

Abstract
Autophagy is a conserved catabolic process that utilizes a defined series of membrane trafficking events to generate a de novo double-membrane vesicle termed the autophagosome, which matures by fusing to the lysosome. Subsequently, the lysosome facilitates the degradation and recycling of the cytoplasmic cargo. In yeast, the upstream signals that regulate the induction of starvation-induced autophagy are clearly defined. The nutrient-sensing kinase Tor inhibits the activation of autophagy by regulating the formation of the Atg1-Atg13-Atg17 complex, through hyperphosphorylation of Atg13. However, in mammals, the ortholog complex ULK1-ATG13-FIP200 is constitutively formed. As such, the molecular mechanism by which mTOR regulates mammalian autophagy is unknown. Here we report the identification and characterization of novel nutrient-regulated phosphorylation sites on ATG13: Ser-224 and Ser-258. mTOR directly phosphorylates ATG13 on Ser-258 while Ser-224 is modulated by the AMPK pathway. In ATG13 knock-out cells reconstituted with an unphosphorylatable mutant of ATG13, ULK1 kinase activity is more potent, and amino acid starvation induced more rapid ATG13 and ULK1 translocation. These events culminated in a more rapid starvation-induced autophagy response. Therefore, ATG13 phosphorylation plays a crucial role in autophagy regulation.
Keywords: AMP-activated kinase (AMPK), autophagy, mammalian target of rapamycin (mTOR), phosphorylation, TOR complex (TORC), Macroautophagy, ULK1
Introduction
Macroautophagy is a catabolic cellular process that facilitates lysosome-dependent degradation. It promotes cellular homeostasis by facilitating nutrient recycling and degrading damaged organelles and protein aggregates. Autophagy is conserved in all eukaryotic cells and is indispensable for normal development (1). The hallmark of autophagy is the autophagosome, a double-membrane vesicle that forms de novo in the cytoplasm and sequesters bulk cytoplasmic components. This vesicle matures by fusing to the lysosome, leading to the degradation of the contents inside (2, 3). Upon nutrient starvation, autophagy is induced to promote cell survival (4, 5). Yeast genetic and follow-up studies identified over 30 autophagy-related genes (ATG)2 (5, 6), some of which facilitate different forms of selective autophagy while others constitute the core machinery of autophagy (7). Components of the core machinery of autophagy can be segmented into multiple separate functional units (8, 9).
In this study, we focus on the Atg1/ULK1 initiator complex. Atg1, and its mammalian counterpart ULK1, are the only protein kinase among the more than 36 ATG proteins. In Saccharomyces cerevisiae, the nutrient-sensing kinase Tor negatively regulates the induction of autophagy by inhibiting the formation of a multiprotein complex containing Atg1, Atg13, and Atg17, through the hyperphosphorylation of Atg13 (10). The binding of Atg13 and Atg1 is essential to induce autophagy because Atg13 stabilizes Atg1 protein and enhances its kinase activity (10, 11). Upon inactivation of Tor through starvation or pharmacological inhibition, Atg13 is rapidly dephosphorylated, allowing for the Atg1-Atg13-Atg17 complex to assemble and promote the recruitment of downstream ATG proteins (10–15). In fact, expression of an unphosphorylatable form of Atg13 is sufficient to induce autophagy in nutrient-replete conditions (16). As such, Atg13 is a vital regulatory component of autophagy that is responsive to environmental cues through a dynamic phosphorylation status (17). In higher eukaryotes, the mechanism by which mTOR negatively regulates autophagy is less understood. The mammalian counterpart of the initiator complex is the ULK1-ATG13-FIP200 complex (18–20). Unlike yeast, the mammalian ULK1 complex is constitutively formed (18). As such, it is not clear if and how mTOR-mediated phosphorylation of ATG13 controls the autophagy initiation function of the ULK1 kinase complex.
In this study, we determined the role of ATG13 phosphorylation status in regulating the autophagy function of the ULK1 kinase complex. We identified two nutrient-regulated phosphorylation sites on ATG13. We found that ATG13 is phosphorylated by multiple kinases, including mTOR, and that these phosphorylation events are sensitive to the nutritional status of the cell. Mechanistically, we established that nutrient-regulated phosphorylation of ATG13 control the induction of autophagy by modulating distinct properties of ULK1, including its enzyme activity and cellular localization.
Experimental Procedures
Reagents and Antibodies
The following antibodies were used for Western blot analysis: rabbit anti-ATG13 (1:10,000, Sigma, no. SAB4200100); rabbit anti-LC3b (1:3,000, Sigma, no. L7543); rabbit anti-ULK1 (1:3,000, Sigma, no. A7481); mouse anti-β actin clone AC-15 (1:15,000, Sigma, no. A1978); rabbit anti-S6K (1:3,000, Cell Signaling, no. 2708); rabbit anti-phospho-S6K (T389) (1:1,000, Cell Signaling, no. 9205); rabbit anti-p62/SQSTM1 (1:10,000, MBL, no. PM045B); rabbit anti-phospho-ATG13 (S318) (1:1,500, Rockland, no. 600-401-C49); rabbit anti-S-Tag (1:3,000, Bethyl Laboratories, no A190–135A); mouse anti-c-Myc (1:500, Santa Cruz Biotechnology, no. sc-40). Rabbit anti-phospho-ATG13 (S224) and rabbit anti-phospho-ATG13 (S258) were generated through GenScript. Rabbit anti-phospho-ULK1 (S637) and rabbit anti-phospho-ULK1 (S757) were a generous gift from Dr. Xiaodong Wang. Rapamycin was purchased from Enzo Life Sciences (A-275). Torin 1 was purchased from Tocris Bioscience (4247). Bafilomycin A1 was purchased from Sigma (B1793). Compound C was purchased from Tocris (3093). PI-103 was purchased from Cayman Chemical (371935–74-9).
Cell Culture
HEK293T and immortalized MEFs were cultured at 37 °C, 5% CO2 in Dulbecco's modified Eagle's medium (DMEM), supplemented with 10% fetal bovine serum, 2 mm l-glutamine and 1× penicillin-streptomycin. For induction of autophagy, cells were washed twice in 1× PBS and incubated in DMEM lacking amino acids and serum (starvation medium), or maintained in complete full-medium as a control, for the indicated amount of time. We performed amino acid starvation by culturing cells in DMEM without amino acids, and supplemented with 10% dialyzed FBS. We performed serum starvation by culturing cells in DMEM supplemented with 2 mm l-glutamine.
DNA Constructs
ATG13 mouse cDNA was purchased from ATCC (clone ID 5359944). Serine to alanine, aspartic acid, and glutamic acid mutation were generated by PCR-mediated site-directed mutagenesis. The residue of interest was mutated alongside adjacent acceptor residues, in order to prevent compensatory phosphorylation: S224A (S222A/S223A/S224A); S258A (S256A/T257A/S258A); S224E (S222E/S223E/S224E); S258E (S256E/T257E/S258E). All mutations were subsequently confirmed by direct sequencing. For expression in MEF and HEK293T cells, ATG13 was subcloned into pBabe/puro containing an N-terminal FLAG-S tag or HA tag.
