

Abstract
Present adoptive immunotherapy strategies are based on the re-targeting of autologous T-cells to recognize tumor antigens. As T-cell properties may vary significantly between patients, this approach can result in significant variability in cell potency that may affect therapeutic outcome. More consistent results could be achieved by generating allogeneic cells from healthy donors. An impediment to such an approach is the endogenous T-cell receptors present on T-cells, which have the potential to direct dangerous off-tumor antihost reactivity. To address these limitations, we assessed the ability of three different TCR-α-targeted nucleases to disrupt T-cell receptor expression in primary human T-cells. We optimized the conditions for the delivery of each reagent and assessed off-target cleavage. The megaTAL and CRISPR/Cas9 reagents exhibited the highest disruption efficiency combined with low levels of toxicity and off-target cleavage, and we used them for a translatable manufacturing process to produce safe cellular substrates for next-generation immunotherapies.
Introduction
T-cell–based immunotherapies utilizing chimeric antigen receptor T-cells (CAR T-cells) hold tremendous potential for the treatment of malignancies and have shown encouraging activity in early clinical trials.1,2 However, CAR approaches have so far been implemented using autologous patient T-cells, rendering them cumbersome to generate for widespread or urgent use, and potentially leading to variable clinical outcomes due to differential functional properties of each patient's starting T-cell populations.
Potential approaches to address the variability of autologous approaches include the use of allogeneic T-cells from healthy donors whose functional properties can be carefully defined prior to administration to a patient. A drawback of this approach is that the endogenous T-cell receptor (TCR) present on therapeutic T cells may direct those cells to produce off-tumor reactivity in the form of graft versus host disease.
As a solution to TCR-driven host tissue reactivity, gene-editing nucleases have been employed in order to disrupt components of the TCR.3,4,5,6 The TCR α chain (TCRa) is encoded by a single TRAC gene and pairs with the TCR β chain encoded by two TCRB genes. As the TCR a/b dimer is essential for a fully functioning TCR complex, disruption of TCRa function has proven the simplest approach to elimination of TCR expression and undesired TCR-driven off-tumor recognition.
Four major classes of gene-editing proteins exist that share a common mode of action in binding a user-defined sequence of DNA and mediating a double-stranded DNA break (DSB). Zinc finger nucleases (ZFN) are heterodimeric arrays that colocalize at a target DNA site. They are comprised of individual finger subunits that bind DNA and are tethered to the FokI nuclease domain that cleaves DNA. Transcription activator-like effector nucleases (TALEN) are comprised of repeating units that bind DNA by virtue of a hypervariable two amino acid sequence (repeat variable diresidue) that governs DNA base recognition.7 Similar to ZFNS, TALENs function as dimeric proteins that are fused to the FokI endonuclease domain for DSB generation. Meganucleases (MN) are monomeric proteins with innate nuclease activity that are derived from bacterial homing endonucleases and engineered for a unique target site.8,9 The clustered regularly interspaced short palindromic repeats (CRISPR) and associated Cas9 nuclease platform comprised a small guide RNA (gRNA) transcript that contacts a target DNA sequence via Watson–Crick base pairing and the Cas9 nuclease that cleaves the DNA.10,11
All but CRISPR/Cas9 has been employed for disruption of the TCR complex by targeting either TRAC or TRBC of the TCR. ZFNs delivered as mRNA resulted in gene disruption rates of 27–37% for TRAC and 4–15% for TRBC.3 Delivering this same pair in either integrase-deficient lentiviral (IDLV) or adenoviral delivery vehicles resulted in TRAC disruption rates of 10% with IDLV and 50% with adenovirus, and ~5% with IDLV and ~40% with adenovirus for TRBC.4 TALEN mRNA delivery, at maximal rates, has resulted in ~60% TRAC and ~40% TRBC gene disruption.5,12 We (A.M.S.) have previously described a fusion protein of a meganuclease to TAL repeats (termed a megaTAL (MT)) and achieved editing rates for TRAC of >60% using a first-generation enzyme codelivered with the Trex2 gene product, an exonuclease that increases gene disruption rates.13
While each of these studies has contributed to the field, no evaluation has been performed that has focused on relative suitability of different nuclease reagents for implementation of TCR disruption in the context of a scalable translatable manufacturing process. Critical components of such an evaluation are reagent optimization, interrogation of off-target (OT) effects, and scalable manufacturing of gene-edited cell populations. Here, we have evaluated a TCRa-targeted TALEN, the previously described TCRa MT, and TCRa-targeted CRISPR/Cas9 nuclease reagents for editing of the highly clinically relevant TRAC gene target. We observed varying degrees of gene disruption among reagents generated from the three platforms, with a paucity of OT effects. The approach was further validated by generating TCR-null cells that expressed a CD19 CAR construct and a translationally compatible manufacturing process to demonstrate the capacity to generate and expand large numbers of engineered cells. This process is therefore a first step toward second-generation T-cells amenable to introduction of potency enhancements and/or as universal donor cells following introduction of CAR and TCR candidates being pursued for translational use.
Results
Gene-editing platform targeting, architecture, and delivery format
We generated nucleases from three different platforms each targeted to overlapping sequences within exon 1 of the TRAC gene (Figure 1a and Supplementary Figure S1). The MT site was nearly identical to the Streptococcus pyogenes (Sp) CRISPR/Cas9 (located on the opposite strand), and the TALEN site covered the same genomic locus (Figure 1a and Supplementary Figure S1) as the previously reported ZFN.3
Figure 1.

TRAC gene targeting and nuclease architecture. (a) Exon 1 of the TRAC locus with the positions of the nuclease target sites shown in relation to one another. (b) TALE recognition code and MT architecture. TALE repeat variable diresidue:DNA base recognition code is represented by colored bars. The amino acid sequences HD recognize DNA base C, NN binds G, NI interacts with A, and NG binds T. Eleven TALE repeat regions are fused to the meganuclease domain by a peptide linker (blue line), and the hybrid protein is termed a megaTAL. The central four bases common to the parental I-OnuI homing endonuclease from which the MT is derived are shown in blue lettering. (c) The dimeric TALEN proteins contain a deletion of 152 amino acids at the N-terminus and maintenance of 63 TAL amino acids at the C-terminus. The individual repeat variable diresidues each bind a single base of DNA and each half array is joined to a subunit of the FokI heterodimeric nuclease. (d) CRISPR/Cas9 architecture. A chimeric gRNA is shown and in purple is the constant portion of the molecule that interacts extensively with the Cas9 protein and the gene-specific component is shown in black letters. The gRNA contacts a target sequence (yellow boxed, black letters) in the context of a–NGG protospacer adjacent motif. The Cas9 protein contains two domains (HNH and RuvC) each responsible for the cleavage of a single strand of DNA. (e) Expression platforms. MT, Cas9, and TALEN mRNA was generated with either a T3 or T7 RNA polymerase promoter and the ‘+/-” refers to the presence or absence of a polyadenylation signal added in vitro. gRNA was produced as an RNA transcript (blue line), a circular plasmid with a human U6 polIII promoter, or a linear fragment generated by PCR containing the U6 promoter and full-length guide RNA sequences (black circle/line).
