-immunoprecipitation or SecA footprinting techniques to readdress this issue. Our findings show that the SecY dimer to monomer ratio is relatively constant regardless of whether translocons are actively engaged with protein substrate or not. Under the former conditions the SecY dimer can be captured associated with a translocon-jammed substrate, indicative of SecY dimer function. Furthermore, SecA ATPase can be cross-linked to two copies of SecY when the complex contains a translocation intermediate. Collectively, our results suggest that SecYEG dimers are functional units of the translocon.
Keywords: cysteine-mediated cross-linking, membrane protein, oligomer, protein chimera, protein translocation, SecY, photo-cross-linking, translocon jamming
Introduction
Up to one-third of bacterial proteomes consist of proteins that must be inserted into or secreted across the plasma membrane. Such proteins are generally synthesized with one or more signal-anchor sequences that determine their ultimate fate and topology. Proteins that contain more hydrophobic signal-anchor sequences (typically membrane proteins) are directed into the co-translational translocation pathway, whereby their ribosome-nascent chain complexes are guided to the SecYEG channel complex by interaction of the signal recognition particle both with the emerging signal-anchor sequence and its membrane receptor located adjacent to the channel complex. After ribosome-translocon docking, it is thought that the elongation phase of translation provides energy for integration of proteins into the membrane along with the thermodynamics of protein folding in this environment (1). On the other hand, proteins that contain moderately hydrophobic signal-anchor sequences (typically secretory preproteins) are directed to the post-translational translocation pathway (2). In this case, incomplete or fully translated, but partially folded preproteins are bound by cytosolic chaperones (SecB in the case of Gram-negative bacteria like Escherichia coli), keeping them competent for transport. SecB then targets the pre-protein to SecYEG-bound SecA protein, which recognizes both the signal peptide and SecB, and promotes the SecB-preprotein release step. In addition to facilitating preprotein targeting to the translocon, SecA is also a motor protein that utilizes its ATP-promoted conformational cycles to drive protein transport by a still poorly defined mechanism (3, 4). A popular model based on the SecA-SecYEG x-ray structure suggests that the SecA two-helix finger serves as a molecular ratchet to processively bind and insert substrate preproteins into the mouth of the SecYEG channel utilizing consecutive rounds of ATP hydrolysis (5, 6). The nature and depth of SecA insertion into the translocon channel remains unclear since its initial discovery over two decades ago (4, 7).
The translocon channel complex is a universally conserved, heterotrimeric protein, Sec61αβγ in eukaryotes or SecYEG in bacteria. It forms an hourglass-shaped structure composed of 10 transmembrane helices of Sec61α/SecY hinged by Sec61γ/SecE on one side, allowing for a clamp-like action that opens and closes the lateral gate region on the side opposite the hinge. Signal-anchor sequences insert into the lateral gate region of the channel (8), allowing for the subsequent release of membrane proteins into the lipid bilayer. The channel complex also contains a constriction pore at its center as well as a small helical plug domain that relocates to the channel exterior to open a path for the translocation of secretory proteins across the membrane (9, 10).
Early biochemical and structural work pointed to the existence of SecYEG dimers and their potential physiological importance in protein transport. For example, the purified SecYEG complex in detergent or reconstituted into proteoliposomes formed dimers and tetramers in a concentration-dependent manner that was enhanced by the presence of translocation partners such as SecA, preprotein, or cardiolipin (11–15). Disulfide cross-linking studies of inverted membrane vesicles demonstrated the presence of a SecYEG dimer complex by the formation of SecY-SecY or SecE-SecE homo-dimers (16–19). Moreover, when purified SecYEG was reconstituted into nanodisks as either monomers or dimers, only the latter unit was capable of activating SecA translocation ATPase activity: a requirement for protein transport in vitro (20). Furthermore, a genetically fused SecY dimer was found to display intragenic complementation both in vivo and in vitro: a result most simply understood by assuming a functional dimer state at some point in the protein translocation cycle (20, 21).
Indeed considerable controversy exists as to whether the translocon channel complex functions as a monomer or a dimer. Recent crystal structures of the SecY complex with or without its associated SecA ATPase protein clearly reveal a protein-conducting channel within a single SecY subunit (5, 9, 22, 23). Although structures of the ribosome-nascent chain-SecYEG complex obtained by cryoelectron microscopy also support this view, they differ in the presence of a second, non-translocating SecYEG protomer within the complex (24–27). Likewise, single molecule studies have also lead to conflicting results on the active state of the translocon. For example, a fluorescence-based study using differentially labeled SecYEG proteins reconstituted into giant unilamellar vesicles failed to detect the presence of SecYEG dimers during SecA or protein substrate-bound conditions (28). However, another study, where SecYEG monomers and artificially stabilized dimers were compared, found that whereas monomers sufficed for SecA and preprotein binding, dimers were required for active transport (29). An in vivo study was performed employing disulfide cross-linking at the SecY dimer interface to assess the oligomeric status of the translocon under translocation conditions that simulate ongoing protein transport (by utilizing a substrate that jammed the translocon channel). The authors found that the SecY dimer level decreased precipitously when channels were jammed with appropriate substrate proteins, suggesting that protein transport occurs from a SecYEG monomer (30). One limitation of this latter study, however, is that substrate jamming may have led to a conformational change of SecY that prevented disulfide cross-linking. These authors also functionally disrupted SecY dimer assembly by mutagenesis at the dimer interface, further supporting the functional monomer view. Adding to the ongoing controversy, a recent study utilizing a highly efficient SecYEG reconstitution system found that preproteins apparently differed in their translocon stoichiometry requirement, where a monomer or dimer was required depending on the preprotein species under study (31).
Studies assessing the functional size of the translocon channel are difficult to reconcile with a functional monomer subunit that contains the observed channel dimensions. For example, a study employing pro-OmpA conjugated to rigid tetraarylmethane derivatives estimated a channel diameter of greater than 22–24 Å (32). Similarly, a fluorescence-based study of the mammalian endoplasmic reticular channel dimensions utilizing quenchers of defined sizes placed the channel diameter within a range of 40–60 Å (33). Both of these findings exceed the maximum expandable size of a single channel, and they would be more consistent with a dimer-based, composite channel.
Here we have utilized both in vivo photo-cross-linking and disulfide cross-linking approaches to investigate the functional oligomeric state of the SecY channel complex, given the conflicting claims present in the literature. The former method was developed by Schultz and co-workers (for review, see Ref. 34). It uses a specially engineered amber suppressor tRNA/tRNA synthetase pair that incorporates the photo-activatable phenylalanine derivative pBpA2 into the target protein of interest. Excitation by light at ∼365 nm generates a triplet diradical that reacts with nearly C-H acceptors (within 5 Å) to form a covalent cross-link. This technique offers a physiological method to study protein-protein interaction. The results presented below indicate that SecY dimers comprise at least part of the active protein translocational unit.