Tandem Affinity Purification
FLAG-S-ATG13 was stably expressed to near endogenous levels in MEF cells. Cells were either starved in amino acid and serum-free DMEM media for 1.5 h or kept in full-medium and stimulated with insulin at 1 μg/ml for 15 min before harvesting. We used 24 × 15-cm plates for each condition. Cells were lysed in 600 μl of IP lysis buffer (50 mm HEPES pH 7.4, 10 mm KCl, 1 mm EDTA, 1 mm MgCl2, 10% glycerol, and 0.5% Triton X-100) per 15-cm plate, supplemented with protease inhibitors and a mixture of phosphatase inhibitors (Sigma, no P5726 and P0044). Lysates were incubated on ice for 20 min and then centrifuged for 15 min at 16,000 × g and then followed by an additional spin at 100,000 × g. An 8 m urea solution was added to the clarified lysates to 1.5 m. The partially denatured clarified lysate was incubated overnight with 100 μl of S-agarose beads (EMD Millipore, no. 69704-3) at 4 °C. The beads were then washed four times with lysis buffer containing 1 m urea. The bound ATG13 was eluted twice with 250 μl of lysis buffer containing 8 m urea. Additional lysis buffer was added to the eluates to dilute urea to 0.25 m and supplemented with protease inhibitors and a mixture of phosphatase inhibitors. Eluates were further incubated with 150 μl of FLAG beads (Sigma, no. A2220) overnight at 4 °C. After four washes in IP lysis buffer, the bound ATG13 was eluted in SDS sample buffer and analyzed by SDS-PAGE and silver staining.
Protein Identification by Nano-Liquid Chromatography Coupled to Tandem Mass Spectrometry (LC-MS/MS) Analysis
Proteins were resolved using SDS-polyacrylamide gel electrophoresis, followed by staining with Coomassie Blue and excision of the separated protein bands; In situ trypsin digestion of polypeptides in each gel slice was performed as described (21). The tryptic peptides were purified using a 2-μl bed volume of Poros 50 R2 (Applied Biosystems) reversed-phase beads packed in Eppendorf gel-loading tips (22). The purified peptides were diluted to 0.1% formic acid and then subjected to nano-liquid chromatography coupled to tandem mass spectrometry (nano-LC-MS/MS) analysis as follows. Peptide mixtures (in 20 μl) were loaded onto a trapping guard column (0.3 × 5 mm Acclaim PepMap 100 C18 cartridge from LC Packings, Sunnyvale, CA) using an Eksigent nano MDLC system (Eksigent Technologies, Inc. Dublin, CA) at a flow rate of 20 μl/min. After washing, the flow was reversed through the guard column and the peptides eluted with a 5–45% acetonitrile gradient over 85 min at a flow rate of 200 nl/min, onto and over a 75-micron x 15-cm fused silica capillary PepMap 100 C18 column (LC Packings, Sunnyvale, CA). The eluent was directed to a 75-micron (with 10-micron orifice) fused silica nano-electrospray needle (New Objective, Woburn, MA). The electrospray ionization needle was set at 1800 V. A linear ion quadrupole trap-Orbitrap hybrid analyzer (LTQ-Orbitrap, ThermoFisher, San Jose, CA) was operated in automatic, data-dependent MS/MS acquisition mode with one MS full scan (450–2000 m/z) in the Orbitrap analyzer at 60,000 mass resolution and up to ten concurrent MS/MS scans in the LTQ for the ten most intense peaks selected from each survey scan. Survey scans were acquired in profile mode and MS/MS scans were acquired in centroid mode. The collision energy was automatically adjusted in accordance with the experimental mass (m/z) value of the precursor ions selected for MS/MS. Minimum ion intensity of 2000 counts was required to trigger an MS/MS spectrum; dynamic exclusion duration was set at 60 s.
Initial protein/peptide identifications from the LC-MS/MS data were performed using the Mascot search engine (Matrix Science, version 2.3.02; www.matrixscience.com) with the Xenopus segment of Uniprot protein database (3,363 sequences; European Bioinformatics Institute, Swiss Institute of Bioinformatics and Protein Information Resource). The search parameters were as follows: (i) two missed cleavage tryptic sites were allowed; (ii) precursor ion mass tolerance = 10 ppm; (iii) fragment ion mass tolerance = 0.8 Da; and (iv) variable protein modifications were allowed for methionine oxidation, cysteine acrylamide derivatization and protein N-terminal acetylation, mono- and di-methylated lysine and arginine, and tri-methylated Lysine. MudPit scoring was typically applied using significance threshold score p < 0.01. Decoy database search was always activated and, in general, for merged LS-MS/MS analysis of a gel lane with p < 0.01, false discovery rate averaged around 1%.
Scaffold (Proteome Software Inc., Portland, OR), version 3_6_1 was used to further validate and cross-tabulate the tandem mass spectrometry (MS/MS)-based peptide and protein identifications. Protein and peptide probability was set at 95% with a minimum peptide requirement of 1.
CRISPR-CAS9 Knock-out MEF Cell Line
CRISPR-CAS9 system of RNA-guided genome editing was used to generate ATG13−/− MEFs. The targeting sequence 5′-ACTGTCCAAGTGATTGTCC-3′ was incorporated into a 60mer oligo, as described previously (23). The forward and reverse oligos were annealed to make a 100 bp double-stranded DNA fragment using Phusion polymerase (NEB, no. M0530S). The DNA fragment was fused by Gibson assembly (NEB, no. E2611S) to an AflII-linearized U6 target gRNA expression vector (Addgene, no. 41824). The targeting sequence was confirmed by sequencing. MEF cells were then electroporated using the Amaxa MEF2 nucleofector kit (VAPD-1005), as per manufacturer's instructions. Cells were electroporated in three consecutive days and then plated for single clones.
Preparation of Cell Lysates and Immunoprecipitation
Cells were washed with ice-cold 1× PBS buffer and then lysed in RIPA buffer (10 mm Tris-HCl, pH 7.5, 100 mm NaCl, 1.0% Triton X-100, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate, 10% glycerol, 1 mm EDTA/EGTA), or IP lysis buffer, supplemented with protease inhibitors and a mixture of phosphatase inhibitors. The lysates were incubated on ice for 20 min and then centrifuged at 12,000 × g for 15 min. For IP, lysates were incubated with 10 μl of S-protein agarose beads for 4 h at 4 °C. The beads were then washed four times in IP lysis buffer and eluted by boiling in SDS sample buffer. The precipitated proteins were resolved by SDS-PAGE and analyzed by Western blotting.