The hybrid MT protein contains the I-OnuI LAGLIDADG homing endonuclease architecture that maintains its “central 4” bp recognition domain8,9 but has been repurposed for TRAC gene binding and is fused to 11 TAL repeat regions (Figure 1b).6 The TALEN candidate target site contained a 5′ T nucleotide and 22 and 18 bp contacting repeat variable diresidues, respectively, with an optimized architecture containing a N-terminal deletion of 152 residues and 63 wild-type TAL sequences at the C-terminus (Figure 1c).14,15 The Sp CRISPR/Cas9 platform employs a guide RNA that contacts a target locus possessing a GN20GG sequence motif that serves to recruit the Cas9 protein to the target site where it induces a DSB (Figure 1d).10,11,16
The TALENs were assembled using the Golden Gate methodology into the RCIscript Goldy TALEN backbone that allows for in vitro mRNA production using T3 polymerase.15 Cas9 can be delivered as a protein or, like the MT, can be in vitro transcribed using a T7 promoter. We also tested the gRNA as DNA (plasmid or linear DNA expression fragment) or RNA, for their efficiency on gene editing (Figure 1e). These RNA species were either transcribed in vitro locally or acquired from a commercial vendor as an unmodified RNA or one whose first and last three bases contained a 2′-O-methyl (2′-OMe) phosphorothioate modification, respectively.
Evaluation of nuclease activity in transformed T lymphocytes
We first tested the MT candidate for activity in Jurkat cells by electroporating 1 µg of mRNA generated following the standard protocol for T7 RNA polymerase that includes addition of an exogenous polyadenylation signal by the E. coli poly (A) polymerase enzyme. Flow cytometry was employed to quantify TRAC gene disruption rates using an anti-CD3 antibody that recognizes the intact TCR complex. CD3 disruption rates, as evidenced by the lack of cell surface CD3 expression, were ~80% in MT-treated cells, while GFP-treated cells with rates of gene transfer of 95% showed >95% CD3 (Figure 2a).
Figure 2.

Nuclease comparison in Jurkat T-ALL cell line. Nucleic acids were delivered by electroporation into Jurkats and 7 days later, the amount of CD3 loss from the cell surface was determined by flow cytometry. (a) The first lane is the GFP transfection control with 95+% CD3 expression levels. Lane two shows the MT disruption rates. Lanes three and four are TALEN optimization conditions. TALEN mRNA was generated from a T3 polymerase promoter without (T3 alone) or with (T3 + pA) a exogenous polyadenylation signal. (b) Cas9 mRNA and protein with varying platforms of gRNA-editing rates. The first lane is GFP followed by a linear DNA fragment encoding the gRNA (Cas9, linear) or circularized gRNA plasmid (Cas9, plasmid) borne expression systems. Following that are a gRNA transcript (Cas9, RNA) produced locally by in vitro transcription and commercially synthesized gRNAs that are unmodified or modified with 2′O-Methyl (2′-OMe) bases that were delivered at doses of 5 or 10 μg by either complexing with Cas9 protein (RNP) or with Cas9 mRNA. Statistical comparisons were done using the Student's t-test. *,***, and **** represent P values (Student's t-test) of <0.05, <0.001, and <0.0001, respectively. Data are from four independent experiments.
The TALEN RCIscript Goldy backbone7,15 contains 5′ and 3′ UTR sequences derived from the Xenopus β-globin gene, included to increase translation efficiency.17 Utilizing the T3 polymerase-based system to generate mRNA resulted in TRAC gene disruption rates of ~30% (Figure 2a). The T3-based procedure does not include a polyadenylation step, and we hypothesized that including this may increase the stability and expression of TALEN mRNA. When we generated T3 mRNA and added a polyA using reagents from the T7 kit, the knockout rates increased to ~60% (Figure 2a). These data show an optimized RNA generation procedure for TALENs using the publically available Goldy backbone plasmid.
Next, we determined the optimal reagents for delivery of the CRISPR/Cas9 material via electroporation. Delivery of a plasmid bearing a U6 promoter driving expression of the gRNA versus a linear PCR product lacking the extraneous plasmid backbone sequences was assessed. Cas9 mRNA with a gRNA expressed from a plasmid resulted in ~90% CD3 disruption rates, while the linear gRNA, similar to TALENs, resulted in 60% disruption rates (Figure 2b). Because of the potential for ectopic DNA integration using plasmid delivery, we next delivered 1 µg of Cas9 mRNA and 500 ng of gRNA transcript that was produced by in vitro transcription in our laboratory, resulting in low gene-editing rates (Figure 2b). These data indicated that electroporated gRNA transcripts may not reach the nucleus at sufficient rates and/or are degraded by intracellular nucleases prior to complexing with the Cas9 protein.18 Recent advances in long synthetic RNA production allowed us to obtain high doses of the gRNA as an unmodified or nuclease-protected species by virtue of 2′-OMe–modified bases.19 The unmodified and modified gRNAs were employed at a dose of 5 µg using either Cas9 protein or Cas9 mRNA. Use of the nuclease-protected modified gRNA resulted in higher editing rates than unmodified guide using both the complexed ribonucleoprotein format as well as with all RNA delivery approach (Figure 2b). A representative fluorescence-activated cell sorting (FACS) plot of Figure 2 is shown as Supplementary Figure S2, and collectively, our results define the optimal delivery vehicles for each class of nuclease. MT functioned at high levels with in vitro transcribed, polyadenylated mRNA and TALEN activity could be enhanced with the addition of an exogenous polyadenylation signal. For CRISPR/Cas9, the delivery of the gRNA as a modified nuclease-resistant gRNA species yielded maximum editing rates. Using these data, the nucleases were assessed in primary human T lymphocytes for TRAC gene modification.
Nuclease activity in primary lymphocytes
Our experimental schema for performing gene editing in primary T-cells is shown in Figure 3a and employs an initial cellular activation step followed by electroporation-based gene transfer. We employed the optimized delivery platform for each candidate determined in the Jurkat studies: MT T7 polymerase generated polyadenylated mRNA, TALEN T3 mRNA with a polyA, and Cas9 mRNA or protein with a modified gRNA. In accordance with previous reports that show enhanced nuclease activity in conjunction with a transient “cold shock” at 30 °C incubation,20 we incubated nuclease-treated cells at 30 °C for 24 hours post gene transfer. The TALEN showed a significant decrease compared to their profile in Jurkat cells; moreover, significant toxicity was associated with TALEN expression in primary T-cells (Figure 3b). Due to their maximal activity in Jurkat cells, we also tested the MT and CRISPR/Cas9 reagents over several doses and achieved maximal MT-editing rates of ~75% with 2 µg of mRNA and ~85% with Cas9 mRNA and 5 µg of nuclease-protected modified gRNA (Figure 3b,c). Collectively, our data show that the highest levels of TRAC gene disruption was accomplished using the MT and CRISPR/Cas9 reagents, and a representative FACS plot for these data is shown in Supplementary Figure S3.
Figure 3.

Nuclease activity in primary T lymphocytes. (a) Experimental schema. T-cells were isolated from peripheral blood, cultured at a 3:1 CD3/CD28 bead:cell ratio followed by bead removal and electroporation with the indicated dose of nuclease. Cells were cultured transiently for 24 hours at 30 °C. At day 7, gene knockout efficiencies and cellular viability were assessed. (b) CD3 disruption rates using TALEN mRNA. (c) MT mRNA doses of 1, 2, 4, or 8 μg. (d) Cas9 RNP or mRNA with nuclease protected gRNA at 5 or 10 μg. Experiments were done using at least three unrelated donors in quadruplicate. Average CD3 disruption rate with SEM are shown. RNP, ribonucleoprotein. 2′OMe, 2′ O-methly RNA. Dashed lines indicate the demarcation of CD3 disruption rates on the left portion of the graph and the cellular viability on the right side.