Experimental Procedures
Chemicals, Media, Strains, and Plasmids
LB (Miller) broth, IPTG, and arabinose were purchased from Fisher Scientific. pBpA and maltose were purchased from Bachem and Difco, respectively. DDM was purchased from Anatrace, whereas anti-GFP beads were from ChromoTek. QuikChangeTM and Wizard Plus SV Miniprep DNA Purification System kits were obtained from Stratagene and Promega, respectively. Restriction enzymes were obtained from New England Biolabs, whereas the WesternBrightTM Sirius enhanced chemiluminescence kit was obtained from Advansta. Protease Inhibitor Mixture was obtained from Sigma. Most other common laboratory chemicals were obtained from the latter supplier or Fisher Scientific and were laboratory grade or better. Mouse anti-c-Myc monoclonal antibody and HRP-conjugated goat anti-mouse antibody were obtained from GenScript, whereas chicken anti-GFP antibody and HRP-conjugated goat anti-rabbit antibody were obtained from Abcam. HRP-conjugated goat anti-chicken antibody was obtained from Jackson ImmunoResearch Laboratories. Peptide affinity-purified SecY antisera was prepared by hyper-immunizing rabbits to a peptide identical to the carboxyl terminus of SecY (CYESALKKANLKGYGR) conjugated to keyhole limpet hemocyanin by maleimide chemistry utilizing Tana Laboratories, LC (Houstin, TX) for peptide synthesis, protein carrier conjugation, immunization, and peptide affinity purification of the antisera. E. coli BLR(λDE3) (F−ompT hsdS (rB−mB−) gal dcm Δ(srl-recA)306::Tn10 (TetR)) was obtained from Stratagene, whereas its isogenic BL26 (λDE3) ΔlacU169 recA+ derivative was obtained from Bill Studier (35). BL26.1(λDE3) is a recA::KAN version of this latter strain made by P1 transduction. MM18.7 is a recA derivative of MM18 containing the malE-lacZ72-47(Hyb) fusion that has been described previously (36). MC4100.2(λDE3) is a recA1 srl::Tn10 derivative of MC4100 (37) that was lysogenized with λDE3, which has been previously described (35). The pSup-pBpARS-6TRN plasmid encoding the Methanococcus jannaschii amber suppressor tRNA-tRNA synthetase pair that efficiently incorporates pBpA in place of an amber codon has been described previously (34). A series of pCDFT7secYEG plasmids with secYEG under control of the T7 promoter with amino-terminal c-Myc tags on secY or secE or a carboxyl-terminal c-Myc tag on secG have been described (38). The pBAD-OmpA-GFP plasmid carrying the arabinose-inducible, OmpA-GFP jamming chimera was obtained from Tom Rapoport (30). pBAD-secA-OmpA-GFP was created by fusing secA onto the N terminus of ompA-GFP. For this purpose, pT7-secA-his (39) was modified using QuikChangeTM mutagenesis by making synonymous codon changes eliminating two internal NcoI sites within secA, and a NdeI site at the secA start codon was changed into a NcoI site. This allowed the entire secA gene to be isolated as an NcoI-XhoI restriction fragment. pBAD-OmpA-GFP was modified using QuikChangeTM mutagenesis to insert a XhoI site after the NcoI site that includes the ompA start codon. This allowed insertion of the secA fragment into the NcoI-XhoI cleaved pBAD-OmpA-GFP vector fragment to form the SecA-OmpA-GFP trimera, which was verified by DNA sequence analysis utilizing the University of Pennsylvania DNA Sequencing Facility. All codon substitutions within plasmid-borne secY, secE, secA, or ompA genes were made by QuikChangeTM mutagenesis and also verified by DNA sequence analysis.
In Vivo Photo-cross-linking
A freshly struck out single colony of BLR(λDE3) containing pSup-BpARS-6TRN and pCDFT7secYmycEG or pCDFT7secYEmycG plasmids with the given secY or secE amber mutation, respectively, was inoculated into LB media supplemented with appropriate antibiotics (25 μg/ml of chloramphenicol, 50 μg/ml of streptomycin, and 100 μg/ml of ampicillin as needed) and grown overnight at 37 °C with shaking at 250 rpm. The overnight culture was diluted 1:50 into LB media supplemented with appropriate antibiotics and 1 mm pBpA and grown until A600 reached 0.3, when SecYEG was induced with IPTG at a final concentration of 1 mm for 2 h. All subsequent steps were done at 4 °C or on ice. To adjust for somewhat different cell densities, 4.8 A600 cell equivalent of each culture was harvested by sedimentation in a microcentrifuge at 14,000 rpm for 5 min, washed with 5 ml of PBS (10 mm sodium phosphate, pH 7.5, 140 mm NaCl), and resuspended in 4 ml of PBS buffer. 2-ml samples were UV irradiated on ice at 365 nm for 20 min using a Rayonet 2000 UV cross-linker (Southern New England Ultraviolet Company), whereas 2 ml of non-irradiated samples served as negative controls. Each sample was sedimented, resuspended in 1 ml of breakage buffer (10 mm Tris-HCl, pH 7.5, 1 mm EDTA, 0.25 mm PMSF, 1 mm DTT, 100 μg/ml of RNase, 100 μg/ml of DNase, 60 μg/ml of lysozyme, 1× Protease inhibitor mixture), and cells were placed in a polycarbonate tube and disrupted with 30-s bursts of a cup horn sonicator (Heat Systems) until near clarity. Unbroken cells were removed by sedimentation in a microcentrifuge at 14,000 rpm for 5 min. Supernatant was isolated and sedimented in a Sorvall S120 AT2 rotor at 82,000 rpm for 30 min at 4 °C to isolate the membrane fraction. Each membrane pellet was solubilized in 60 μl of ABB buffer (5% SDS, 10 mm Tris-Cl, pH 8, 1 mm EDTA) with constant stirring for 1 h at 37 °C, when 20 μl of 4× sample buffer (8% SDS, 500 mm Tris-HCl, pH 6.8, 20% 2-mercaptoethanol, 60% glycerol, 0.02% bromphenol blue) was added, and stirring continued for an additional 10 min. 15-μl samples were loaded onto a 15% SDS-PAGE gel, which was run at 100 V at 4 °C until the dye front reached the bottom. Western transfers were performed at 100 V for 1 h, and nitrocellulose membranes were blocked overnight with 10 ml of TBS buffer (20 mm Tris-HCl, pH 7.5, 140 mm NaCl, 0.25% Tween 20) supplemented with 10% nonfat dry milk. The membrane was probed with mouse anti-c-Myc monoclonal antibody, followed by HRP-conjugated goat anti-mouse antibody, both at a 1:5,000 dilution. SecYY was probed separately with rabbit anti-c-term-SecY antibody and HRP-conjugated goat anti-rabbit antibody at a 1:5,000 dilution. Proteins were visualized using a WesternBrightTM Sirius kit as described by the manufacturer.
Translocon Jamming
An overnight culture of MC4100.2(λDE3) containing pSup-pBpARS-6TRN, pCDFT7secYmycEG with a given secY amber mutation, and pBAD-OmpA-GFP plasmids was grown in LB containing appropriate antibiotics, 30 μm IPTG, and 0.2% maltose as described above under “In Vivo Photo-cross-linking.” It was then subcultured 1:100 into LB supplemented similarly except for addition of 1 mm pBpA and omission of chloramphenicol. The culture was grown until A600 reached 0.15, when arabinose was added to a final concentration of 0.2%. For each time point analyzed, 6 A600 cell equivalents of culture was harvested and prepared for UV irradiation and subsequent analysis as described above. For detection of the OmpA-GFP chimera, 5 μl of the final non-irradiated sample was analyzed by SDS-PAGE, and Western blots were probed with chicken anti-GFP antibody and HRP-conjugated goat anti-chicken antibody each at a 1:5,000 dilution. The extent of translocon jamming was assessed by MBP fractionation. For each time point analyzed, 0.6 A600 cell equivalent was harvested by sedimentation in the microcentrifuge at 14,000 rpm for 5 min, and resuspended in 200 μl of 20% sucrose, 0.03 m Tris-HCl, pH 8. Half the sample was saved as the total cell control, whereas the other half was spheroplasted by addition of 10 μl of 1 mg/ml of lysozyme in 0.1 m EDTA, pH 8, for 15 min. Treated cells were sedimented in the microcentrifuge at 5,500 rpm for 5 min, and the supernatant (i.e. periplasmic fraction) was removed, whereas the cell pellet (cytoplasm/membrane fraction) was resuspended in 100 μl of buffer A (10 mm Tris-HCl, pH 7.5, 150 mm NaCl, 5 mm MgSO4). All samples were boiled for 10 min in the presence of sample buffer, and 5-μl aliquots were analyzed by SDS-PAGE and Western blotting with rabbit anti-MBP antibody and HRP-conjugated goat anti-rabbit antibody, both at 1:5,000 dilution. For quantification of SecY and MBP, ImageJ was used. Average pixel count of SecY monomer to dimer ratio (D/M) or cytoplasm/membrane MBP to total MBP ratio (C/T) was used and plotted with standard error measurement.
In Vivo Cysteine Cross-linking
An overnight culture of MC4100.2(λDE3) containing pCDFT7secYmycEG with a given secY Cys mutation and pBAD-OmpA-GFP plasmids was grown in LB containing appropriate antibiotics, 10 or 30 μm IPTG, and 0.2% maltose, subcultured in the absence of pBpA, and arabinose induced for 45 min as described above under “Translocon Jamming.” 3 A600 cell equivalents was harvested, resuspended in 2 ml of protoplast buffer (100 mm sodium phosphate, pH 7.5, 5 mm EDTA, 10 mm phenanthroline), and treated with copper phenanthroline (180 mm phenanthroline, 60 mm CuSO4, 50 mm NaH2PO4) to a final concentration of 300 μm (40) for 10 min at 30 °C as indicated. The reaction was quenched by addition of N-ethylmaleimide and EDTA to final concentrations of 1 mg/ml and 5 μm, respectively, for 10 min. For treatment with reducing agent, DTT was added to a final concentration of 60 mm for 10 min. Membrane was isolated and subsequently analyzed as described above for “In Vivo Photo-cross-linking” except that the final sample was solubilized in 40 μl of ABB buffer and an equal volume of 2× non-reducing sample buffer (40 mm Tris-HCl, pH 7.8, 16 mm NaH2PO4, 2% (w/v) SDS, 50 μg/ml of bromphenol blue, 20 mm EDTA, 2 mg/ml N-ethylmaleimide, and 0.10 g/ml of sucrose).
