

Abstract
Genome editing based on sequence-specific designer nucleases, also known as programmable nucleases, seeks to modify in a targeted and precise manner the genetic information content of living cells. Delivering into cells designer nucleases alone or together with donor DNA templates, which serve as surrogate homologous recombination (HR) substrates, can result in gene knockouts or gene knock-ins, respectively. As engineered replication-defective viruses, viral vectors are having an increasingly important role as delivery vehicles for donor DNA templates and designer nucleases, namely, zinc-finger nucleases (ZFNs), transcription activator-like effector nucleases (TALENs) and clustered, regularly interspaced, short palindromic repeats (CRISPR)-associated Cas9 (CRISPR−Cas9) nucleases, also known as RNA-guided nucleases (RGNs). We review this dual role played by engineered viral particles on genome editing while focusing on their main scaffolds, consisting of lentiviruses, adeno-associated viruses, and adenoviruses. In addition, the coverage of the growing body of research on the repurposing of viral vectors as delivery systems for genome editing tools is complemented with information regarding their main characteristics, pros, and cons. Finally, this information is framed by a concise description of the chief principles, tools, and applications of the genome editing field as a whole.
Genome editing based on sequence-specific designer nucleases, also known as, programmable nucleases (Figure 1) is opening a vast array of scientific and technological possibilities. Its broad range of action stems from granting researchers the means to modify, in a targeted and precise manner, the genetic make-up of cells from an increasing number of higher eukaryotes, including those of humans and other mammals.1,2,3 In general, this is achieved by inducing double-stranded DNA breaks (DSBs) at predefined chromosomal sequences after designer nuclease delivery into target cells. The delivery of designer nucleases alone (Figure 2) or together with so-called donor DNA (Figure 3) can result in different targeted genome modification outcomes, each of which resulting from the repair of site-specific DSBs by nonhomologous end-joining (Figure 2) or homologous recombination (HR) (Figure 3), respectively.
Figure 1.

Diagrams of the three principal designer nuclease platforms. (a) Zinc-finger nucleases (ZFNs). ZFNs are based on artificial zinc-finger motifs in which two cysteine residues in a β-sheet hairpin and two histidines in a α-helix are tetrahedrally coordinated by a zinc ion. ZF, zinc-finger motif dictating the interaction with a specific nucleotide triplet; FokI, nuclease domain of the type IIS restriction enzyme FokI. (b) Transcription activator-like effector nucleases (TALENs). TALENs are based on type III secretory systems of phytopathogenic bacteria (e.g., Xanthomonas sp.). TALE, transcription activator-like effector comprising a central DNA-binding domain consisting of an highly conserved 33–34 residue-long repetitive motif; FokI, nuclease domain of the type IIS restriction enzyme FokI; RVDs, repeat variable di-residues located at positions 12 and 13 of each TALE repeat governing the interaction with a particular nucleotide. (c) RNA-guided nucleases (RGNs). RGNs are based on prokaryotic type II clustered, regularly interspaced, short palindromic repeat (CRISPR)-associated (Cas) systems (e.g., Cas9 from Streptococcus pyogenes). sgRNA, chimeric single-guide RNA consisting of a fusion between a sequence-tailored CRISPR RNA (crRNA) and a scaffolding trans-activating crRNA (tracrRNA). PAM, protospacer adjacent motif (NGG, in the case of S. pyogenes); RuvC and HNH, the two nuclease domains of the Cas9 nuclease protein.
Figure 2.

Genome-editing approaches based on nonhomologous end-joining (NHEJ)–mediated repair of chromosomal DSBs using designer nucleases. NHEJ DNA repair mechanisms acting at site-specifically generated DSBs can result in different genome editing outcomes. (a) NHEJ-mediated introduction of small insertions and deletions (indels) at the target site often leads to DNA sequence frame shifting, which in turn, can yield gene-specific knockouts whenever protein or RNA coding sequences are targeted (left panel). Conversely, targeted DSB-induced frame shifting can restore the proper reading frame usage (right panel). Of note, targeted mutagenesis resulting from the activity of designer nucleases might also generate protein variants whose mode of action involve dominant negative, or positive, effects (not drawn). (b) DNA-level exon skipping can be achieved by targeting genomic sequences corresponding to key splice acceptor elements. (c) Coordinated DSB formation by designer nuclease pairs (multiplexing) can yield specific deletions or inversions if the target sites are located in a particular chromosome (left panel) or translocations if they are present in different chromosomes (right panel). Solid boxes and horizontal lines, exons and introns, respectively; DSB, double-stranded DNA break (open vertical arrowheads); Broken arrows, cis-acting gene regulatory elements including promoters/enhancers. Indels, small insertions and deletions (open vertical bars); j, chromosomal DNA junctions formed by non-homologous recombination events triggered by designer nuclease multiplexing. SD and SA, splice donor and splice acceptor, respectively.
Figure 3.

Genome-editing approaches based on HR-mediated repair of chromosomal double-stranded DNA breaks (DSBs) using donor DNA templates and designer nucleases. Robust homology-directed gene targeting, or knock-in, is achievable by combining designer nuclease-induced DSB formation with the delivery of donor DNA templates whose sequences share identity to regions flanking the targeted chromosomal lesion. These genome-editing procedures, based on the activation and recruitment of the homologous recombination (HR) DNA repair machinery, are particularly useful for the precise modification of predefined chromosomal target sequences of choice. Indeed, these DNA-modifying approaches offer the possibility to (a) repair, mutate or modify endogenous genes in a site-specific manner, (b) introduce entire recombinant transcriptional units (transgenes) into predefined positions in the genome, such as at so-called “safe harbours”, and (c) endow endogenous proteins with new domains or tag endogenous genes, such as for tracing their expression patterns or isolating their encoded products. Asterisks, nucleotide change, such as, addition or deletion of a single nucleotide polymorphism (SNP) or mutation; Large and small open boxes, recombinant transcriptional unit (transgene) and heterologous motif (e.g., a tag), respectively. For a description of the other symbols and abbreviations see the legend of Figure 2.
Therefore, a crucial aspect pertaining to the application of genome editing strategies is that of introducing into target cells designer nucleases (Figure 2) and, whenever new genetic information needs to be added, surrogate HR substrates in the form of exogenous donor DNA templates (Figure 3). Viral vectors are particularly suitable options to introducing genome editing reagents into target cells because, while being replication-defective, they retain the efficient cell entry mechanisms evolved by their wild-type counterparts.2,4,5 Indeed, as engineered replication-deficient viruses, viral vectors have been extensively used in academia and industry to deliver foreign genetic payloads into virtually any cell type of interest. Moreover, besides nucleic acids they are also starting to be adapted for the direct transduction of recombinant proteins into target cells, including designer nucleases. In these cases, designer nucleases are fused to structural components of vector particles (for a recent review, see ref. 6).