Microscopy
For fluorescence analysis, cells were either stably transfected with GFP-LC3 or processed for ULK1(1:400, Sigma, no. A7481) or F/S-ATG13 (1:400, EMD Millipore, no. 71549) immunofluorescence. Cells were plated on coverslips in a 6-well plate format. The subsequent day, cells were either placed in full-medium or starvation medium for the indicated amount of time. Coverslips were then fixed with 3.7% paraformaldehyde in 20 mm HEPES pH 7.5 for 20 min at room temperature. For immunofluorescence, the fixed cells were permeabilized with 0.1% Triton X-100 in 1× PBS for 5 min. After washing, the coverslips were then incubated with ULK1 antibody or S-Tag antibody in blocking buffer (1% BSA/1× PBS) for 30 min at room temperature. After washing three times for 10-min intervals, slides were incubated with Alexa Fluor secondary antibody (1:1,000, ThermoFisher, no. A11012or A11029) for 30 min at room temperature. After washing, coverslips were mounted on microscope slides using ProLong Gold antifade reagent with DAPI (Life Technologies, no. P36935). GFP-LC3, ATG13 and ULK1 punctae were visualized with a Nikon Eclipse TE2000-U confocal microscope using the 60× objective. Images were acquired using Nikon EZ-C1 image acquisition software. Subsequently, the images were processed using Photoshop.
ULK1 Kinase Assay
FLAG-S-ATG13 (WT or S224A/S258A) and HA-ULK1 were co-transfected in HEK293T cells with Lipofectamine 2000, as per manufacturer's instructions. We transfected 2 × 10-cm plates for each ATG13. After 48 h, cells were lysed in lysis buffer (50 mm Tris, pH 7.5, 150 mm NaCl, 10% glycerol, 1% Triton X-100, 1 mm EDTA/EGTA, 0.5 mm DTT) supplemented with protease inhibitors and phosphatase inhibitors (Sigma, P0044 and P5726). Lysates were centrifuged at 20,000 × g for 15 min. An equal amount of total protein was then incubated with 15 μl of S-protein agarose beads (EMD Millipore, 69704–3) overnight with rotation at 4 °C. After washing with lysis buffer, the S-protein agarose beads were resuspended in ULK1 kinase buffer (25 mm HEPES, pH 7.5, 50 mm NaCl, 10 mm MgCl2, 0.1% Tween-20, 1 mm DTT, 0.5 mg/ml BSA 10 μm cold ATP, 0.5 μCi of [γ32-P]ATP (PerkinElmer, BLU002A250UC) and 0.25 mg/ml myelin basic protein (MBP) (EMD Millipore, 13-110). The reaction was incubated at 30 °C for 30 min. The reaction was terminated by the addition of sample buffer. The samples were run in a 15% SDS-PAGE gel, transferred to nitrocellulose and signal was acquired with x-ray film.
mTOR Kinase Assay
For the kinase assay, Myc-mTOR and FLAG-S-ATG13 were separately transfected into HEK293T cells using Lipofectamine 2000, as per manufacturer's instructions. Cells transfected with Myc-mTOR were maintained in full-medium while cells transfected with F/S-ATG13 were starved for 4 h prior to harvesting, to remove any phosphorylations mediated by endogenous mTOR. Myc-mTOR and F/S-ATG13 were immunoprecipitated using c-Myc agarose beads (Clontech, no. 631208) or S-protein agarose beads (EMD Millipore, 69704-3), respectively. Cells were lysed in lysis buffer and incubated with 20 μl of beads while rotating at 4 °C for 5 h. The beads were washed four times with lysis buffer, aliquoted, and then resuspended in mTOR kinase buffer (20 mm HEPES, pH7.5, 100 mm NaCl, 5 mm MnCl2, 1 mm DTT, 0.5 mg/ml BSA, 20 μm cold ATP, and 0.5 μCi of [γ32-P]ATP). This reaction was performed on the beads. The reaction was incubated at 30 °C for 45 min. The reaction was terminated by the addition of sample buffer. The samples were run in a 7% SDS-PAGE gel, transferred to nitrocellulose and signal was acquired with x-ray film.
In Vitro Phosphatase Assay
Cells were lysed in RIPA buffer supplemented with protease with or without phosphatase inhibitors. A total of 60 μg of lysate was incubated in 1× NEB3 buffer and with or without 10 units of calf intestinal phosphatase (CIP) (New England BioLabs, M0290S) for 1 h at 37 °C. The reaction was terminated by the addition of sample buffer and analyzed by immunoblotting.
Statistical Analysis
The statistical significance of differences between means was calculated by two-tailed Student's t test. Values of p < 0.05 were considered significant.
Results
ATG13 Is a Phosphoprotein with Two Nutrient-regulated Phosphorylation Sites
Recent studies have suggested that mTOR negatively regulates autophagy in mammalian cells through impinging inhibitory phosphorylation on the ULK kinase complex. Specifically, mTOR was demonstrated to phosphorylate mouse-derived ULK1 at Ser-637 and Ser-757 (24, 25). However, no studies have explored the role of mTOR-driven phosphorylation of the ULK1 binding partners, such as ATG13, in regulating the induction of autophagy. Studies in yeast have implicated Tor-mediated phosphorylation of Atg13 as the main regulatory component of the initiator complex. Therefore, we hypothesized that mammalian ATG13 might also have a vital role in regulating autophagy. However, the phosphorylation residues on mammalian ATG13 by mTOR have not been identified. In this study, we sought to identify these sites and characterize the function of these phosphorylation events.
In cultured mouse embryonic fibroblasts (MEFs), amino acid starvation or pharmacological inhibition of mTOR resulted in the appearance of a fast-migrating form of ATG13 assayed by Western blot (Fig. 1a). Treatment of the cell lysates with calf intestinal alkaline phosphatase (CIP) yielded faster migrating bands, indicating that the gel-shifts are due to altered phosphorylation. This result suggests that ATG13 is phosphorylated when cells are cultured in a nutrient-rich condition, likely in an mTOR-dependent manner, and is dephosphorylated upon starvation. To identify these nutrient-regulated phosphorylation sites, we conducted tandem affinity tag immunoprecipitation and nanoscale liquid chromatography coupled to tandem mass spectrometry (nano-LC-MS/MS). We generated a MEF cell line stably expressing ATG13 with an N-terminal FLAG and S-tag (F/S-ATG13). We expressed F/S-ATG13 to near endogenous levels. This ectopically expressed F/S-ATG13 retained the ability to undergo a starvation-induced electrophoretic mobility shift (Fig. 1b). We purified F/S-ATG13 from cells under nutrient-rich medium or upon amino acid/serum starvation. The MS analysis covered nearly 80% of the ATG13 protein sequence. Under nutrient rich conditions, multiple distinct phosphorylated sites were identified, including Ser-224 and Ser-258 (Fig. 1, c and d). We further found that Ser-224 and Ser-258 sites are dephosphorylated upon starvation; we compared the integrated selected ion current chromatogram (monoisotopic mass ± 5 ppm) of phosphorylated peptide versus the integrated selected ion current chromatogram (monoisotopic mass ± 5 ppm) of the nonphosphorylated peptide under full medium or starvation media (an internal ATG13 peptide was used to normalize the integrated peak areas across the different conditions). In doing so, we detected a 56% reduction in phosphorylated Ser-224 and a 93% reduction in phosphorylated Ser-258, under starvation conditions (Fig. 1e).