OmpA-GFP-SecY Co-immunoprecipitation
MC4100.2(λDE3) containing the pCDFT7secYmycEG with the secY Q212C mutation and pBAD-OmpA-GFP plasmids was utilized. Cell growth, cysteine cross-linking, and membrane isolation were as described for “In Vivo Cysteine Cross-linking.” Each membrane pellet was solubilized in 60 μl of TSGM buffer (20 mm Tris-HCl, pH 8, 130 mm NaCl, 10% glycerol, and 2 mm MgCl2) supplemented with 2% (w/v) DDM for 3 h at 4 °C with constant stirring. Insoluble material was removed by sedimentation at 82,000 rpm for 30 min. Anti-GFP beads were pre-equilibrated by washing three times consecutively with 700 μl of dilution buffer (10 mm Tris-HCl, pH 7.5, 150 mm NaCl, 0.5 mm EDTA) by successive sedimentation steps in a microcentrifuge at 5,500 rpm for 2 min. Solubilized membrane was diluted 1:20 into TSGM buffer, when 30 μl of anti-GFP beads were added, followed by incubation at 4 °C for 2 h. Beads were sedimented and washed three times consecutively with 1 ml of TSGM buffer supplemented with 0.6 mm DDM. For elution, 40 μl of elution buffer (0.2 m glycine, pH 2.5) was added to the washed beads, which were agitated for 2 min and re-sedimented. The eluate was removed and quickly neutralized with 6 μl of 1 m Tris base. The purification was monitored by taking samples before, during, and after the purification, which were analyzed by SDS-PAGE and Western blotting with c-Myc or GFP antibodies.
Translocon Clearing
An overnight culture of BL26.1(λDE3) containing pSup-pBpARS-6TRN, pCDFT7secYmycEG with the secY Lys20 amber mutation, and pBAD-lacZ plasmids was grown as described above for “In Vivo Photo-cross-linking.” The culture was grown until A600 reached 0.20 when kasugamycin was added to a final concentration of 2 mg/ml. For each time point analyzed, 3 A600 cell equivalents of culture was harvested and prepared for UV irradiation and subsequent analysis of protein as described above under “In Vivo Photo-cross-linking.”
SecA Footprinting
MC4100.2(λDE3) containing pSup-pBpARS-6TRN, pCDFT7secYmycEG with the secY Cys68 mutation, and pBAD-SecA-OmpA-GFP with the secA 59 Amber and OmpA Cys21 mutations was grown and arabinose induced as described above under “Translocon Jamming.” 20 ml of culture was harvested 45 min after arabinose addition and subjected to photo-cross-linking or cysteine cross-linking as described under “In Vivo Photo-cross-linking” or “In Vivo Cysteine Cross-linking.” For double cross-linking, cysteine cross-linking was performed first, cells were sedimented in the microcentrifuge at 14,000 rpm for 5 min, decanted, and resuspended in 2 ml of PBS prior to photo-cross-linking. After photo-cross-linking, final sample treatments were performed as indicated under “In Vivo Cysteine Cross-linking.” Western blots were probed with either anti-GFP antibody or anti-c-Myc antibody as described above.
Results
In Vivo Photo-cross-linking Methodology
SecYEG protein is known to exist as monomers and dimers. Two types of SecYEG dimers have been proposed to date based largely on previous disulfide cross-linking studies: a back-to-back dimer largely stabilized through interactions between SecE, and a front-to-front dimer stabilized by interactions around the lateral gate region of SecY (16–19). Less evidence exists in support of the front-to-front dimer orientation (30). We investigated the formation of both dimers using in vivo photo-cross-linking by engineering the incorporation of pBpA at various locations at the dimer interface (Fig. 1). SecYEG dimer structures were obtained by manually docking two SecYEG monomers together based on the previous cysteine cross-linking data (16, 17).
FIGURE 1.

Placement of pBpA at the dimer interface for in vivo photo-cross-linking. A–C, two protomers of Thermotoga maritima SecYEG were manually docked into a front-to-front conformation based on cysteine cross-linking studies (16, 17). SecY, SecE, and SecG are colored green, red, and cyan, respectively. TM2b and 7 of the lateral gate are colored orange. Sites with amber mutations for incorporation of pBpA are shown as magenta spheres. Views from the A, cytoplasm; B, along the plane of membrane; and C, periplasm are shown. D–F, similar to A–C except protomers were docked into a back-to-back conformation. Magenta and blue spheres indicate the amber mutations on SecY and SecE subunits, respectively. Two negative controls, shown in purple spheres, are amber mutations engineered into regions distant to either dimer interface.
We performed in vivo photo-cross-linking on secY amber mutant Lys20 to demonstrate the ability of this technique to provide credible information on SecY dimer status. Several criteria were used to assess the success of this approach. First, we noted that the production of full-length SecY was dependent on inclusion of pBpA in the media (Fig. 2, compare lanes 2 and 7). The low residual level of SecY production in the absence of pBpA was probably due to a minor level of mis-incorporation of other amino acids by the engineered tRNA synthetase/amber suppressor tRNA. Second, we noted that SecY dimer formation was largely dependent on UV exposure (compare lanes 1 and 2). Third, the presence of the amber codon was necessary for efficient UV-dependent cross-linking, because the low residual level of SecY dimer observed in the wild-type strain was not particularly UV dependent (compare lanes 1 and 2 with lanes 10 and 11). Fourth, the identity of SecY monomers and dimers was confirmed by their heat-dependent aggregation (lane 3 and 12) (42), the failure of an isogenic strain lacking the c-Myc tag on SecY to react with c-Myc antibody in the Western blot (lanes 15 and 16), co-migration of a genetically fused SecY dimer with our cross-linked species (lanes 1 and 17), and finally the susceptibility of disulfide cross-linked mono-cysteine SecY dimer to DTT treatment to produce monomers (see “Disulfide Cross-linking Studies” below). We conclude that our in vivo photo-cross-linking methodology can detect bona fide SecY interprotomer interactions.
FIGURE 2.

Verification of in vivo photo-cross-linking. Each strain was grown as indicated in the presence or absence of 1 mm pBpA until A600 reached 0.3, when SecYEG was induced or not with IPTG at a final concentration of 1 mm for 2 h. Cells were harvested, resuspended in PBS buffer, and treated with UV irradiation (350–365 nm) for 20 min where indicated. Cell membranes were isolated and analyzed by Western blotting using c-Myc antibody as described under “Experimental Procedures.” Samples in lanes 3 and 12 were heated at 100 °C for 5 min prior to loading on the gel to induce SecY aggregation. YY indicates a genetically fused SecY dimer used as a marker (48). Anti-SecY peptide antiserum was used to detect this latter species.
SecY Forms Both Front-to-front and Back-to-back Dimers
After confirming the feasibility of our approach, we proceeded to confirm the existence of front-to-front and back-to-back SecY dimers by in vivo photo-cross-linking of appropriate mutant strains. Both dimer forms were detectable by this approach (Fig. 3). In addition, certain secY or secE mutants produced two or even three possible secY or secE dimers (e.g. Fig. 3, A, Gly296 and D, Gly110, respectively) that ran as tightly spaced bands, consistent with the promiscuous cross-linking chemistry of pBpA and the separation by SDS-PAGE of dimers with differing degrees of asymmetry in the position of their cross-link. Alternatively, one or more of these species could represent SecY cross-linked to another membrane protein that associates peripherally with the translocon. Consistent with previously identified sites within the SecY-SecY interface, we found strong cross-links that support the existence of the front-to-front dimer when amber codons were positioned at residues Ser111, Asn151, and Phe154 (Fig. 3A) (30). An additional 8 novel sites supporting this dimer orientation were also found. We detected an additional major cross-linked species (Fig. 3A, indicated by a star) migrating between the monomer and dimer forms of SecY, which likely corresponds to SecY interacting with either SecE or SecG because both proteins are part of the heterotrimeric complex. To investigate this possibility, we created c-Myc-tagged versions of either SecE (SecYEmycG) or SecG (SecYEGmyc) in a secY Tyr122 mutant. Photo-cross-linking of the relevant strains demonstrated that this novel species was in fact a SecY-SecG complex (Fig. 3B). Indeed, when docking SecY in a front-to-front orientation, SecG is the nearest neighbor to some of the engineered amber sites (Fig. 1, A–C). For other secY mutations at this interface, we cannot exclude the possibility of SecY interacting with additional protein partners outside of the heterotrimeric complex, although a previously documented SecY-YidC interaction (43) is unlikely here given the large size predicated for this complex (∼95 kDa).