The on-going adaptation of viral vectors to genome editing settings builds upon a vast amount of knowledge gained from their development for “classical” gene therapy or gene replacement approaches in which the delivered foreign nucleic acids remain mostly in an episomal state or integrate randomly or semi-randomly throughout the target cell's genome.2,4,5
Clearly, inserting instead transgenes, or any exogenous DNA for that matter, into specific genomic sequences reduces the chance for various problematic events sometimes emergent whenever using systems that lead to the uncontrolled chromosomal integration of foreign nucleic acids (e.g., retroviral vectors and transposons/transposases). These unwarranted outcomes include positional-effect variegation, transgene silencing and, in some cases, insertional mutagenesis caused by transcriptional deregulation or physical disruption of endogenous target-cell genes.7 The more defined genome modification outcomes resulting from the aforementioned designer nuclease-assisted genome editing strategies, are having a clear impact in many fields. For instance, in functional genomics by helping deciphering the role of cis- and trans-acting nucleotide sequences, in transgenesis by speeding-up animal model generation via direct zygote engineering and in disease modeling by mimicking the origins of certain cancers through the deliberate induction of specific mutations or oncogenic rearrangements. Likewise related to disease modeling, and besides its potential role in future cell therapies, the integration of genome editing with induced pluripotent stem cell (iPSC) technologies is already helping in establishing genotype–phenotype relationships underlying not only monogenic but also polygenic or complex illnesses.8
In addition, genome editing strategies are being investigated for developing new treatment modalities aiming at tackling infectious diseases and advancing gene- and cell-based therapies. A first example of the former already exists in the shape of clinical studies testing whether designer nuclease-induced knockout of the HIV-1 coreceptor gene CCR5 confers therapeutic benefit to acquired immune deficiency syndrome patients.9,10 In parallel, the investigation of many other candidate gene therapies based on designer nuclease-induced gene knockout and gene knock-in approaches proceeds at the experimental and preclinical levels. These “genome surgery” research lines include deploying designer nucleases for disrupting alleles linked to dominant disorders and triggering homology-directed DNA targeting for repairing or complementing defective genes. The former entails the direct in situ correction of endogenous loci; the latter encompasses the targeted insertion of therapeutic DNA at ectopic “safe harbour” loci such as the AAVS1 (19q13.42). Transgene insertion at such loci results in much higher probabilities for stable and homogeneous expression levels while lessening the chances for the deregulation of target-cell endogenous genes.11
In view of the many common goals and substantial overlap between “classical” gene therapy and therapeutic gene-editing research, the co-option of viral vector technologies for the latter purpose is logical and multifaceted in that they are being investigated for delivering not only designer nucleases but also donor DNA templates. Related to this, different types of viral vectors are, in some cases, combined in individual gene-editing transduction protocols. Here we review the roles that the main classes of viral vectors are having on improving the performance of and expanding the scope for genome-editing technologies.
Viral Vectors as Gene-Editing Tools
Lentiviral vectors
Conventional lentiviral vectors based on HIV-1 establish permanent genetic modification of target cells owing to the fact that their integrase-dependent mechanisms ensure semirandom chromosomal insertion of the transported foreign nucleic acids.12 In “classical” gene therapy settings, these mechanisms are a crucial feature for achieving stable complementation of genetic defects in proliferating target cells and effector progenies.13 In the context of genome editing approaches, however, the lentiviral DNA insertion mechanisms should best be disabled in order to ensure that the resulting episomal vector templates are available as substrates for HR or for transient designer nuclease expression. As previously mentioned, the short-term presence of designer nucleases in target cells is important for reducing the chances that deleterious effects caused by off-target activity arise. Therefore, by using trans-complementing packaging constructs harboring specific point-mutations in the HIV-1 pol region, researchers can assemble lentiviral particles whose integrase moiety contains disabling amino acid substitutions at crucial positions within its catalytic pocket (i.e., D64, D116, and/or E152) (Figure 4).12,14,15 Importantly, these so-called class I integrase mutations are nonpleiotropic in that they interfere specifically with proviral establishment and not with any other of the viral transduction steps, such as, receptor binding, uncoating and nuclear import of the reverse-transcribed linear double-stranded vector genomes. Hence, integrase-defective lentiviral vectors (IDLVs), made with the aid of such packaging constructs serve as valuable vehicles for delivering nucleic acid templates for gene targeting and/or transient designer nuclease gene expression. Of note, similarly to their integration-proficient counterparts, the tropism of IDLV particles are normally altered by endowing them with envelop proteins derived from viruses whose cell surface receptors are different from those engaged by HIV-1. Accordingly, such pseudotyping manoeuvres permit narrowing or expanding the range of cell types transduced by vector particles.16 For instance, to confer broad host range and high physical particle stability to lentiviral vectors, the vesicular stomatitis virus glycoprotein-G (VSV-G) is often selected as the heterologous envelop moiety (Figure 4).
Figure 4.

Schematics of the main HIV-1-based vector systems. HIV-1-based vectors are assembled by cotransfecting producer cells (e.g., HEK293T cells) with transfer, envelop, and packaging constructs with the resulting particles being collected and purified after budding from producer cells. Transfer plasmids harbor foreign nucleic acid sequences flanked by HIV-1 5' and 3' long terminal repeats (LTRs). To confer HIV-1 Tat independence to vector genome expression, third-generation transfer plasmids have hybrid 5' LTRs composed of HIV-1 and heterologous transcriptional elements (e.g., cytomegalovirus and Rous sarcoma virus). Self-inactivating (SIN) vectors are deleted from specific LTR enhancers (ΔU3) to abrogate unwarranted transcriptional activity. Additional HIV-1 cis-acting elements include the packaging signal (Ψ), for vector genome encapsidation, the Rev-responsive element (RRE), for nuclear export of unspliced and singly spliced transcripts, and the central polypurine tract (cPPT), for transduction enhancement. The in trans-acting envelope plasmids typically encode the pseudotyping VSV-G moiety to confer a pantropic host range to vector particles. The also in trans-acting packaging constructs drive expression of HIV-1 Gag and Pol alone (third-generation) or together with Tat and Rev (second-generation). Owing to the Tat removal and the splitting of Rev from Gag-Pol templates, the former systems display a superior biosafety profile. The proteolytic processing of precursor Pol sequences yields mature reverse transcriptase and integrase (IN) molecules. Crucially, integrase-defective lentiviral vectors are assembled by using packaging constructs encoding IN moieties with substitutions of one or more amino acids of the DDE triad; D64, D116, and E152. These so-called class I mutations (e.g., D116N) abrogate specifically proviral establishment.
IDLVs were the first viral vectors to be tested in the context of designer nuclease-assisted genome editing experiments.17 These experiments, involving ZFN technology and various human target cell types (e.g., K562 erythromyeloblastoid leukemia cells, lymphoblastoid cells, and embryonic stem cells), provided an initial proof-of-concept for using IDLVs in designer nuclease-induced gene addition and gene repair studies. The former and latter experiments comprised, respectively, inserting recombinant DNA at specific genomic sequences (i.e., CCR5) and correcting IL2RG mutations underpinning X-linked severe combined immunodeficiency (X-SCID). These data revealed that IDLV genomes can serve as efficient HR substrates yielding, in some cell types, homology-directed DNA targeting frequencies exceeding 10% of the total target cell population with the majority of cells harboring mono-allelic insertions. These initial data has been followed-up by various other studies in which IDLV transfer of donor DNA templates resulted in the addition of reporter and therapeutically relevant transgenes into “safe harbour” loci in a diverse set of target cells, including human myocytes,18,19 human epithelial stem cells,20 and iPSC lines.21 Examples of these experiments are the site-specific chromosomal insertion of microdystrophin and FANCA transgenes into the “safe harbour” CCR5 locus in, respectively, human muscle progenitor cells18 and iPSCs from reprogrammed fibroblasts of Fanconi anemia patients.21 It is worth mentioning however that, in common with any other HR-based genome editing approaches, the recruitment of IDLV donor DNA for gene addition or for gene repair purposes is limited in non-dividing or quiescent cells due to the fact that HR occurs preferentially during the G2/S phase of the cell cycle, when endogenous repairing templates are available.22 Hence, the cellular DNA of quiescent primary cells, of which many display a high therapeutic relevance, is particularly difficult to edit through HR. An outstanding example of such cells is provided by primitive CD34+ human hematopoietic stem cells (HSCs). HSCs are defined as cells capable of long-term multilineage repopulation of the hematopoietic compartment in conditioned immune-deficient mice.23 Of note, only genome modification at the HSC level is expected to ensure life-long correction of genetic disorders affecting components of the hematopoietic system. Aiming at improving HR-based genome editing of these cells, Genovese et al.24 have developed a protocol in which donor DNA and ZFN delivery into HSCs is carried out by IDLV transduction and synthetic mRNA electroporation, respectively. Crucially, this transduction/electroporation protocol is combined with exposing target cells not only to cytokines but also to 16,16-dimethyl-prostaglandin E2 (dmPGE2) mixed with the aryl-hydrocarbon receptor protein antagonist, StemRegenin 1 (SR1). The rationale for including dmPGE2 and SR1 was to interfere with the loss of stem cell properties resulting from HSC exposure to extended ex vivo culture conditions and cell cycle-activating cytokines. By using these methods, the authors report that homology-directed gene targeting frequencies at AAVS1 and IL2RG in bona fide HSCs are increased, as stringently demonstrated by serial transplantation of human CD34+ cells from primary to secondary NSG (NOD-SCID-Il2rg-/-) mice. In a subsequent study, Hoban et al.25 have also tested an ex vivo protocol based on the transfer of ZFN-encoding mRNA and IDLV donor templates into bone marrow-derived CD34+ cells for correcting the A-to-T transversion in β-globin alleles causing sickle cell anemia.