FIGURE 1.

ATG13 is a nutrient-regulated phosphoprotein. a, mTOR-mediated phosphorylations on ATG13 are dephosphorylated during autophagy. Wild-type MEF cells were placed in full-medium, starved of amino acids and serum for 2 h, or treated with 1 μm Torin 1 for 2.5 h. The lysate were subjected to a phosphatase assay and analyzed by Western blotting. b, exogenously expressed FLAG-S-ATG13 exhibits starvation-induced electrophoretic mobility. F/S-ATG13 was stably expressed in MEF wild-type cells using a retroviral packaging system, followed by puromycin selection. Cells were placed in full-medium or starvation medium for 1.5 h. c and d, selected ion current chromatograms of putative phosphorylation sites at Ser-224 and Ser-258. Tandem affinity tag immunoprecipitation and nanoscale liquid chromatography coupled to tandem mass spectrometry (nano-LC-MS/MS) were used to identify ATG13 phosphorylation sites under full media or starvation medium. e, change in the percentage of phosphorylation at Ser-224 and Ser-258, under starvation media relative to full medium. f, analyzing the contribution of phosphorylation at Ser-224 and Ser-258 to the electrophoretic mobility of ATG13. ATG13−/− MEFs were stably reconstituted with ATG13 wild-type, S224A or S258A using a retroviral packaging system. Cells were placed in full-media or starvation media, followed by an analysis of ATG13 shift patterns by Western blotting. g, ClustalW alignment of ATG13 homologs. The sequences used are: human-NP_001192048.1; mouse-XP_006499950.1; rat-NP_001258141.1; chicken-XP_421116.3; frog-NP_001096420.1; zebrafish-NP_956727.1; fly-NP_649796.1; worm-P34379; yeast-A6ZX59. The blue asterisk denotes Ser-224. The red asterisk denotes Ser-258.
Although yeast Tor-mediated Atg13 phosphorylation sites had been identified, it was impractical to extrapolate that information to higher eukaryotes because of the extremely limited homology between yeast Atg13 and that of mammals. Indeed, using a ClustalW multiple sequence alignment analysis, we failed to detect any yeast serine or threonine conserved with Ser-224 or Ser-258. Both Ser-224 and Ser-258 are conserved in higher eukaryotes (Fig. 1f). Ser-258 is also conserved in Caenorhabditis elegans.
To further determine whether the putative sites are nutrient-regulated, we tested whether we could abrogate the starvation-induced increase in gel migration mobility of ATG13 by mutating the residues to alanine. Interestingly, even when cells were cultured in full-medium, the Ser-258 alanine mutant migrated as fast as starvation-treated wild-type ATG13, whereas substitution of Ser-224 to alanine had no effect on gel migration shift regardless culture condition (Fig. 1g). These results indicate that the phosphorylation status of Ser-258 but not Ser-224 accounts for the migration shift of ATG13.
ATG13 Is a Phosphoprotein that Senses Amino Acid Starvation
To further analyze ATG13 phosphorylation, we generated phospho-antibodies specific for pSer224 and pSer258. We validated these antibodies by ectopically expressing ATG13 alanine and glutamic acid mutants and monitoring the phosphorylation under full-medium or starved conditions. The pSer258 and pSer224 antibodies recognized the wild-type protein under full-media conditions, and showed a drastic decrease in signal upon starvation (Fig. 2a). Mutation of Ser-258 to alanine or glutamic acid abrogated detection by the pSer258 antibody. Similarly, mutation of Ser-224 to alanine or glutamic acid abrogated detection by the pSer224 antibody. Importantly, mutation of Ser-224 did not affect detection by the pSer258 antibody, and vice versa, indicating that these two phosphorylation events are not inter-dependent. These results validate that ATG13 is phosphorylated in vivo at Ser-224 and Ser-258 under full-medium conditions, and upon starvation these sites are dephosphorylated. To further dissect the trigger that induces dephosphorylation, we performed either serum starvation or amino acid starvation. Both sites demonstrate a drastic dephosphorylation upon amino acid starvation and there is no perceivable dephosphorylation under serum starvation alone (Fig. 2b), indicating these two phosphorylation events are solely dictated by amino acid status but not able to sense growth factor signaling.
FIGURE 2.
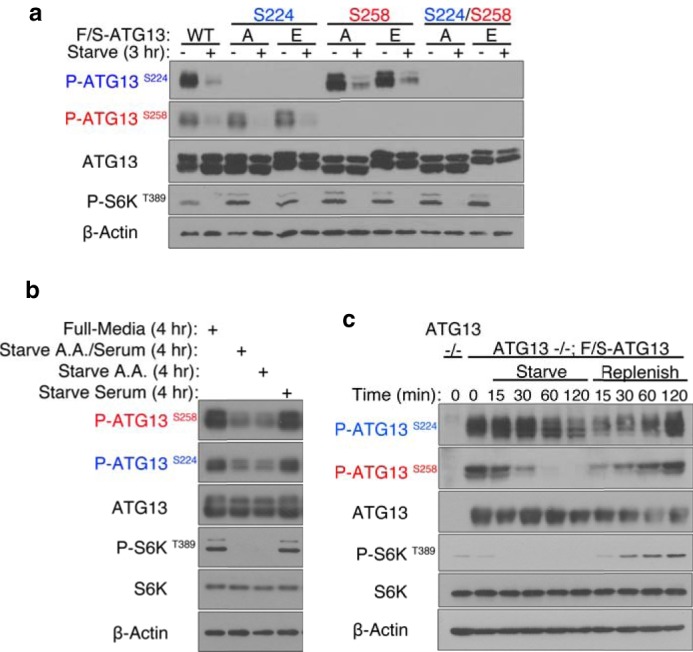
ATG13 dynamic phosphorylation at Ser-224 and Ser-258 senses amino acid starvation. a, phospho-antibodies were generated against ATG13 pSer224 and pSer258. To establish their specificity, HEK293T cells were transiently transfected with F/S-ATG13 wild-type, S224A, S224E, S258A, S258E, S224A/S258A, or S224E/S258E. The cells were placed in full-medium or starvation medium for 3 h. F/S-ATG13 was affinity S-tag immunoprecipitated using S-protein agarose beads and analyzed by Western blotting. b, ATG13 is dephosphorylated at Ser-224 and Ser-258 upon amino acid starvation. HEK293T cells were transiently transfected with F/S-ATG13 wild-type. The cells were placed in full-medium, amino acid and serum-free medium, amino acid-free medium or serum-free medium for 4 h. F/S-ATG13 was affinity S-tag immunoprecipitated using S-protein agarose beads and analyzed by Western blotting. c, phosphorylation of ATG13 at Ser-224 and Ser-258 is dynamic. ATG13 knock-out MEF cells were reconstituted by the stable expression of F/S-ATG13. The cells were starved for up to 120 min, after which they were replenished by switching to full-medium, for the indicated amount of time.