FIGURE 3.

Detection of distinct SecYEG dimer states using in vivo photo-cross-linking. Each strain carrying the indicated amber mutation was grown in the presence of 1 mm pBpA until A600 reached 0.3, when SecYEG was induced with IPTG at a final concentration of 1 mm for 2 h. Cells were harvested, resuspended in PBS buffer, and treated with UV irradiation for 20 min as indicated. Cell membranes were isolated and analyzed by Western blotting using c-Myc antibody as described under “Experimental Procedures.” Analysis of A and B, secY amber mutations in the front-to-front (FTF) dimer interface; C and D, secY and secE mutations, respectively, in the back-to-back (BTB) dimer interface; and E, secY mutations that lie at neither interface. Certain samples (boil) were heated at 100 °C for 5 min to induce SecY aggregation. YY indicates a genetically fused SecY dimer used as a marker (48). The star indicates a SecY-SecG cross-linked complex. Single letter amino acids are indicated.
We found two novel sites within SecY that support the back-to-back interface, Lys20 and Lys434, which formed strong cross-links (Fig. 3C). Amber mutations at previously identified secE sites also resulted in strong SecE-SecE cross-links (Fig. 3D), consistent with the back-to-back dimer (16, 17). When amber mutations were engineered at two sites within secY that are remote from either dimer interface, no cross-linked SecY dimer was observed (Fig. 3E), consistent with the specificity of our analysis. Together these results show the utility of our approach for detecting both types of SecYEG dimers in vivo.
SecY Dimer Levels Are Refractory to Translocon Jamming
We next set out to determine whether SecY dimers are involved in ongoing protein translocation or not. We first investigated how cellular SecY dimer levels change when they are engaged in active protein transport. Because protein translocation occurs at a very rapid speed (on the order of one preprotein translocated per s/translocon based on E. coli secretory protein content, doubling time, and translocon number), we made use of an inducible OmpA-GFP chimera that contains a rapidly folding GFP variant fused to an OmpA signal sequence to jam translocon channels to mimic a translocation intermediate state (30). If SecY functions exclusively as a monomer during protein transport, then we would expect to see a decrease in SecY dimer levels over time as more translocon channels become jammed with the OmpA-GFP substrate. Translocon jamming was assessed by the cytoplasmic accumulation of MBP precursor (Fig. 4). Furthermore, to minimize the potential for artificial dimerization due to SecY overexpression, we titrated SecY levels using different concentrations of IPTG to find a low expression level that still allowed us to visualize our starting and potentially diminishing SecY dimer signal throughout the experiment. We found that 30 μm IPTG, which modestly overexpressed SecY (Fig. 5A), meets this criteria, and it was used for all subsequent experiments.
FIGURE 4.

Accumulation of MBP in the cytoplasm as a result of translocon jamming. Under normal translocation conditions (A) pre-MBP is targeted to the translocon and rapidly secreted into the periplasm as mature MBP. When the translocon is jammed by either OmpA-GFP (B) or MalE-LacZ (C), pre-MBP builds up in the cytoplasm.
FIGURE 5.

Analysis of the secY Lys434 mutant during OmpA-GFP-induced translocon jamming by in vivo photo-cross-linking. A, titration of SecY expression with IPTG. The secY Lys434 mutant was grown in the presence of 1 mm pBpA until A600 reached 0.15, when IPTG was added to the indicated final concentration and growth continued for another 60 min. Cells were harvested and membranes were isolated and analyzed by Western blotting using anti-SecY peptide antiserum. B, view of T. maritima SecYEG docked in the back-to-back conformation with the same color scheme as described in the legend to Fig. 1. Residue Lys434 is shown as magenta spheres. C–F, the secY Lys434 mutant was grown in the presence of 1 mm pBpA, 30 μm IPTG, and 0.2% maltose until A600 reached 0.15, when the OmpA-GFP chimera was induced by adding arabinose to a final concentration of 0.2%. Cells were harvested at the indicated time points post jamming, and exposed to UV irradiation as indicated. A wild-type (WT) strain was used in parallel as a control. C, Western blot of cell membranes probed with c-Myc antibody. D, Western blot of cell membranes probed with GFP antibody. E, Western blot of cells divided into cytoplasm/membrane (C) and periplasmic (P) fractions, compared with total (T) cell input, probed with MBP antibody. F, quantification of the SecY dimer to monomer ratio (D/M) or cytoplasm/membrane MBP to total MBP ratio (C/T) during jamming. The average results from three experiments are plotted with standard error measurements. The error bars for the D/M points are too small to be seen on this scale.
We picked mutants that produced the strongest photo-cross-linking signals for the back-to-back (Lys434 and Lys20) or front-to-front (Ser111) dimers and jammed them over a 60-min time course. Samples were collected at different time points to access the change in SecY dimer levels, as well as the extent of translocon jamming. For assessment of the former quantity, we utilized the SecY dimer to monomer ratio (D/M) to control for minor variations in SecY amounts or detection. Similarly for assessment of the latter quantity, we utilized the cytosolic/membrane-associated MBP to total MBP ratio (C/T) for the same reason. To perform this latter measurement cells were spheroplasted with lysozyme/EDTA and sedimented, and the supernatant (containing periplasmic proteins including secreted MBP) and pellet (containing cytosolic/membrane proteins including retained preMBP/MBP) were isolated and quantified by Western blotting. We found that the cellular SecY D/M ratio remained unchanged during the course of jamming for the secY Lys434 mutant (Fig. 5, C and F). Such jamming was very effective as evident by the rapid buildup of MBP precursor in the cytoplasm (Fig. 5E), which was saturated between 30 and 40 min (Fig. 5F). This correlated well with the build-up of OmpA-GFP during this time frame (Fig. 5D). A similar pattern was also observed for other secY mutants, including Lys20 (data not shown) and Ser111 (Fig. 6) that report on the back-to-back or front-to-front dimer conformation, respectively. Interestingly, there was a subtle increase in the SecY D/M ratio for the latter mutant (Fig. 6E), possibility due to more efficient photo-cross-linking at this position in the translocon-jammed conformation. To make sure that our results were not unique to the OmpA-GFP fusion, we also carried out similar experiments using a strain that carries a chromosomal MalE-LacZ fusion (Fig. 4C). This chimera contains the first third of pre-MBP fused to β-galactosidase, and its ability to jam protein transport has been previously demonstrated (44). We found that the SecY D/M ratio remained unchanged under conditions of MalE-LacZ jamming as well (Fig. 7).
FIGURE 6.

Analysis of the secY Ser111 mutant during OmpA-GFP-induced translocon jamming by in vivo photo-cross-linking. A, view of T. maritima SecYEG docked in the front-to-front conformation with the same color scheme as described in the legend to Fig. 1. Residue Ser111 is shown as magenta spheres. B–E, the secY Ser111 mutant was grown in the presence of 1 mm pBpA, 30 μm IPTG, and 0.2% maltose until A600 reached 0.15, when the OmpA-GFP chimera was induced by adding arabinose to a final concentration of 0.2%. Cells were harvested at the indicated time points post jamming, and exposed to UV irradiation as indicated. A wild-type (WT) strain was used in parallel as a control. B, Western blot of cell membranes probed with c-Myc antibody. C, Western blot of cell membranes probed with GFP antibody. D, Western blot of cells divided into cytoplasm/membrane (C) and periplasmic (P) fractions, compared with total (T) cell input, probed with MBP antibody. E, quantification of SecY dimer to monomer ratio (D/M) or cytoplasm/membrane MBP to total MBP ratio (C/T) during jamming. The average results from three experiments are plotted with standard error measurements.
FIGURE 7.