Hitherto, the delivery of designer nucleases by IDLVs remains mostly restricted to ZFNs. Considering that the yields of functional lentiviral vector particles decrease sharply with increasing transgene size,26 it is possible that generating IDLVs containing the 4.1-kb Cas9 open reading frame (ORF) from Streptococcus pyogenes results in IDLV preparations with low functional particle titers. In addition, experimental results indicate that the genetic instability resulting from frequent reverse transcriptase template switching events within TALE repeats in lentiviral vector genomes leads to defective particles.27 This makes the assembly of TALEN-encoding IDLVs dependent on substantial ORF optimization for minimizing the frequency and length of unstable repetitive tracts.28 Of note, the same principle of sequence identity reduction has permitted to package and deliver transcriptional units encoding two ZFN monomers in single IDLV particles.29,30 This two-in-one approach is especially useful at low transduction rates since it ensures that each transduced cell is exposed to both members of a working ZFN pair at the proper 1:1 stoichiometry.
An issue pertaining to the optimal use of IDLVs as designer nuclease expression platforms is that of the susceptibility of their genomes to epigenetic silencing mechanisms in transduced cells.31,32,33 These mechanisms involve the action of cellular histone deacetylases and have been shown to curtail DSB-induced targeted mutagenesis after IDLV-mediated transfer of ZFN expression units.34 Finally, another issue regards the susceptibility of free-ended double-stranded IDLV genomes to “illegitimate” recombination processes such as nonhomologous end-joining. As a result, IDLV templates can become “captured” at off-target or spontaneous DSBs and form undesirable DNA structures such as concatemers and non-HR-derived junctions involving target or off-target sequences.17,19 These adverse genome-modifying events contribute to reduce the fidelity of the genome editing process as a whole.2
Adeno-associated viral vectors
In contrast to lentiviral vectors, recombinant adeno-associated viral vectors (rAAVs) lack an integration machinery (Figure 5).35,36 As a result, once in target cell nuclei, their genomes remain mostly in an episomal status with only a small fraction of them becoming incorporated in the cellular DNA (0.1–0.5 integrations per infectious unit)37 presumably upon nonhomologous end-joining–mediated repair of sporadic chromosomal DSBs.36 These vectors entered the scene of homology-directed gene targeting during the late 1990's, after the demonstration that viral particle transduction of single-stranded rAAV donor DNA yields more than 1,000-fold higher frequencies of gene repair (up to 1% of the total target-cell population) when compared to those achieved by transfecting conventional donor plasmids.38 Despite the feasibility of this approach, including in in vivo settings,39 the dominance of off-target insertions combined with the high dependency on large multiplicities of infection (>104 total vector particles per cell) and cell selection schemes,40,41 has contributed to the initiation of research lines based on designer nuclease-assisted rAAV donor DNA targeting.
Figure 5.

Schematics of the main recombinant adeno-associated viral vector (rAAV) production system. Recombinant AAV vectors are typically assembled by cotransfecting producer cells (e.g., HEK293T cells) with transfer, packaging and helper constructs with the resulting particles being purified after producer cell lysis. Transfer plasmids harbor foreign nucleic acid DNA flanked by 145 bp-long palindromic AAV-2 inverted terminal repeats (ITRs) whose primary sequence and T-shaped secondary structure form the origins of replication. The in trans-acting packaging plasmids contain the three AAV ORFs, rep, cap, and AAP. Transcription of rep from two different promoters and splicing of each of the resulting transcripts yields four proteins. Rep78 and Rep68 participate in DNA replication; Rep52 and Rep40 are necessary for DNA packaging into preformed empty capsids. The cap ORF is transcribed from a single promoter with alternative splicing resulting in two mRNA templates for the synthesis of three viral capsomers (VP1, VP2, and VP3). VP2 and VP3 share the same mRNA template with the former being translated from a “weak” start codon (ACG) located upstream of that for the latter product (AUG). Moreover, another “weak” start codon (CUG) present within the VP2-VP3 mRNA marks the beginning of a third reading frame, which codes for the assembly-activating protein (AAP). Productive wild-type AAV infections depend on the presence of an unrelated virus for providing AAV helper gene functions (e.g., HAdV-5 E1A-E1B, E2A, VAI-VAII, and E4ORF6). In the context of rAAV production, these functions are supplied by transfecting E1A-E1B-expressing cells (e.g., HEK293) with a helper construct containing E2A, VAI-VAII and E4ORF6. Often, the packaging and helper functions are combined in a single plasmid.
Like previous data had shown for HR substrates delivered in the context of standard plasmids,42,43 experiments based on inducing DSBs at chromosomally integrated reporter genes by the homing endonuclease I-SceI, provided a proof-of-concept for combining sequence-specific nucleases with rAAV donor DNA in gene-targeting settings. Indeed, these initial studies revealed that rAAV-based gene targeting can be enhanced by approximately 100-fold if a DSB is generated at a predefined target locus.44,45 In this realm, and similarly to IDLVs, rAAVs have been mostly used so far for delivering donor DNA templates and ZFNs. Of note, when compared to those of Cas9 and TALEN, ZFN ORFs are the smallest (i.e., ~1.2 kb per monomer versus ~4.1 kb and ~3 kb for S. pyogenes Cas9 and TALEN ORFs, respectively). This permits the flexible construction and packaging of transcriptional units encoding one or even two ZFNs in single rAAV particles46 whose effective maximum capacity is only ~4.5 kb (Figure 5). Clearly, in addition to TALEN and Cas9 nuclease delivery, the low packaging capacity of rAAV also introduces some limitations on the designing of HR substrates for the purpose of site-specific addition of whole transcriptional units. In any case, the combination of ZFN and rAAV technologies has clearly proven its potential for not only targeted gene disruption and deletion46 but also for gene repair strategies. In what the latter genome editing approaches are concerned, these experiments involved the targeting of both reporter and endogenous loci after the delivery of ZFNs and gene correcting templates into a diverse panel of human cell types. These different cell types included, U2OS osteosarcoma cells,47,48 HEK 293 cells,46 HeLa cervix carcinoma cells,48 HT-1080 fibrosarcoma cells,48 and bona fide human embryonic stem cells (ESCs) as well as iPSCs.49 Noticeably, due to the very diverse range of tools, experimental models and conditions, the gene-targeting frequencies in both absolute and relative terms (i.e., targeted versus random insertion events), varied substantially. As an example, Asuri et al.49 compared ZFN-induced gene repair levels after transducing ESCs with a HR template packaged either in natural or variant AAV capsids. The latter capsid type, isolated by sequential cycles of biopanning of libraries of cap-mutant viruses on target cells, confers high-level rAAV transduction of hard-to-transfect ESCs and iPSCs. The authors showed, by using a highly quantitative readout system based on the rescue of defective reporter gene expression, that the transfer of corrective donor DNA by the molecularly evolved rAAV variant (R459G) yielded significantly higher (~10-fold) ZFN-induced gene repair levels in ESCs (~1.3% of the total target cell population) when compared to those resulting from using a prototypic, serotype 2-based, rAAV. Importantly, the proportion of random rAAV DNA chromosomal insertions was not augmented by the presence of active ZFNs in the transduced cells. Collectively, this and the above-mentioned studies established that site-specific DSB formation serves as a potent trigger for homology-directed gene targeting of donor DNA delivered in the context of single-stranded rAAV genomes.
