

Abstract
The dorsal root ganglion (DRG) is a highly vulnerable site in diabetic neuropathy. Under diabetic conditions, the DRG is subjected to tissue ischemia or lower ambient oxygen tension that leads to aberrant metabolic functions. Metabolic dysfunctions have been documented to play a crucial role in the pathogenesis of diverse pain hypersensitivities. However, the contribution of diabetes-induced metabolic dysfunctions in the DRG to the pathogenesis of painful diabetic neuropathy remains ill-explored. In this study, we report that pyruvate dehydrogenase kinases (PDK2 and PDK4), key regulatory enzymes in glucose metabolism, mediate glycolytic metabolic shift in the DRG leading to painful diabetic neuropathy. Streptozotocin-induced diabetes substantially enhanced the expression and activity of the PDKs in the DRG, and the genetic ablation of Pdk2 and Pdk4 attenuated the hyperglycemia-induced pain hypersensitivity. Mechanistically, Pdk2/4 deficiency inhibited the diabetes-induced lactate surge, expression of pain-related ion channels, activation of satellite glial cells, and infiltration of macrophages in the DRG, in addition to reducing central sensitization and neuroinflammation hallmarks in the spinal cord, which probably accounts for the attenuated pain hypersensitivity. Pdk2/4-deficient mice were partly resistant to the diabetes-induced loss of peripheral nerve structure and function. Furthermore, in the experiments using DRG neuron cultures, lactic acid treatment enhanced the expression of the ion channels and compromised cell viability. Finally, the pharmacological inhibition of DRG PDKs or lactic acid production substantially attenuated diabetes-induced pain hypersensitivity. Taken together, PDK2/4 induction and the subsequent lactate surge induce the metabolic shift in the diabetic DRG, thereby contributing to the pathogenesis of painful diabetic neuropathy.
Keywords: diabetes, dorsal root ganglia, lactic acid, metabolism, neuron, pyruvate dehydrogenase kinase (PDC kinase), glia, glycolytic metabolic shift, painful neuropathy
Introduction
Painful neuropathy is one of the most common complications of diabetes. Patients with diabetes frequently exhibit a variety of aberrant sensations, including pain hypersensitivity (1, 2). Interrelation and mutual perpetuation of distinct aberrations of specific metabolic pathways cause painful diabetic neuropathy. Furthermore, painful diabetic neuropathy probably results from a combination of metabolic and immune factors (3, 4). Metabolic aberrations are thought to be early events in painful diabetic neuropathy, leading to biochemical, structural, and functional changes in the dorsal root ganglion (DRG)3 and its nerve trunk (5, 6). Likewise, hyperglycemia-induced immune activation creates an inflammatory microenvironment surrounding the influenced nerves (7).
The DRG is pathologically important in diabetes presenting with painful neuropathic states, which patients with early polyneuropathy commonly experience (8). Ganglionic sensory neurons are devoid of any special protection by the blood-brain or blood-nerve barrier and have higher metabolic requirements than the nerve trunk, which makes the ganglion a vulnerable site in the pathogenesis of diabetic neuropathy (9, 10). Furthermore, the diabetic DRG, compared with the nerve trunk or ending, is highly prone to metabolic abnormalities (11) and is subjected to lower ambient oxygen tensions during hyperglycemia, which may lead to aberrant metabolic functions (12). Emerging evidence indicates that vacuolar degeneration, sensory neuronal apoptosis (13), nerve demyelination (11), inflammatory infiltration (14), and activation of satellite glial cells (SGCs) (15) in the DRG are common presentations in diabetes and are considered as crucial prerequisites for the onset of painful diabetic neuropathy (16). Diabetes-induced DRG tissue ischemia may also lead to metabolic dysfunctions (17). Mitochondrial dysfunction-associated outcomes have been suggested to play a crucial role in the pathogenesis of pain hypersensitivities (18). However, the contribution of hyperglycemia-induced metabolic aberrations to the pathogenesis of painful diabetic neuropathy remains elusive.
The pyruvate dehydrogenase (PDH) kinases (PDKs) are key regulators of the mitochondrial gatekeeping enzyme PDH complex that plays a central role in glucose metabolism. PDH plays an important role in glucose metabolism by linking the citric acid cycle and oxidative phosphorylation with glycolysis and gluconeogenesis (19, 20). In addition, the reversible phosphorylation of PDH by PDKs and dephosphorylation by PDH phosphatases is primarily responsible for the regulation of PDH complex activity (21). PDH complex activity is inhibited by the phosphorylation of PDH-E1 by four different PDK isoforms (PDK1–4) that are expressed in diverse peripheral and central tissues (22). PDK-mediated phosphorylation and subsequent inactivation of PDH result in a metabolic shift toward glycolysis and produce lactate as an end product (23). This glycolytic metabolic shift has been outlined in diverse pathological conditions, including cancer (24), obesity (25), cardiovascular disease (26), liver disease (27), and most importantly diabetes (28). Although lactate is the foremost energy source for neurons (29), augmented lactate accumulation or lactic acidosis has been implicated in neuronal damage and cytotoxic brain edema (30, 31). An acidic microenvironment due to accumulation of lactic acid has been documented to contribute to the development of pathological pain, including tactile allodynia, via nociceptor activation (32). However, the role of the DRG PDKs and glycolytic metabolic shift in the pathogenesis of painful diabetic neuropathy remains to be explored. In the present study, we investigated PDK expression in the streptozotocin (STZ)-induced diabetic DRG. We also compared the characteristics of the DRG, histopathological features of peripheral nerves, nerve conduction velocities, and nociceptive behaviors in wild-type and Pdk2/4-deficient mice. Role of the PDK-lactic acid axis was further investigated by pharmacological inhibition of PDKs and lactic acid production. Cultured DRG neurons were employed to investigate the mechanistic relationship among hyperglycemia, PDK, and lactic acid in the pathogenesis of painful neuropathy.
Experimental Procedures
Mouse Breeding and Maintenance
All experiments were conducted in accordance with approved animal protocols and guidelines established by the Animal Care Committee of Kyungpook National University (Approval KNU-2012-73/66). All efforts were made to minimize the number of animals used and animal suffering. Male wild-type (WT, Pdk2+/+ Pdk4+/+) and Pdk2/4 double knock-out (DKO, Pdk2−/− Pdk4−/−) mice aged 8–10 weeks were used. Pdk2 KO and Pdk4 KO mice were generated as described previously (33). Pdk2 KO mice were crossed with Pdk4 KO mice to produce Pdk2/4 DKO mice. Age-matched WT mice were produced from the C57BL/6J mice (Jackson Laboratory, Bar Harbor, ME), which were used to stabilize the genetic backgrounds of the Pdk2 KO and Pdk4 KO mice. Genotypes were confirmed by PCR of the genomic DNA as described previously (34). Animals were housed under a 12-h light/dark cycle (lights on 07:00–19:00) at a constant ambient temperature of 23 ± 2 °C with food and water provided ad libitum. Each individual animal was used for a single experimental purpose.
Diabetes Induction
Age-matched Pdk2/4 DKO and WT mice of the same background strain (C57BL/6J) were used for the induction of diabetes. The mouse model of diabetes was generated as described previously (35). Briefly, type-1 diabetes was induced by an intraperitoneal injection of STZ (Sigma-Aldrich; 150 mg/kg body weight) in 0.1 m citrate buffer (pH 4.5). For pharmacological studies, type-1 diabetes was induced in male Sprague-Dawley rats (8 weeks old) by intraperitoneal injection of the STZ (65 mg/kg body weight). Blood samples were collected from the tail vein after 3 days of the injection, and glycemia was determined by using an SD CodeFreeTM glucometer (SD Biosensor Inc., Suwon, Korea). Animals with fasting blood glucose values of >260 mg/dl were considered diabetic. In this study, all STZ-injected animals of both genotypes were confirmed to be diabetic.
Behavioral Testing
After arrival in the animal care unit, mice were allowed to acclimate to the testing room, equipment, and experimenter for 1 week and before the actual testing on the same testing day. One experimenter, who was unaware of the animal genotypes or treatment conditions, handled and tested all of the animals. Before the assessment of any pain behavior, we performed the open field test as described previously (36) and confirmed that the deletion of Pdk2/4 genes did not cause motor impairment, which is an important prerequisite for proper pain behavioral testing. Mechanical allodynia associated with painful diabetic neuropathy was assessed by measuring the paw withdrawal threshold (PWT). PWT was evaluated before STZ injection and at different time points afterward. The mechanical sensitivity was tested using calibrated Von Frey filaments (BiosebTM, Chaville, France), as described previously (37). In brief, mice were acclimated for 20 min in inverted individual acrylic boxes with wire mesh floors to provide access to the ventral side of hind paws. Von Frey filaments were presented perpendicularly to the plantar surface and held in position for ∼5 s with enough force to cause a slight bend. Two trials per paw were conducted with an interval of at least 3 min. A positive response was defined as abrupt paw withdrawal. When there was a positive response, the next lower filament was applied, and when there was no response, the next higher filament was used. PWT was quantified from five consecutive responses using Dixon's up-down method (38).
Measurement of Nerve Conduction Velocity
The measurement of nerve conduction velocity (NCV) was performed as described previously (39, 40). Briefly, mice were anesthetized with ketamine. Animals' body temperature was automatically maintained at a mean rectal temperature of 37 ± 0.5 °C with a heated water circulating system (T/Pump, model TP500, Gaymar, Orchard Park, NY). For motor NCV, the sciatic nerve was stimulated (5–10 mA, 0.05-ms single square-wave pulses with low and high filter settings of 0.3 and 3 kHz) proximally at the level of the sciatic notch and distally at the level of the ankle with bipolar electrodes. Compound muscle action potentials were recorded from the first interosseous muscle (between digits 1 and 2) of the hind paw, amplified (16-channel microelectrode amplifier, model 3600, A-M Systems Inc.), stored, displayed, and digitized by a personal computer using AxoScope software (Axon Instruments, Inc., Foster City, CA). Motor latencies were measured from the stimulus to the onset of the negative M-wave deflection of the compound muscle action potentials. Distal and proximal motor latencies from 20 separate recordings were averaged. Motor NCV (in m/s) was calculated by dividing the distance between stimulating electrodes by the average latency difference. Similarly, for sensory NCV, the digital nerve was stimulated with a square-wave pulse of 0.05-ms duration. The compound muscle action potentials were recorded distally at the level of the ankle and proximally at the level of the knee. Distal and proximal sensory latencies from 20 separate recordings were averaged. Sensory NCV (in m/s) was calculated by dividing the distance between recording electrodes by the average latency difference.
Immunohistochemistry and Histopathology
Mice were deeply anesthetized and then perfused through the aorta with 0.1 m phosphate-buffered saline (PBS) followed by 4% paraformaldehyde fixative. The lumbar spinal cord segments and DRGs from the level of L4, L5, and L6 and 1 cm of sciatic nerve were dissected out, post-fixed in the same paraformaldehyde fixative overnight, and cryo-protected in 30% sucrose in 0.1 m PBS overnight at 4 °C. A cryostat was used to prepare 30-μm-thick sections for the spinal cord tissue and 10-μm-thick sections for the DRG tissue. The sectioned DRG tissues were mounted on gelatin-coated slides, and spinal cord sections were placed in 0.1 m PBS. Sections were then blocked with 4% normal serum in 0.3% Triton X-100 for 90 min at room temperature. For immunofluorescence staining, sections were incubated with primary antibodies against PDK2 (rabbit, 1:200; Acris Antibodies, San Diego, CA), PDK4 (rabbit, 1:200; Atlas Antibodies AB, Stockholm, Sweden), phospho-Ser293-PDH-E1α (pyruvate dehydrogenase E1α) (rabbit, 1:200; Calbiochem), phospho-Ser300-PDH-E1α (pyruvate dehydrogenase E1α) (rabbit, 1:200; Calbiochem), MAP2 (mouse, 1:200; Sigma-Aldrich), Iba-1 (goat, 1:200; Novus Biologicals, Littleton, CO), or GFAP (mouse, 1:500; BD Biosciences), overnight at 4 °C, and then incubated with FITC- or Cy3-conjugated secondary antibodies (1:200; Jackson ImmunoResearch, West Grove, PA). Slides were washed, coverslipped with Vectashield mounting medium (Vector Laboratories, Burlingame, CA), and visualized under a fluorescence microscope. DRG and sciatic nerve tissues for hematoxylin and eosin (H&E) staining and sciatic nerve tissues for toluidine blue staining were dehydrated and embedded in paraffin after fixation in 4% paraformaldehyde overnight; a rotary slicer (LEICA RM2135, Wetzlar, Germany) was used to prepare 3-μm thick sections. H&E and toluidine blue staining were performed and analyzed under a light microscope (DP70, Olympus, Tokyo, Japan). The total number of normal or vacuolated DRG neurons was manually counted in six randomly selected microscopic fields captured at high magnification. For quantitative assessment, the percentage of the vacuolated DRG neurons relative to the total number of cells was presented (41).