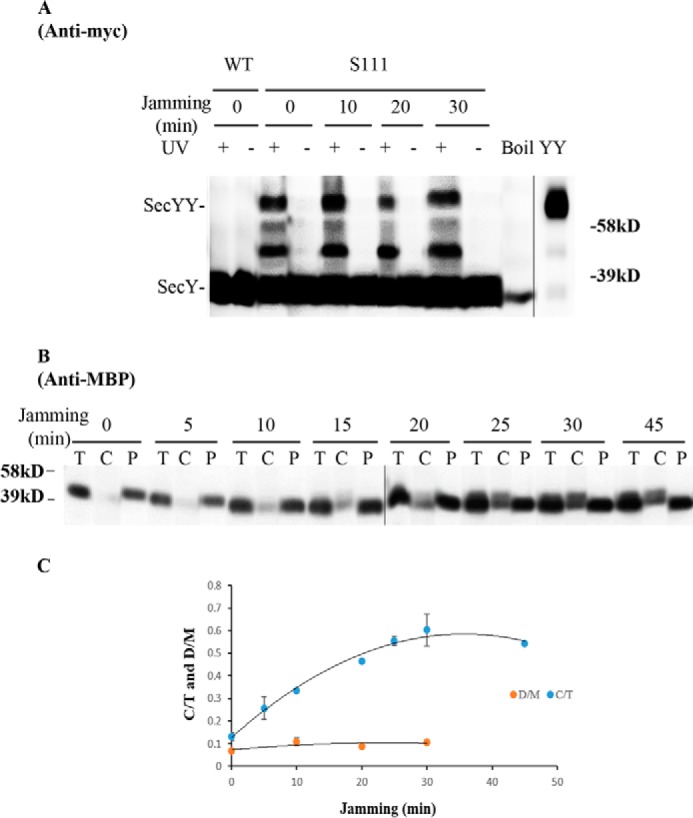
Analysis of the secY Ser111 mutant during MalE-LacZ-induced translocon jamming by in vivo photo-cross-linking. A–C, MM18.7 containing the pSup-pBpARS-6TRN and pCDFT7secYmycEG plasmids with the secY Ser111 amber mutation was grown in the presence of 1 mm pBpA and 30 μm IPTG until A600 reached 0.15, when the MalE-LacZ chimera was induced by adding maltose to a final concentration of 0.2%. Cells were harvested at the indicated time points post jamming, and exposed to UV irradiation as indicated. A wild-type (WT) strain was used in parallel as a control. A, Western blot of cell membranes probed with c-Myc antibody. B, Western blot of cells divided into cytoplasm/membrane (C) and periplasmic (P) fractions, compared with total (T) cell input, probed with MBP antibody. C, quantification of SecY dimer to monomer ratio (D/M) or cytoplasmic/membrane MBP to total MBP ratio (C/T) during jamming. The average results from three experiments are plotted with standard error measurements. The error bars for the D/M points are too small to be seen on this scale.
Disulfide Cross-linking Studies
To determine whether the foregoing results were unique to photo-cross-linking, we performed similar experiments utilizing cysteine disulfide cross-linking. For this purpose we used a cysteine at secY Gln212 (Fig. 8A): a known hot spot for forming SecY back-to-back dimers by disulfide cross-linking (30). SecY dimer levels were assessed in the secY Q212C mutant before and after translocon jamming utilizing the OmpA-GFP chimera as before. We obtained a result similar to our in vivo photo-cross-linking analysis, namely SecY dimer levels remained relatively constant independent of translocon jamming (Fig. 8B). This result was not caused by artificial dimerization of SecY due to overexpression, because we obtained a similar result when SecY was expressed around chromosomal levels using 10 μm IPTG (Figs. 5A and 8B). It was also not caused by ineffective OmpA-GFP jamming because jamming led to an efficient buildup of MBP in the cytoplasm (Fig. 8C). Collectively, these results suggest a common phenomenon, namely the retention of the SecY dimer during translocation-arrested conditions.
FIGURE 8.

Analysis of the secY Q212C mutant during OmpA-GFP-induced translocon jamming by disulfide cross-linking. A, view of T. maritima SecYEG docked in the back-to-back conformation with the same color scheme as described in the legend to Fig. 1. Magenta spheres indicate the Gln212 residue mutated to cysteine. B and C, the secY Q212C mutant was grown in the presence of 10 or 30 μm IPTG and 0.2% maltose until A600 reached 0.15, when the OmpA-GFP chimera was induced by adding arabinose to a final concentration of 0.2%. Cells were harvested 45 min post jamming and treated with CuPhe3 and DTT as indicated. B, Western blot of cell membranes probed with c-Myc antibody. C, Western blot of cells divided into cytoplasm/membrane (C) and periplasmic (P) fractions, compared with total (T) cell input, probed with MBP antibody.
Our data thus far suggests that SecY dimers are retained and likely participate in active protein translocation. However, because we examined total cellular SecY protein in the foregoing analysis, one could argue that these experiments simply captured a residual pool of inactive SecY dimer. Given the rapidity and efficiency of our translocon-jamming methodology, we do not feel that this explanation is likely. However, to directly address this issue, we performed a co-immunoprecipitation experiment to directly examine the nature of the OmpA-GFP-jammed SecY pool. Accordingly, a jamming experiment was performed with the secY Q212C mutant, followed by in vivo disulfide cross-linking. Next, cells were broken and total membrane proteins were isolated after solubilization with DDM, which has been shown to preserve at least some translocon associations (13). Anti-GFP beads were utilized to specifically bind OmpA-GFP-associated membrane proteins. Controls indicated the specificity of the system, because SecY protein was not observed in the anti-GFP bead eluate when the strain remained uninduced (Fig. 9, lanes 15–21). By contrast, during arabinose induction, both SecY dimers and monomers were observed in the eluate fraction along with OmpA-GFP (Fig. 9, lanes 1–7). The identity of the disulfide-linked SecY dimer was confirmed by its sensitivity to reducing agent either within the total solubilized cell membranes or within the eluate itself (Fig. 9, compare lanes 1 and 8, or 6 and 7, respectively). These results indicate that a significant fraction of total cellular SecY dimer pool is associated with OmpA-GFP. However, the experiment was limited by SecY recovery, because OmpA-GFP dissociated from the majority of both forms of SecY, presumably due to its instability in detergent (Fig. 9, lanes 2 and 9). In several repetitions of this experiment we found that SecY dimers were more readily pulled down than monomers, suggesting that OmpA-GFP forms a more stable complex with the former species (data not shown). The possibility that SecY dimers formed in vitro under our conditions appeared unlikely based on a parallel study (13). Together, these results strongly suggest a direct involvement of SecY dimers in active translocation.
FIGURE 9.

Analysis of OmpA-GFP-jammed secY complex by co-immunoprecipitation. The secY Q212C mutant was grown in the presence of 30 μm IPTG to an A600 of 0.15, when arabinose was added as indicated to a final concentration of 0.2%, and growth was continued for an additional 45 min. Cells were harvested and treated with CuPhe3 to cross-link SecY prior to purification. Membranes were isolated and solubilized at 4 °C for 3 h in TSGM buffer containing 2% (w/v) DDM without (w/o βME) or with (w/βME) 5% β-mercaptoethanol as indicated, and insoluble material was removed by high-speed sedimentation. Solubilized membrane protein was mixed with anti-GFP beads at 4 °C for 2 h, when beads were sedimented, washed, and the sample eluted as described by the supplier. Western blot of the total input (I) of solubilized membrane protein, the flow-through (FT) fraction, three consecutive washes (W1, W2, and W3), and specifically eluted protein without (E) or with (EβME) treatment with 5% β-mercaptoethanol are shown. Western blots were probed with c-Myc or GFP antibody as indicated. Given the rapidly folding GFP domain, OmpA-GFP can run as a doublet (30).
SecY Dimer Levels Are Refractory to Translocon Clearing
The foregoing results contradict a recent study indicating that SecY monomerizes under translocon-jamming conditions (30). This latter finding would predict that SecYEG dimers would tend to accumulate when the translocon channel is idle. To test this prediction we utilized the translation initiation inhibitor kasugamycin to clear channels of any substrate proteins. This antibiotic inhibits translation initiation by perturbing the mRNA codon-tRNA anticodon interaction, thus preventing the binding of tRNAfMet to the P-site of the 30S ribosomal subunit (45). We first titrated kasugamycin under our standard growth conditions in an isogenic strain that contained an arabinose-inducible lacZ gene to determine an effective concentration for use. We found that treatment with 2 mg/ml of kasugamycin for 2 min was sufficient to inhibit 99% of subsequently induced β-galactosidase activity (Fig. 10A). Given known translation elongation rates under these growth conditions (12–17 amino acids/s (46)), even the longest nascent polypeptide chains should be released from ribosomes within the first couple of minutes after this treatment. Furthermore, given the rapid in vivo protein translocation rates observed in E. coli with a wild-type secretion machinery (47), translocon channels should be cleared of all substrates after 5 min of kasugamycin treatment.
FIGURE 10.