Owing to a favorable set of characteristics, rAAVs are particularly suited for testing genome-editing strategies in vivo. These characteristics include low immunogenicity in immunocompetent animal models and amenability to tissue tropism modification methodologies based on engineered capsids generated by rational or directed evolution approaches.50 Moreover, reminiscent of the above-described tropism engineering strategies involving enveloped lentiviral vectors; nonenveloped rAAVs can also be pseudotyped. In this case, rAAV genomes consisting of foreign DNA flanked by prototypic AAV serotype 2 inverted terminal repeats, are packaged within the capsids of other natural AAV isolates such as those of serotypes 1, 5, 6, 8, or 9.50 These novel capsid-modified rAAVs are powerful gene delivery tools in that they can bypass pre-existing immunity associated with the presence of neutralizing antibodies against particular rAAV serotype(s) and can overcome transductional blocks linked to the absence of viral receptor(s) on the surface of specific cell types or tissues. In addition to the previously mentioned work in which a molecularly evolved rAAV was used,49 another case in point is provided by the body-wide transduction of murine tissues by rAAV2/6 vectors, that is, AAV serotype 2-derived rAAV genomes pseudotyped by packaging in AAV serotype 6 capsids.51 Moreover, it has been shown that rAAV2/8 particles achieve frequencies of murine liver cell transduction that are 10- to 100-fold higher than those obtained by using vectors based on other serotypes.52 Importantly, these experiments equally revealed that the rAAV2/8 gene delivery activity was not hindered in animals preimmunized by exposure to other AAV serotypes.
The relevance and utility of rAAVs in in vivo settings is also underscored by the fact that a first proof-of-principle for designer nuclease-induced genome editing in vivo involved the use of these vectors in a murine model of hemophilia B, a blood coagulation disorder caused by factor IX deficiency.53 In particular, rAAV2/8 particles containing a corrective cDNA spanning exons 2 through 8 of human F9 were administered to new-born hemophilia B mice together with rAAV2/8 particles encoding donor-matched ZFNs targeting intron-1 of a defective human F9 transgenic allele. Gene targeting was detected and meaningful in that it resulted in 3–7% of normal levels of circulating factor IX that led to the improvement of the disease phenotype as measured by clot-formation kinetic assays. Of note, molecular analysis of genomic DNA from treated mice revealed that therapeutic construct insertions at the intended target site occurred through both homologous and non-HR.53 The latter, vector genome capture events, were likely caused by end-to-end nonhomologous end-joining of broken chromosome and AAV inverted terminal repeat sequences. A subsequent study extended these findings of AAV/ZFN-mediated in vivo therapeutic genome editing to adult hemophilia B mice.54
The in vitro and in vivo transfer of RGN components by rAAVs, has also been initiated. After constructing and validating shortened expression units encoding Cas9 and sgRNAs, Senís et al.55 were able to demonstrate delivery of Cas9 alone or together with a sgRNA by single vector particles built on chimeric AAV-DJ capsids. The latter “all-in-one” rAAV construction achieved approximately 8% indel formation at a target miRNA locus in HEK 293T cells when applied at a multiplicity of infection of 106 particles per cell. However, in mouse livers, RGN-induced indel formation at the conserved miRNA target locus by different rAAV constructs was invariably below 1% at 2 weeks postadministration. These in vivo results have been complemented by other animal model experiments in which rAAV-mediated delivery of RGN components served as a direct, transgenesis-free, approach for studying gene function in the mammalian brain.56 These initial studies together with the advent of shorter Cas9 variants bode well for the implementation of rAAV/RGN tools in different in vitro and in vivo systems. Indeed, Ran et al.57 have recently used a comparative genomic analysis to isolate and characterize a Staphylococcus aureus Cas9 protein whose relatively small size permits flexible rAAV design, including copackaging of both RGN components within single vector particles. The delivery of these tools into the livers of C57BL/6 mice by rAAV2/8 particles led 1 week after intravenous administration to approximately 5 and 40% indel formation at Apob and Pcsk9 sequences, respectively.57
Adenoviral vectors
The sizable packaging capacity of adenoviral vectors (AdVs) combined with their high-titers and efficiency in transducing dividing and nondividing cells, makes them a broadly applicable option for in vitro and in vivo delivery of designer nucleases and donor DNA templates (Figure 6). Similarly to rAAVs, AdVs started to be deployed in the context of homology-directed gene targeting experiments that did not involve designer nuclease-induced DSB formation. In these experiments, helper-dependent AdVs, also known as “gutless” AdVs, were chosen owing to their lack of viral genes, permitting the use of high multiplicities of infection, and high capacity, allowing for large donor DNA packaging and delivery. Indeed, Ohbayashi et al.58 utilized helper-dependent AdVs with 18.6 kb homology arms to correct a mutation in HPRT through HR without the involvement of artificial DSB formation in mouse ES cells. With the emergence of iPSCs, helper-dependent AdVs were also shown to be useful for correcting disease-related mutations in these pluripotent stem cells. In particular, they were used to repair several mutations in LMNA alleles associated with laminopathies, thus expanding the application of this gene delivery system to human disease modeling and targeted gene repair.59 A follow-up study by Aizawa et al.60, demonstrated that regardless of the transcriptional status of the target gene, helper-dependent AdVs can mediate both gene knock-ins and gene knockouts by HR with high fidelity in both iPSCs and ESCs of human origin. Of note, however, the absolute gene targeting levels achieved by helper-dependent AdVs are rather low requiring as a result the use of drug-based selection pressure for isolating the desired targeted clones.
Figure 6.

Schematics of the principal adenoviral vector (AdV) systems. The genome structures of the main AdV classes are drawn in relation to that of the prototypic HAdV-5 from species C. The 103 bp-long “left” and “right” inverted terminal repeats (L-ITR and R-ITR, respectively) contain the origins of replication, with the viral DNA packaging signal (Psi) being located adjacent to the L-ITR. The early (E) and late (L) regions are expressed before and after the onset of viral DNA replication, respectively. The former regions (i.e., E1A-E2A, E2A-E2B, E3, and E4) encode proteins involved in gene regulation (viral and host) and viral DNA replication; the later encode gene products primarily responsible for virion maturation and assembly (L1-L5). Expression units corresponding to small RNAs (VAI-VAII) and intermediate gene products (IX and IVa2) are also shown. First-generation AdVs lack E1A-E1B or E1A-E1B plus E3. Since E3 is dispensable during in vitro replication, all these vectors can be produced in packaging cell lines expressing exclusively the E1 functions (e.g., HEK293 or PER.C6). Second-generation AdVs have deletions in additional early regions (e.g., E2A and/or E4) being, as a result, produced in their respective complementing cell lines. Third-generation AdVs (also known as “gutless” or high-capacity) lack all viral DNA sequences except for the cis-acting ITRs and packaging signal. These vectors are produced in E1-complementing cells in the presence of a first-generation helper AdV which furnishes in trans all the viral gene products necessary for the replication and assembly of “gutless” AdV particles. The helper has its packaging elements framed by target sites for a site-specific recombinase (e.g., Cre or FLP) so that in recombinase-expressing producer cells is rendered packaging-defective in a selective manner.