Quantitative Real-time Reverse Transcription-PCR
Deeply anesthetized mice were perfused through the aorta with 0.1 m PBS to remove the blood, and the lumbar spinal cord and DRG tissues were rapidly dissected. Samples were then immediately frozen in liquid nitrogen and instantly homogenized in TRIzol reagent (Life Technologies, Inc.) for total RNA isolation. Total RNA (2 μg) from each sample was reverse-transcribed into cDNA using a first strand cDNA synthesis kit (MBI Fermentas, Hanover, Germany). Real-time RT-PCR was performed using the one-step SYBR®PrimeScriptTM RT-PCR kit (Perfect Real-Time; Takara Bio Inc., Tokyo, Japan) and the ABI Prism® 7000 sequence detection system (Applied Biosystems, Foster City, CA), according to the manufacturer's instructions. The 2−ΔΔCT method was used to calculate the relative changes in gene expression (42), and Gapdh was used as an internal control. The nucleotide sequences of the primers used in the real-time RT-PCR were as follows: Pdk1, 5′-CAC CAC GCG GAC AAA GG-3′ (forward) and 5′-GCC CAG CGT GAC GTG AA-3′ (reverse); Pdk2, 5′-CCC CGT CCC CGT TGT C-3′ (forward) and 5′-TCG CAG GCA TTG CTG GAT-3′ (reverse); Pdk3, 5′-GGA GCA ATC CCA GCA GTG AA-3′ (forward) and 5′-TGA TCT TGT CCT GTT TAG CCT TGT-3′ (reverse); Pdk4, 5′-CCA TGA GAA GAG CCC AGA AGA-3′ (forward) and 5′-GAA CTT TGA CCA GCG TGT CTA CAA-3′ (reverse); TNF-α, 5′-ATG GCC TCC TCA TCA GTT C-3′ (forward) and 5′-TTG GTT TGC TAC GAC GTG-3′ (reverse); IL-1β, 5′-AAG TTG ACG GAC CCC AAA AGA T-3′ (forward) and 5′-TGT TGA TGT GCT GCT GCG A-3′ (reverse); IL-6, 5′-AGT TGC CTT CTT GGG ACT GA-3′ (forward) and 5′-TCC ACG ATT TCC CAG AGA AC-3′ (reverse); Trpv1, 5′-ACC ACG GCT GCT TAC TAT CG-3′ (forward) and 5′-TCC CCA ACG GTG TTA TTC AG-3′ (reverse); Asic3, 5′-ACA TTG GGG GAC AGA TGG-3′ (forward) and 5′-CAC TGG GAG CGG TAG GAG-3′ (reverse); Gapdh, 5′-TGG GCT ACA CTG AGC ACC AG-3′ (forward) and 5′-GGG TGT CGC TGT TGA AGT CA-3′ (reverse).
Western Blotting Analysis
DRGs (L4–L6) were isolated and washed in ice-cold PBS and placed in 300 μl of lysis buffer (150 mm sodium chloride, 1% Triton X-100, 1% sodium deoxycholate, 0.1% SDS, 50 mm Tris-HCl (pH 7.5), 2 mm EDTA) (GenDEPOT, Barker, TX) containing HaltTM protease inhibitor (1×) and phosphatase protease inhibitor mixtures (1×) (Thermo Scientific). Specimens were individually homogenized and then centrifuged at 13,400 × g at 4 °C for 15 min. Protein concentration was determined with a Bio-Rad protein assay kit using bovine serum albumin as a standard. Proteins (20–30 μg) from each sample were separated on 8 or 15% SDS-polyacrylamide gels and transferred to PVDF membranes (Bio-Rad) by the semidry electroblotting method. The membranes were blocked with 5% skim milk and sequentially incubated with primary antibodies against PDK2 (rabbit, 1:1000 monoclonal antibody; Acris Antibodies), PDK4 (rabbit, 1:1000; Atlas Antibodies AB), phospho-Ser293-PDH-E1α (pyruvate dehydrogenase E1α) (rabbit monoclonal antibody, 1:1000; Calbiochem), phospho-Ser300-PDH-E1α (pyruvate dehydrogenase E1α) (rabbit monoclonal antibody, 1:1000; Calbiochem), PDH-E1 (rabbit monoclonal antibody, 1:1000; Cell Signaling), or α-tubulin (mouse monoclonal antibody, 1:2000; Sigma-Aldrich) and horseradish peroxidase-conjugated secondary antibodies (anti-rabbit and anti-mouse IgG antibody; Amersham Biosciences, Buckinghamshire, UK), followed by enhanced chemiluminescence detection (Amersham Biosciences). Western blotting was repeated three times (n = 3) for each condition.
Lactate Measurement
Mice were sacrificed by cervical dislocation, and the DRGs (L4–L6) were isolated quickly. All of the tissue samples were snap-frozen in liquid nitrogen. On the day of the experiment, tissues were homogenized into 300 μl of lactate assay buffer (Lactate Colorimetric kit, Abcam) and centrifuged at 4 °C at 10,000 × g for 4 min. Supernatants were filtered through a molecular weight 10,000 spin filter to remove all proteins. Samples were tested according to the manufacturer's protocol. Lactate levels were normalized to controls.
Collection and Culture of DRG Neurons
DRG neurons were prepared from adult mice as described previously (43). Briefly, the DRGs from all spinal levels were collected in ice-cooled DMEM (Gibco/Life Technologies, Darmstadt, Germany) and treated with enzyme solution containing 4 mg/ml collagenase, 60 units of papain, and 5 mg/ml dispase (Sigma-Aldrich) for 10 min at 37 °C. Following trituration and centrifugation, the dispersed cells were resuspended in DMEM/F-12 culture media (Sigma-Aldrich) containing 10% FBS (nerve growth factor or glial cell line-derived neurotropic factor was not included in the culture medium) and were plated in 35-mm poly-d-lysine-coated glass-bottomed dishes. Cultures were maintained at 37 °C in a 5% CO2 incubator and assayed after 16–20 h.
Evaluation of DRG Neuronal Viability and Survival
DRG neurons were plated into poly-d-lysine-coated 96-well plate with a density of 5 × 104 cells/well. After 24 h of incubation at different experimental conditions, DRG neurons were treated with glucose (25 mm) for 24 and 48 h and lactic acid (1, 6, 10, and 15 mm with pH 7.38, 7.19, 6.89, and 6.73 media, respectively) for 24 h and were processed for quantification of cell viability by a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT, 0.5 mg/ml; Sigma-Aldrich) assay (44). Briefly, MTT was added to each well and incubated at 37 °C for 2 h in a 5% CO2 incubator. Insoluble formazan crystals were completely dissolved in DMSO, and absorbance at 570 nm was measured using a microplate reader (VersaMax, Molecular Devices, Sunnyvale, CA). Similarly, the same culture conditions were used to perform trypan blue staining. Briefly, DRG neurons plated in 35-mm poly-d-lysine-coated glass-bottomed dishes were exposed to lactic acid (6, 10, and 15 mm) for 24 h. Medium was removed, and cells were stained with 0.2% trypan blue solution for 1 min. Cell survival was quantified and expressed as a percentage relative to the control cells.
Intraganglionic Injection
To ascertain the effect of the pharmacological inhibition of the DRG PDKs and lactic acid production on painful diabetic neuropathy, dichloroacetate (DCA; a PDK inhibitor, 10 mg/kg DRG weight, 5 μl) or vehicle (saline, 5 μl) and 2,3-dihydroxy-6-methyl-7-(phenylmethyl)-4-propyl-1-naphthalenecarboxylic acid (FX11; a small molecule inhibitor of lactate dehydrogenase A, 2, 5, or 10 mg/kg DRG weight, 5 μl) or vehicle (2% (v/v) DMSO, 5 μl) were delivered into the right side L5 DRG of rats using methods similar to those described previously for the intraganglionic injection in rats (45, 46). Briefly, an incision was made along the midline of the back to expose the right para-vertebral region. The right para-spinal muscles were separated from the transverse process at the L5 spinal level, and connective tissues and muscles were removed by iris scissors until the right L5 intervertebral foramen was identified. The L5 DRG was exposed using a micro bone rongeur. A 29-gauge needle with a slightly bent beveled tip was then advanced 2–4 mm into the L5 DRG to deliver the DCA or FX11 using a microinjector. The intraganglionic injection was performed at an angle of 60º relative to the spinal cord. Rats were used for the intraganglionic injection experiments due to the large size of the DRG and surgical convenience compared with mice. Employment of rats in this study also offered an opportunity to validate our findings in another species. It is worthy of mention that DCA at a dose of 25 mg/kg/day has been found to exert peripheral nerve toxicity (47), which is the common side effect during chronic DCA treatment (48). Several previous studies have advocated that 10 mg/kg/day of DCA is non-toxic to nerve directly at least for short term application (49, 50). Lactate dehydrogenase A kinetically favors the conversion of pyruvate to lactate coupled with the recycling of NAD+ (32, 51). Nonetheless, it should be noted that the inhibition of lactate dehydrogenase A also inhibits the production of pyruvate by indirectly suppressing glycolysis at the level of glyceraldehyde-3-phosphate dehydrogenase.
Quantification and Statistical Analysis
For the immunohistochemical analysis, microscopic images of the DRG were obtained (using a ×40 objective lens) in 3–4 tissue sections/animal. Similarly, microscopic images of the dorsal horn of the spinal cord from the L4–L6 region were obtained in 3–4 sections/animal (using a ×20 objective lens). Images of the immunostained tissues were captured with an Olympus DP70 camera and DP Controller software (Olympus). At least three microscopic images were selected randomly for statistical analysis to obtain a better representation of the whole tissue. For the determination of immunofluorescence intensity, the area of the whole image was selected, and the mean intensity was measured using ImageJ software (National Institutes of Health, Bethesda, MD). Likewise, intensities of bands obtained by Western blotting were also quantified using ImageJ. The background intensity of the band was also determined and deducted from the values obtained. The graphs represent the average of all images. All of the results are presented as the means ± S.E. Statistical comparisons were performed by either Student's t test or one-way analysis of variance (ANOVA) with Dunnett's multiple-comparison or two-way ANOVA test by using SPSS version 22.0K (SPSS Inc., Chicago, IL). The Mann-Whitney test and the one-way ANOVA with Dunnett's multiple-comparison tests were used to compare the PWTs measured by the von Frey filaments with the up-down paradigm at a single interval. Differences in the probability values of less than 0.05 (p < 0.05) were considered statistically significant.