Culture, expansion, phenotyping, and CAR transduction of T-cells
Toward achieving our goal of generating a pool of cells lacking the TCR, we developed a manufacturing process for CD3-negative cells in which we electroporated T-cells at a low, mid, and high scale with either MT or CRISPR/Cas9 reagents. The experimental schema is shown in Figure 4a and employs the use of IL2, IL-7, and IL-15 and 9 days of culture followed by negative selection using the column free EasySep system that resulted in a purity of 85% that was improved to 99% by repeating the procedure (Figure 4b). TRAC gene-edited cells were then maintained in IL-7 and IL-15 alone. At the low scale with an initial input (i.e., electroporation) of 200,000 cells, we reintroduced the TRAC gene mRNA by electroporation at day 15 and then restimulated cells with CD3/CD28 activation beads. Control cells cultured in IL-7 and IL-15 cytokines alone maintained a steady state with little proliferation (Figure 4c, no TRAC no stim). Addition of the CD3/CD28 beads to cells that did not receive TRAC mRNA resulted in a slight proliferative increase, most likely due to CD28 stimulation5 or activation of the <1% of CD3+ cells that remain after negative depletion (Figure 4b,c, no TRAC+stim). In contrast, transient recapitulation of the CD3 complex by TRAC mRNA electroporation and stimulation with CD3/28 activation beads allowed for a further fivefold expansion (Figure 4c, TRAC+stim), thus providing the rapid and efficient generation of TCR-deficient T-cells from a small starting population. Because reactivation of the cells may impact their phenotype, we pursued higher order scaling. The CRISPR/Cas9 system required 5 µg of modified gRNA and is a dose that may not be conducive for large-scale application, and therefore, we used an input cell number of 500,000 cells. Nuclease treatment, negative selection, and culture of CD3-null cells in homeostatic cytokines allowed for the recovery of 10 times the input number at day 15 of the procedure (Figure 4d). At a high scale with a 2 × 106 initial cell number using the MT nuclease, we obtained ~108 TRAC-null cells at day 15 (Figure 4e), and we utilized these cells for a comprehensive flow cytometry–based phenotyping panel (Figure 5). The majority of the cells were CD4+ (Figure 5a) that, importantly, retained effector function (Figure 5b) (e.g., secretion of IL-2, IFNg, GzmA, GzmB). Furthermore, the in vitro culture period needed for TRAC ablation did not affect differentiative state, as these cells did not demonstrate a senescent (e.g., loss of CD27, CD28, or CCR7; expression of CD57 or KLRG1) or exhausted (expression of PD-1, Tim-3, or CTLA4) phenotype (Figure 5c).21 To validate that the culture conditions and TCR disruption would allow for CAR transduction and antigen-specific killing, we used the schema shown in Figure 6a in which CD19-CAR lentiviral transduction is performed followed by gene editing. CD3 negative CD4+ or CD8+ CAR transduced cells showed the ability to form lytic granules in response to K562 cells expressing the CD19 antigen (Figure 6b). These data demonstrate that our engineering process does not drive the cells toward exhaustion and the cells maintain functionality when expressing an antigen-specific CAR.
Figure 4.

Expansion and scaling of CD3-negative cells. (a) Experimental schema. T-cells were harvested, activated, electroporated at 48 hours, and transiently cold shocked. During the first 9 days, the cells were grown in the presence of IL-2, IL-7, and IL-15. Following CD3 cell depletion, the cells were maintained in IL-7 and IL-15 until day 15 when they were enumerated and, if indicated, electroporated with TRAC mRNA and restimulated with CD3/CD28 beads in the presence of IL-2. (b) CD3-negative selection. Post-nuclease treated cells were depleted of CD3-positive cells by completion of one (left) or two (right) treatments with the EasySep procedure. (c) Reintroduction of TRAC mRNA. At day 0, 200,000 cells were treated with MT, and at day 15 post sorted, CD3-negative cells were cultured in IL-7 and IL-15 alone (labeled no TRAC no stim) or with a 3:1 CD3/CD28 bead: cell ratio (labeled no TRAC + stim). A third group received 1 μg of TRAC mRNA via electroporation followed by CD3/CD28 bead stimulation (labeled + TRAC + stim). Cell counts were performed at 2, 4, 6, and 8 days post gene transfer. (d) 500,000 cells were treated with 1 μg of Cas9 mRNA and 5 μg of modified gRNA or (e) 2 × 106 cells were treated with 10 μg of MT. Cells were grown in bulk to day 9 when they were enumerated. Negative depletion was performed, the CD3-null cells were replated in media with IL-7 and IL-15, and cultured to day 15 when they were counted. Experiments are from three donors and are the total from three experiments with four experimental replicates with averages and SEM shown. Arrow indicates CD3 depletion step with subsequent plating of CD3-null cells.
Figure 5.

T-cell phenotyping. Cells treated in a manner as those from Figure 4c were harvested at day 15 for flow cytometric analysis for (a) subset composition, (b) cytokines, and (c) surface exhaustion markers. The numbers of CD4 and CD8 cells positive for the indicated marker are shown in each column. In parentheses are the SEM from the individual experiments (n = 3 donors).
Figure 6.

CAR transduction and antitumor properties of gene-modified cells. (a) Experimental schema. T-cells were isolated, activated, and cultured in the presence of IL-2, IL-7, and IL-15. CD19 CAR lentiviral transduction was performed on day 0 with a self inactivating (SIN) lentiviral construct encoding the CD19R single-chain variable fragment with the CD28 transmembrane domain (CD28 tm), the 4-1BB costimulatory domain (41BB), the CD3-zeta costimulatory signaling domain (ζ), a self-cleaving T2A picornaviral peptide sequence (T2A), and a non–ligand-binding truncated epidermal growth factor receptor (tEGFR). CD3-negative depletion was performed on day 9 with culture overnight followed by incubation of T-cells with K562 or CD19 transgenic K562 cells. (b) Antitumor activity of engineered T-cells. Equal numbers of T-cells and K562 transgenic cells expressing human CD19 (CD19 Tg K562) or CD19-null K562 were incubated and analyzed for degranulation. Shown is a representative FACS analysis on cells from two donors for CD107a with gating on CD4 and CD8 subsets.
Unbiased genome level off-target assessment
Having optimized the conditions to achieve very high levels of on-target genome editing in primary human T-cells, we next turned to the potential for off-target activity at other sites across the genome. To achieve such an assessment in an unbiased manner at the genome level, we utilized the IDLV gene trapping, linear amplification–mediated PCR (LAM-PCR), and nonrestrictive LAM-PCR (nrLAM-PCR) methodology22,23 in Jurkat cells (Figure 7a). Insertion of the IDLV vector into the genome can occur in the presence of DSBs, permanently marking the locus and enabling mapping by LAM and nrLAM PCR (Figure 7a).22,24 DNA breaks can occur naturally in the absence of engineered nucleases due to normal cellular physiology25 or genomic fragile spots,26 and these non–nuclease-associated events, in the parlance of the assay nomenclature, are termed integration sites (IS).22 IDLVs delivered in tandem with a nuclease results in multiple insertions at either on-target or OT sites and are termed clustered integration sites (CLIS).22 To confirm on-target capture, we screened the Jurkat cells by PCR using an LTR and TRAC-specific PCR primer set that showed IDLV presence at the TRAC locus only in cells treated with a nuclease (Figure 7b). Subsequent LAM and nrLAM PCR with MiSeq deep sequencing were performed, and at least 178,000 sequence reads were analyzed for each treatment group (Tables 1 and 2). There were 1,491 IS identified for IDLV only treated cells and, importantly, these did not cluster at loci with sequence homology to the nuclease candidates (Tables 1 and 2). At the TRAC locus in nuclease-treated cells, we mapped 36, 15, and 31 CLIS for CRISPR/Cas9, TALEN, and MT, respectively (Table s 1 and 2). Surprisingly, we were not able to detect OT CLIS in the TALEN or CRISPR/Cas9-treated cells. In the MT treatment group, using saturating nuclease expression, we identified a number of CLIS that represented 10 putative OT sites, and these were analyzed further. The 10 OT sites and their proximity to genes are shown as a percentage of the total OT CLIS in Table 2 columns 3 and 4. Importantly, none of the OT CLIS occurred in exons and four occurred within introns of genes (DR1, KAT2B, PDE11A, and HIAT1 (Tables 1 and 2)). To ascertain whether these CLIS showed evidence of nuclease activity and repair by nonhomologous endjoining (NHEJ), we deep sequenced the TRAC and 10 OT target sites in Jurkat cells to quantify insertion/deletion (indel) frequency. These data showed that four of the sites had indel frequencies of greater than 1% at a resolution of at least 7,000 sequence reads (Supplementary Table S1). We next assessed whether these OT sites identified with indel frequencies >1% and/or those that occurred within introns in Jurkat cells would be an accurate indicator for OT effects in primary T-cells. A pure population of CD3-null cells (by virtue of treatment with 4 µg of MT; Figure 4b) were harvested and analyzed by Surveyor analysis that showed OT activity at the KAT2B locus (Figure 8a), some potential cleavage products for a minimal sequence homology site at GBP5 (Figure 8b), and no OT activity at the other loci (Figure 8c). To determine whether OT activity in the KAT2B intron disrupted splicing of the gene, we cloned and sequenced the cDNA and observed an intact open-reading frame, indicating that OT activity did not alter KAT2B splicing (Supplementary Figure S4). Overall, these data highlight the ability of the IDLV LAM PCR-based assay in Jurkat cells to identify OT sites relevant to gene editing processes performed in primary T-cells.