Similarly to lentiviral and adeno-associated viral vector systems, AdVs are equally amenable to tropism modification and Good Manufacturing Practice methodologies. The former strategies include exchanging the apical fiber motifs of prototypic species C serotypes, which interact with the Coxsackie B virus and adenovirus receptor (CAR), with those of other natural serotypes (e.g., species B adenoviruses), which interact with other primary receptors. This “fiber swapping” genetic retargeting strategy allows by-passing the absence of CAR on the surface of human cells with scientific and therapeutic value such as hematopoietic stem/progenitor cells,61,62 mesenchymal stromal cells,63,64,65 and muscle progenitor cells.65,66 Alternative AdV retargeting methods include capsid modifications by genetic fusion of fiber or pIX capsid proteins to heterologous ligands67 or by chemical binding of capsid components to targeting moieties.68 In this regard, it is noteworthy mentioning that the first testing of a therapeutic approach based on genome editing entails ZFN-mediated CCR5 knockout in CD4+ T-cells from acquired immune deficiency syndrome patients after their ex vivo transduction with fiber-modified AdV particles.9,10 Examples of other genome-editing studies based on the integration of AdV and ZFN technologies include the targeted mutagenesis of endogenous T-cell receptor genes in lymphocytes69 and of CCR5 and β-globin alleles in hematopoietic stem/progenitor cells.70,71 Moreover, homology-directed gene targeting induced after AdV-mediated delivery of ZFNs, is equally being pursued in various cell types such as myoblasts, epithelial stem cells, and keratinocytes.18,20
Highlighting their versatility, AdV systems have in addition to ZFNs been validated for delivering TALENs and RGN complexes into human somatic cells regardless of their transformation status.27,72 Concerning the former research it was found that, in striking contrast to lentiviral vector systems, the direct repeat arrays coding for the DNA-binding domains of TALENs are stable during AdV production in complementing packaging cell lines.27,73 Importantly, the resulting vector preparations led to dose-dependent and high-level (up to 67%) targeted DSB formation in exposed cells (e.g., muscle progenitor cells and mesenchymal stromal cells). The genetic stability of AdVs is also underscored by the fact that transcriptional units encoding ZFN9,74 or TALEN dimers can be packaged intact in single vector particles.71,75 Due to the sizable length of TALEN ORFs (~3.0 kb per monomer), the latter studies deployed the high-capacity “gutless” AdV platform (Figure 6). In addition to the aforementioned muscle progenitor cells and mesenchymal stromal cells, the combination of AdV and TALEN technologies has served for inducing site-specific DSB formation in iPSCs75 as well as in CD34+ cells isolated from G-CSF-mobilized peripheral blood mononuclear cells.71
Recently, various research groups started exploiting the efficient transduction of particular murine tissues by AdVs for studying genetic lesions underlying the emergence of specific cancers and, subsequently, modeling their progression in vivo. Such approaches based on the direct induction of targeted genomic changes in vivo (e.g., mutations, inversions, and translocations) are more expeditious than those based on transgenic mice and mimic more accurately the stochastic mosaicism characteristic of many tumors. For instance, Zhang et al.76 succeeded in inducing higher rates of Apc mutations in the murine liver after tail vein injection of TALEN-encoding AdVs than those achieved after plasmid hydrodynamic injections (33 versus 7–19%, respectively). Maddalo et al.77 have in turn deployed RGN-encoding AdVs for inducing an approximately 11 Mb chromosomal inversion involving the Alk and Eml4 loci to model the development of non–small-cell lung cancer in vivo.
Besides cancer modeling, other experiments sought to mutagenize Cebpα78 and Pcsk9 (ref. 79) in murine livers after the administration of AdVs encoding RGN complexes. The former gene is a transcriptional factor involved in the activation of metabolic target genes; the latter is associated with low-density lipoprotein cholesterol levels, with its loss-of-function correlating with reduced risk of coronary heart disease development. Collectively, these experiments strengthened the view that, together with rAAVs, AdVs serve as a valuable platform for introducing designer nucleases in vivo. However, with the expansion and finer follow-up of in vivo genome editing procedures, one can expect encountering the immunological hurdles identified previously in countless gene transfer studies in animals. These hurdles include the activation of innate and adaptive immune responses against viral particle components and foreign antigens derived from transgenic and, in the case of helper-independent AdVs, viral ORFs.80,81,82 Moreover, the long-term presence of designer nucleases in target tissues adds yet another hurdle that needs to be tackled by, for instance, incorporating regulatory devices for minimizing the risks of chromosomal mutations and/or rearrangements.
In addition to the introduction of designer nucleases into target cells, AdVs are also being exploited as a source of donor DNA templates for homology-directed gene editing after site-specific chromosomal DSB formation by ZFNs,20,74 TALENs,19,75 and RGNs.19,75 In this regard, it has been shown that combining designer nucleases (i.e., TALENs and RGNs) and AdV-mediated donor DNA transfer induces homology-directed gene targeting that is more specific and accurate than that resulting from delivering donor DNA templates through conventional nonviral vectors or IDLVs.19 The finding of precise genome editing resulting from designer nuclease-induced AdV donor DNA targeting (“Ad.iting”, in short) could be attributed to the capping of linear AdV DNA by the 5' covalently-attached viral terminal protein which, presumably, reduces non-HR events. The resulting targeted, single-copy, donor DNA integrants lead to uniform transgene expression in gene-modified cell populations.19
A synopsis of the main characteristics of the viral vector systems being repurposed as gene-editing devices is presented in Table 1, whereas their principal pros and cons are summarized in Table 2. On the basis of this review and on the information gathered in Table 1 and Table 2, there is no evidence for an “ideal” one-fits-all combination of gene delivery and gene-editing tools. Instead, one can put forward the view that a specific arrangement(s) of these tools is best suited to achieve a particular goal.
Table 1. Overview of the main viral vector systems being repurposed as gene-editing tools.
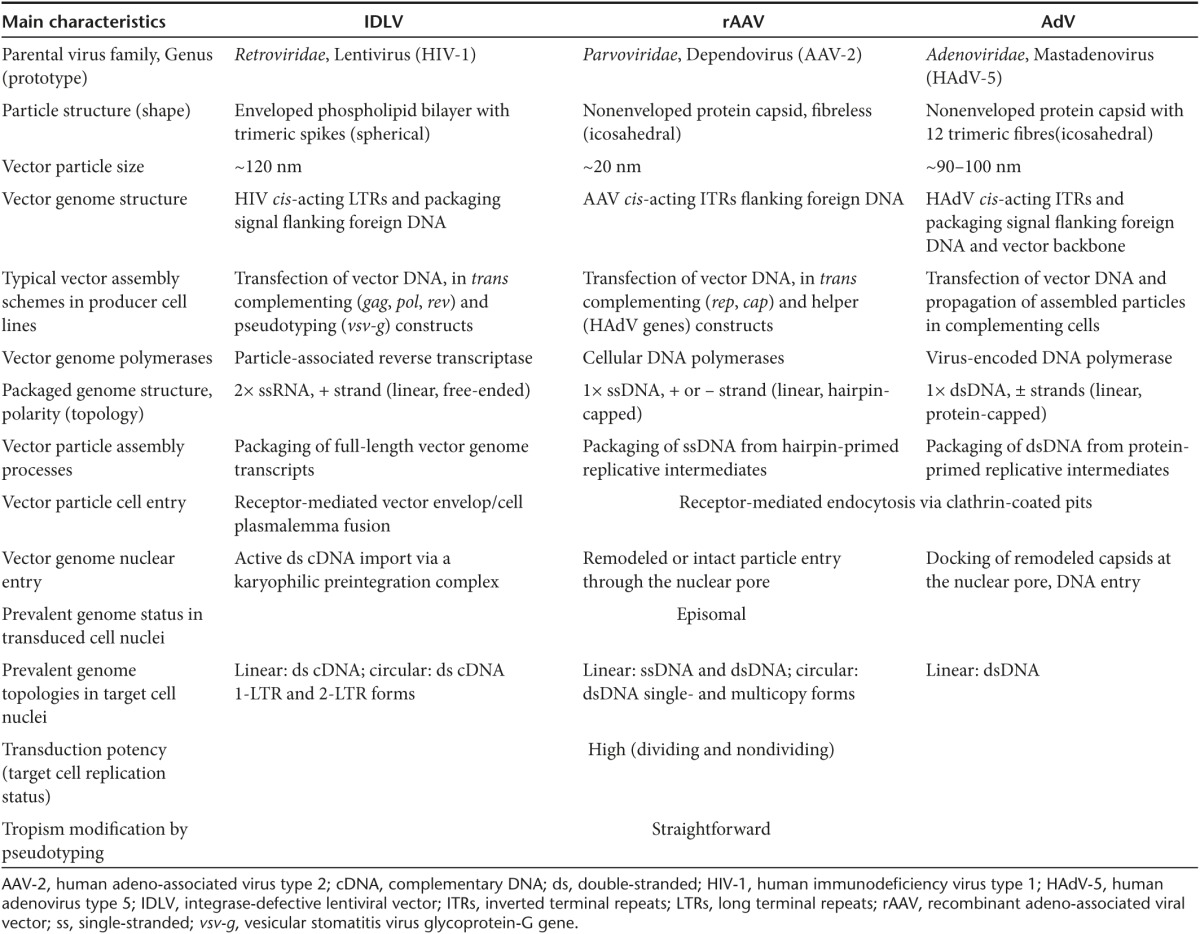
Table 2. Overview of the main pros and cons of IDLV, rAAV, and AdV systems.