Results
Enhanced Expression and Activity of PDK2 and PDK4 in the Diabetic DRG
To investigate the role of PDKs in the pathogenesis of diabetic neuropathic pain, we first examined the expression and activity of PDK isoforms (PDK1–4) in the DRG tissues following STZ injection by using real-time RT-PCR, Western blotting analysis, and immunostaining. A substantially increased expression of Pdk2 mRNA at 3 weeks and Pdk4 mRNA at 2, 3, and 6 weeks (Fig. 1A), but not Pdk1 or Pdk3 mRNA (data not shown), was found in the DRG tissues following STZ injection. The expression of Pdk2 and Pdk4 mRNAs peaked at 3 weeks post-STZ injection. Similarly, the levels of PDK2 and PDK4 proteins (Fig. 1B) and phosphorylated PDH proteins (p-S293-PDH and p-S300-PDH) (Fig. 1, C and D) were markedly increased in the DRG tissues at 1–3 weeks post-STZ injection compared with those of vehicle-injected control animals. Next, we conducted immunostaining to identity the cell types in the DRG that express PDKs (Fig. 2). Double immunostaining of PDK2/4 and MAP2 (a neuronal marker) in the DRG at 3 weeks after STZ injection showed that PDK2 and PDK4 were primarily co-localized with the ganglionic neurons (Fig. 2A), indicating that neurons are the major cell type expressing PDKs in the DRG from diabetic animals. Furthermore, increased immunoreactivities for Iba-1 (a macrophage marker in the periphery (52)) (Fig. 2B) and GFAP (a SGC marker in the DRG (15)) (Fig. 2C) were observed in the DRG tissues at 3 weeks post-STZ injection, which is the time point representing the maximal expression of PDK2 and PDK4 mRNAs and proteins. PDK2 and PDK4 were also found to be partially co-localized with macrophages and SGCs. No considerable immunoreactivities for Iba-1 or GFAP were observed in the vehicle-injected control animals (Fig. 2). These data suggest that hyperglycemia enhanced the expression and activity of PDKs, especially PDK2 and PDK4, in the ganglionic neurons, SGCs, and infiltrated macrophages.
FIGURE 1.

Expression of PDK2, PDK4, and phosphorylated PDH in the DRG post-STZ injection. The expression of Pdk2 and Pdk4 mRNAs in the DRG at 1, 2, 3, and 6 weeks following STZ injection was assessed by real-time RT-PCR (A). Results were obtained from three different animals for each condition. Protein levels of PDK2 and PDK4 at 3 weeks post-STZ injection (B) and phosphorylated PDH (p-S293-PDH and p-S300-PDH) at 1 week (C) and 3 weeks (D) after STZ injection in the DRG were assessed by Western blotting analysis. Quantification of the band intensities is shown in the adjacent graphs. *, p < 0.05 versus the vehicle-treated control animals. Quantification was based on normalization to PDH-E1 for C and D, Student's t test, n = 3; mean ± S.E. (error bars). w, week(s).
FIGURE 2.

Expression of PDK2 and PDK4 in neurons, infiltrated macrophages, and activated satellite glial cells in the diabetic DRG. Immunofluorescence analyses show the expression of PDK2 and PDK4 in neurons (A), infiltrated macrophages (B), and activated SGCs (C) in the DRG at 3 weeks post-STZ injection. PDK2 and PDK4 co-localized with MAP2 (a neuronal marker), Iba-1 (a macrophage marker), and GFAP (a SGC marker). Arrows indicate the representative double-labeled cells. Scale bars, 100 μm. The number of cells expressing PDK2 or PDK4 is presented in adjacent graphs. Images show the representative results of at least three independent experiments. *, p < 0.05 versus the vehicle-treated control animals, Student's t test, n = 3; mean ± S.E. (error bars). ND, not detected; MAP2, microtubule-associated protein 2.
Attenuation of Diabetes-induced Pain Hypersensitivity in Pdk2/4-deficient Animals
Based on the enhanced expression and activity of PDK2 and PDK4 in the DRG, we hypothesized that PDK2 and PDK4 up-regulation in the DRG may play an important role in the pathogenesis of painful diabetic neuropathy. This hypothesis was tested using Pdk2/4 DKO mice. We compared the pain responses of WT and Pdk2/4 DKO mice before and up to 15 weeks after STZ injection (Fig. 3). Pain hypersensitivity (mechanical allodynia) was induced from 1 to 4 weeks post-STZ injection. The diabetes-induced pain hypersensitivity was substantially attenuated by Pdk2/4 deficiency. Mechanical hypersensitivity was observed in diabetic mice at an early phase (1–4 weeks post-STZ injection). However, diabetes-induced mechanical allodynia was not seen after 4 weeks post-STZ injection. This finding suggests that PDK2/4 play a crucial role in the pathogenesis of diabetes-induced pain hypersensitivity.
FIGURE 3.
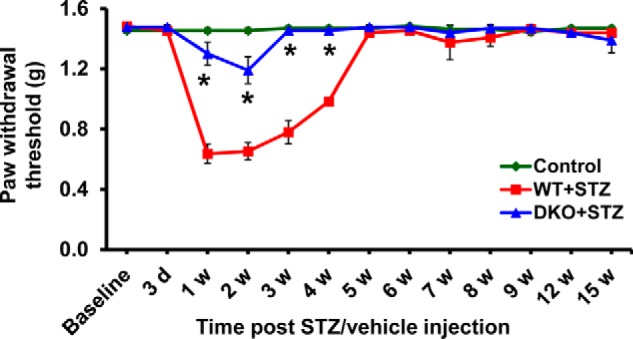
Pdk2/4 DKO mice display attenuated pain response following diabetes induction. STZ injection reduced paw withdrawal threshold to force. The STZ-induced mechanical hypersensitivity is attenuated in Pdk2/4 DKO mice compared with WT animals up to 4 weeks. *, p < 0.05 versus the WT animals with diabetes, Mann-Whitney and one-way ANOVA with Dunnett's multiple-comparison tests for paw withdrawal threshold, n = 10 (up to 6 weeks) or n = 4 (after 6 weeks); mean ± S.E. (error bars). w, week(s).
Pdk2/4 Deficiency Diminished Macrophage Infiltration, Activation of Satellite Glial Cells, Proinflammatory Cytokine Release, Lactate Surge, and Expression of Pain-related Ion Channels in the Diabetic DRG
Immunofluorescence analyses showed the infiltration of macrophages and activation of SGCs in the DRG at 3 weeks post-STZ injection. Infiltration of Iba-1-positive macrophages in the diabetic DRG was significantly reduced in the Pdk2/4-deficient mice (Fig. 4A). Similarly, the DRGs from diabetic mice showed activation of SGCs, defined by increased immunoreactivity for GFAP and structural hypertrophy, compared with that of vehicle-injected control animals. Interestingly, these hallmarks of activated SGCs were reduced in the DRG from Pdk2/4-deficient diabetic mice (Fig. 4A). Furthermore, diabetes-induced expression of proinflammatory cytokines, such as TNF-α, IL-1β, and IL-6, in the DRGs was substantially diminished by Pdk2/4 deficiency (Fig. 4B). The fact that the diabetic DRG is subjected to lower ambient oxygen tensions during hyperglycemia (12), which favors the conversion of pyruvate to lactate, encouraged us to assess the production of lactic acid in the diabetic DRGs. As expected, the lactate production was substantially augmented in the DRGs of diabetic animals compared with that of control animals. In this study, DRG lactate concentration in control animals has been found to be 0.74 mmol/liter, which is consistent with the previous study showing a lactate level of less than 2 mmol/liter in normal hind paw tissue (53). Pdk2/4 deficiency lessened the hyperglycemia-induced lactate surge (Fig. 4C). The results are consistent with the up-regulation of PDK2 and PDK4 in the diabetic DRG (Fig. 1, A and B). Up-regulated PDKs may inhibit PDH, thereby inducing a metabolic shift toward lactic acid production. It is well documented that the acidic microenvironment favors the expression of the transient receptor potential vanilloid (TRPV) (54) and acid-sensing ion channel (ASIC) (55). TRPV1, which is widely distributed in the peripheral nervous system, has been reported to play a crucial role in the detection, transduction/transmission, and regulation of pain in diverse pathologies (56, 57). Similarly, ASIC3, which is widely distributed in the ganglionic sensory neurons, is highly sensitive to acidic pH and is activated by extracellular acidosis, causing pain under several pathological conditions (58). Thus, we next assessed the expression of TRPV1 and ASIC3 in the diabetic DRGs and found their substantial up-regulation at the level of mRNAs (Fig. 4D). Genetic deletion of Pdk2/4 significantly lessened the up-regulation of Trpv1 and Asic3 mRNAs expression in the DRGs (Fig. 4D), indicating a mechanistic role played by PDK2/4 in the peripheral sensitization and the subsequent pain transduction. Taken together, these findings suggest that PDK2/4 in the diabetic DRG play a crucial role in the induction of inflammatory infiltration, activation of SGCs, augmented lactate production, and ultimately the induction of peripheral sensitization, a prerequisite for the development of pain hypersensitivity.
FIGURE 4.

Pdk2/4 deficiency attenuates macrophage infiltration, activation of satellite glial cells, proinflammatory cytokines expression, lactate surge, and expression of Trpv1 and Asic3 in the diabetic DRG. A, Iba-1(a macrophage marker) and GFAP (an SGC marker) immunostainings reveal their increased immunoreactivity in the DRG of WT animals at 3 weeks post-STZ injection, whereas Pdk2/4 deficiency significantly attenuates such an increase in immunoreactivities. Quantifications and statistical analyses of stained images are presented in adjacent graphs. Scale bars, 100 μm. *, p < 0.05 versus the vehicle-treated control animals; #, p < 0.05 between the indicated groups, Student's t test, n = 6; mean ± S.E. IR, immunoreactivity. B, the relative expression of TNF-α, IL-1β, and IL-6 mRNAs in the DRGs after 3 weeks of STZ injection as evaluated by real-time RT-PCR. C, lactate assay was performed to measure the lactate accumulation in the DRG at 3 weeks after STZ injection. Results of lactate levels are presented as the -fold change relative to control. D, the expression of Trpv1 and Asic3 mRNAs in the DRGs at 3 weeks after STZ injection was assessed by real-time RT-PCR. Results for mRNA expression are displayed as the -fold increase of gene expression normalized to GAPDH. *, p < 0.05 versus the vehicle-treated control animals; #, p < 0.05 between the indicated groups, Student's t test, n = 6 (for B and D) or n = 3 (for C); mean ± S.E. (error bars).
Effect of Hyperglycemia and Lactic Acid on Cultured DRG Neurons
Enhanced expression of PDK2/4, augmented production of lactic acid, and up-regulated expression of Trpv1 as well as Asic3 in the diabetic DRGs led us to investigate the mechanistic relationship among hyperglycemia, PDKs, lactic acid, ion channels, and viability of sensory neurons in the development of pain hypersensitivity using cultured DRG neurons. Exposure of cultured DRG neurons to glucose (25 mm), but not lactic acid (10 mm) and recombinant mouse TNF-α protein (rmTNF-α; 100 ng/ml), for 24 h substantially enhanced the expression of Pdk2 and Pdk4 mRNAs (Fig. 5A). These results are consistent with a previous study reporting that treatment of primary islet cells with a high concentration of glucose substantially increased the expression of Pdk2 and Pdk4 mRNAs (59). However, the expression of Pdk1 and Pdk3 mRNAs was unaltered compared with those in untreated control neurons (data not shown). These findings suggest that hyperglycemia directly enhances the expression of PDK2 and PDK4 in the DRG. Moreover, following 24-h exposure to lactic acid, but not glucose, cultured DRG neurons showed enhanced expression of Trpv1 and Asic3 mRNAs (Fig. 5B). In addition, rmTNF-α-stimulated cultured neurons revealed significantly enhanced expression of Trpv1 but not Asic3. In a separate set of experiments, hyperglycemic exposure for 24 and 48 h induced a small but significant decrease in the viability of DRG neuron cultures, which was not observed in the DRG neurons isolated from Pdk2/4-deficient mice (Fig. 5C). Furthermore, following 24 h of exposure to lactic acid at concentrations of 10 and 15 mm, but not of 1 or 6 mm, cultured DRG neurons exhibited slightly but significantly compromised viability (Fig. 5D). These findings outline the role of hyperglycemia as a direct enhancer of PDK2/4 expression in the DRG, the role of lactic acid as an inducer of pain-related ion channel expression, and the neurotoxic effect of glucose-lactic acid in the diabetic DRG. These results also indicate that the metabolic aberrations associated with the glucose-PDK-PDH-lactic acid axis in the diabetic DRG cause neuronal toxicity and pain sensitization, which ultimately drive the pathogenesis of painful diabetic neuropathy. This assumption was supported by histological analysis of DRG. The histological study of DRG from diabetic animals revealed that the DRG tissues exhibited loose, disorganized, and vacuolar-like defects along with degenerated and dispersed sensory neurons compared with those of vehicle-injected control animals; however, such morphological changes in the diabetic DRG were minimized by the Pdk2/4 deficiency (Fig. 5E). These results suggest that PDK2/4 are closely involved in the progression of diabetes-induced histopathological alterations in the DRG and subsequent development of pain hypersensitivity.