Figure 7.

Off-target genome mapping. (a) Experimental schema for IDLV gene trapping. Nuclease mRNA for MT and TALEN and Cas9 mRNA and gRNA plasmid were introduced into Jurkats followed by transduction with an IDLV expressing GFP and puromycin. The IDLV is integrated into loci where a DNA break has occurred. LAM and nRLAM PCR experimental schema. LTR priming results in linear fragments that are converted to double-stranded DNA products that are barcoded, deep sequenced, and interrogated against the genome for off-target sites. (b) TRAC IDLV gene trapping confirmation at on-target TRAC locus. A PCR using LTR- and TRAC-specific primers (red and blue arrows shown in a) revealed presence of the IDLV cargo at the TRAC locus for all of the nuclease reagents visualized by agarose gel.
Table 1. On- and off-target summary: TRAC CLIS.
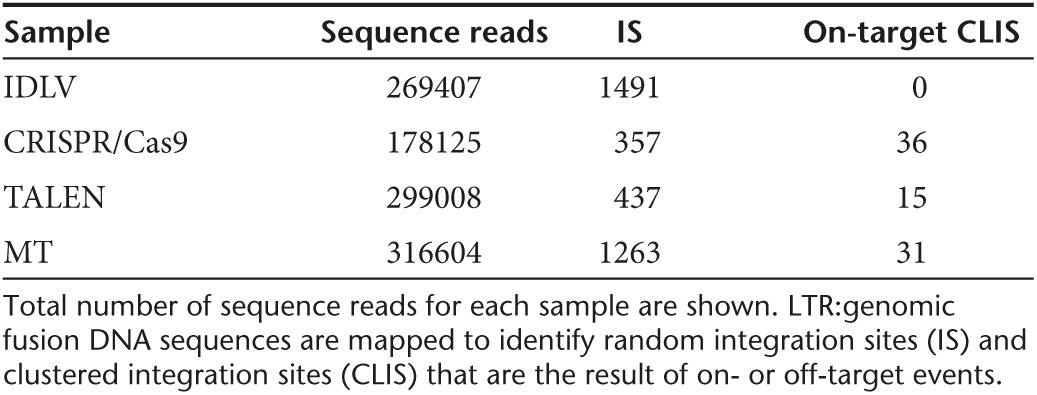
Table 2. MT OT sites with number of CLIS shown as a percentage of all recovered sites.
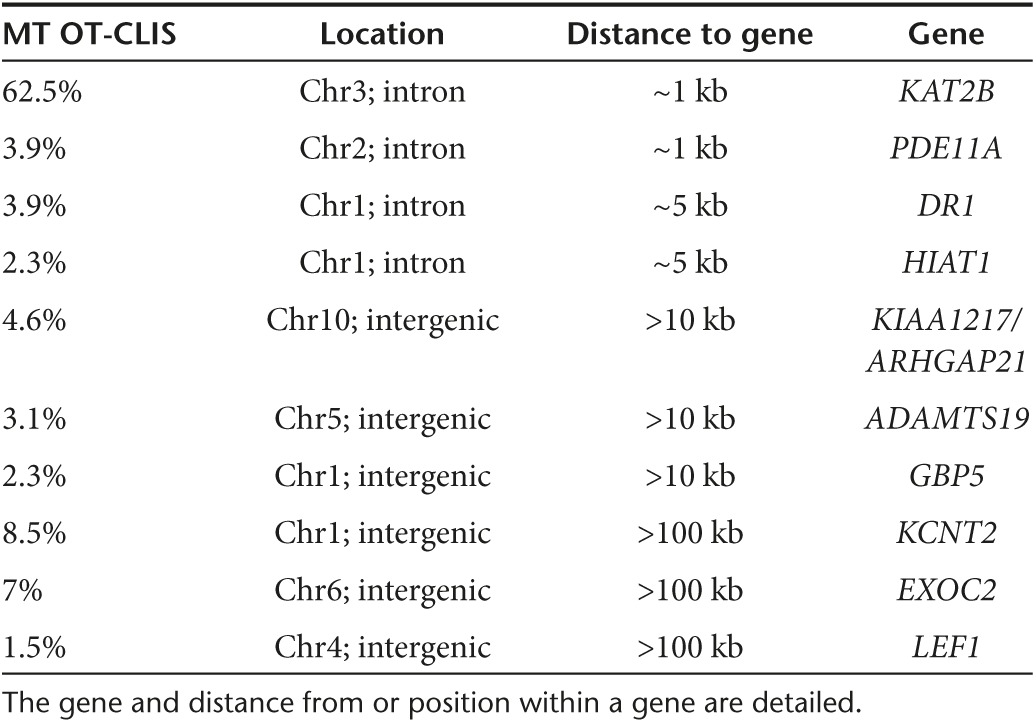
Figure 8.

Off-target assessment in primary T-cells. Target loci were amplified from 100% CD3-null cells and analyzed by the Surveyor assay for evidence of nuclease activity. (a) KAT2B Surveyor cleavage products and sequence alignment to TRAC. Cleavage products are indicated by arrows and quantitative gel analysis indicating is shown at bottom of gel. (b) GBP5 Surveyor image and genomic sequence in relation to TRAC. (c) PDE11A, DR1, HIAT1, KIAA1217, and EXOC2 Surveyor analysis that did not reveal MT OT cleavage. DR1, downregulator of transcription 1; EXOC2, exocyst complex component 2, TBP-binding (negative cofactor); GBP5, guanylate-binding protein 5; HIAT1, hippocampus abundant transcript 1; KAT2B, K(Lysine) acetyltransferase 2B; KIAA1217, sickle tail; PDE11A, phosphodiesterase 11A. Black arrows indicate cleavage products following Surveyor/CELII enzyme digest. The sequence alignments are in relation to the TAL repeat variable diresidues, the intervening spacer sequence separating the TAL from the meganuclease domain, and the meganuclease domain with the I-OnuI homing endonuclease “central four” ATTC sequence. Mismatches between the TRAC and OT sites are lighter shaded letters.
Discussion
At present, multiple clinical trials are open for patient enrolment or are in development that employ adoptive T-cell immunotherapies achieving tumor targeting based on single-chain variable fragment–based antibodies. In this rapidly evolving field, the elimination of the TCR by virtue of engineered nuclease-mediated gene disruption would represent a synergistic approach to amplifying the antitumor effects of T-cells while removing their propensity for initiating off-tumor reactivity. Here, we focused on developing a process for the generation of TCR-deficient T-cells through TRAC gene disruption. Within the scope of this study, we tested several nuclease candidates with overlapping target sequences in exon 1 of the TRAC gene (Figure 1), allowing us to assess and achieve TCRa gene disruption without confounding positional effects.