In conclusion, viral vectors can serve a dual role in genome engineering efforts by delivering into virtually any human cell type, templates for not only designer nuclease expression but also for targeted chromosomal integration of foreign DNA. These features, combined with their well-established production systems and regulatory history build-up, are expected to foster and expand their application in genome editing settings, including in the realm of translational research.
Acknowledgments
We thank Ignazio Maggio, Josephine M. Janssen, Rob C. Hoeben (Department of Molecular Cell Biology, Leiden University Medical Center, the Netherlands), and Otto-Wilhelm Merten (Généthon, France) for their critical reading of the manuscript. X.C. is the recipient of a PhD research fellowship from the China Scholarship Council-Leiden University Joint Scholarship Programme. M.A.F.V. Gonçalves is coinventor in a patent application on the use of capped donor DNA constructs for targeted DSB-induced genome modifications. The funds for the research carried out in our laboratory include grants from the Dutch Prinses Beatrix Spierfonds (W.OR11-18) and the French AFMTéléthon (16621).
References
- Gaj, T, Gersbach, CA and Barbas, CF 3rd (2013). ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol 31: 397–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maggio, I and Gonçalves, MA (2015). Genome editing at the crossroads of delivery, specificity, and fidelity. Trends Biotechnol 33: 280–291. [DOI] [PubMed] [Google Scholar]
- Kim, H and Kim, JS (2014). A guide to genome engineering with programmable nucleases. Nat Rev Genet 15: 321–334. [DOI] [PubMed] [Google Scholar]
- Kay, MA, Glorioso, JC and Naldini, L (2001). Viral vectors for gene therapy: the art of turning infectious agents into vehicles of therapeutics. Nat Med 7: 33–40. [DOI] [PubMed] [Google Scholar]
- Gonçalves, MA (2005). A concise peer into the background, initial thoughts and practices of human gene therapy. Bioessays 27: 506–517. [DOI] [PubMed] [Google Scholar]
- Skipper, KA and Mikkelsen, JG (2015). Delivering the goods for genome engineering and editing. Hum Gene Ther 26: 486–497. [DOI] [PubMed] [Google Scholar]
- Biasco, L, Baricordi, C and Aiuti, A (2012). Retroviral integrations in gene therapy trials. Mol Ther 20: 709–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sterneckert, JL, Reinhardt, P and Schöler, HR (2014). Investigating human disease using stem cell models. Nat Rev Genet 15: 625–639. [DOI] [PubMed] [Google Scholar]
- Perez, EE, Wang, J, Miller, JC, Jouvenot, Y, Kim, KA, Liu, O et al. (2008). Establishment of HIV-1 resistance in CD4+ T cells by genome editing using zinc-finger nucleases. Nat Biotechnol 26: 808–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tebas, D, Stein, WW, Tang, I, Frank, SQ, Wang, G, Lee, SK et al. (2014). Gene editing of CCR5 in autologous CD4 T cells of persons infected with HIV. N Engl J Med 370: 901–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadelain, M, Papapetrou, EP and Bushman, FD (2012). Safe harbours for the integration of new DNA in the human genome. Nat Rev Cancer 12: 51–58. [DOI] [PubMed] [Google Scholar]
- Mátrai, J, Chuah, MK and VandenDriessche, T (2010). Recent advances in lentiviral vector development and applications. Mol Ther 18: 477–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naldini, L (2011). Ex vivo gene transfer and correction for cell-based therapies. Nat Rev Genet 12: 301–315. [DOI] [PubMed] [Google Scholar]
- Philpott, NJ and Thrasher, AJ (2007). Use of nonintegrating lentiviral vectors for gene therapy. Hum Gene Ther 18: 483–489. [DOI] [PubMed] [Google Scholar]
- Wanisch, K and Yáñez-Muñoz, RJ (2009). Integration-deficient lentiviral vectors: a slow coming of age. Mol Ther 17: 1316–1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cronin, J, Zhang, XY and Reiser, J (2005). Altering the tropism of lentiviral vectors through pseudotyping. Curr Gene Ther 5: 387–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lombardo, A, Genovese, P, Beausejour, CM, Colleoni, S, Lee, YL, Kim, KA et al. (2007). Gene editing in human stem cells using zinc finger nucleases and integrase-defective lentiviral vector delivery. Nat Biotechnol 25: 1298–1306. [DOI] [PubMed] [Google Scholar]
- Benabdallah, BF, Duval, A, Rousseau, J, Chapdelaine, P, Holmes, MC, Haddad, E et al. (2013). Targeted Gene Addition of Microdystrophin in Mice Skeletal Muscle via Human Myoblast Transplantation. Mol Ther Nucleic Acids 2: e68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holkers, M, Maggio, I, Henriques, SF, Janssen, JM, Cathomen, T and Gonçalves, MA (2014). Adenoviral vector DNA for accurate genome editing with engineered nucleases. Nat Methods 11: 1051–1057. [DOI] [PubMed] [Google Scholar]
- Coluccio, A, Miselli, F, Lombardo, A, Marconi, A, Malagoli Tagliazucchi, G, Gonçalves, MA et al. (2013). Targeted gene addition in human epithelial stem cells by zinc-finger nuclease-mediated homologous recombination. Mol Ther 21: 1695–1704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rio, P, Baños, R, Lombardo, A, Quintana-Bustamante, O, Alvarez, L, Garate, Z et al. (2014). Targeted gene therapy and cell reprogramming in Fanconi anemia. EMBO Mol Med 6: 835–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kass, EM and Jasin, M (2010). Collaboration and competition between DNA double-strand break repair pathways. FEBS Lett 584: 3703–3708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doulatov, S, Notta, F, Laurenti, E and Dick, JE (2012). Hematopoiesis: a human perspective. Cell Stem Cell 10: 120–136. [DOI] [PubMed] [Google Scholar]
- Genovese, P, Schiroli, G, Escobar, G, Di Tomaso, T, Firrito, C, Calabria, A et al. (2014). Targeted genome editing in human repopulating haematopoietic stem cells. Nature 510: 235–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoban, MD, Cost, GJ, Mendel, MC, Romero, Z, Kaufman, ML, Joglekar, AV et al. (2015). Correction of the sickle cell disease mutation in human hematopoietic stem/progenitor cells. Blood 125: 2597–2604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar, M, Keller, B, Makalou, N and Sutton, RE (2001). Systematic determination of the packaging limit of lentiviral vectors. Hum Gene Ther 12: 1893–1905. [DOI] [PubMed] [Google Scholar]
- Holkers, M, Maggio, I, Liu, J, Janssen, JM, Miselli, F, Mussolino, C et al. (2013). Differential integrity of TALE nuclease genes following adenoviral and lentiviral vector gene transfer into human cells. Nucleic Acids Res 41: e63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, L, Guell, M, Byrne, S, Yang, JL, De Los Angeles, A, Mali, P et al. (2013). Optimization of scarless human stem cell genome editing. Nucleic Acids Res 41: 9049–9061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joglekar, AV, Hollis, RP, Kuftinec, G, Senadheera, S, Chan, R and Kohn, DB (2013). Integrase-defective lentiviral vectors as a delivery platform for targeted modification of adenosine deaminase locus. Mol Ther 21: 1705–1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abarrategui-Pontes, C, Créneguy, A, Thinard, R, Fine, EJ, Thepenier, V, Fournier, le RL et al. (2014). Codon swapping of zinc finger nucleases confers expression in primary cells and in vivo from a single lentiviral vector. Curr Gene Ther 14: 365–376. [DOI] [PubMed] [Google Scholar]