To further establish the validity of these sites, we monitored the phosphorylation status of ATG13 stably expressed in ATG13 knock-out MEFs. We monitored dynamic phosphorylation changes at Ser-224 and Ser-258, in response to nutrient availability (Fig. 2c). pSer258 exhibited an apparent dephosphorylation within only 15 min in starvation medium, and no phosphorylation was detected by 60 min of starvation. On the other hand, pSer224 exhibited a slower response to nutrient withdrawal. pSer224 exhibited an apparent dephosphorylation by 60 min and there was still some residual phosphorylation by 120 min. Similarly, when cells were replenished with full-medium, Ser-258 was rapidly phosphorylated starting at 15 min while phosphorylation at Ser-224 only increased after 60 min. The different kinetics of the phosphorylation between Ser-224 and Ser-258 is suggestive that these two sites are differentially regulated.
ATG13 Is Phosphorylated on Ser-258 by mTOR and on Ser-224 by the AMPK Pathway
Since phosphorylation of Ser-224 and Ser-258 was sensitive to amino acid starvation, we examined whether the sites are phosphorylated by mTOR. We attenuated the mTOR signaling pathway using the following kinase inhibitors, Rapamycin, PI-103, and Torin 1. We found that pSer258 was sensitive to inhibition by Rapamycin, an allosteric inhibitor of mTORC1, as well as to PI-103 and Torin 1, direct inhibitors of mTOR kinase activity (Fig. 3a). Importantly, ATG13 pSer258 exhibited the same sensitivity to mTOR inhibition as ULK1 pSer757, another target of mTOR. However, pSer224 was unresponsive to mTOR inhibition. Interestingly, when we treated cells with the AMPK antagonist Compound C, pSer224 was selectively dephosphorylated while pSer258 was insensitive (Fig. 3a). AMPK functions as a nutrient and energy sensor in the cell. It is known to phosphorylate ULK1 on multiple sites (24–26).
FIGURE 3.

ATG13 is phosphorylated by mTOR at Ser258 while the AMPK pathway modulates phosphorylation at Ser-224. a, ATG13 pSer258 is sensitive to pharmacological inhibition of mTOR. ATG13 knock-out MEF cells were reconstituted by the stable expression of HA-ATG13. The cells were either placed in full-medium, starved, or placed in full-medium with either 1 μm Rapamycin, 2 μm PI-103, 0.5 μm Torin 1, or 25 μm Compound C. b, endogenous ATG13 phosphorylation at Ser-258 in TSC2−/− versus +/+ MEF cells. The black arrow indicates a nonspecific band. The red arrow indicates pSer258. c, mTOR directly phosphorylates ATG13 at Ser-258. MYC-mTOR and F/S-ATG13 were separately transiently transfected in HEK293T cells, after 48 h, the cells were placed in full-medium or 4 h of starvation medium, respectively. The proteins were separately purified by affinity tag immunoprecipitation, and subjected to an mTOR kinase assay. d, MEF wild-type cells were placed in full-medium or amino acid and serum-starvation medium for 1 h. e, HEK293T cells were transiently transfected with F/S-ATG13 wild-type. The cells were either placed in full-medium or the appropriate starvation medium for the indicated amount of time. F/S-ATG13 was affinity S-tag immunoprecipitated using S-protein agarose beads and analyzed by Western blotting. f, AMPK−/− MEFs were stably transduced with exogenous F/S-ATG13. The cells were placed in full-medium or amino acid and serum-starvation medium for up to 2 h. F/S-ATG13 was affinity S-tag immunoprecipitated using S-protein agarose beads and analyzed by Western blotting.
To further establish that phosphorylation of Ser-258 is modulated by the mTOR signaling pathway, we compared the phosphorylation level of pSer258 between TSC2-null (TSC2−/−) and wild-type (TSC2+/+) MEFs, under starvation or Torin 1 treatment (Fig. 3b). We observed that TSC2−/− MEFs, which have increased mTOR signaling, had higher levels of basal pSer258, which persisted upon starvation. In the TSC2−/− MEFs, dephosphorylation of pSer258 was only achieved by direct inhibition of mTOR through Torin 1 treatment. In contrast, in TSC2+/+ MEFs, pSer258 was sensitive to both starvation and Torin 1 treatment.
To distinguish if mTOR-regulated sites may be phosphorylated directly by mTOR or by kinases downstream of mTOR, we established an in vitro kinase assay. Ectopically overexpressed mTOR and ATG13 were immunoprecipitated from HEK293T cells and subjected to an in vitro mTOR kinase assay. We demonstrated that ATG13 was directly phosphorylated by mTOR on Ser-258, and this phosphorylation is inhibited by Torin 1 treatment (Fig. 3c).
We then examined whether ATG13 phosphorylation at Ser-224 is modulated by the AMPK pathway. First, we found that upon amino acid and serum starvation, AMPK activity was diminished (Fig. 3d). This decrease in AMPK activity correlates with a concomitant decrease in pSer224. This observation is consistent with previous publications showing that amino acid and serum-starvation cause a decrease of AMPK activity (25). We also modulated AMPK activity with glucose starvation, a known activator of AMPK activity (24). Upon glucose starvation, we observed an increase in phosphorylation of Ser-224 but not Ser-258 (Fig. 3e). To directly establish a requirement for the AMPK pathway in modulating the phosphorylation at Ser-224, we overexpressed exogenous ATG13 in AMPKα1/2-null MEFs. We found that immunoprecipitated F/S-ATG13 was phosphorylated at Ser-258 but not at Ser-224 (Fig. 3f). Therefore, the AMPK pathway selectively modulates phosphorylation at ATG13 Ser-224.
Although ATG13 phosphorylation at Ser-224 is enhanced during glucose starvation, this event is probably not relevant to glucose starvation-induced autophagy functionally, because starvation-induced autophagy is independent of the ULK1 complex (27, 28).