The TALEN pair we designed overlapped the previously reported ZFN site and when the TALEN dimers were delivered as mRNA gave similar, albeit slightly lower, rates of editing compared to ZFNs (Figure 3).3 The study by Torikai et al.3 using ZFNs showed TRAC disruption rates of ~30% using 2.5 µg of mRNA, and we observed ~10% using 1 µg (2.5 times less) of TALEN. We did not pursue higher doses of TALEN as we observed toxicity associated with TALEN expression (Figure 3b). Our TALEN disruption rates were significantly lower than that reported by Berdien et al.5 and Poirot et al.12 who achieved editing rates of >55% using mRNA. Interestingly, for each study, their mRNA generation procedure included the addition of a polyadenylation signal that we also found to be highly beneficial to increasing TALEN rates of activity in Jurkat cells (Figure 2). Of further interest is that the TALEN target sites of the study by Berdien et al.5 and Poirot et al.12 and our CRISPR/Cas9 and MT target sites were either directly overlapping or separated by <10 bp (Figure 1 and Supplementary Figure S1), and in each study, targeting this small stretch of the TRAC gene produced the highest rates of gene editing (Figures 2, 3, and 4).5,12
The CRISPR/Cas9 system is a highly flexible and user-friendly system that in our optimization studies showed the highest levels of TRAC gene disruption (Figures 2 and 3). We observed poor editing rates when employing gRNA transcripts produced by in vitro transcription in our laboratory. Of note, Agilent analysis confirmed full-length gRNA transcripts of high quality using our procedure; however, our yields were routinely <1 μg/μl, and we theorize that this was responsible for our observed poor editing rates. Indeed, using commercially synthesized gRNA at a dose of 5 μg/μl resulted in ~60+% CD3 disruption rates using Cas9 mRNA (Figures 2b and 3d). Complexing of the gRNA with Cas9 protein has proven to be an effective delivery strategy as well,27 and we observed efficient editing rates in Jurkat and primary T-cells using the ribonucleoprotein approach (Figures 2b and 3d). A recent study has further shown that that gRNAs containing modified RNA bases that protect from nuclease degradation results in higher editing rates for the IL2RG, HBB, and CCR5.19 In agreement with this, we achieved extremely high gene knockout rates in Jurkat and primary T-cells using nuclease-protected gRNA (Figures 2, 3, and 4). A potential limitation of this application, at present, is the high dose required and associated cost and specialized manufacturing process required to generate and modify RNA oligos of ≥100 bp in length. As a solution to this, we developed a strategy whereby low or midrange numbers of cells could be employed for CD3 disruption and expansion (Figure 4). We were able to isolate TCR/CD3-negative cells with >99% purity (Figure 4b)—an extremely high level of purity that we believe is important to achieve for clinical translation, as even a very small number of residual TCR-expressing cells may be capable of causing significant TCR-driven inflammation and tissue damage. We attempted to expand the purified cells in homeostatic cytokines IL-7 and IL-15 that have been shown to promote maintenance of CD3-null cells (Figure 4c)4,28; however, while the cells maintained a high degree of viability (>95%) in these conditions, they did not robustly expand (Figure 4c). To provide for expansion of the TCR/CD3-deficient cells to clinically relevant numbers, we reintroduced the TRAC gene mRNA into the cells via electroporation and stimulated them with CD3/CD28 beads, achieving a consistent and rapid fivefold further expansion (Figure 4c). These cells were archived and when re-thawed showed only a slight decrement in viability (viability = 99% pre-cryopreservation versus 85% post-thaw as assessed by trypan blue exclusion). A potential hurdle to this approach is that gene transfer of unactivated T-cells can be low (unpublished observations), and/or reexpansion may alter the cellular phenotype. As such, we utilized CRISPR/Cas9 with an input number of 500,000 cells and were able to obtain a 10-fold higher number of CD3-negative cells (Figure 4d). Using the MT reagent at a nuclease to cell dose of 2 µg of MT:100,000 cells with an input of 2 × 106 cells, we were able to obtain 108 TRAC-null cells (Figure 4e). The small, highly active, easily generated MT thus allows for high-level gene disruption at a comparatively low dose that does not require specialized modifications or contracted/specialized generation. Importantly, the cells modified in this manner retained functionality and did not exhibit a senescent phenotype (Figure 5). Moreover, when transduced with a CAR that recognizes the CD19 surface antigen, the cells were able to form lytic granules at a near-identical rate as reported by Berdien et al.,5 showing the ability of CAR and nuclease-modified cells to fully function (Figure 6). CAR T-cells, importantly, have shown an ability to expand in the presence of antigen that is provided via artificial antigen presenting and retain their ability to lyse cells.3 Our studies complement this approach and represent a manner in which cells can be generated without artificial antigen-presenting cells.
In this work, we employed a genome-level methodology to assess nuclease off-target effects for MT, TALEN, and CRISPR/Cas9 head to head for a single, clinically relevant gene (Figure 7). Nuclease specificity has been assessed by in silico and genomic level assays in relevant cells; however, the computer-based modeling algorithms can be limiting.29,30 A new methodology, Digenome-seq,31 employs an in vitro, DNA-cleavage methodology to produce Cas9-cleaved identical 5′ ends that are sequenced and computationally aligned.31 A potential limitation is that the purified, cell-free, DNA that is exposed to the nuclease may not faithfully represent the intracellular architecture in regard to structure and/or chromatin condensation. Our data using IDLV capture and LAM PCR showed the importance of this consideration as the Jurkat IDLV data for MT demonstrated that KAT2B was a bona fide OT site in Jurkat and primary T-cells, while the other Jurkat OT sites were absent or minimally prevalent in primary T-cells (Tables 1 and 2 and Figure 8). We hypothesize that this may be directly related to the aneuploid, and likely highly euchromatic, genome of Jurkat cells that would be more permissive to nuclease access to extended portions of the genome. We further hypothesize that along with nuclease activity and expression levels at or near saturation (Figure 2), the OT effects should be amplified in Jurkat cells making them ideally suited for OT analysis. A caveat to the IDLV approach and other cell-based assays like GUIDE-seq,32 and LAM PCR high-throughput, genome-wide, translocation sequencing (HTGTS)33 is that they all require viable cells for analysis. LAM PCR HTGTS utilizes LAM PCR to identify OT sites by virtue of translocation events that occur between on-target and OT cleavage ends.33 GUIDE-seq uses an oligonucleotide “bait” and single-tail adapter/tag PCR to generate fragments that are deep sequenced and mapped within the genome.32 As such, LAM IDLV, GUIDE-seq, or HTGTS will not be able to identify the full spectrum of OT events if they were to occur within genes that are essential for survival or that generate toxic translocation events. Thus, we cannot definitively rule out viability-incompatible OT events for TALEN and CRISPR/Cas9, particularly with the observed TALEN toxicity in primary T-cells (Figure 3b). Importantly, the MT and CRISPR/Cas9-treated primary T-cells showed high level TRAC gene disruption with correspondingly high viability and expansion (Figures 3 and 4). This indicates that the KAT2B OT site that represented 62.5% of the OT CLIS is well tolerated in T-cells. Moreover, the OT site is in the fifth intron of the gene, and the MT-induced indel did not perturb KAT2B splicing (Supplementary Figure S4). Furthermore, we note that OT effects with genome-editing reagents are not unprecedented, as evidenced by CCR5 ZFN therapies that have entered the clinic with significant rates of OT cleavage at the CCR2 locus.34 Moreover, our identification of this previously unknown OT site6 is important, as it will guide additional protein engineering of the homing endonuclease cleavage head domain to eliminate OT effects in second-generation megaTAL enzymes.22,34 Additionally, by design, our CAR construct also coexpresses a truncated, non–ligand-binding form of the epidermal growth factor gene (tEGFR) (Figure 6) that serves as a safety mechanism to allow for preferential eradication of CAR T-cells by administration of Erbitux. Thus, potential adverse events related to OT effects, lentiviral transduction, or cellular infusion can be tightly regulated by using a suicide construct.