FIGURE 5.

Effect of glucose and lactic acid on DRG neurons. Cultured DRG neurons were treated with glucose (25 mm), lactic acid (10 mm), or recombinant mouse TNF-α protein (rmTNF-α; 100 ng/ml) for 24 h. The expression of Pdk2 and Pdk4 (A) and Trpv1 and Asic3 (B) mRNAs was assessed by real-time RT-PCR. Cultured DRG neurons were treated with 25 mm glucose for 24–48 h (C) or with 1, 6, 10, and 15 mm lactic acid for 24 h (D). An MTT assay (left) or trypan blue staining (right) were performed to assess the neuronal viability at the specified time points following the treatment. For trypan blue staining, the number of neurons in the whole image was counted. The average number of neurons in control conditions was 400.33 ± 6.23. The experiment was done in duplicate. E, H&E staining of paraffin sections of the DRG was performed to assess diabetes-induced alterations in the DRG tissue integrity at 3 weeks post-STZ injection. Quantification of vacuolated DRG neurons (in percent) is presented in the adjacent graph. Scale bar, 50 μm. *, p < 0.05 versus the untreated controls; #, p < 0.05 between the indicated groups, one-way ANOVA with Dunnett's multiple-comparison test (for A, B, and D); two-way ANOVA (for C). NS, not significant; n = 3 (for A–D) or n = 6 (for E); mean ± S.E. (error bars).
Pharmacological Inhibition of DRG PDKs and Lactic Acid Production Attenuated Diabetes-induced Pain Hypersensitivity
The crucial contribution of PDKs and lactic acid production in the DRG to the pathogenesis of painful diabetic neuropathy was ascertained by pharmacological inhibition of DRG PDKs and lactate dehydrogenase A. Intraganglionic injection of the pharmacological inhibitors was done in rats, because the size of mouse DRG is too small for the direct needle injection. DCA was used to inhibit PDK, whereas lactate dehydrogenase A inhibitor FX11 was used to suppress lactic acid production. Single intraganglionic injection of DCA (10 mg/kg DRG weight) (Fig. 6A) or FX11 (10 mg/kg DRG weight) (Fig. 6B) in rats having painful diabetic neuropathy (2 weeks after STZ administration) significantly attenuated the diabetes-induced mechanical allodynia within 6 h to 1 day following injection. We also assessed the effect of FX11 on mechanical allodynia at the doses of 2 and 5 mg/kg DRG weight: FX11 exerted dose-dependent effects on diabetes-induced mechanical allodynia (data not shown). However, vehicle or surgical operation alone did not alter the PWTs. We confirmed that STZ-induced diabetes in rats was also accompanied by a substantial enhancement of PDK4 expression in the DRG at 3 weeks post-STZ injection (Fig. 6C).
FIGURE 6.
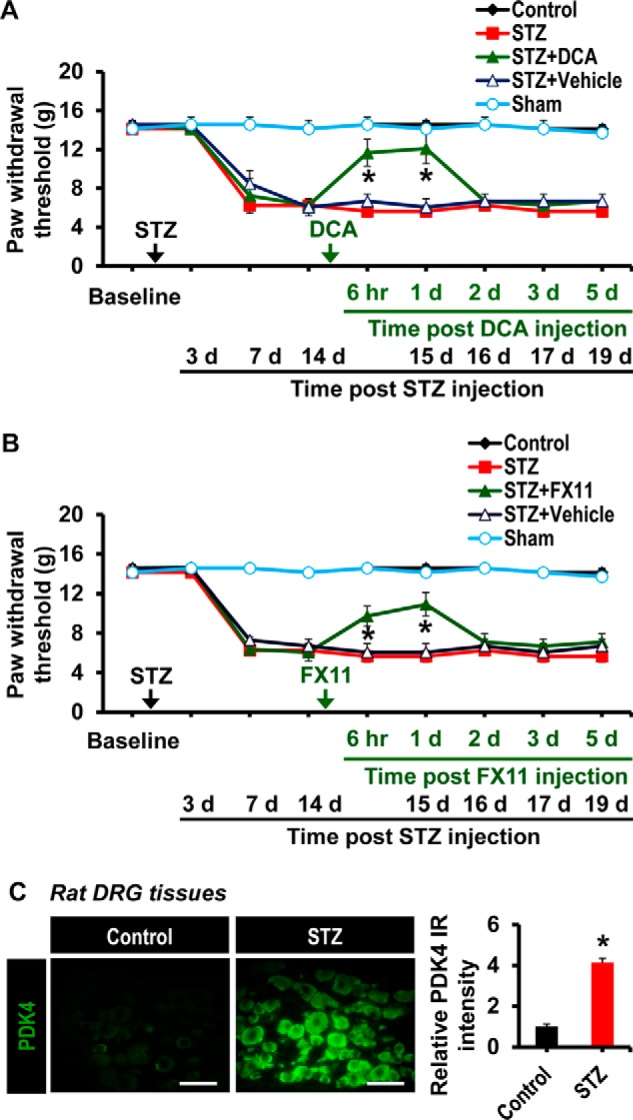
Effect of the pharmacological inhibition of the DRG PDKs and lactic acid production on diabetes-induced mechanical allodynia. To determine the role of the DRG PDKs and lactic acid in the diabetes-induced pain hypersensitivity, DCA (10 mg/kg DRG, 5 μl) or vehicle (A) and FX11 (10 mg/kg DRG weight, 5 μl) or vehicle (B) were administered to rats via the intraganglionic route after 2 weeks of STZ injection. Paw withdrawal threshold to force was assessed at 6 h, 1 day, 2 days, 3 days, and 5 days following DCA and FX11 administration. Arrows, time points of STZ, DCA, FX11, or vehicle administration. C, immunofluorescence analysis shows the expression of PDK4 in rat DRG tissues at 3 weeks post-STZ injection. *, p < 0.05 versus the animals with diabetes, Mann-Whitney and one-way ANOVA with Dunnett's multiple-comparison tests for paw withdrawal thresholds, n = 4 (for A and B) or n = 3 (for C); mean ± S.E. (error bars). d, day(s).
Pdk2/4 Deficiency Attenuated Diabetes-induced Deficit in Nerve Conduction Velocity and Loss of Sciatic Nerve Integrity
The pivotal role of PDK2/4 in diabetes-induced histopathological alterations in the DRG encouraged us to investigate the diabetes-induced functional and structural changes in its nerve trunk. We first examined the peripheral nerve function by measuring the NCVs. Animals with diabetes exhibited substantial deficits in the sensory and motor NCVs compared with control animals. Pdk2/4 deficiency significantly attenuated the diabetes-induced NCV deficit (Fig. 7A). We performed H&E and toluidine blue staining of the sciatic nerve to further examine the diabetes-induced histopathological changes in the peripheral nerve. Diabetic mice exhibited loose, thin, and disorganized myelinated nerve fibers. Some nerve fibers in the sciatic nerve appeared demyelinated, lamellar spaces were expanded and separated from each other, and visible signs of axonal atrophy were evident at 3 weeks post-STZ injection compared with the vehicle-injected control animals. On the other hand, the sciatic nerve from Pdk2/4-deficient animals was partially resistant to the diabetes-induced structural and functional alterations (Fig. 7B). These findings suggest that PDK2/4 are implicated in the structural and functional alterations of the diabetic nerve trunk that originates from the DRG and thereby contribute to the pathogenesis of painful diabetic neuropathy.
FIGURE 7.
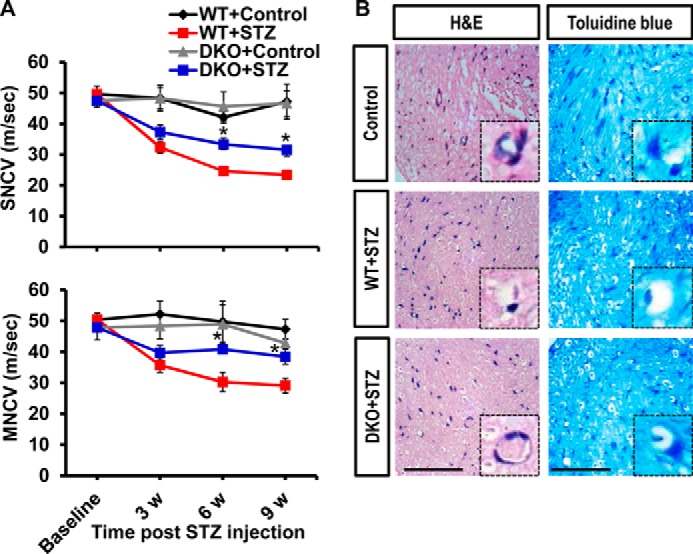
Pdk2/4 deficiency attenuates nerve conduction velocity deficit and sciatic nerve damage in diabetic mice. A, sensory and motor nerve conduction velocities (sensory NCV and motor NCV) assessed before and at 3, 6, and 9 weeks after STZ injection. B, H&E and toluidine blue staining of paraffin-sectioned sciatic nerve performed at 3 weeks post-STZ injection. The high magnification image (insets in B; original magnification, ×400) shows a single nerve fiber. Scale bar, 50 μm. Images show the representative results of at least three independent experiments. *, p < 0.05 between WT and DKO animals with diabetes, Student's t test, and one-way ANOVA with Dunnett's multiple-comparison test. NS, not significant; n = 3; mean ± S.E. w, week(s).
Role of PDK2/4 in Spinal Glial Activation, Expression of Proinflammatory Cytokines, and Induction of Central Sensitization
We next examined the expression and role of PDK2/4 in the spinal cord at 3 weeks post-STZ injection. Although STZ-induced hyperglycemia did not cause any significant change in the expression of Pdk1-4 mRNAs in the lumbar segment of the spinal cord (data not shown), a significant increase in the number of Iba-1-positive microglial cells (60) was observed in its dorsal horn, where microglia displayed enhanced Iba-1 immunoreactivity with reactive morphological changes. However, the Iba-1 immunoreactivity and microglial morphological changes were attenuated in the DKO mice (Fig. 8A). Likewise, the number of GFAP-positive astrocytes (61) was markedly increased in the spinal cord dorsal horn of WT mice, which was accompanied by the increase of GFAP immunoreactivity and hypertrophic morphology with thick processes. On the other hand, DKO animals with diabetes showed a significantly decreased spinal GFAP immunoreactivity (Fig. 8A). Moreover, WT mice with diabetes showed a significantly increased expression of TNF-α, IL-1β, and IL-6 mRNAs in the lumbar segment of the spinal cord at 3 weeks of post-STZ injection, but such an increase was substantially lessened in Pdk2/4-deficient animals (Fig. 8B). Furthermore, we also assessed the expression of spinal phosphorylated ERK (p-ERK), which is a dynamic and canonical marker for central sensitization in spinal neurons (62). Phosphorylation of ERK in the spinal cord dorsal horn, especially in superficial laminas, was strongly induced in diabetic animals, which was inhibited by Pdk2/4 deficiency (Fig. 8C). These findings indicate that the activation of glia, enhanced expression of proinflammatory cytokines, and induction of central sensitization in the spinal cord might be a direct consequence of PDK2/4-associated pathological alterations in the DRG.
FIGURE 8.