Analysis of the secY Lys20 mutant during kasugamycin treatment by in vivo photo-cross-linking. A, kinetics of kasugamycin inhibition. BL26.1(λDE3) containing pSup-pBpARS-6TRN, pCDFT7secYmycEG, and pBAD-lacZ was grown to an A600 of 0.2, when kasugamycin was added to a final concentration of 2 mg/ml at time 0; arabinose was added to a final concentration of 0.2% at the indicated times, and 40 min later β-galactosidase activity was measured. The average Miller units (41) of β-galactosidase activity of duplicate or triplicate samples are shown. The basal level of β-galactosidase activity in the untreated culture lacking both kasugamycin and arabinose was subtracted as background from all assays. B, the secY Lys20 mutant was grown in the presence of 1 mm pBpA and 30 μm IPTG until A600 reached 0.2, when kasugamycin was added to a final concentration of 2 mg/ml. Cells were harvested at the indicated times and exposed to UV irradiation as indicated. Cell membranes were isolated and analyzed by Western blotting using c-Myc antibody.
We compared SecY dimer levels before and after kasugamycin treatment by in vivo photo-cross-linking of the secY Lys20 mutant. The SecY dimer level remained relatively constant in cells whether they were treated with kasugamycin for 5 or 10 min or not at all (Fig. 10B). This result, along with the results presented above, indicate that SecY dimer levels remain relatively constant in cells and appear to be mostly refractory to changes in protein secretion physiology.
SecA Interacts with Two Copies of SecY During Translocon Jamming
Given the general controversy surrounding SecY oligomeric function, we sought an independent method to address this issue. In particular, we have previously utilized a form of in vivo photo-cross-linking termed SecA footprinting, where distinct halves of the SecA molecule (NBD-1-NBD-2 or PPXD-HSD-THF-HWD-CTL) were found to associate with individual protomers of the SecY dimer (38). We reasoned that this method could be modified to address the present issue if SecA could be captured in a translocationally active complex with SecY. To accomplish this task, we fused SecA at its non-essential carboxyl terminus to the amino terminus of OmpA-GFP, to form a SecA-OmpA-GFP trimera. Because this trimera should be capable of efficient SecA-dependent jamming, it was placed under the tightly regulated araBAD promoter system. We next engineered cross-linking sites for both halves of SecA to test for interactions with individual protomers of the SecY dimer. For this purpose, an amber mutation was made at residue 59 within NBD-1 of SecA, and a cysteine was introduced at residue 21 (Cys21) of the OmpA signal sequence, adjacent to the distal half of SecA within the trimera (Fig. 11A). The former position has been shown to be a hot spot for SecA-SecY photo-cross-linking, whereas the latter position has been shown to efficiently cross-link to a cysteine at residue 68 (Cys68) of the plug domain of SecY (30, 38). This latter interaction would also ensure that only SecA-OmpA-GFP translocation intermediates that had inserted the OmpA signal sequence into the lateral gate of a translocationally active SecY protomer would be detected by our system.
FIGURE 11.

SecA footprinting study of SecY dimer status. A, schematic of SecA footprinting study, where interaction of the NBD-1-NBD-2 (N domain) half of SecA with SecY1 or PPXD-HSD-THF-HWD-CTL (C domain) half of SecA with SecY2 are depicted along with the OmpA-GFP chimera attached to SecA. Sites for photo-cross-linking the SecA N domain (59) with SecY1 or for disulfide cross-linking the OmpA signal sequence (C21) to the plug domain (C68) of SecY2 are shown. B-D, the secY Cys68 mutant carrying the pBAD-secA-OmpA-GFP plasmid with the ompA Cys21 and secA 653 Amber (panel B) or secA 59 Amber (panels C and D) allele was grown in the presence of 1 mm pBpA and 30 μm IPTG until A600 reached 0.15, when arabinose was added to a final concentration of 0.2%. 45 min later, cells were harvested and either fractionated (panel B) or subjected to cross-linking (panels C and D). B, Western blot of cells divided into cytoplasm/membrane (C) and periplasmic (P) fractions, compared with total (T) cell input, probed with MBP antibody. C and D, Western blot of isolated membranes from cells treated with UV irradiation, CuPhe3 and/or DTT as indicated probed with GFP or c-Myc antibody as indicated. A large degradation fragment is visible immediately below the SecA-OmpA-GFP trimera.
We performed a jamming experiment with the strain containing the SecA-OmpA-GFP trimera, and we found effective translocon jamming based on cytoplasmic MBP accumulation (Fig. 11B). We then performed a SecA footprinting experiment, where SecA-SecY interaction was probed during jamming conditions by utilizing photo-cross-linking and/or cysteine disulfide cross-linking. When single cross-linking was utilized, we found that the SecA-OmpA-GFP trimera was cross-linked to a single SecY protomer (Fig. 11, panels C and D, lane 1-4). The identity of the cross-linked species was verified by its UV or copper phenanthroline (CuPhe3) dependence, its sensitivity to reduction in the latter case, and its reactivity to either GFP or c-Myc antibody. The intensity of the cross-linked species was significantly greater with disulfide cross-linking, consistent with the generally higher efficiency of this method compared with pBpA-induced photo-cross-linking. We also verified that the Cys21-Cys68 interaction of the OmpA signal sequence with the SecY plug domain was specific, because when Cys21 was mutated to serine, no cross-linked complex was generated (data not shown). Importantly, when double cross-linking was performed, we detected the SecA-OmpA-GFP-SecYY triple complex migrating above the double SecA-OmpA-GFP-SecY complex (Fig. 11, panels C and D, lane 5). The former complex completely disappeared, and the latter complex was substantially diminished as well, when the doubly cross-linked sample was treated with DTT, consistent with the majority of cross-linking coming from the disulfide method (Fig. 11, panels C and D, compare lanes 5 and 6). Because we used a disulfide pair that captures a translocation intermediate at the trans side of the channel, we were confident that the two cross-linked complexes represent ones engaged in active protein translocation. In short, the results of the SecA footprinting study indicate that a SecA protomer can interact with two copies of SecY when it is fused to a translocation intermediate: indicative of an active role for SecY dimers in ongoing protein transport. However, given the limitations inherent to our footprinting method, this approach does not address whether some protein translocation also occurs from SecA-bound, SecY monomers, or whether the passive SecY copy stays associated with the active one throughout the entire translocation cycle.
Discussion
Although the earliest studies suggested that the protein-conducting channel might reside at the subunit interface of SecYEG dimers or tetramers (14, 15), later studies visualized a protein-conducting channel within a single SecYEG protomer that has been suggested to be fully functional (5, 9, 22–26). However, a variety of other structural and functional studies during the past decade have also pointed to the existence of a second SecYEG subunit that appears to play a critical role in facilitating overall translocon function (16–21, 27, 29).
Thus the primary focus of the present study was to investigate the oligomeric state of the SecYEG protein during ongoing protein translocation utilizing several different approaches that might yield a consistent picture in this matter. Toward this end we utilized both in vivo photo-cross-linking and disulfide cross-linking methods along with a previously developed SecA footprinting approach to address this question. We found that the SecY dimer pool remained relatively constant whether translocons were cleared of substrate by kasugamycin pre-treatment, or whether they were engaged in active protein translocation utilizing the translocon jamming technique. This latter result was not simply due to more limited sampling of a subset of inactive translocons, because the SecY dimer could be isolated associated with the OmpA-GFP translocation intermediate. Likewise, the existence of a substrate-engaged SecY dimer was also demonstrated in our SecA footprinting study, where cross-linking footprints were seen with both the translocating and non-translocating SecY subunits. Collectively, these results argue strongly for a role of the SecYEG dimer in ongoing protein transport.
Our results contradict a parallel study that also utilized disulfide cross-linking and substrate jamming to study the functional state of the SecYEG subunit within cells (30). Indeed, one reason that we compared our photo-cross-linking method with disulfide cross-linking, including utilizing some identical mutations with the same jamming OmpA-GFP chimera, was because of the different results noted. We also compared jamming promoted by the shorter OmpA-GFP chimera with that promoted by the longer MalE-LacZ chimera, and we found similar results in both cases. In addition, we performed our study at moderate or even near chromosomal SecY levels to minimize the chance of artificial dimerization due to SecYEG overproduction. Indeed, one of the strengths of our in vivo photo-cross-linking approach is its relatively low cross-linking efficiency (∼10%) that minimally distorts the natural monomer-dimer equilibrium present within the cell. Although it remains possible that different degrees of jamming were obtained in the two studies due to the differing stains or growth physiologies employed, our co-immunoprecipitation experiment indicated that at least a subset of the SecY dimer pool detected was derived from jammed translocons associated with substrate. The exact cause of discrepancy between the two studies is not clear at present, although we note different strains and somewhat different protocols were utilized for this portion of our study. Beyond the good agreement obtained between our photo-cross-linking and disulfide cross-linking studies, our SecA footprinting study provided yet another independent method to evaluate this issue. Here again we obtained a clear indication of the existence of a translocationally active SecY dimer, although we were unable to address the more complicated question as to whether the dimer was stable throughout the entire translocation process given the limitations inherent in our approach.