This study along with the studies by Poirot et al.12 and Berdien et al.5 document the highest reported TRAC gene disruption rates using TALENs. In contrast, our TALEN pair exhibited high toxicity and low activity. Conversely, the MT and CRISPR/Cas9 reagents showed robust TRAC knockout rates. Ostensibly, the differential targeting sites and their associated accessibility to nucleases in regard to epigenetic factors (e.g., chromatin state) will factor in nuclease design and application for the most complete targeting strategy. An important component of this is the ability to assess OT events and to engineer, maintain, and expand the cells in a manner that is clinically viable.
Herein, we document the first genome-wide OT screen comparing candidates from three of the major classes of nucleases. The MT reagent exhibited OT effects that do not appear to disrupt gene expression and the MT's small size and high rate of editing at low dose makes it an attractive reagent. CRISPR/Cas9 is user friendly and allowed for the generation of a reagent that yielded high TRAC disruption rates and did not exhibit OT activity; however, large doses of synthetic RNA were required for which many laboratories will require commercial acquisition. These considerations are important, and we extended this to further optimize the engineering process by either expanding gene edited cells by transiently reexpressing TRAC and restimulating the cells or by controlling the initial number of cells as part of a scalable process. Under either condition, we were successful in obtaining adequate numbers of cells such that putative cell or dosing hurdles can be surmounted resulting in a cell population whose dose is within the range of those being used for T-cell therapy.35 Collectively, our data delineate an approach to determine the critical properties of gene-editing reagents for unique gene targets.
Materials and Methods
Nucleic acid constructs. The MT was constructed as previously described.6 The TALEN site was selected using the TAL effector Nucleotide Targeter 2.036 and were assembled using the GoldenGate7 methodology and cloned into the RCIscript Goldy backbone.15 CRISPR/Cas9 target site selection and in silico predictive off-target assessment were accomplished using the CRISPR Design Tool (crispr.mit.edu)37 and constructed using a synthetic G-block gene fragment (IDT DNA Technologies, Coralville, IA). Cas9 plasmid was obtained from Addgene (Cambridge, MA).11
RNA production. The MT plasmid was linearized with SwaI, TALEN plasmid linearization was accomplished with SacI, and Cas9 was linearized with NheI. TRAC (NC_000014.9) cDNA was a linear PCR fragment. One microgram of DNA was used for either T3 (TALEN) or T7 (MT, Cas9, TRAC) promoter generated mRNA in vitro transcription using the mMESSAGE Machine T3 or mMESSAGE Machine T7 Ultra kits (Thermo Fisher Scientific, Grand Island, NY). For T3 transcripts that were polyadenylated, the mMESSAGE Machine T3 procedure was followed by the polyadenylation step from the mMESSAGE Machine T7 Ultra kit. Guide RNA transcript was performed by amplifying a single-stranded oligonucleotide (target site underlined): (TRAC): AGCGCTCTCGTACAGAGTTGGCATTATAATACGACTCACTATAGGGGAGAATCAAAATCGGTGAATGTTTTAGAGCTAGA AATAGCAAGTTAAAATAAGGCTAGTCCGTTATCAACTTGAAAAAGTGGCACCGAGTCGGTGCTTTTTTT
using the primers: forward: AGCGCTCTCGTACAGAGTTGG and reverse: AAAAAAAGCACCGACTCGGTGCC with Q5 Hot Start High-Fidelity 2X Master Mix (New England BioLabs, Ipswich, MA). mRNA and gRNA transcripts were DNase treated and then purified using the RNeasy MinElute Cleanup Kit (Qiagen, Valencia, CA) with elution in water. Commercially synthesized gRNAs/Cas9 were obtained from TriLink Biotechnologies (San Diego, CA), and the sequences are: unmodified gRNA: 5′- GAG AAU CAA AAU CGG UGA AUG UUU UAG AGC UAG AAA UAG CAA GUU AAA AUA AGG CUA GUC CGU UAU CAA CUU GAA AAA GUG GCA CCG AGU CGG UGC UUU U -3. Modified: 5′- 2′OMe(G(ps)A(ps)G(ps)) AAU CAA AAU CGG UGA AUG UUU UAG AGC UAG AAA UAG CAA GUU AAA AUA AGG CUA GUC CGU UAU CAA CUU GAA AAA GUG GCA CCG AGU CGG UGC 2′OMe(U(ps)U(ps)U(ps) U -3′. 2′OMe=2′O)-methyl RNA and ps=phosphorothioate. Guide RNAs were reconstituted at 5 μg/μl in Neon Buffer T.
T-cell isolation and negative selection. Eight microliters of whole blood was obtained by phlebotomy and heparinized with 15 USP of heparin and T-cells were isolated using the RosetteSep Human T-cell enrichment cocktail (StemCell Technologies, Vancouver, BC). Negative selection was performed by subjecting the T-cells to either one or two rounds of depletion using the EasySep Human CD3 Positive Selection Kit (StemCell Technologies, Vancouver, British Columbia, Canada).
Cell activation and culture. T-cells were cultured in X-VIVO 20 media (Lonza, Basel, Switzerland) supplemented with 20% AB human serum (Thermo Fisher Scientific). For activation, the cells were further supplemented with recombinant IL-2 (Chiron, Emeryville, CA) at a concentration of 300 IU/ml. Anti-CD3/CD28 Dynabeads (Thermo Fisher Scientific) were added at a 3:1 bead:cell ratio, and cells were cultured at 37 °C and 5% CO2 at a concentration of 500,000/ml. Where indicated, recombinant human IL-7 or IL-15 (PeproTech, Rocky Hill, NJ) was included at 5 ng/ml.28
Gene transfer. Forty-eight hours after activation, the CD3/CD28 beads were magnetically removed, and the cells were cultured in the absence of beads for 6–12 hours. 200,000 cells were electroporated with the indicated amounts of nucleic acid. For the ribonucleoprotein conditions, either 5 or 10 μg of gRNA was incubated with 1 μg of Cas9 protein (Thermo Fisher Scientific) in a total volume of 5 μl for 15 minutes at room temperature. The 10 or 100 µl tip of the Neon Transfection System (Thermo Fisher Scientific, Grand Island, NY) was used for gene transfer with the following conditions: primary T-cells (in Buffer T): 1,400 V, 10 ms, 3 pulses. Jurkats (In Buffer R): 1,325 V, 10 ms, 3 pulses. Cells were plated in 200 µl of antibiotic-free media and cultured at 30 °C for 24 hours. Cells were maintained at 500,000/ml in media containing antibiotics after the initial plating in antibiotic free media.
Surveyor assay. Genomic DNA was isolated using the PureLink Genomic DNA Mini Kit (Thermo Fisher Scientific) and amplified with the primers listed below using AccuPrime Taq DNA polymerase, high fidelity (Thermo Fisher Scientific) as follows: 95 °C × 2 minutes, and 40 cycles of 95 °C × 40 seconds, 59 °C × 40 seconds, and 68 °C × 1 minute. Nine microliters of the PCR product was denatured/renatured with 1× PCR buffer and then incubated for 20 minutes with the Enhancer and Surveyor enzyme38 (IDT DNA Technologies). Products were then resolved on a 10% TBE PAGE gel (Thermo Fisher Scientific, Grand Island, NY) at 200V and stained with ethidium bromide.