- Kantor, B, Ma, H, Webster-Cyriaque, J, Monahan, PE and Kafri, T (2009). Epigenetic activation of unintegrated HIV-1 genomes by gut-associated short chain fatty acids and its implications for HIV infection. Proc Natl Acad Sci USA 106: 18786–18791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelascini, LP, Janssen, JM and Gonçalves, MA (2013). Histone deacetylase inhibition activates transgene expression from integration-defective lentiviral vectors in dividing and non-dividing cells. Hum Gene Ther 24: 78–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joglekar, AV, Stein, L, Ho, M, Hoban, MD, Hollis, RP and Kohn, DB (2014). Dissecting the mechanism of histone deacetylase inhibitors to enhance the activity of zinc finger nucleases delivered by integrase-defective lentiviral vectors. Hum Gene Ther 25: 599–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelascini, LP, Maggio, I, Liu, J, Holkers, M, Cathomen, T and Gonçalves, MA (2013). Histone deacetylase inhibition rescues gene knockout levels achieved with integrase-defective lentiviral vectors encoding zinc-finger nucleases. Hum Gene Ther Methods 24: 399–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonçalves, MA (2005). Adeno-associated virus: from defective virus to effective vector. Virol J 2: 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller, DG, Petek, LM and Russell, DW (2004). Adeno-associated virus vectors integrate at chromosome breakage sites. Nat Genet 36: 767–773. [DOI] [PubMed] [Google Scholar]
- McCarty, DM, Young, SM Jr and Samulski, RJ (2004). Integration of adeno-associated virus (AAV) and recombinant AAV vectors. Annu Rev Genet 38: 819–845. [DOI] [PubMed] [Google Scholar]
- Russell, DW and Hirata, RK (1998). Human gene targeting by viral vectors. Nat Genet 18: 325–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller, DG, Wang, PR, Petek, LM, Hirata, RK, Sands, MS and Russell, DW (2006). Gene targeting in vivo by adeno-associated virus vectors. Nat Biotechnol 24: 1022–1026. [DOI] [PubMed] [Google Scholar]
- Hirata, R, Chamberlain, J, Dong, R and Russell, DW (2002). Targeted transgene insertion into human chromosomes by adeno-associated virus vectors. Nat Biotechnol 20: 735–738. [DOI] [PubMed] [Google Scholar]
- Vasileva, A and Jessberger, R (2005). Precise hit: adeno-associated virus in gene targeting. Nat Rev Microbiol 3: 837–847. [DOI] [PubMed] [Google Scholar]
- Lukacsovich, T, Yang, D and Waldman, AS (1994). Repair of a specific double-strand break generated within a mammalian chromosome by yeast endonuclease I-SceI. Nucleic Acids Res 22: 5649–5657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouet, P, Smih, F and Jasin, M (1994). Expression of a site-specific endonuclease stimulates homologous recombination in mammalian cells. Proc Natl Acad Sci USA 91: 6064–6068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller, DG, Petek, LM and Russell, DW (2003). Human gene targeting by adeno-associated virus vectors is enhanced by DNA double-strand breaks. Mol Cell Biol 23: 3550–3557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porteus, MH, Cathomen, T, Weitzman, MD and Baltimore, D (2003). Efficient gene targeting mediated by adeno-associated virus and DNA double-strand breaks. Mol Cell Biol 23: 3558–3565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis, BL, Hirsch, ML, Porter, SN, Samulski, RJ and Porteus, MH (2013). Zinc-finger nuclease-mediated gene correction using single AAV vector transduction and enhancement by Food and Drug Administration-approved drugs. Gene Ther 20: 35–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Händel, EM, Gellhaus, K, Khan, K, Bednarski, C, Cornu, TI, Müller-Lerch, F et al. (2012). Versatile and efficient genome editing in human cells by combining zinc-finger nucleases with adeno-associated viral vectors. Hum Gene Ther 23: 321–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman, SH, Bobis-Wozowicz, S, Chatterjee, D, Gellhaus, K, Pars, K, Heilbronn, R et al. (2013). The nontoxic cell cycle modulator indirubin augments transduction of adeno-associated viral vectors and zinc-finger nuclease-mediated gene targeting. Hum Gene Ther 24: 67–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asuri, P, Bartel, MA, Vazin, T, Jang, JH, Wong, TB and Schaffer, DV (2012). Directed evolution of adeno-associated virus for enhanced gene delivery and gene targeting in human pluripotent stem cells. Mol Ther 20: 329–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotterman, MA and Schaffer, DV (2014). Engineering adeno-associated viruses for clinical gene therapy. Nat Rev Genet 15: 445–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregorevic, P, Blankinship, MJ, Allen, JM, Crawford, RW, Meuse, L, Miller, DG et al. (2004). Systemic delivery of genes to striated muscles using adeno-associated viral vectors. Nat Med 10: 828–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao, GP, Alvira, MR, Wang, L, Calcedo, R, Johnston, J and Wilson, JM (2002). Novel adeno-associated viruses from rhesus monkeys as vectors for human gene therapy. Proc Natl Acad Sci USA 99: 11854–11859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H, Haurigot, V, Doyon, Y, Li, T, Wong, SY, Bhagwat, AS et al. (2011). In vivo genome editing restores haemostasis in a mouse model of haemophilia. Nature 475: 217–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anguela, XM, Sharma, R, Doyon, Y, Miller, JC, Li, H, Haurigot, V et al. (2013). Robust ZFN-mediated genome editing in adult hemophilic mice. Blood 122: 3283–3287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senís, E, Fatouros, C, Große, S, Wiedtke, E, Niopek, D, Mueller, AK et al. (2014). CRISPR/Cas9-mediated genome engineering: an adeno-associated viral (AAV) vector toolbox. Biotechnol J 9: 1402–1412. [DOI] [PubMed] [Google Scholar]