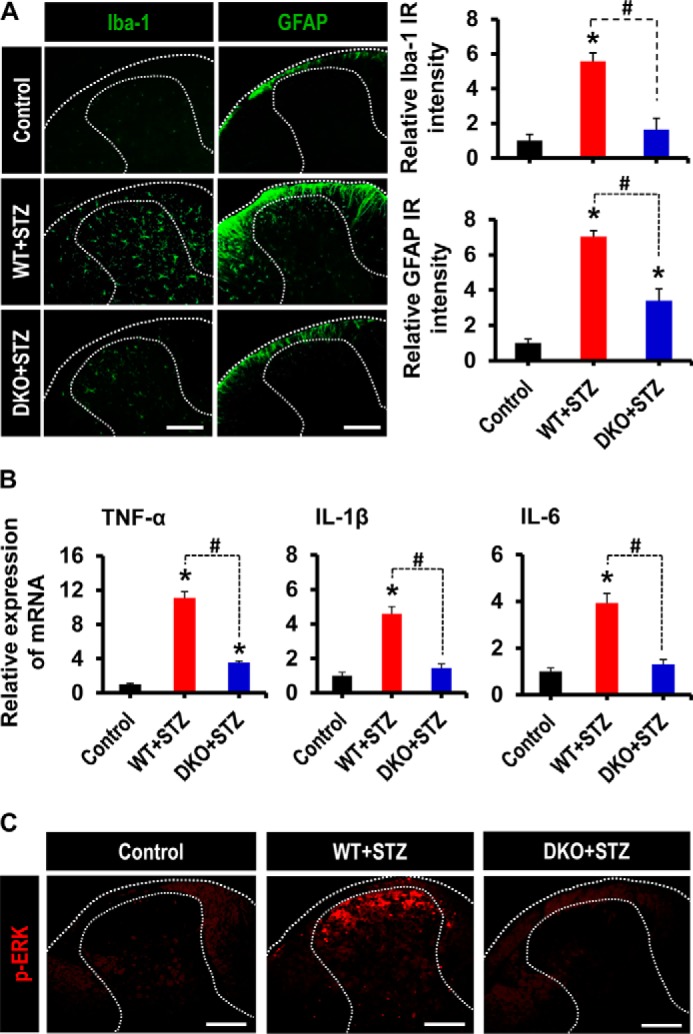
Role of PDK2/4 in glial activation, expression of proinflammatory cytokines, and phosphorylation of ERK in the spinal cord of diabetic mice. A, Iba-1 (a microglia marker) and GFAP (an astrocyte marker) immunoreactivities significantly increased in the dorsal horn of the lumbar segment of the spinal cord of WT mice at 3 weeks post-STZ injection, whereas these immunoreactivities significantly diminished in the diabetic DKO mice. Dotted lines demarcate the white and gray matters in the spinal cord dorsal horn. Quantifications and statistical analyses of stained images are presented in adjacent graphs. B, the relative mRNA expression of TNF-α, IL-1β, and IL-6 in the lumbar segment of the spinal cord (L4–6) at 3 weeks after STZ injection as assessed by real-time RT-PCR. Results for mRNA expression are displayed as the -fold increase of gene expression normalized to GAPDH. C, immunofluorescence staining was performed to detect the expression of p-ERK in the lumbar segment of the spinal cord from vehicle/STZ-injected animals at 3 weeks post-injection. Scale bar, 200 μm. Images show the representative results of at least three independent experiments. *, p < 0.05 versus the vehicle-treated control animals; #, p < 0.05 between the indicated groups, Student's t test, n = 6 (for A and B) and n = 3 (for C); mean ± S.E. (error bars).
Discussion
The present study demonstrates that PDK2/4-mediated glycolytic metabolic shift in the DRG plays an important role in the pathogenesis of diabetes-induced pain hypersensitivity. STZ-induced diabetes significantly up-regulated the expression and activity of PDK2 and PDK4 in the DRG. These PDKs were found to be expressed in DRG sensory neurons, activated SGCs, and infiltrated macrophages. Our study employing the Pdk2/4-deficient mice revealed that the hyperglycemia-induced pain hypersensitivity was substantially attenuated but not completely abolished by the genetic ablation of Pdk2/4. Furthermore, hyperglycemia-induced lactate surge, activation of SGCs, infiltration of macrophages, and expression of pain-related ion channels in the DRG, prerequisites for peripheral sensitization, were significantly attenuated in Pdk2/4-deficient mice. Besides, ablation of Pdk2/4 genes inhibited hyperglycemia-induced spinal glial activation and phosphorylation of ERK, potential markers for central sensitization. We also found that Pdk2/4 deficiency alleviated the diabetes-induced structural and functional deficits in the peripheral nerve. In addition, cultured DRG neurons exposed to a high concentration of glucose or lactic acid showed a reduction in the viability, and the lactic acid-induced acidic microenvironment favored the enhanced expression of Trpv1 and Asic3. Moreover, these ganglionic neurons exhibited substantially enhanced expression of Pdk2 and Pdk4 when exposed to high glucose concentration. Furthermore, the pharmacological inhibition of the DRG PDKs or lactic acid production substantially attenuated the diabetes-induced pain hypersensitivity. These in vivo and in vitro findings suggest that hyperglycemia-triggered increase in the expression and activity of PDK2/4 mediates the glycolytic metabolic shift in the DRG and drives the pathogenesis of painful diabetic neuropathy (Fig. 9).
FIGURE 9.

A proposed schematic outlining the implications of PDK2/4 in the pathogenesis of painful diabetic neuropathy. Hyperglycemia induces the expression of PDK2/4 in DRG neurons, SGCs, and infiltrated macrophages. Glucose is thought to directly enhance the expression of the PDKs in DRG sensory neurons, besides exerting direct neurotoxic effects. Consequently, the hyperglycemia-induced heightened expression of the PDKs facilitates the conversion of pyruvate into lactic acid via inhibition of PDH. The infiltrated macrophages and reactive SGCs also participate in the production of lactic acid. The glycolytic metabolic shift in the DRG and ensuing production of lactic acid induces neuronal damage and lowers pH. The acidic microenvironment favors the enhanced expression of Trpv1 and Asic3 in the ganglionic sensory neurons, which causes neuronal hyperexcitability (peripheral sensitization). Furthermore, reactive SGCs and infiltrated macrophages in the DRG also release diverse proalgesic mediators, including inflammatory cytokines. The peripheral nerve trunk is also affected by hyperglycemia, contributing to the induction of peripheral sensitization. Finally, the glucose-PDK-PDH-lactic acid axis in the DRG induces central sensitization involving spinal glial activation, up-regulation of proinflammatory cytokines, and neuronal ERK phosphorylation. Thus, PDK2/4 up-regulation in the diabetic DRG plays a critical role in inducing peripheral as well as central sensitizations, eventually causing diabetic neuropathic pain.
Pain is a manifestation of aberrant function of the nervous system in patients with diabetes (63, 64). In this study, the expression and activity of PDK2/4 were found to be substantially increased at 1–6 weeks and peaking at 3 weeks in the DRG following diabetes induction. STZ-induced diabetic rodents are known to display robust mechanical hypersensitivity from the very beginning of diabetes induction that persists for 4–5 weeks (65–67). The STZ-induced hyperglycemia invites alterations in nerve fibers and subsequent abnormal nerve signals/functions (68). Further, the hyperglycemia adversely interferes with the functioning of blood vessels supplying to nerves (69). Hyperglycemic conditions in concert with nerve abnormalities and dysfunctional blood supply result in the release of proalgesic mediators and activation of nociceptors, thereby leading to the development of pain hypersensitivity (70). However, the STZ-induced alterations, such as axonopathy, deficit in nerve conduction velocity, nerve demyelination, and neurodegeneration, are progressive and irreversible, which results in the attenuation of pain perception (71). The progressive loss of distal fibers and subsequent lessening of epidermal small fiber density also suggest the transient nature of the STZ-induced pain hypersensitivity. Moreover, altered pain behavioral responses in the STZ-induced diabetic rodents might be due to ketoacidosis, weight loss, and physical decline (72). Expectedly, in the present study, the diabetes-induced mechanical hypersensitivity was observed from 1 to 4 weeks post-STZ injection. In addition, deficiency of Pdk2/4 substantially inhibited the development of diabetes-induced mechanical hypersensitivity compared with WT animals. DCA administration into the DRG attenuated diabetes-induced pain hypersensitivity, suggesting that the DRG PDKs play a critical role in the pathogenesis of diabetic pain hypersensitivity. DCA, a PDK inhibitor, has been well documented to manage lactic acidosis (73), inhibit activation of immune cells, and obstruct the inflammatory progression (74). As discussed previously, neuron is the major cell type that expresses PDKs. It is worth mentioning that diabetic DRG is subjected to irreversible nerve damage, which might be related to the transient expression of PDKs. The diabetic DRGs also display activation of satellite glial cells, an upsurge of lactic acid, and an inflammatory microenvironment. Such diabetes-induced pathological changes are key drivers for the ontology of painful neuropathy in experimental animals and patients. In line with preclinical studies, patients with painful diabetic neuropathy also experience irreversible nerve damage, lactic acidosis, and diminished nerve functions (71), indicating that the critical role of PDKs found in preclinical models might also be relevant to patients. Moreover, PDKs are markedly increased in skeletal muscle of rats exposed to a high fat diet (75, 76). Furthermore, Tao et al. (77) have recently reported that inactivation of Pdk genes improves hepatic insulin resistance-induced diabetes. In addition, skeletal muscle expression of PDK4 and related genes regulating mitochondrial function has reflected alterations in substrate utilization and clinical features associated with type 2 diabetes (78). PDK inhibitors binding at the pyruvate or lipoyl binding sites have been shown to lower blood glucose level in insulin-resistant animals (79). Combining our results with these findings, it is speculated that PDKs might play a pivotal role in both type 1 and 2 diabetes-driven pain hypersensitivity. These results, taken together, suggest an important role of PDK2/4 in the pain complication of patients with diabetes.
Peripheral as well as central glial cells are crucial players in the maintenance of homeostasis of the nervous system, providing neuronal metabolic support, pH regulation, and neuronal survival (80). SGC, a glial cell type in the DRG, has been recently implicated in pain pathobiology. Emerging evidence has amply documented the pathological contributions of SGCs in diverse experimental models of chronic pain, including axotomy, inflammation, and chemical-induced neuropathy as well as diabetic neuropathy (15). Furthermore, SGCs, similar to their central counterparts (81), are considered to be activated in many preclinical pain models, including the painful diabetic neuropathy model (15, 82). Activated SGCs produce proinflammatory mediators, including TNF-α and IL-1β, under diverse pathological conditions (83). In this study, Pdk2/4 deficiency remarkably lessened the hyperglycemia-induced activation of SGCs, demonstrating that PDK2/4 play a crucial role in the pathological alterations of SGCs. Hyperglycemia also drives the inflammatory infiltration of macrophages in the DRG, which subsequently contributes to nerve demyelination and release of different proinflammatory mediators having an important role in the pathogenesis of painful neuropathy (14, 84, 85). Macrophage-derived proinflammatory cytokines, such as TNF-α, IL-1β, and IL-6, are documented to contribute to the pathogenesis of painful diabetic neuropathy (86, 87). In the current study, Pdk2/4 deficiency markedly attenuated the diabetes-induced macrophage infiltration and the subsequent expression of proinflammatory cytokines in the DRG. These findings suggest that the PDKs play a pivotal role in the induction of reactive SGCs and the creation of proinflammatory microenvironment in the diabetic DRG, the prerequisites for pain pathogenesis.