A number of explanations have been given for the formation of SecYEG dimers. It has been suggested that the non-translocating SecYEG subunit could provide an additional binding platform for SecA or the ribosome (29). In the case of SecA, for example, association of the NBD-1-NBD-2 half of SecA with the inactive copy of SecYEG would provide a stator during cycles of SecA-dependent insertion of the preprotein into the translocationally active SecYEG subunit promoted by the PPXD-HSD-THF-HWD-CTL half of SecA. Indeed the NBD-1-NBD-2 half of SecA is the portion that displays high affinity binding specificity for SecYEG (49). This hypothesis would also be consistent with in vivo or in vitro complementation results obtained with strains that contain two different secY alleles (20, 21), as well as the recently observed processive action of SecA ATPase during protein transport (6). It would also provide a mechanism to switch between ribosome and SecA-promoted transport for integral membrane proteins with periplasmic domains that require both pathways for their biogenesis. Perhaps, dimerization allows SecYEG to transit from a resting state to a translocation-ready state, characterized by greater SecA binding affinity (20) and more flexibility of the plug domain to accommodate preprotein (10). Interestingly, a recent study hints at a more subtle type of dimer function where the nature of the preprotein substrate may determine whether transport occurs via a SecYEG monomer or dimer (31). This phenomenon points to a possible structural signal within particular substrate proteins that facilitates dimer formation. Finally, it could be that the dimer could be a storage form of SecYEG protein, in keeping with its accumulation under conditions of overproduction. However, our data does not favor this hypothesis, because we saw little change in the dimer pool whether translocons were empty of protein substrate during kasugamycin treatment or whether they were largely occupied during translocon jamming conditions. This hypothesis is also inconsistent with reports that SecA and substrate promote SecYEG dimer assembly in vitro, suggesting a functional role of dimers rather than merely a storage form (11, 13).
The extensive set of cross-linking residues utilized in our photo-cross-linking study provides additional confirmation for the existence of the two SecYEG dimer forms in vivo, particularly for the front-to-front dimer that is supported by relatively few cross-links (30). Of course the physiological relevance of either dimer form remains unclear. The front-to-front dimer with the lateral gates facing one another could allow protomers to fuse to form a larger composite channel, which would permit expansion of the channel to dimensions more consistent with two previous estimates (32, 33). Such a fused dimer would only be useful for the transit of partially folded, secretory proteins, because it would lack any lateral gate opening for the exit of membrane proteins into the lipid bilayer. On the other hand, the back-to-back dimer would be suitable for translocation of either integral membrane or secretory proteins, but it would be more limited in its potential for channel expansion. Its two outward facing lateral gates would be in an ideal position for interaction with YidC or other chaperones involved in membrane protein folding and assembly (43). Positing different functions for the two observed SecYEG dimer forms seems appealing given the large diversity of substrate proteins in bacterial proteomes, which differ greatly in their disposition of topogenic sequences as well as rate of delivery to the translocon.
Given the quantity and quality of seemingly opposing findings in this area, it seems reasonable to conclude that both views have merit. Thus the SecYEG monomer may indeed be the minimal functional unit of the translocon for many protein substrates, whereas the dimer may be a more optimal functional unit under a variety of conditions including those examined in the present study. Thus studies reporting exclusive dimer function may simply be reflective of the higher specific binding and transport activity of the dimer form of the translocon compared with its monomer counterpart, or they may relate to the type of substrate protein under investigation. A more complicated view posits that both forms of the translocon are required at different stages of the translocation cycle such that the monomer and dimer switch off with one another as needed. Clearly the initiation or elongation phases of protein transport place different requirements on the translocon as does the translocation of proteins with differing topologies or destinations. Thus additional approaches with a higher degree of sophistication and resolution will be required to address these two opposing viewpoints. State of the art techniques such as real time FRET and fluorescence microscopy may enable us to watch how SecYEG monomers and dimers work during active protein translocation in model systems or perhaps even within living cells.
Author Contributions
Z. Z. conducted most of the experiments, and Z. Z. and D. O. wrote the paper. A. B. conducted in vivo photo-cross-linking for the back-to-back dimer and the channel clearing experiments. T. B. performed experiments on translocon jamming with the MalE-LacZ chimera and created the secA-OmpA-GFP construct. Q. W. and V. D. validated the feasibility of the in vivo photo-cross-linking method and participated in in vivo photo-cross-linking for the front-to-front SecYEG dimer.
Acknowledgments
We thank Stephanie Eben for help in making certain strains and carrying out in vivo photo-cross-linking and Dr. Tom Rapoport for providing the OmpA-GFP plasmid. We also thank Lorry Grady for initial editing. We acknowledge Manju Hingorani, Rich Olson, and Ishita Mukerji for intellectual input.
This work was supported, in whole or in part, by National Institutes of Health Grant GM110552 (to D. O.). The authors declare that they have no conflict of interest with the content of this article.
- pBpA
- ρ-benzoyl-phenylalanine
- CTL
- carboxyl-terminal linker domain of SecA
- DDM
- n-dodecyl-β-d-maltopyranoside
- HSD
- helical scaffold domain of SecA
- HWD
- helical wing domain of SecA
- IPTG
- isopropyl β-d-1-thiogalactopyranoside
- NBD
- nucleotide-binding domain of SecA
- PPXD
- preprotein-cross-linking domain of SecA
- THF
- two helix-finger sub-domain of SecA
- MBP
- maltose-binding protein.
References
- 1. Cymer F., and von Heijne G. (2013) Cotranslational folding of membrane proteins probed by arrest-peptide-mediated force measurements. Proc. Natl. Acad. Sci. U.S.A. 110, 14640–14645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. du Plessis D. J., Nouwen N., and Driessen A. J. (2011) The Sec translocase. Biochim. Biophys. Acta 1808, 851–865 [DOI] [PubMed] [Google Scholar]
- 3. Hartl F.-U., Lecker S., Schiebel E., Hendrick J. P., and Wickner W. (1990) The binding cascade of SecB to SecA to SecY/E mediates preprotein targeting to the E. coli plasma membrane. Cell 63, 269–279 [DOI] [PubMed] [Google Scholar]
- 4. Economou A., and Wickner W. (1994) SecA promotes preprotein translocation by undergoing ATP-driven cycles of membrane insertion and deinsertion. Cell 78, 835–843 [DOI] [PubMed] [Google Scholar]
- 5. Zimmer J., Nam Y., and Rapoport T. A. (2008) Structure of a complex of the ATPase SecA and the protein-translocation channel. Nature 455, 936–943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bauer B. W., Shemesh T., Chen Y., and Rapoport T. A. (2014) A “push and slide” mechanism allows sequence-insensitive translocation of secretory proteins by the SecA ATPase. Cell 157, 1416–1429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kim Y. J., Rajapandi T., and Oliver D. (1994) SecA proteins is exposed to the periplasmic surface of the E. coli inner membrane in its active state. Cell 78, 845–853 [DOI] [PubMed] [Google Scholar]
- 8. Plath K., Wilkinson B. M., Stirling C. J., and Rapoport T. A. (2004) Interactions between Sec complex and prepro-α-factor during posttranslational protein transport into the endoplasmic reticulum. Mol. Biol. Cell 15, 1–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Van den Berg B., Clemons W. M. Jr., Collinson I., Modis Y., Hartmann E., Harrison S. C., and Rapoport T. A. (2004) X-ray structure of a protein-conducting channel. Nature 427, 36–44 [DOI] [PubMed] [Google Scholar]
- 10. Tam P. C, Maillard A. P., Chan K. K., and Duong F. (2005) Investigating the SecY plug movement at the SecYEG translocon channel. EMBO J. 24, 3380–3388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Scheuring J., Braun N., Nothdurft L., Stumpf M., Veenendaal A. K., Kol S., van der Does C., Driessen A. J., and Weinkauf S. (2005) The oligomeric distribution of SecYEG is altered by SecA and translocation ligands. J. Mol. Biol. 354, 258–271 [DOI] [PubMed] [Google Scholar]
