

Abstract
RNA-guided nucleases (RGNs) based on the type II CRISPR-Cas9 system of Streptococcus pyogenes (Sp) have been widely used for genome editing in experimental models. However, the nontrivial level of off-target activity reported in several human cells may hamper clinical translation. RGN specificity depends on both the guide RNA (gRNA) and the protospacer adjacent motif (PAM) recognized by the Cas9 protein. We hypothesized that more stringent PAM requirements reduce the occurrence of off-target mutagenesis. To test this postulation, we generated RGNs based on two Streptococcus thermophilus (St) Cas9 proteins, which recognize longer PAMs, and performed a side-by-side comparison of the three RGN systems targeted to matching sites in two endogenous human loci, PRKDC and CARD11. Our results demonstrate that in samples with comparable on-target cleavage activities, significantly lower off-target mutagenesis was detected using St-based RGNs as compared to the standard Sp-RGNs. Moreover, similarly to SpCas9, the StCas9 proteins accepted truncated gRNAs, suggesting that the specificities of St-based RGNs can be further improved. In conclusion, our results show that Cas9 proteins with longer or more restrictive PAM requirements provide a safe alternative to SpCas9-based RGNs and hence a valuable option for future human gene therapy applications.
Introduction
Genome engineering using designer nucleases has become increasingly popular for applications ranging from basic research, biotechnology, disease modeling, to human gene therapy.1 Double-strand breaks induced by customized nucleases are harnessed to introduce precise and permanent genetic modifications by activating one of the two main DNA repair mechanisms in the target cells, the error-prone nonhomologous end-joining (NHEJ) pathway, or the accurate homology-directed repair.2,3 Traditionally, customizable endonucleases based on zinc finger nucleases (ZFNs) or transcriptional activator-like effector nucleases (TALENs) have been used to modify the genomes of several model organisms, including mouse, rat, zebrafish, and monkeys.4,5,6,7 A recent addition to the genome editing toolbox are engineered nucleases based on the clustered regularly interspaced short palindromic repeat (CRISPR)-CRISPR-associated protein 9 (Cas9) system, also known as RNA-guided nucleases (RGNs).8 They are based on the type II CRISPR-Cas9 system of bacteria and consist of the Cas9 endonuclease protein bound to a dual crRNA:tracrRNA9 molecule or a single RNA molecule, the so-called guide RNA (gRNA).10,11,12 Unlike ZFNs and TALENs, which rely on protein–DNA interactions for target site recognition, RGNs bind to their cognate targets by RNA–DNA base pairing between the ribonucleotide sequence located at the 5′-end of the gRNA, the spacer, and the complementary DNA target site, the protospacer.13,14 The second element conferring specificity is located immediately downstream of the protospacer. It is referred to as protospacer adjacent motif (PAM) that is directly recognized by the Cas9 protein.15,16,17 The PAM is essential for both acquisition of novel spacer sequences into the bacterial CRISPR locus and their orientation within the repeat array.15 If the PAM is not adjacent to a target site, Cas9-mediated cleavage is completely abolished. Cas9 proteins from different species have been isolated, each with distinct PAM requirements.18,19 For example, the commonly used Cas9 protein of Streptococcus pyogenes (SpCas9) recognizes a 5′-NGG trinucleotide, while Cas9 of other bacteria, like Staphylococcus aureus (SaCas9) or Neisseria meningitidis (NmCas9) bind to 5′-NNGRRT or 5′-NNNNGATT, respectively.20,21 Interestingly, as compared to SpCas9, these Cas9 proteins are smaller in size although they recognize longer PAMs. Given the large size of SpCas9, this aspect has important implications in terms of vectorization of the CRISPR-Cas9 system, as a smaller size allows easier viral packaging.21
Although SpCas9-based RGNs have been successfully applied in a wide range of organisms and cell types, several studies have reported high frequencies of off-target mutagenesis, particularly in investigations aimed at developing novel therapeutics for human disorders.22,23 Off-target cleavage can occur at DNA sequences which harbor up to five mismatches compared to the intended target site22; interestingly, most of the off-target sites are mismatched in the 5′-end of the target site,22 confirming the previously described knowledge that the 3′-end “seed” sequence is crucial for proper interaction of the CRISPR-Cas9 complex and the protospacer.24,25 A number of improvements have led to a substantial increase in the fidelity of the SpCas9 system. While truncated gRNAs were shown to reduce the tolerance to mismatched nucleotides, dimeric RGNs, which either consist of two Cas9-based nickases or two catalytically dead Cas9 (dCas9) proteins fused to the FokI nuclease domain, use expanded target sites.26,27,28 However, reduced on-target activities of these three systems can be a major drawback.26
We hypothesized that the PAM is another major determinant of CRISPR-Cas9 specificity. To assay whether a requirement for more stringent PAMs will improve overall specificity in the human genome, we focused on Cas9 proteins that recognize longer PAM sequences as compared to the SpCas910,14,20,21, such as Cas9 proteins derived from Streptococcus thermophilus (St). St1Cas9 and St3Cas9 are encoded by the St CRISPR1 or CRISPR3 loci and require 5′-NNAGAAW or 5′-NGGNG PAMs, respectively.29,30 Since these PAM sequences are different from the canonical Sp-PAM, StCas9-based RGNs will also expand the targeting range of the CRISPR-Cas technology. Our results demonstrate that expression of St1Cas9 and St3Cas9 proteins was well tolerated by human cells. Importantly, the cleavage activities of the StCas9 nucleases at two human loci was comparable to the established SpCas9-based RGNs and similar to previously reported cleavage frequencies for targeting endogenous human genes.10,31,32 Importantly, as compared to the Sp-derived CRISPR-Cas9 system, the cleavage activities at predicted off-target sites was considerably lower for both St1Cas9 and St3Cas9-based RGNs. These St CRISPR-Cas9 systems therefore represent a valid alternative for expanding the targeting range of RGNs in general, and for safe human genome editing in particular.