Lactate, the end product of glycolysis (23), serves as an oxidative fuel for neurons (88). Glycolytic metabolic shift has been demonstrated to be a key player in prevalent diseases, including brain edema, inflammation, tissue injury, ischemic stroke, and cancer (31, 89, 90). Neurons also produce lactate in pathological conditions. This lactate is involved in the pathogenesis of diverse neurodegenerative diseases (91–93). Similarly, augmented lactate accumulation has been identified as one of the most important deleterious effects of hyperglycemia (94). It has been well documented that patients with diabetes frequently exhibit increased blood lactic acid levels with decreased pH (95, 96). In the present study, STZ-induced hyperglycemia increased the lactate production in the DRG, which was significantly diminished in Pdk2/4-deficient animals. Neurons are reported to be greatly susceptible to swelling at certain non-physiologic pH levels, and they are found to be more vulnerable than glial cells (97, 98). Glia and macrophages are also sources of lactic acid under diverse pathological conditions (99–101). A lactic acid-induced acidic microenvironment provokes the activation of glial cells and macrophages and thereby favors the release of diverse proinflammatory mediators, suggesting that the decreased pH due to accumulation of lactic acid might serve as a potential inducer of proinflammatory cytokine production (102). In addition, lactic acid influences every cell type that expresses ASIC. Especially, ASIC3 is of interest, because it is commonly expressed at extremely high levels in virtually all DRG sensory neurons (103). Likewise, TRPV1 is a well documented ion channel associated with the progression of pain hypersensitivities (104, 105). ASIC3 and TRPV1 are highly sensitive to acidic microenvironment, acting as transducers for nociceptive responses. Accumulating evidence suggests that the expression of these ion channels is increased in the DRG of experimental diabetic animals (106, 107). In this study, the induction of Trpv1 and Asic3 expression was confirmed in the diabetic DRG, and Pdk2/4 deficiency significantly attenuated the hyperglycemia-induced heightened expression of the ion channels. Furthermore, cultured DRG neurons following exposure to lactic acid exhibited enhanced expression of Trpv1 and Asic3 along with a significant degree of irreversible neurotoxic effects. The crucial role of lactic acid in diabetes-induced nociceptive behavior was confirmed by pharmacological inhibition of lactic acid production using lactate dehydrogenase A inhibitor, which substantially attenuated diabetes-induced pain hypersensitivity. In addition, electrophysiological and histopathological investigations revealed a substantial difference between WT and Pdk2/4-deficient diabetic mice in terms of the structural and functional characteristics of the DRG nerve trunk. Diabetes-induced NCV deficit and compromised sciatic nerve integrity were significantly attenuated by Pdk2/4 deficiency. These findings substantiate the crucial role of PDK/PDH-regulated lactic acid accumulation in the diabetic DRG in the development of peripheral neuropathy and pain hypersensitivity.
Neuronal sensitization in the spinal cord dorsal horn is considered as a central mechanism for the induction and maintenance of pain hypersensitivity (108). The central sensitization causes substantial prolongation and enhancement of response to both noxious and innocuous stimuli (hyperalgesia and allodynia, respectively) under diabetic conditions (109, 110). Although the expression of PDKs in the spinal cord, unlike the DRG, was not induced in diabetic animals, hyperglycemia-induced pathological alterations in the periphery caused the activation of spinal microglia and astrocytes, which was significantly attenuated in the Pdk2/4-deficient mice. Hyperglycemia-induced expression of TNF-α, IL-1β, and IL-6 in the lumbar segment of the spinal cord was also inhibited by Pdk2/4 deficiency. Furthermore, diabetes-induced expression of p-ERK in the superficial laminas of the spinal cord dorsal horn was remarkably lessened by Pdk2/4 deficiency. Previously, the phosphorylation of ERK within spinal neurons, a potential marker for central sensitization, has been commonly observed in diverse experimental chronic pain models (111). Rodents with diabetic neuropathy also exhibit remarkably increased expression of p-ERK in the spinal cord dorsal horn (112). These findings suggest that hyperglycemia-triggered up-regulation of PDK2/4 expression, the subsequent glycolytic metabolic shift, and finally, the lactic acid surge, may sequentially lead to pathophysiological consequences in the diabetic DRGs, such as the induction of reactive gliosis, macrophage infiltration, acidic and proinflammatory microenvironment, and neuronal sensitization in the periphery. This in turn triggers spinal events that ultimately cause central sensitization and pain hypersensitivities.
In conclusion, our findings suggest that PDK2 and PDK4 are imperatively involved in the development of painful diabetic neuropathy. Hyperglycemia-induced expression and activity of PDK2/4 lead to a glycolytic metabolic shift in the diabetic DRG. The PDK2/4-mediated metabolic dysfunctions, activation of SGCs, and infiltration of macrophages in the diabetic DRG concurrently augment the production of lactic acid and other proinflammatory mediators, which favors the sensitization of the peripheral nociceptive pathway. In addition, the augmented lactic acid production adversely affects the neuronal viability in the DRG. Diabetes-induced pathological consequences in the DRG and its nerve trunk trigger the central sensitization and thereby contribute to the development of diabetes-induced pain hypersensitivities. Our findings identify the glucose-PDK2/4-PDH-lactic acid axis in the DRG as a promising therapeutic target for painful diabetic neuropathy.
Author Contributions
M. H. R. and M. K. J. designed and performed the research, analyzed the data, and prepared the manuscript; J. H. K. assisted with biochemical assays; Y. N. assisted with animal studies; M. G. L., Y. G., R. A. H., D. H. P., H. K., and I.-K. L. contributed new reagents/analytical tools and analyzed data; and K. S. directed the study and was involved in all aspects of the experimental design, data analysis, and manuscript preparation.
This work was supported by Korea Healthcare Technology R&D Project, Ministry of Health and Welfare, Republic of Korea, Grant A111345. This work was also supported by the Basic Science Research Program through the National Research Foundation funded by the Korean Ministry of Education, Science, and Technology, Grants 2008-0062282 and 2015R1A2A1A10051958. The authors declare that they have no conflicts of interest with the contents of this article.
- DRG
- dorsal root ganglion
- PDH
- pyruvate dehydrogenase
- PDK
- PDH kinase
- STZ
- streptozotocin
- DKO
- double knock-out
- SGC
- satellite glial cell
- PWT
- paw withdrawal threshold
- GFAP
- glial fibrillary acidic protein
- MAP2
- microtubule-associated protein 2
- MTT
- 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- NCV
- nerve conduction velocity
- TRPV
- transient receptor potential vanilloid
- ASIC
- acid-sensing ion channel
- p-ERK
- phosphorylated ERK
- ANOVA
- analysis of variance
- DCA
- dichloroacetate
- FX11
- 2,3-dihydroxy-6-methyl-7-(phenylmethyl)-4-propyl-1-naphthalenecarboxylic acid.
References
- 1. Polonsky K. S. (2012) The past 200 years in diabetes. N. Engl. J. Med. 367, 1332–1340 [DOI] [PubMed] [Google Scholar]
- 2. Phillips L. K., Deane A. M., Jones K. L., Rayner C. K., and Horowitz M. (2015) Gastric emptying and glycaemia in health and diabetes mellitus. Nat. Rev. Endocrinol. 11, 112–128 [DOI] [PubMed] [Google Scholar]
- 3. Yagihashi S. (1995) Pathology and pathogenetic mechanisms of diabetic neuropathy. Diabetes Metab. Rev. 11, 193–225 [DOI] [PubMed] [Google Scholar]
- 4. Shakeel M. (2015) Recent advances in understanding the role of oxidative stress in diabetic neuropathy. Diabetes Metab. Syndr. 9, 373–378 [DOI] [PubMed] [Google Scholar]
- 5. Sima A. A., and Sugimoto K. (1999) Experimental diabetic neuropathy: an update. Diabetologia 42, 773–788 [DOI] [PubMed] [Google Scholar]
- 6. Tavee J., and Zhou L. (2009) Small fiber neuropathy: a burning problem. Cleve. Clin. J. Med. 76, 297–305 [DOI] [PubMed] [Google Scholar]
- 7. Gibbons C. H., and Freeman R. (2015) Treatment-induced neuropathy of diabetes: an acute, iatrogenic complication of diabetes. Brain 138, 43–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zochodne D. W., Verge V. M., Cheng C., Sun H., and Johnston J. (2001) Does diabetes target ganglion neurones? Progressive sensory neurone involvement in long-term experimental diabetes. Brain 124, 2319–2334 [DOI] [PubMed] [Google Scholar]
- 9. Greene D. A., Winegrad A. I., Carpentier J. L., Brown M. J., Fukuma M., and Orci L. (1979) Rabbit sciatic nerve fascicle and “endoneurial” preparations for in vitro studies of peripheral nerve glucose metabolism. J. Neurochem. 33, 1007–1018 [DOI] [PubMed] [Google Scholar]
- 10. Kadekaro M., Crane A. M., and Sokoloff L. (1985) Differential effects of electrical stimulation of sciatic nerve on metabolic activity in spinal cord and dorsal root ganglion in the rat. Proc. Natl. Acad. Sci. U.S.A. 82, 6010–6013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kishi M., Tanabe J., Schmelzer J. D., and Low P. A. (2002) Morphometry of dorsal root ganglion in chronic experimental diabetic neuropathy. Diabetes 51, 819–824 [DOI] [PubMed] [Google Scholar]
- 12. Zochodne D. W., and Ho L. T. (1991) Unique microvascular characteristics of the dorsal root ganglion in the rat. Brain Res. 559, 89–93 [DOI] [PubMed] [Google Scholar]
- 13. Schmeichel A. M., Schmelzer J. D., and Low P. A. (2003) Oxidative injury and apoptosis of dorsal root ganglion neurons in chronic experimental diabetic neuropathy. Diabetes 52, 165–171 [DOI] [PubMed] [Google Scholar]
- 14. Galloway C., and Chattopadhyay M. (2013) Increases in inflammatory mediators in DRG implicate in the pathogenesis of painful neuropathy in Type 2 diabetes. Cytokine 63, 1–5 [DOI] [PubMed] [Google Scholar]
- 15. Hanani M., Blum E., Liu S., Peng L., and Liang S. (2014) Satellite glial cells in dorsal root ganglia are activated in streptozotocin-treated rodents. J. Cell. Mol. Med. 18, 2367–2371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sasaki H., Schmelzer J. D., Zollman P. J., and Low P. A. (1997) Neuropathology and blood flow of nerve, spinal roots and dorsal root ganglia in longstanding diabetic rats. Acta Neuropathol. 93, 118–128 [DOI] [PubMed] [Google Scholar]
- 17. Brock F. E., Abri O., Baitsch G., Bechara G., Beck K., Corovic D., Diehm C., Marshall M., Rahmel B., and Scheffler P. (1990) Iloprost in the treatment of ischemic tissue lesions in diabetics: results of a placebo-controlled multicenter study with a stable prostacyclin derivative. Schweiz. Med. Wochenschr. 120, 1477–1482 [PubMed] [Google Scholar]
- 18. Baloh R. H. (2008) Mitochondrial dynamics and peripheral neuropathy. Neuroscientist 14, 12–18 [DOI] [PubMed] [Google Scholar]
- 19. Patel M. S., and Roche T. E. (1990) Molecular biology and biochemistry of pyruvate dehydrogenase complexes. FASEB J. 4, 3224–3233 [DOI] [PubMed] [Google Scholar]