Nutrient-regulated ATG13 Phosphorylation RegulatesStarvation-induced Autophagy
To assess the functional significance of ATG13 phosphorylation in regulating starvation-induced autophagy, we generated ATG13−/− MEFs by CRISPR-CAS9 approach. As expected, the resulted ATG13−/− MEFs failed to undergo autophagy upon starvation (Fig. 4a). We subsequently stably expressed GFP-LC3 and the wild-type ATG13 or its phospho-mutants in these cells. We first evaluated glutamic acid and aspartic acid mutants of Ser-224 and Ser-258. We did not observe any change in the autophagy flux as compared with ATG13-null cells reconstituted with wild-type ATG13 (data not shown). Since the phospho-antibodies could not recognize their respective “phospho-mimetic” mutants (Fig. 2a), these mutants do not recapitulate the properties of these specific phosphorylation events. We also evaluated the alanine mutants. We observed significantly more GFP-LC3 punctae when both phosphorylation sites were mutated to alanine (Fig. 4b). On the other hand, single mutation of either site did not affect the autophagy flux, suggesting that ATG13 integrates signals of nutritional deficiency from multiple pathways into a coordinated autophagic response. These results were corroborated by Western blotting of endogenous LC3: a greater depletion of LC3-I was observed when both phosphorylation sites were mutated to alanine whereas single mutation of either site did not appreciably affect the autophagy flux (Fig. 4c). We also observed a greater depletion of p62 in cells expressing the combined alanine mutant. Additionally, we monitored the phosphorylation of ATG13 at Ser-318 with a phospho-antibody. ULK1 phosphorylates ATG13 on Ser-318, thus phosphorylation of this residue can be used as a read-out of ULK1 kinase activity in cells (29, 30). Cells expressing the combined alanine mutant had higher levels of ATG13 pSer318, as compared with cells expressing wild-type or single alanine mutant of ATG13 (Fig. 4c). Based on these results, we conclude that phosphorylation of ATG13 on Ser-224 and Ser-258 inhibits autophagy initiation, thus an unphosphorylatable mutant of ATG13 renders the ULK1 kinase complex to be more readily activated upon starvation, instead of waiting for these inhibitory phosphorylations to dissipate. Interestingly, unlike starvation-induced autophagy, when Torin 1 was used to trigger autophagy, the combined mutant did not further potentiate autophagy compared with wild-type ATG13 (Fig. 3d). This result suggests that upon mTOR inhibition, the ATG13 mutant needs to work together with certain additional cellular events to potentiate autophagy, and such events are inducible by amino acid starvation but not by pharmacological inhibition of mTOR. Indeed, it has been recently demonstrated that amino acid starvation triggers both stimulation of protein phosphatase 2A (PP2A) and inhibition of mTOR; these two events coordinately lead to potent activation of ULK1 complex-dependent autophagy (31).
FIGURE 4.

ATG13 phosphorylation at Ser-224 and Ser-258 negatively regulates the induction of autophagy. a, ATG13 knock-out MEFs were generated by CRISPR-CAS9. The cells exhibit a lack of starvation-induced autophagy. b, ATG13−/− MEFs stably expressing GFP-LC3 were reconstituted by the stable expression of F/S-ATG13 wild-type, S224A/S258A, S224A, or S258A. The cells were placed in full-medium or starvation medium for 1 to 2 h. The GFP-LC3 punctae were scored. There are significantly more GFP-LC3 punctae in cells expressing S224A/S258A compared with wild-type at one and two hour starvation. *, p < 0.001. c, ATG13−/− MEFs were reconstituted by the stable expression of F/S-ATG13 wild-type, S224A/S258A, S224A, or S258A. Starvation-induced autophagy was assessed by LC3-I to LC3-II conversion and p62 depletion. In vivo ULK1 kinase activity was assessed by phosphorylation of ATG13 at Ser-318. d, reconstituted ATG13−/− MEFs were subjected to 1 μm Torin 1-induced autophagy.
Nutrient-regulated Phosphorylation of ATG13 Modulates the Autophagic Activity of ULK1
To understand the molecular mechanism by which nutrient-dependent phosphorylation on ATG13 regulates starvation-induced autophagy, we evaluated distinct properties of ULK1 that are regulated by ATG13, such as protein stability, cellular localization and enzyme activity. ULK1 is destabilized in ATG13−/− MEFs. When we reconstituted these cells by expressing wild-type ATG13, or its double site or single site alanine mutants, ULK1 was stabilized to comparable levels (Fig. 4c). Next, we analyzed the cellular localization of ULK1. Previous work has shown that upon starvation ULK1 translocates to the isolation membrane in an ATG13-dependent fashion (18). We analyzed endogenous ULK1 cellular localization using ULK1 immunofluorescence in ATG13−/− MEFs reconstituted with either wild-type ATG13 or its mutants. We observed that starvation induced a greater number of ULK1 punctae in cells expressing the double alanine mutants, as compared with cells expressing wild-type or single site alanine mutants of ATG13 (Fig. 5a). This observation is also mirrored by the translocation pattern of ATG13 (Fig. 5b). Therefore, phosphorylation of ATG13 on Ser-224 and Ser-258 inhibits translocation of the ULK1 kinase complex. To further characterize the role of these phosphorylation events in modulating the ULK1 kinase complex, we evaluated their effect on protein interactions. Phosphorylation at Ser-224 and Ser-258 did not affect binding of ATG13 to ULK1 or ATG101 (Fig. 5, c and d). This result is expected, as previous work has demonstrated that the ULK1 kinase complex is constitutively formed (18). We additionally probed the relationship between the ULK1-catalyzed phosphorylations on Ser-318 and the novel phosphorylations on Ser-224 and Ser-258. Abrogating phosphorylation of Ser-318 by alanine mutation did not affect phosphorylation at Ser-224 or Ser-258 (Fig. 5e).
FIGURE 5.

ATG13 phosphorylation at Ser-224 and Ser-258 regulates functional properties of ULK1. a, ATG13−/− MEFs were reconstituted by the stable expression of ATG13 wild-type, S224A/S258A, S224A, or S258A. The cells were placed in full-medium or starvation medium for 1 to 2 h. ULK1 immunofluorescence was used to image ULK1 punctae. There are significantly more ULK1 punctae at one and two hour starvation in cells stably expressing S224A/S258A as compared with wild-type. *, p < 0.001. b, S-Tag immunofluorescence was used to image F/S-ATG13 punctae. There are significantly more ATG13 punctae at one and two hour starvation in cells stably expressing S224A/S258A as compared with wild-type. *, p < 0.001. c, F/S-ATG13 was co-transfected in HEK293T alongside HA-ULK1 or d, Myc-ATG101. The cells were placed in full-media or starvation medium for 4 h, then complexes were S-tag immunoprecipitated using S-protein agarose beads and analyzed by Western blotting. e, F/S-ATG13 wild-type or S318A mutant were transiently transfected into HEK293T cells and subjected to an amino acid and serum starvation time course. F/S-ATG13 was then immunoprecipitated using S-protein agarose beads and analyzed by Western blotting. f and g, ectopically expressed HA-ULK1 and FLAG-S-ATG13 complexes were affinity purified by S-protein agarose beads and subjected to an ULK1 kinase assay using 32P-labeled MBP as a readout. ULK1 bound to S224A/S2458A is significantly more active than when its bound to ATG13 wild-type. *, p < 0.001.