Surveyor primers. The following surveyor primers were used:
TRAC F: ATCACGAGCAGCTGGTTTCT
TRAC R: CCCGTGTCATTCTCTGGACT
KAT2B F: AGCTCTAGGACGATGAGGGA
KAT2b R: CCTATCCCACCAGCATCCAA
PDE11A F: CAGCATCAGCACTGCACTTT
PDE11A R: TGCTGTAGGGCCTGGTTTAC
HIAT1 F: CTGCTTCCATCTTGCCTCAC
HIAT1 R: TATCCCCAGCACCCAGTAAG
DR1 F: TCCATTGTGTAATGAAGATGATTTG
DR1 R: TGCAACAAAATAGGAACACCTTT
KIAA1217 F: TGCCTATATTTTCTTCTGTGAGA
KIAA1217 R: AGACCTAGAATTGCCAAAACA
GBP5 F: TGGTCAAGTGTCGAGTTTGT
GBP5 R: ATCCAGTCACCTTCCACCAG
EXOC2 F: AGGCCATAGTCACCCAAACA
EXOC2 R: TTGGGTTCTTGGTCACGAAG
Flow cytometry. For TCR disruption rate assessment, Jurkats and primary T-cells were cultured for 7 or 9 days post electroporation, respectively, and analyzed for the presence of CD3 by flow cytometry. Similarly, phenotypic analysis was performed on day 9 T-cells using the antibodies indicated in Figure 5. For FACS, cells were washed with phosphate-buffered saline and stained at 1:100 dilution with the appropriate antibody (all antibodies obtained from eBiosciences, San Diego, CA) for 60 minutes at 4 °C in fluorescence-activated cell sorting buffer consisting of phosphate-buffered saline + 1% fetal bovine serum + 1 mmol/l ethylenediaminetetraacetic acid. Cells were then washed three times in fluorescence-activated cell sorting buffer and resuspended in fluorescence-activated cell sorting buffer containing Sytox Blue (Thermo Fisher Scientific) for dead cell exclusion. Acquisition was performed on a BD FACSCanto (BD Biosciences, San Jose, CA), and data was subsequently analyzed using FlowJo (Tree Star, Ashland, OR).
Off-target analysis, deep sequencing, and KAT2B cloning. Six samples for CRISPR/Cas9 and MT and four for TALEN were generated for OT analysis by electroporating Jurkat cells with 1 μg of MT or TALEN mRNA or 500 ng each of Cas9 mRNA and gRNA plasmid. The cells were immediately cocultured with a GFP-2A-puromycin IDLV at a multiplicity of infection of 5 with subsequent spinning transduction. Forty-eight hours post gene transfer, 400 ng/ml of puromycin was added, and the cells were expanded for 7 additional days followed by genomic DNA isolation. nrLAM PCR or LAM PCR with MseI or MluCI22,23,39 was performed, and deep sequencing data were generated with the Illumina MiSeq platform (San Diego, CA). The high-throughput insertion site analysis pipeline40 was utilized for vector trimming and genome alignment, and IS/CLIS identification was determined.
Total RNA was isolated from MT-treated CD3-negative cells that were sorted to purity using the RNeasy MinElute Cleanup Kit (Qiagen) and reverse transcribed with SuperScript Vilo (Thermo Fisher Scientific) and PCR amplified with primers: F:5′-GCCGAGGAGTCTTGTAAATGTAATGG-3′ and R:5′- TCACTTGTCAATTAATCCAGCTTCC-3′ at 98 °C × 2 minutes, 98 °C × 40 seconds, 58 °C × 40 seconds, 68 °C × 2 minutes and 30 seconds for 40 cycles. PCR products were TA cloned into the pCR-4-TOPO Vector and Sanger sequenced with M13 Forward, M13 Reverse, and KAT2B1 internal: 5′-TACCTCGGTACGAAACCACACAGG-3′. Sequence products were aligned to NM_003884.4.
Cytotoxicity assay. VSV-G pseudotyped CD19 CAR-tEGFR lentivirus was produced in 293T cells, and 50 ml of supernatant was concentrated with Lenti-X Concentrator, and the viral pellet resuspended in 2 ml of T-cell culture media. Five hundred microliters of viral supernatant was added to a retronectin-coated plate and spun for 3 hours at 1,200 rpm. 1 × 106 T-cells were added with CD3/CD28 beads and cultured for 48 hours and then treated with the appropriate nuclease. At day 15, equal numbers (100,000 each) of CD3-negative T-cells and K562 or K562-CD19 transgenic cells were incubated together for 1 hour at 37 °C. Monensin (eBiosciences, San Diego, CA) was then added to the cells followed by an additional 1-hour incubation. The cells were permeabilzed (Affymetrix, Santa Clara, CA), stained with anti-CD107a antibody (eBiosciences), and fixed (Affymetrix) for flow cytometric analysis.
Statistics. Student's t-test was utilized with P < 0.05 considered significant.
SUPPLEMENTARY MATERIAL Figure S1. Individual nuclease target sequences in the TRAC gene. Figure S2. Representative FACS plots for data used in Figure 2. Figure S3. Representative FACS plots for data in primary T-cells used in Figure 3. Figure S4. KAT2B indel and cDNA analysis in primary T-cells. Table S1. Deep sequencing of on and off target loci.
Acknowledgments
The authors are grateful to Kelsey Vigoren for editorial help with this article. The authors are also grateful to Jordan Jarjour and Alexander Astrakhan for helpful comments and edits. The authors appreciate funding support from The Children's Cancer Research Fund, the Lindahl Family & the Corrigan Family, and the Masonic Cancer Center Cancer Experimental Therapeutics Initiative. B.R.W. is supported by NIH T32- HL007062. J.T. is supported in part by R01 AR063070 and P01 CA065493. M.J.O. is supported by 8UL1TR000114-02. Research reported in this publication was supported by the National Center for Advancing Translational Sciences of the National Institutes of Health Award Number UL1TR000114 (M.J.O.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Supplementary Material
Individual nuclease target sequences in the TRAC gene.
Representative FACS plots for data used in Figure 2.
Representative FACS plots for data in primary T-cells used in Figure 3.
KAT2B indel and cDNA analysis in primary T-cells.
Deep sequencing of on and off target loci.