- 20. Takubo K., Nagamatsu G., Kobayashi C. I., Nakamura-Ishizu A., Kobayashi H., Ikeda E., Goda N., Rahimi Y., Johnson R. S., Soga T., Hirao A., Suematsu M., and Suda T. (2013) Regulation of glycolysis by Pdk functions as a metabolic checkpoint for cell cycle quiescence in hematopoietic stem cells. Cell Stem Cell 12, 49–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Harris R. A., Bowker-Kinley M. M., Huang B., and Wu P. (2002) Regulation of the activity of the pyruvate dehydrogenase complex. Adv. Enzyme Regul. 42, 249–259 [DOI] [PubMed] [Google Scholar]
- 22. Jha M. K., Jeon S., and Suk K. (2012) Pyruvate dehydrogenase kinases in the nervous system: their principal functions in neuronal-glial metabolic interaction and neuro-metabolic disorders. Curr. Neuropharmacol. 10, 393–403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Rogatzki M. J., Ferguson B. S., Goodwin M. L., and Gladden L. B. (2015) Lactate is always the end product of glycolysis. Front. Neurosci. 9, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Jha M. K., and Suk K. (2013) Pyruvate dehydrogenase kinase as a potential therapeutic target for malignant gliomas. Brain Tumor Res. Treat. 1, 57–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ramos-Nino M. E. (2013) The role of chronic inflammation in obesity-associated cancers. ISRN Oncol. 2013, 697521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Nisoli E., Clementi E., Carruba M. O., and Moncada S. (2007) Defective mitochondrial biogenesis: a hallmark of the high cardiovascular risk in the metabolic syndrome? Circ. Res. 100, 795–806 [DOI] [PubMed] [Google Scholar]
- 27. Luft F. C. (2001) Lactic acidosis update for critical care clinicians. J. Am. Soc. Nephrol. 12, S15–S19 [PubMed] [Google Scholar]
- 28. Weisberg L. S. (2015) Lactic acidosis in a patient with type 2 diabetes mellitus. Clin. J. Am. Soc. Nephrol. 10, 1476–1483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wyss M. T., Jolivet R., Buck A., Magistretti P. J., and Weber B. (2011) In vivo evidence for lactate as a neuronal energy source. J. Neurosci. 31, 7477–7485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kempski O., Staub F., Jansen M., Schödel F., and Baethmann A. (1988) Glial swelling during extracellular acidosis in vitro. Stroke 19, 385–392 [DOI] [PubMed] [Google Scholar]
- 31. Kraig R. P., Petito C. K., Plum F., and Pulsinelli W. A. (1987) Hydrogen ions kill brain at concentrations reached in ischemia. J. Cereb. Blood Flow Metab. 7, 379–386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Laferrière A., Millecamps M., Xanthos D. N., Xiao W. H., Siau C., de Mos M., Sachot C., Ragavendran J. V., Huygen F. J., Bennett G. J., and Coderre T. J. (2008) Cutaneous tactile allodynia associated with microvascular dysfunction in muscle. Mol. Pain 4, 49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Jeoung N. H., Rahimi Y., Wu P., Lee W. N., and Harris R. A. (2012) Fasting induces ketoacidosis and hypothermia in PDHK2/PDHK4-double-knockout mice. Biochem. J. 443, 829–839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Herbst E. A., MacPherson R. E., LeBlanc P. J., Roy B. D., Jeoung N. H., Harris R. A., and Peters S. J. (2014) Pyruvate dehydrogenase kinase-4 contributes to the recirculation of gluconeogenic precursors during postexercise glycogen recovery. Am. J. Physiol. Regul. Integr. Comp. Physiol. 306, R102–R107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wu K. K., and Huan Y. (2008) Streptozotocin-induced diabetic models in mice and rats. Curr. Protoc. Pharmacol. 10.1002/0471141755.ph0547s40 [DOI] [PubMed] [Google Scholar]
- 36. Jang E., Lee S., Kim J. H., Kim J. H., Seo J. W., Lee W. H., Mori K., Nakao K., and Suk K. (2013) Secreted protein lipocalin-2 promotes microglial M1 polarization. FASEB J. 27, 1176–1190 [DOI] [PubMed] [Google Scholar]
- 37. Mansikka H., Zhao C., Sheth R. N., Sora I., Uhl G., and Raja S. N. (2004) Nerve injury induces a tonic bilateral μ-opioid receptor-mediated inhibitory effect on mechanical allodynia in mice. Anesthesiology 100, 912–921 [DOI] [PubMed] [Google Scholar]
- 38. Jha M. K., Jeon S., Jin M., Ock J., Kim J. H., Lee W. H., and Suk K. (2014) The pivotal role played by lipocalin-2 in chronic inflammatory pain. Exp. Neurol. 254, 41–53 [DOI] [PubMed] [Google Scholar]
- 39. McGuire J. F., Rouen S., Siegfreid E., Wright D. E., and Dobrowsky R. T. (2009) Caveolin-1 and altered neuregulin signaling contribute to the pathophysiological progression of diabetic peripheral neuropathy. Diabetes 58, 2677–2686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Naruse K., Sato J., Funakubo M., Hata M., Nakamura N., Kobayashi Y., Kamiya H., Shibata T., Kondo M., Himeno T., Matsubara T., Oiso Y., and Nakamura J. (2011) Transplantation of bone marrow-derived mononuclear cells improves mechanical hyperalgesia, cold allodynia and nerve function in diabetic neuropathy. PLoS One 6, e27458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Marshall J., Ashe K. M., Bangari D., McEachern K., Chuang W. L., Pacheco J., Copeland D. P., Desnick R. J., Shayman J. A., Scheule R. K., and Cheng S. H. (2010) Substrate reduction augments the efficacy of enzyme therapy in a mouse model of Fabry disease. PLoS One 5, e15033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Livak K. J., and Schmittgen T. D. (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(−ΔΔC(T)) method. Methods 25, 402–408 [DOI] [PubMed] [Google Scholar]
- 43. Kim S. J., Park G. H., Kim D., Lee J., Min H., Wall E., Lee C. J., Simon M. I., Lee S. J., and Han S. K. (2011) Analysis of cellular and behavioral responses to imiquimod reveals a unique itch pathway in transient receptor potential vanilloid 1 (TRPV1)-expressing neurons. Proc. Natl. Acad. Sci. U.S.A. 108, 3371–3376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Mosmann T. (1983) Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J. Immunol. Methods 65, 55–63 [DOI] [PubMed] [Google Scholar]
- 45. Puljak L., Kojundzic S. L., Hogan Q. H., and Sapunar D. (2009) Targeted delivery of pharmacological agents into rat dorsal root ganglion. J. Neurosci. Methods 177, 397–402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Jelicic Kadic A., Boric M., Kostic S., Sapunar D., and Puljak L. (2014) The effects of intraganglionic injection of calcium/calmodulin-dependent protein kinase II inhibitors on pain-related behavior in diabetic neuropathy. Neuroscience 256, 302–308 [DOI] [PubMed] [Google Scholar]
- 47. Kaufmann P., Engelstad K., Wei Y., Jhung S., Sano M. C., Shungu D. C., Millar W. S., Hong X., Gooch C. L., Mao X., Pascual J. M., Hirano M., Stacpoole P. W., DiMauro S., and De Vivo D. C. (2006) Dichloroacetate causes toxic neuropathy in MELAS: a randomized, controlled clinical trial. Neurology 66, 324–330 [DOI] [PubMed] [Google Scholar]
- 48. Kurlemann G., Paetzke I., Möller H., Masur H., Schuierer G., Weglage J., and Koch H. G. (1995) Therapy of complex I deficiency: peripheral neuropathy during dichloroacetate therapy. Eur. J. Pediatr. 154, 928–932 [DOI] [PubMed] [Google Scholar]
- 49. Stacpoole P. W., Henderson G. N., Yan Z., and James M. O. (1998) Clinical pharmacology and toxicology of dichloroacetate. Environ. Health Perspect. 106, 989–994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Stacpoole P. W., Henderson G. N., Yan Z., Cornett R., and James M. O. (1998) Pharmacokinetics, metabolism and toxicology of dichloroacetate. Drug Metab. Rev. 30, 499–539 [DOI] [PubMed] [Google Scholar]
- 51. Le A., Cooper C. R., Gouw A. M., Dinavahi R., Maitra A., Deck L. M., Royer R. E., Vander Jagt D. L., Semenza G. L., and Dang C. V. (2010) Inhibition of lactate dehydrogenase A induces oxidative stress and inhibits tumor progression. Proc. Natl. Acad. Sci. U.S.A. 107, 2037–2042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kwon M. J., Kim J., Shin H., Jeong S. R., Kang Y. M., Choi J. Y., Hwang D. H., and Kim B. G. (2013) Contribution of macrophages to enhanced regenerative capacity of dorsal root ganglia sensory neurons by conditioning injury. J. Neurosci. 33, 15095–15108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kim T. J., Freml L., Park S. S., and Brennan T. J. (2007) Lactate concentrations in incisions indicate ischemic-like conditions may contribute to postoperative pain. J. Pain 8, 59–66 [DOI] [PubMed] [Google Scholar]
- 54. Qi J., Buzas K., Fan H., Cohen J. I., Wang K., Mont E., Klinman D., Oppenheim J. J., and Howard O. M. (2011) Painful pathways induced by TLR stimulation of dorsal root ganglion neurons. J. Immunol. 186, 6417–6426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Wemmie J. A., Taugher R. J., and Kreple C. J. (2013) Acid-sensing ion channels in pain and disease. Nat. Rev. Neurosci. 14, 461–471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Chung M. K., Güler A. D., and Caterina M. J. (2008) TRPV1 shows dynamic ionic selectivity during agonist stimulation. Nat. Neurosci. 11, 555–564 [DOI] [PubMed] [Google Scholar]
- 57. Brenneis C., Kistner K., Puopolo M., Segal D., Roberson D., Sisignano M., Labocha S., Ferreirós N., Strominger A., Cobos E. J., Ghasemlou N., Geisslinger G., Reeh P. W., Bean B. P., and Woolf C. J. (2013) Phenotyping the function of TRPV1-expressing sensory neurons by targeted axonal silencing. J. Neurosci. 33, 315–326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Chen C. C., Zimmer A., Sun W. H., Hall J., Brownstein M. J., and Zimmer A. (2002) A role for ASIC3 in the modulation of high-intensity pain stimuli. Proc. Natl. Acad. Sci. U.S.A. 99, 8992–8997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Arumugam R., Horowitz E., Noland R. C., Lu D., Fleenor D., and Freemark M. (2010) Regulation of islet beta-cell pyruvate metabolism: interactions of prolactin, glucose, and dexamethasone. Endocrinology 151, 3074–3083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Bocarsly M. E., Fasolino M., Kane G. A., LaMarca E. A., Kirschen G. W., Karatsoreos I. N., McEwen B. S., and Gould E. (2015) Obesity diminishes synaptic markers, alters microglial morphology, and impairs cognitive function. Proc. Natl. Acad. Sci. U.S.A. 112, 15731–15736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Ashton R. S., Conway A., Pangarkar C., Bergen J., Lim K. I., Shah P., Bissell M., and Schaffer D. V. (2012) Astrocytes regulate adult hippocampal neurogenesis through ephrin-B signaling. Nat. Neurosci. 15, 1399–1406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Gao Y. J., and Ji R. R. (2009) c-Fos and pERK, which is a better marker for neuronal activation and central sensitization after noxious stimulation and tissue injury? Open Pain J. 2, 11–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Greig M., Tesfaye S., Selvarajah D., and Wilkinson I. D. (2014) Insights into the pathogenesis and treatment of painful diabetic neuropathy. Handb. Clin. Neurol. 126, 559–578 [DOI] [PubMed] [Google Scholar]