It has been demonstrated that ATG13 stimulates the kinase activity of ULK1 (18). Using ectopically expressed FLAG-S-ATG13 and HA-ULK1 complex affinity-purified by S-Tag agarose beads, we confirmed that the complex containing wild type ULK1 but not K46I (kinase inactive mutant) can phosphorylate the artificial substrate myelin basic protein (MBP) in vitro (Fig. 5f). To further evaluate the role of ATG13 phosphorylation in regulating ULK1 kinase activity, we compared the activation of ULK1 kinase activity when incubated with ATG13 wild-type or the combined alanine mutant. ULK1 bound with the alanine mutant exhibited higher kinase activity against MBP than ULK1 bound with wild-type ATG13, as determined by the incorporation of [γ-32P]phosphate to MBP (Fig. 5g). This result and that of cellular Ser-318 ATG13 phosphorylation (Fig. 4c) indicate that the nutrient-regulated phosphorylation of ATG13 on Ser-224 and Ser-258 suppresses ULK1 kinase activity.
Discussion
In this study, we describe the identification and characterization of nutrient-regulated phosphorylation events on ATG13, pSer224, and pSer258. These sites are dephosphorylated upon amino acid starvation, with pSer258 showing faster kinetics. Upon nutrient replenishment, these sites are re-phosphorylated. Interestingly, these sites are differentially regulated with pSer258 being a direct substrate of mTORC1, while pSer224 modulated by the AMPK pathway. As such, phosphorylation of Ser-224 and Ser-258 on ATG13 function as molecular sensors of nutrient availability that integrate signals from multiple signaling pathways. Phosphorylation of ATG13 at Ser-224 and Ser-258 exerts an inhibitory effect on the autophagic function of ULK1, as detected by monitoring both ULK1 kinase activity and its cellular translocation.
Although the autophagic process is conversed from yeast to humans, its regulatory mechanisms have diverged. In yeast, Tor negatively regulates the induction of autophagy through regulating the formation of the Atg1-Atg13-Atg17 complex by hyperphosphorylating Atg13. Upon Tor inactivation, Tor-mediated phosphorylation on Atg13 is inhibited, allowing for the formation of the initiator complex and the subsequent recruitment of downstream players. However, in mammals, although inhibition of mTOR is also sufficient to unleash the autophagy function of the ULK1-ATG13-FIP200 complex (which is the counterpart of the yeast Atg1 complex), the ULK1 complex is always assembled even under nutrient-rich, mTOR-active conditions. The lack of the dynamic association-dissociation between ULK1 and ATG13 stipulates that mammalian autophagy must be initiated differently from that in yeast.
How does the phosphorylation status of ULK1 and ATG13 regulate the ULK1 complex without controlling complex assembly? We ruled out the possibility that phosphorylation of ATG13 at Ser-224 and Ser-258 regulates the binding of ATG13 with ATG101, a regulatory protein of the ULK1 complex that has no homologous counterpart in baking yeast. Other possible mechanisms include that phosphorylation of ULK1 and ATG13 controls the conformation of the ULK1 complex or interaction of the complex with an unknown novel regulator. Further investigation is required to define the precise mechanism by which these phosphorylation events regulate the autophagy function of the ULK1 complex.
Intriguingly, this study also demonstrates that the energy-sensing kinase AMPK can regulate autophagy via phosphorylating ATG13. The role of AMPK in autophagy appears to be more complicated than that of mTOR. On the one hand, AMPK has been shown to be a positive regulator of autophagy, especially under energy-depriving, AMPK-stimulating conditions such as glucose starvation (24, 32, 33). On the other hand, AMPK can also negatively regulate autophagy by phosphorylating ULK1 at Ser-638 site, thus tuning down the autophagy activity of ULK1 (25). Here we show that AMPK phosphorylates ATG13 at Ser224 and this event also reduces the autophagy activity of the ULK1 complex. Notably, the role of AMPK on the ULK1 complex is dispensable and of a “fine-tuning” nature: regardless of the status of AMPK activity or expression, ULK1 complex-dependent autophagy can proceed once mTOR activity is suppressed. It is most likely that phosphorylation of ATG13 at Ser-224 by AMPK provides an additional layer of mechanism for controlling autophagy in a more accurate and efficient manner.
Author Contributions
C. P. and X. J. conceived the study, designed the experiments, analyzed the data, and wrote the paper. C. P. performed the experiments. R. H. guided the mass spectrometry (MS) analysis and analyzed the MS results.
Acknowledgments
We thank Hediye Erdjument-Bromage for performing the mass spectrometry analysis. We would also like to thank all members of the Jiang Laboratory for active discussion and advice.
This work was supported in part by National Institutes of Health NCI Cancer Center Support Grant P30 CA008748 to Microchemistry and Proteomics Core Laboratory, Memorial Sloan-Kettering Cancer Center, NIH F31GM103248 (to C. P.), NIH R01GM113013 (to X. J.), and NIH R01CA166413 (to X. J.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- ATG
- autophagy-related gene
- AMPK
- AMP-activated kinase
- mTOR
- mammalian target of rapamycin
- TORC
- TOR complex.