References
- Porter, DL, Levine, BL, Kalos, M, Bagg, A and June, CH (2011). Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med 365: 725–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudley, ME, Wunderlich, JR, Shelton, TE, Even, J and Rosenberg, SA (2003). Generation of tumor-infiltrating lymphocyte cultures for use in adoptive transfer therapy for melanoma patients. J Immunother 26: 332–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torikai, H, Reik, A, Liu, PQ, Zhou, Y, Zhang, L, Maiti, S et al. (2012). A foundation for universal T-cell based immunotherapy: T cells engineered to express a CD19-specific chimeric-antigen-receptor and eliminate expression of endogenous TCR. Blood 119: 5697–5705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Provasi, E, Genovese, P, Lombardo, A, Magnani, Z, Liu, PQ, Reik, A et al. (2012). Editing T cell specificity towards leukemia by zinc finger nucleases and lentiviral gene transfer. Nat Med 18: 807–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berdien, B, Mock, U, Atanackovic, D and Fehse, B (2014). TALEN-mediated editing of endogenous T-cell receptors facilitates efficient reprogramming of T lymphocytes by lentiviral gene transfer. Gene Ther 21: 539–548. [DOI] [PubMed] [Google Scholar]
- Boissel, S, Jarjour, J, Astrakhan, A, Adey, A, Gouble, A, Duchateau, P et al. (2014). megaTALs: a rare-cleaving nuclease architecture for therapeutic genome engineering. Nucleic Acids Res 42: 2591–2601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cermak, T, Doyle, EL, Christian, M, Wang, L, Zhang, Y, Schmidt, C et al. (2011). Efficient design and assembly of custom TALEN and other TAL effector-based constructs for DNA targeting. Nucleic Acids Res 39: e82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baxter, S, Lambert, AR, Kuhar, R, Jarjour, J, Kulshina, N, Parmeggiani, F et al. (2012). Engineering domain fusion chimeras from I-OnuI family LAGLIDADG homing endonucleases. Nucleic Acids Res 40: 7985–8000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoddard, BL (2011). Homing endonucleases: from microbial genetic invaders to reagents for targeted DNA modification. Structure 19: 7–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong, L, Ran, FA, Cox, D, Lin, S, Barretto, R, Habib, N et al. (2013). Multiplex genome engineering using CRISPR/Cas systems. Science 339: 819–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mali, P, Yang, L, Esvelt, KM, Aach, J, Guell, M, DiCarlo, JE et al. RNA-guided human genome engineering via Cas9. Science 339: 823–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poirot, L, Philip, B, Schiffer-Mannioui, C, Le Clerre, D, Chion-Sotinel, I, Derniame, S et al. (2015). Multiplex genome-edited T-cell manufacturing platform for “off-the-shelf” adoptive T-cell immunotherapies. Cancer Res 75: 3853–3864. [DOI] [PubMed] [Google Scholar]
- Certo, MT, Gwiazda, KS, Kuhar, R, Sather, B, Curinga, G, Mandt, T et al. (2012). Coupling endonucleases with DNA end-processing enzymes to drive gene disruption. Nat Methods 9: 973–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Pinera, P, Ousterout, DG, Brunger, JM, Farin, AM, Glass, KA, Guilak, F et al. (2013). Synergistic and tunable human gene activation by combinations of synthetic transcription factors. Nat Methods 10: 239–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson, DF, Tan, W, Lillico, SG, Stverakova, D, Proudfoot, C, Christian, M et al. (2012). Efficient TALEN-mediated gene knockout in livestock. Proc Natl Acad Sci U S A 109: 17382–17387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinek, M, East, A, Cheng, A, Lin, S, Ma, E and Doudna, J (2013). RNA-programmed genome editing in human cells. Elife 2: e00471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruys, V, Wathelet, M, Poupart, P, Contreras, R, Fiers, W, Content, J et al. (1987). The 3' untranslated region of the human interferon-beta mRNA has an inhibitory effect on translation. Proc Natl Acad Sci USA 84: 6030–6034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran, FA, Hsu, PD, Lin, CY, Gootenberg, JS, Konermann, S, Trevino, AE et al. (2013). Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell 154: 1380–1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendel, A, Bak, RO, Clark, JT, Kennedy, AB, Ryan, DE, Roy, S et al. (2015). Chemically modified guide RNAs enhance CRISPR-Cas genome editing in human primary cells. Nat Biotechnol 33: 985–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyon, Y, Choi, VM, Xia, DF, Vo, TD, Gregory, PD and Holmes, MC (2010). Transient cold shock enhances zinc-finger nuclease-mediated gene disruption. Nat Methods 7: 459–460. [DOI] [PubMed] [Google Scholar]
- Wherry, EJ and Kurachi, M (2015). Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol 15: 486–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabriel, R, Lombardo, A, Arens, A, Miller, JC, Genovese, P, Kaeppel, C et al. (2011). An unbiased genome-wide analysis of zinc-finger nuclease specificity. Nat Biotechnol 29: 816–823. [DOI] [PubMed] [Google Scholar]
- Paruzynski, A, Arens, A, Gabriel, R, Bartholomae, CC, Scholz, S, Wang, W et al. (2010). Genome-wide high-throughput integrome analyses by nrLAM-PCR and next-generation sequencing. Nat Protoc 5: 1379–1395. [DOI] [PubMed] [Google Scholar]
- Vink, CA, Gaspar, HB, Gabriel, R, Schmidt, M, McIvor, RS, Thrasher, AJ et al. (2009). Sleeping beauty transposition from nonintegrating lentivirus. Mol Ther 17: 1197–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieber, MR (2010). The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu Rev Biochem 79: 181–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma, K, Qiu, L, Mrasek, K, Zhang, J, Liehr, T, Quintana, LG et al. (2012). Common fragile sites: genomic hotspots of DNA damage and carcinogenesis. Int J Mol Sci 13: 11974–11999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, S, Kim, D, Cho, SW, Kim, J and Kim, JS (2014). Highly efficient RNA-guided genome editing in human cells via delivery of purified Cas9 ribonucleoproteins. Genome Res 24: 1012–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cieri, N, Camisa, B, Cocchiarella, F, Forcato, M, Oliveira, G, Provasi, E et al. (2013). IL-7 and IL-15 instruct the generation of human memory stem T cells from naive precursors. Blood 121: 573–584. [DOI] [PubMed] [Google Scholar]
- Hendel, A, Fine, EJ, Bao, G and Porteus, MH (2015). Quantifying on- and off-target genome editing. Trends Biotechnol 33: 132–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng, L, Blazar, B, High, K and Porteus, M (2011). Zinc fingers hit off target. Nat Med 17: 1192–1193. [DOI] [PubMed] [Google Scholar]
- Kim, D, Bae, S, Park, J, Kim, E, Kim, S, Yu, HR et al. (2015). Digenome-seq: genome-wide profiling of CRISPR-Cas9 off-target effects in human cells. Nat Methods 12: 237–43, 1 p following 243. [DOI] [PubMed] [Google Scholar]
- Tsai, SQ, Zheng, Z, Nguyen, NT, Liebers, M, Topkar, VV, Thapar, V et al. (2015). GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nat Biotechnol 33: 187–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frock, RL, Hu, J, Meyers, RM, Ho, YJ, Kii, E and Alt, FW (2015). Genome-wide detection of DNA double-stranded breaks induced by engineered nucleases. Nat Biotechnol 33: 179–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tebas, P, Stein, D, Tang, WW, Frank, I, Wang, SQ, Lee, G et al. (2014). Gene editing of CCR5 in autologous CD4 T cells of persons infected with HIV. N Engl J Med 370: 901–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maude, SL, Frey, N, Shaw, PA, Aplenc, R, Barrett, DM, Bunin, NJ et al. (2014). Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med 371: 1507–1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle, EL, Booher, NJ, Standage, DS, Voytas, DF, Brendel, VP, Vandyk, JK et al. (2012). TAL Effector-Nucleotide Targeter (TALE-NT) 2.0: tools for TAL effector design and target prediction. Nucleic Acids Res 40: W117–W122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu, PD, Scott, DA, Weinstein, JA, Ran, FA, Konermann, S, Agarwala, V et al. (2013). DNA targeting specificity of RNA-guided Cas9 nucleases. Nat Biotechnol 31: 827–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guschin, DY, Waite, AJ, Katibah, GE, Miller, JC, Holmes, MC and Rebar, EJ (2010). A rapid and general assay for monitoring endogenous gene modification. Methods Mol Biol 649: 247–256. [DOI] [PubMed] [Google Scholar]
- Osborn, MJ, Starker, CG, McElroy, AN, Webber, BR, Riddle, MJ, Xia, L et al. (2013). TALEN-based gene correction for epidermolysis bullosa. Mol Ther 21: 1151–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arens, A, Appelt, JU, Bartholomae, CC, Gabriel, R, Paruzynski, A, Gustafson, D et al. (2012). Bioinformatic clonality analysis of next-generation sequencing-derived viral vector integration sites. Hum Gene Ther Methods 23: 111–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Individual nuclease target sequences in the TRAC gene.
Representative FACS plots for data used in Figure 2.
Representative FACS plots for data in primary T-cells used in Figure 3.
KAT2B indel and cDNA analysis in primary T-cells.
Deep sequencing of on and off target loci.