- 64. Aslam A., Singh J., and Rajbhandari S. (2014) Pathogenesis of painful diabetic neuropathy. Pain Res. Treat. 2014, 412041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Bishnoi M., Bosgraaf C. A., Abooj M., Zhong L., and Premkumar L. S. (2011) Streptozotocin-induced early thermal hyperalgesia is independent of glycemic state of rats: role of transient receptor potential vanilloid 1(TRPV1) and inflammatory mediators. Mol. Pain 7, 52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Hwang H. S., Yang E. J., Lee S. M., Lee S. C., and Choi S. M. (2011) Antiallodynic effects of electroacupuncture combined with MK-801 treatment through the regulation of p35/p25 in experimental diabetic neuropathy. Exp. Neurobiol. 20, 144–152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Zhao W. C., Zhang B., Liao M. J., Zhang W. X., He W. Y., Wang H. B., and Yang C. X. (2014) Curcumin ameliorated diabetic neuropathy partially by inhibition of NADPH oxidase mediating oxidative stress in the spinal cord. Neurosci. Lett. 560, 81–85 [DOI] [PubMed] [Google Scholar]
- 68. Kennedy J. M., and Zochodne D. W. (2000) The regenerative deficit of peripheral nerves in experimental diabetes: its extent, timing and possible mechanisms. Brain 123, 2118–2129 [DOI] [PubMed] [Google Scholar]
- 69. Naruse K., Hamada Y., Nakashima E., Kato K., Mizubayashi R., Kamiya H., Yuzawa Y., Matsuo S., Murohara T., Matsubara T., Oiso Y., and Nakamura J. (2005) Therapeutic neovascularization using cord blood-derived endothelial progenitor cells for diabetic neuropathy. Diabetes 54, 1823–1828 [DOI] [PubMed] [Google Scholar]
- 70. Doupis J., Lyons T. E., Wu S., Gnardellis C., Dinh T., and Veves A. (2009) Microvascular reactivity and inflammatory cytokines in painful and painless peripheral diabetic neuropathy. J. Clin. Endocrinol. Metab. 94, 2157–2163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Rahman M. H., Jha M. K., and Suk K. (2016) Evolving insights into the pathophysiology of diabetic neuropathy: implications of malfunctioning glia and discovery of novel therapeutic targets. Curr. Pharm. Des. 22, 738–757 [DOI] [PubMed] [Google Scholar]
- 72. Fox A., Eastwood C., Gentry C., Manning D., and Urban L. (1999) Critical evaluation of the streptozotocin model of painful diabetic neuropathy in the rat. Pain 81, 307–316 [DOI] [PubMed] [Google Scholar]
- 73. Morten K. J., Caky M., and Matthews P. M. (1998) Stabilization of the pyruvate dehydrogenase E1α subunit by dichloroacetate. Neurology 51, 1331–1335 [DOI] [PubMed] [Google Scholar]
- 74. Ohashi T., Akazawa T., Aoki M., Kuze B., Mizuta K., Ito Y., and Inoue N. (2013) Dichloroacetate improves immune dysfunction caused by tumor-secreted lactic acid and increases antitumor immunoreactivity. Int. J. Cancer 133, 1107–1118 [DOI] [PubMed] [Google Scholar]
- 75. Rinnankoski-Tuikka R., Silvennoinen M., Torvinen S., Hulmi J. J., Lehti M., Kivela R., Reunanen H., and Kainulainen H. (2012) Effects of high-fat diet and physical activity on pyruvate dehydrogenase kinase-4 in mouse skeletal muscle. Nutr. Metab. (Lond.) 10.1186/1743-7075-9-53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Hwang B., Jeoung N. H., and Harris R. A. (2009) Pyruvate dehydrogenase kinase isoenzyme 4 (PDHK4) deficiency attenuates the long-term negative effects of a high-saturated fat diet. Biochem. J. 423, 243–252 [DOI] [PubMed] [Google Scholar]
- 77. Tao R., Xiong X., Harris R. A., White M. F., and Dong X. C. (2013) Genetic inactivation of pyruvate dehydrogenase kinases improves hepatic insulin resistance induced diabetes. PLoS One 8, e71997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Kulkarni S. S., Salehzadeh F., Fritz T., Zierath J. R., Krook A., and Osler M. E. (2012) Mitochondrial regulators of fatty acid metabolism reflect metabolic dysfunction in type 2 diabetes mellitus. Metabolism 61, 175–185 [DOI] [PubMed] [Google Scholar]
- 79. Roche T. E., and Hiromasa Y. (2007) Pyruvate dehydrogenase kinase regulatory mechanisms and inhibition in treating diabetes, heart ischemia, and cancer. Cell. Mol. Life Sci. 64, 830–849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Jha M. K., Kim J. H., and Suk K. (2014) Proteome of brain glia: the molecular basis of diverse glial phenotypes. Proteomics 14, 378–398 [DOI] [PubMed] [Google Scholar]
- 81. Jasmin L., Vit J. P., Bhargava A., and Ohara P. T. (2010) Can satellite glial cells be therapeutic targets for pain control? Neuron Glia Biol. 6, 63–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Hanani M. (2005) Satellite glial cells in sensory ganglia: from form to function. Brain Res. Brain Res. Rev. 48, 457–476 [DOI] [PubMed] [Google Scholar]
- 83. Ohtori S., Takahashi K., Moriya H., and Myers R. R. (2004) TNF-α and TNF-α receptor type 1 upregulation in glia and neurons after peripheral nerve injury: studies in murine DRG and spinal cord. Spine 29, 1082–1088 [DOI] [PubMed] [Google Scholar]
- 84. Ton B. H., Chen Q., Gaina G., Tucureanu C., Georgescu A., Strungaru C., Flonta M. L., Sah D., and Ristoiu V. (2013) Activation profile of dorsal root ganglia Iba-1(+) macrophages varies with the type of lesion in rats. Acta Histochem. 115, 840–850 [DOI] [PubMed] [Google Scholar]
- 85. Nukada H., McMorran P. D., Baba M., Ogasawara S., and Yagihashi S. (2011) Increased susceptibility to ischemia and macrophage activation in STZ-diabetic rat nerve. Brain Res. 1373, 172–182 [DOI] [PubMed] [Google Scholar]
- 86. Chopra K., Tiwari V., Arora V., and Kuhad A. (2010) Sesamol suppresses neuro-inflammatory cascade in experimental model of diabetic neuropathy. J. Pain 11, 950–957 [DOI] [PubMed] [Google Scholar]
- 87. Liang H., Guan D., Gao A., Yin Y., Jing M., Yang L., Ma W., Hu E., and Zhang X. (2014) Human amniotic epithelial stem cells inhibit microglia activation through downregulation of tumor necrosis factor-α, interleukin-1β and matrix metalloproteinase-12 in vitro and in a rat model of intracerebral hemorrhage. Cytotherapy 16, 523–534 [DOI] [PubMed] [Google Scholar]
- 88. Dienel G. A. (2014) Lactate shuttling and lactate use as fuel after traumatic brain injury: metabolic considerations. J. Cereb. Blood Flow Metab. 34, 1736–1748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Nedergaard M., Kraig R. P., Tanabe J., and Pulsinelli W. A. (1991) Dynamics of interstitial and intracellular pH in evolving brain infarct. Am. J. Physiol. 260, R581–R588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Siesjö B. K., Bendek G., Koide T., Westerberg E., and Wieloch T. (1985) Influence of acidosis on lipid peroxidation in brain tissues in vitro. J. Cereb. Blood Flow Metab. 5, 253–258 [DOI] [PubMed] [Google Scholar]
- 91. Chih C. P., and Roberts E. L. Jr. (2003) Energy substrates for neurons during neural activity: a critical review of the astrocyte-neuron lactate shuttle hypothesis. J. Cereb. Blood Flow Metab. 23, 1263–1281 [DOI] [PubMed] [Google Scholar]
- 92. Dienel G. A., and Hertz L. (2001) Glucose and lactate metabolism during brain activation. J. Neurosci. Res. 66, 824–838 [DOI] [PubMed] [Google Scholar]
- 93. Macauley S. L., Stanley M., Caesar E. E., Yamada S. A., Raichle M. E., Perez R., Mahan T. E., Sutphen C. L., and Holtzman D. M. (2015) Hyperglycemia modulates extracellular amyloid-β concentrations and neuronal activity in vivo. J. Clin. Invest. 125, 2463–2467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Sala F., Menna G., Bricolo A., and Young W. (1999) Role of glycemia in acute spinal cord injury: data from a rat experimental model and clinical experience. Ann. N.Y. Acad. Sci. 890, 133–154 [DOI] [PubMed] [Google Scholar]
- 95. Watkins P. J., Smith J. S., Fitzgerald M. G., and Malins J. M. (1969) Lactic acidosis in diabetes. Br. Med. J. 1, 744–747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Krzymień J., and Karnafel W. (2013) Lactic acidosis in patients with diabetes. Pol. Arch. Med. Wewn. 123, 91–97 [DOI] [PubMed] [Google Scholar]
- 97. Staub F., Mackert B., Kempski O., Haberstok J., Peters J., and Baethmann A. (1994) Swelling and damage to nerves and glial cells by acidosis. Anasthesiol. Intensivmed. Notfallmed. Schmerzther. 29, 203–209 [DOI] [PubMed] [Google Scholar]
- 98. Staub F., Mackert B., Kempski O., Peters J., and Baethmann A. (1993) Swelling and death of neuronal cells by lactic acid. J. Neurol. Sci. 119, 79–84 [DOI] [PubMed] [Google Scholar]
- 99. Loike J. D., Kaback E., Silverstein S. C., and Steinberg T. H. (1993) Lactate transport in macrophages. J. Immunol. 150, 1951–1958 [PubMed] [Google Scholar]
- 100. Sotelo-Hitschfeld T., Niemeyer M. I., Mächler P., Ruminot I., Lerchundi R., Wyss M. T., Stobart J., Fernández-Moncada I., Valdebenito R., Garrido-Gerter P., Contreras-Baeza Y., Schneider B. L., Aebischer P., Lengacher S., San Martín A., Le Douce J., Bonvento G., Magistretti P. J., Sepúlveda F. V., Weber B., and Barros L. F. (2015) Channel-mediated lactate release by K(+)-stimulated astrocytes. J. Neurosci. 35, 4168–4178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Tang F., Lane S., Korsak A., Paton J. F., Gourine A. V., Kasparov S., and Teschemacher A. G. (2014) Lactate-mediated glia-neuronal signalling in the mammalian brain. Nat. Commun. 5, 3284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Hayashi T. (2011) Functional connectivity analysis of the brain network using resting-state FMRI. Brain Nerve 63, 1307–1318 [PubMed] [Google Scholar]
- 103. Chu X. P., and Xiong Z. G. (2013) Acid-sensing ion channels in pathological conditions. Adv. Exp. Med. Biol. 961, 419–431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Lee Y., Lee C. H., and Oh U. (2005) Painful channels in sensory neurons. Mol. Cells 20, 315–324 [PubMed] [Google Scholar]
- 105. Caterina M. J., Schumacher M. A., Tominaga M., Rosen T. A., Levine J. D., and Julius D. (1997) The capsaicin receptor: a heat-activated ion channel in the pain pathway. Nature 389, 816–824 [DOI] [PubMed] [Google Scholar]
- 106. Pabbidi R. M., Yu S. Q., Peng S., Khardori R., Pauza M. E., and Premkumar L. S. (2008) Influence of TRPV1 on diabetes-induced alterations in thermal pain sensitivity. Mol. Pain 4, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Radu B. M., Dumitrescu D. I., Marin A., Banciu D. D., Iancu A. D., Selescu T., and Radu M. (2014) Advanced type 1 diabetes is associated with ASIC alterations in mouse lower thoracic dorsal root ganglia neurons. Cell Biochem. Biophys. 68, 9–23 [DOI] [PubMed] [Google Scholar]
- 108. Latremoliere A., and Woolf C. J. (2009) Central sensitization: a generator of pain hypersensitivity by central neural plasticity. J. Pain 10, 895–926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Tsuda M., Ueno H., Kataoka A., Tozaki-Saitoh H., and Inoue K. (2008) Activation of dorsal horn microglia contributes to diabetes-induced tactile allodynia via extracellular signal-regulated protein kinase signaling. Glia 56, 378–386 [DOI] [PubMed] [Google Scholar]
- 110. Wang D., Couture R., and Hong Y. (2014) Activated microglia in the spinal cord underlies diabetic neuropathic pain. Eur. J. Pharmacol. 728, 59–66 [DOI] [PubMed] [Google Scholar]
- 111. Ji R. R., Baba H., Brenner G. J., and Woolf C. J. (1999) Nociceptive-specific activation of ERK in spinal neurons contributes to pain hypersensitivity. Nat. Neurosci. 2, 1114–1119 [DOI] [PubMed] [Google Scholar]
- 112. Georgadze A. K., Permiakov N. K., Vinogradov V. A., Penin V. A., and Titova G. P. (1985) Effect of an analog of endogenous opioids, dalargin, on the structure and function of the exocrine tissue of the pancreas in experimental acute pancreatitis. Farmakol. Toksikol. 48, 101–104 [PubMed] [Google Scholar]