References
- 1. Mizushima N., and Levine B. (2010) Autophagy in mammalian development and differentiation. Nat. Cell Biol. 12, 823–830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Meléndez A., and Neufeld T. P. (2008) The cell biology of autophagy in metazoans: a developing story. Development 135, 2347–2360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Mizushima N. (2007) Autophagy: process and function. Genes Dev. 21, 2861–2873 [DOI] [PubMed] [Google Scholar]
- 4. Takeshige K., Baba M., Tsuboi S., Noda T., and Ohsumi Y. (1992) Autophagy in yeast demonstrated with proteinase-deficient mutants and conditions for its induction. J. Cell Biol. 119, 301–311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Tsukada M., and Ohsumi Y. (1993) Isolation and characterization of autophagy-defective mutants of Saccharomyces cerevisiae. FEBS Lett. 333, 169–174 [DOI] [PubMed] [Google Scholar]
- 6. Thumm M., Egner R., Koch B., Schlumpberger M., Straub M., Veenhuis M., and Wolf D. H. (1994) Isolation of autophagocytosis mutants of Saccharomyces cerevisiae. FEBS Lett. 349, 275–280 [DOI] [PubMed] [Google Scholar]
- 7. Ohsumi Y. (2014) Historical landmarks of autophagy research. Cell Res. 24, 9–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mizushima N., Yoshimori T., and Ohsumi Y. (2011) The role of Atg proteins in autophagosome formation. Annu. Rev. Cell Dev. Biol. 27, 107–132 [DOI] [PubMed] [Google Scholar]
- 9. Feng Y., He D., Yao Z., and Klionsky D. J. (2014) The machinery of macroautophagy. Cell Res. 24, 24–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kamada Y., Funakoshi T., Shintani T., Nagano K., Ohsumi M., and Ohsumi Y. (2000) Tor-mediated induction of autophagy via an Apg1 protein kinase complex. J. Cell Biol. 150, 1507–1513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kabeya Y., Kamada Y., Baba M., Takikawa H., Sasaki M., and Ohsumi Y. (2005) Atg17 functions in cooperation with Atg1 and Atg13 in yeast autophagy. Mol. Biol. Cell 16, 2544–2553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Suzuki K., Kubota Y., Sekito T., and Ohsumi Y. (2007) Hierarchy of Atg proteins in pre-autophagosomal structure organization. Genes Cells 12, 209–218 [DOI] [PubMed] [Google Scholar]
- 13. Kawamata T., Kamada Y., Kabeya Y., Sekito T., and Ohsumi Y. (2008) Organization of the pre-autophagosomal structure responsible for autophagosome formation. Mol. Biol. Cell 19, 2039–2050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cheong H., Nair U., Geng J., and Klionsky D. J. (2008) The Atg1 kinase complex is involved in the regulation of protein recruitment to initiate sequestering vesicle formation for nonspecific autophagy in Saccharomyces cerevisiae. Mol. Biol. Cell 19, 668–681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cheong H., Yorimitsu T., Reggiori F., Legakis J. E., Wang C. W., and Klionsky D. J. (2005) Atg17 regulates the magnitude of the autophagic response. Mol. Biol. Cell 16, 3438–3453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kamada Y., Yoshino K., Kondo C., Kawamata T., Oshiro N., Yonezawa K., and Ohsumi Y. (2010) Tor directly controls the Atg1 kinase complex to regulate autophagy. Mol. Cell. Biol. 30, 1049–1058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Fujioka Y., Suzuki S. W., Yamamoto H., Kondo-Kakuta C., Kimura Y., Hirano H., Akada R., Inagaki F., Ohsumi Y., and Noda N. N. (2014) Structural basis of starvation-induced assembly of the autophagy initiation complex. Nat. Struct. Mol. Biol. 21, 513–521 [DOI] [PubMed] [Google Scholar]
- 18. Ganley I. G., Lam du H., Wang J., Ding X., Chen S., and Jiang X. (2009) ULK1.ATG13.FIP200 complex mediates mTOR signaling and is essential for autophagy. J. Biol. Chem. 284, 12297–12305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hara T., Takamura A., Kishi C., Iemura S., Natsume T., Guan J. L., and Mizushima N. (2008) FIP200, a ULK-interacting protein, is required for autophagosome formation in mammalian cells. J. Cell Biol. 181, 497–510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Jung C. H., Jun C. B., Ro S. H., Kim Y. M., Otto N. M., Cao J., Kundu M., and Kim D. H. (2009) ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol. Biol. Cell 20, 1992–2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sebastiaan Winkler G., Lacomis L., Philip J., Erdjument-Bromage H., Svejstrup J. Q., and Tempst P. (2002) Isolation and mass spectrometry of transcription factor complexes. Methods 26, 260–269 [DOI] [PubMed] [Google Scholar]
- 22. Erdjument-Bromage H., Lui M., Lacomis L., Grewal A., Annan R. S., McNulty D. E., Carr S. A., and Tempst P. (1998) Examination of micro-tip reversed-phase liquid chromatographic extraction of peptide pools for mass spectrometric analysis. J. Chromatogr. A. 826, 167–181 [DOI] [PubMed] [Google Scholar]
- 23. Mali P., Yang L., Esvelt K. M., Aach J., Guell M., DiCarlo J. E., Norville J. E., and Church G. M. (2013) RNA-guided human genome engineering via Cas9. Science 339, 823–826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kim J., Kundu M., Viollet B., and Guan K. L. (2011) AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 13, 132–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Shang L., Chen S., Du F., Li S., Zhao L., and Wang X. (2011) Nutrient starvation elicits an acute autophagic response mediated by Ulk1 dephosphorylation and its subsequent dissociation from AMPK. Proc. Natl. Acad. Sci. U.S.A. 108, 4788–4793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Egan D. F., Shackelford D. B., Mihaylova M. M., Gelino S., Kohnz R. A., Mair W., Vasquez D. S., Joshi A., Gwinn D. M., Taylor R., Asara J. M., Fitzpatrick J., Dillin A., Viollet B., Kundu M., Hansen M., and Shaw R. J. (2011) Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science 331, 456–461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gammoh N., Florey O., Overholtzer M., and Jiang X. (2013) Interaction between FIP200 and ATG16L1 distinguishes ULK1 complex-dependent and -independent autophagy. Nat. Struct. Mol. Biol. 20, 144–149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Cheong H., Lindsten T., Wu J., Lu C., and Thompson C. B. (2011) Ammonia-induced autophagy is independent of ULK1/ULK2 kinases. Proc. Natl. Acad. Sci. U.S.A. 108, 11121–11126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Petherick K. J., Conway O. J., Mpamhanga C., Osborne S. A., Kamal A., Saxty B., and Ganley I. G. (2015) Pharmacological inhibition of ULK1 kinase blocks mammalian target of rapamycin (mTOR)-dependent autophagy. J. Biol. Chem. 290, 11376–11383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Joo J. H., Dorsey F. C., Joshi A., Hennessy-Walters K. M., Rose K. L., McCastlain K., Zhang J., Iyengar R., Jung C. H., Suen D. F., Steeves M. A., Yang C. Y., Prater S. M., Kim D. H., Thompson C. B., Youle R. J., Ney P. A., Cleveland J. L., and Kundu M. (2011) Hsp90-Cdc37 chaperone complex regulates Ulk1- and Atg13-mediated mitophagy. Mol. Cell 43, 572–585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wong P. M., Feng Y., Wang J., Shi R., and Jiang X. (2015) Regulation of autophagy by coordinated action of mTORC1 and protein phosphatase 2A. Nat. Commun. 6, 8048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ha J., Guan K. L., and Kim J. (2015) AMPK and autophagy in glucose/glycogen metabolism. Mol. Aspects Med. 46, 46–62 [DOI] [PubMed] [Google Scholar]
- 33. Kim J., Kim Y. C., Fang C., Russell R. C., Kim J. H., Fan W., Liu R., Zhong Q., and Guan K. L. (2013) Differential regulation of distinct Vps34 complexes by AMPK in nutrient stress and autophagy. Cell 152, 290–303 [DOI] [PMC free article] [PubMed] [Google Scholar]