- Swiech, L, Heidenreich, M, Banerjee, A, Habib, N, Li, Y, Trombetta, J et al. (2015). In vivo interrogation of gene function in the mammalian brain using CRISPR-Cas9. Nat Biotechnol 33: 102–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran, FA, Cong, L, Yan, WX, Scott, DA, Gootenberg, JS, Kriz, AJ et al. (2015). In vivo genome editing using Staphylococcus aureus Cas9. Nature 520: 186–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohbayashi, F, Balamotis, MA, Kishimoto, A, Aizawa, E, Diaz, A, Hasty, P et al. (2005). Correction of chromosomal mutation and random integration in embryonic stem cells with helper-dependent adenoviral vectors. Proc Natl Acad Sci USA 102: 13628–13633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, GH, Suzuki, K, Qu, J, Sancho-Martinez, I, Yi, F, Li, M et al. (2011). Targeted gene correction of laminopathy-associated LMNA mutations in patient-specific iPSCs. Cell Stem Cell 8: 688–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aizawa, E, Hirabayashi, Y, Iwanaga, Y, Suzuki, K, Sakurai, K, Shimoji, M et al. (2012). Efficient and accurate homologous recombination in hESCs and hiPSCs using helper-dependent adenoviral vectors. Mol Ther 20: 424–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shayakhmetov, DM, Papayannopoulou, T, Stamatoyannopoulos, G and Lieber, A (2000). Efficient gene transfer into human CD34(+) cells by a retargeted adenovirus vector. J Virol 74: 2567–2583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knaän-Shanzer, S, Van Der Velde, I, Havenga, MJ, Lemckert, AA, De Vries, AA and Valerio, D (2001). Highly efficient targeted transduction of undifferentiated human hematopoietic cells by adenoviral vectors displaying fiber knobs of subgroup B. Hum Gene Ther 12: 1989–2005. [DOI] [PubMed] [Google Scholar]
- Knaän-Shanzer, S, van de Watering, MJ, van der Velde, I, Gonçalves, MA, Valerio, D and de Vries, AA (2005). Endowing human adenovirus serotype 5 vectors with fiber domains of species B greatly enhances gene transfer into human mesenchymal stem cells. Stem Cells 23: 1598–1607. [DOI] [PubMed] [Google Scholar]
- Gonçalves, MA, de Vries, AA, Holkers, M, van de Watering, MJ, van der Velde, I, van Nierop, GP et al. (2006). Human mesenchymal stem cells ectopically expressing full-length dystrophin can complement Duchenne muscular dystrophy myotubes by cell fusion. Hum Mol Genet 15: 213–221. [DOI] [PubMed] [Google Scholar]
- Janssen, JM, Liu, J, Skokan, J, Gonçalves, MA and de Vries, AA (2013). Development of an AdEasy-based system to produce first- and second-generation adenoviral vectors with tropism for CAR- or CD46-positive cells. J Gene Med 15: 1–11. [DOI] [PubMed] [Google Scholar]
- Gonçalves, MA, Holkers, M, Cudré-Mauroux, C, van Nierop, GP, Knaän-Shanzer, S, van der Velde, I et al. (2006). Transduction of myogenic cells by retargeted dual high-capacity hybrid viral vectors: robust dystrophin synthesis in duchenne muscular dystrophy muscle cells. Mol Ther 13: 976–986. [DOI] [PubMed] [Google Scholar]
- Vellinga, J, Van der Heijdt, S and Hoeben, RC (2005). The adenovirus capsid: major progress in minor proteins. J Gen Virol 86(Pt 6): 1581–1588. [DOI] [PubMed] [Google Scholar]
- Kreppel, F and Kochanek, S (2008). Modification of adenovirus gene transfer vectors with synthetic polymers: a scientific review and technical guide. Mol Ther 16: 16–29. [DOI] [PubMed] [Google Scholar]
- Provasi, E, Genovese, P, Lombardo, A, Magnani, Z, Liu, PQ, Reik, A et al. (2012). Editing T cell specificity towards leukemia by zinc finger nucleases and lentiviral gene transfer. Nat Med 18: 807–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, L, Krymskaya, L, Wang, J, Henley, J, Rao, A, Cao, LF et al. (2013). Genomic editing of the HIV-1 coreceptor CCR5 in adult hematopoietic stem and progenitor cells using zinc finger nucleases. Mol Ther 21: 1259–1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saydaminova, K, Ye, X, Wang, H, Richter, M, Ho, M, Chen, H et al. (2015). Efficient genome editing in hematopoietic stem cells with helper-dependent Ad5/35 vectors expressing site-specific endonucleases under microRNA regulation. Mol Ther Methods Clin Dev 1: 14057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maggio, I, Holkers, M, Liu, J, Janssen, JM, Chen, X and Gonçalves, MA (2014). Adenoviral vector delivery of RNA-guided CRISPR/Cas9 nuclease complexes induces targeted mutagenesis in a diverse array of human cells. Sci Rep 4: 5105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holkers, M, Cathomen, T and Gonçalves, MA (2014). Construction and characterization of adenoviral vectors for the delivery of TALENs into human cells. Methods 69: 179–187. [DOI] [PubMed] [Google Scholar]
- Zhang, W, Chen, H, Zheng, X, Wang, D, Ji, H, Xia, H et al. (2014). Targeted genome correction by a single adenoviral vector simultaneously carrying an inducible zinc finger nuclease and a donor template. J Biotechnol 188C: 1–6. [DOI] [PubMed] [Google Scholar]
- Suzuki, K, Yu, C, Qu, J, Li, M, Yao, X, Yuan, T et al. (2014). Targeted gene correction minimally impacts whole-genome mutational load in human-disease-specific induced pluripotent stem cell clones. Cell Stem Cell 15: 31–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, S, Li, L, Kendrick, SL, Gerard, RD and Zhu, H (2014). TALEN-mediated somatic mutagenesis in murine models of cancer. Cancer Res 74: 5311–5321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maddalo, D, Manchado, E, Concepcion, CP, Bonetti, C, Vidigal, JA, Han, YC et al. (2014). In vivo engineering of oncogenic chromosomal rearrangements with the CRISPR/Cas9 system. Nature 516: 423–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng, R, Peng, J, Yan, Y, Cao, P, Wang, J, Qiu, C et al. (2014). Efficient gene editing in adult mouse livers via adenoviral delivery of CRISPR/Cas9. FEBS Lett 588: 3954–3958. [DOI] [PubMed] [Google Scholar]
- Ding, Q, Strong, A, Patel, KM, Ng, SL, Gosis, BS, Regan, SN et al. (2014). Permanent alteration of PCSK9 with in vivo CRISPR-Cas9 genome editing. Circ Res 115: 488–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonçalves, MA and de Vries, AA (2006). Adenovirus: from foe to friend. Rev Med Virol 16: 167–186. [DOI] [PubMed] [Google Scholar]
- Louis Jeune, V, Joergensen, JA, Hajjar, RJ and Weber, T (2013). Pre-existing anti-adeno-associated virus antibodies as a challenge in AAV gene therapy. Hum Gene Ther Methods 24: 59–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mingozzi, F and High, KA (2013). Immune responses to AAV vectors: overcoming barriers to successful gene therapy. Blood 122: 23–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotin, RM (2011). Large-scale recombinant adeno-associated virus production. Hum Mol Genet 20(R1): R2–R6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duffy, S, Shackelton, LA and Holmes, EC (2008). Rates of evolutionary change in viruses: patterns and determinants. Nat Rev Genet 9: 267–276. [DOI] [PubMed] [Google Scholar]
- Gabriel, R, Lombardo, A, Arens, A, Miller, JC, Genovese, P, Kaeppel, C et al. (2011). An unbiased genome-wide analysis of zinc-finger nuclease specificity. Nat Biotechnol 29: 816–823. [DOI] [PubMed] [Google Scholar]
- Seregin, SS and Amalfitano, A (2009). Overcoming pre-existing adenovirus immunity by genetic engineering of adenovirus-based vectors. Expert Opin Biol Ther 9: 1521–1531. [DOI] [PubMed] [Google Scholar]