- 12. Gold V. A., Robson A., Bao H., Romantsov T., Duong F., and Collinson I. (2010) The action of cardiolipin on the bacterial translocon. Proc. Natl. Acad. Sci. U.S.A. 107, 10044–10049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bessonneau P., Besson V., Collinson I., and Duong F. (2002) The SecYEG preprotein translocation channel is a conformationally dynamic and dimeric structure. EMBO J. 21, 995–1003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Meyer T. H., Ménétret J.-F., Breitling R., Miller K. R., Akey C. W., and Rapoport T. A. (1999) The bacterial SecY/E translocation complex forms channel-like structures similar to those of the eucaryotic Sec61p complex. J. Mol. Biol. 285, 1789–1800 [DOI] [PubMed] [Google Scholar]
- 15. Manting E. H., van Der Does C., Remigy H., Engel A., and Driessen A. J. (2000) SecYEG assembles into a tetramer to form the active protein translocation channel. EMBO J. 19, 852–861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kaufmann A., Manting E. H., Veenendaal A. K., Driessen A. J., and van der Does C. (1999) Cysteine-directed cross-linking demonstrates that helix 3 of SecE is close to helix 2 of SecY and helix 3 of a neighboring SecE. Biochemistry 38, 9115–9125 [DOI] [PubMed] [Google Scholar]
- 17. Veenendaal A. K., Van Der Does C., and Driessen A. J. (2002) The core of the bacterial translocase harbors a tilted transmembrane segment 3 of SecE. J. Biol. Chem. 277, 36640–36645 [DOI] [PubMed] [Google Scholar]
- 18. Veenendaal A. K., van der Does C., and Driessen A. J. (2001) Mapping the sites of interaction between SecY and SecE by cysteine scanning mutagenesis. J. Biol. Chem. 276, 32559–32566 [DOI] [PubMed] [Google Scholar]
- 19. van der Sluis E. O., Nouwen N., and Driessen A. J. (2002) SecY-SecY and SecY-SecG contacts revealed by site-specific cross-linking. FEBS Lett. 527, 159–165 [DOI] [PubMed] [Google Scholar]
- 20. Dalal K., Chan C. S., Sligar S. G., and Duong F. (2012) Two copies of the SecY channel and acidic lipids are necessary to activate the SecA translocation ATPase. Proc. Natl. Acad. Sci. U.S.A. 109, 4104–4109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Osborne A. R., and Rapoport T. A. (2007) Protein translocation is mediated by oligomers of the SecY complex with one SecY copy forming the channel. Cell 129, 97–110 [DOI] [PubMed] [Google Scholar]
- 22. Tsukazaki T., Mori H., Fukai S., Ishitani R., Mori T., Dohmae N., Perederina A., Sugita Y., Vassylyev D. G., Ito K., and Nureki O. (2008) Conformational transition of Sec machinery inferred from bacterial SecYE structures. Nature 455, 988–991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Egea P. F., and Stroud R. M. (2010) Lateral opening of a translocon upon entry of protein suggests the mechanism of insertion into membranes. Proc. Natl. Acad. Sci. U.S.A. 107, 17182–17187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Frauenfeld J., Gumbart J., Sluis E. O., Funes S., Gartmann M., Beatrix B., Mielke T., Berninghausen O., Becker T., Schulten K., and Beckmann R. (2011) Cryo-EM structure of the ribosome-SecYE complex in the membrane environment. Nat. Struct. Mol. Biol. 18, 614–621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Park E., Ménétret J. F., Gumbart J. C., Ludtke S. J., Li W., Whynot A., Rapoport T. A., and Akey C. W. (2014) Structure of the SecY channel during initiation of protein translocation. Nature 506, 102–106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hizlan D., Robson A., Whitehouse S., Gold V. A., Vonck J., Mills D., Kühlbrandt W., and Collinson I. (2012) Structure of the SecY complex unlocked by a preprotein mimic. Cell Rep. 1, 21–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mitra K., Schaffitzel C., Shaikh T., Tama F., Jenni S., Brooks C. L. 3rd, Ban N., and Frank J. (2005) Structure of the E. coli protein-conducting channel bound to a translating ribosome. Nature 438, 318–324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kedrov A., Kusters I., Krasnikov V. V., and Driessen A. J. (2011) A single copy of SecYEG is sufficient for preprotein translocation. EMBO J. 30, 4387–4397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Deville K., Gold V. A., Robson A., Whitehouse S., Sessions R. B., Baldwin S. A., Radford S. E., and Collinson I. (2011) The oligomeric state and arrangement of the active bacterial translocon. J. Biol. Chem. 286, 4659–4669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Park E., and Rapoport T. A. (2012) Bacterial protein translocation requires only one copy of SecY complex in vivo. J. Cell Biol. 198, 881–893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Mao C., Cheadle C. E., Hardy S. J., Lilly A. A., Suo Y., Sanganna Gari R. R., King G. M., and Randall L. L. (2013) Stoichiometry of SecYEG in the active translocase of E. coli varies with precursor species. Proc. Natl. Acad. Sci. U.S.A. 110, 11815–11820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bonardi F., Halza E., Walko M., Du Plessis F., Nouwen N., Feringa B. L., and Driessen A. J. (2011) Probing the SecYEG translocation pore size with preproteins onjugated with sizable rigid spherical molecules. Proc. Natl. Acad. Sci. U.S.A. 108, 7775–7780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hamman B. D., Chen J. C., Johnson E. E., and Johnson A. E. (1997) The aqueous pore through the translocon has a diameter of 40–60 Å during cotranslational protein translocation at the ER membrane. Cell 89, 535–544 [DOI] [PubMed] [Google Scholar]
- 34. Wang L., Xie J., and Schultz P. (2006) Expanding the genetic code. Annu. Rev. Biophys. Biomol. Struct. 35, 225–249 [DOI] [PubMed] [Google Scholar]
- 35. Studier F. W., Rosenberg A. H., Dunn J. J., and Dubendorff J. W. (1990) Use of T7 RNA polymerase to direct expression of cloned genes. Methods Enzymol. 185, 60–89 [DOI] [PubMed] [Google Scholar]
- 36. Ito K., Akiyama Y., Yura T., and Shiba K. (1986) Diverse effects of the MalE-LacZ hybrid protein on Escherichia coli cell physiology. J. Bacteriol. 167, 201–204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Casadaban M. J. (1976) Transposition and fusion of the lac genes to selected promoters in Escherichia coli using bacteriophage λ and μ. J. Mol. Biol. 104, 541–555 [DOI] [PubMed] [Google Scholar]
- 38. Das S., and Oliver D. (2011) Mapping of the SecA-SecY and SecA-SecG interfaces by site-directed in vivo photocross-linking. J. Biol. Chem. 286, 12371–12380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Jilaveanu L. B., Zito C. R., and Oliver D. (2005) Dimeric SecA is essential for protein translocation. Proc. Natl. Acad. Sci. U.S.A. 102, 7511–7516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bunn M. W., and Ordal G. W. (2003) Transmembrane organization of the Bacillus subtilis chemoreceptor McpB deduced by cysteine disulfide cross-linking. J. Mol. Biol. 331, 941–949 [DOI] [PubMed] [Google Scholar]
- 41. Miller J. H. (1972) Experiments in Molecular Genetics, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 42. Ito K. (1984) Identification of the secY (prlA) gene product involved in protein export in Escherichia coli. Mol. Gen. Genet. 197, 204–208 [DOI] [PubMed] [Google Scholar]
- 43. Sachelaru I., Petriman N. A., Kudva R., Kuhn P., Welte T., Knapp B., Drepper F., Warscheid B., and Koch H. G. (2013) YidC occupies the lateral gate of the SecYEG translocon and is sequentially displaced by a nascent membrane protein. J. Biol. Chem. 288, 16295–16307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Oliver D. B., and Beckwith J. (1981) E. coli mutant pleiotropically defective in the export of secreted proteins. Cell 25, 765–772 [DOI] [PubMed] [Google Scholar]
- 45. Schluenzen F., Takemoto C., Wilson D. N., Kaminishi T., Harms J. M., Hanawa-Suetsugu K., Szaflarski W., Kawazoe M., Shirouzu M., Shirouzo M., Nierhaus K. H., Yokoyama S., and Fucini P. (2006) The antibiotic kasugamycin mimics mRNA nucleotides to destabilize tRNA binding and inhibit canonical translation initiation. Nat. Struct. Mol. Biol. 13, 871–878 [DOI] [PubMed] [Google Scholar]
- 46. Young R., and Bremer H. (1976) Polypeptide chain elongation rate in Escherichia coli B-R as a function of growth rate. Biochem. J. 160, 185–194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Pogliano J. A., and Beckwith J. (1994) SecD and SecF facilitate protein export in Escherichia coli. EMBO J. 13, 554–561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Duong F. (2003) Binding, activation, and dissociation of the dimeric SecA ATPase at the dimeric SecYEG translocase. EMBO J. 22, 4375–4384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Dapic V., and Oliver D. (2000) Distinct membrane-binding properties of N- and C-terminal domains of Escherichia coli SecA ATPase. J. Biol. Chem. 275, 25000–25007 [DOI] [PubMed] [Google Scholar]