Results
Activity and cytotoxicity of St CRISPR-Cas9 systems in human cells
The most widely used type II CRISPR-Cas9 system derives from Streptococcus pyogenes (Sp). For DNA cleavage, the corresponding Cas9 protein depends on binding to its cognate 5′-NGG trinucleotide PAM. Since SpCas9-based RGNs have been associated with a certain degree of genotoxicity due to unwanted cleavage events at off-target sites in the target genome, we hypothesized that RGNs based on alternative Cas9 proteins with more restrictive PAM requirements may reduce the occurrence of off-target mutagenesis. To this end, we examined two known type II CRISPR-Cas9 systems from St CRISPR1 and CRISPR3 (St1 and St3)19 (Supplementary Table S1). To be able to compare these systems side-by-side and to minimize target site variability due to epigenetic modifications, the following considerations were taken into account: (i) the Cas9 open reading frames were codon optimized for expression in human cells and expressed from the same plasmid backbone. Comparable steady-state levels of the three Cas9 proteins were demonstrated by western blot (Supplementary Figure S1). (ii) The various gRNAs were expressed from the same U6 promoter containing plasmid backbone. (iii) The CRISPR-Cas9 systems were designed such that they recognized closely overlapping target sites in the human genome, which will minimize the variability that can arise by targeting different genes in distinct chromatin architectures. To this end, we screened the human genome to identify matching target sites that respect the PAM requirements of all three Cas9 orthologs. We have selected two sites that are located on chromosome 8 in either exon 73 of the PRKDC (protein kinase, DNA-activated, catalytic polypeptide) locus or exon 3 of CARD11 (caspase recruitment domain family, member 11), respectively (Figure 1a).
Figure 1.

Activity and cytotoxicity of RGNs. (a) Protospacers of RGNs and episomal GFP reporter. The target sites of the six RGNs used in this study targeting the human PRKDC and CARD11 genes are shown with the corresponding PAM sequences in bold. The indicated PRKDC or CARD11 sequences were cloned in between the start codon and a dsEGFP gene to create a GFP reporter plasmid used to monitor RGN activity. (b) RGN cleavage activity. HEK293T cells were co-transfected with plasmids expressing the RGNs targeted to PRKDC (upper panel) or CARD11 (lower panel), respectively, and the corresponding reporter plasmid. As a negative control, guide RNA (gRNA) plasmids were omitted or the cells were transfected with a plasmid expressing catalytically inactive Cas9 variants (dCas9). The graph shows the amount of GFP-positive cells determined by flow cytometry 48 hours posttransfection as compared to mock transfected cells (−; n = 3). As a positive control, cells were transfected with a GFP-specific zinc finger nuclease pair (ZFNGFP). Statistically significant differences are indicated by an asterisk (*P < 0.05). (c) Episomal cleavage of the reporter plasmid. The reporter plasmid DNA from cells transfected with PRKDC- or CARD11-specific RGNs was extracted and analyzed for evidence of NHEJ-induced indels via the mismatch-sensitive T7 endonuclease I (T7EI) assay. The arrow indicates the expected position of the digestion products. Numbers at the bottom show the average percentage of modified alleles (n = 3). (d) RGN-associated cytotoxicity. Cells were transfected with the same plasmids as in (c) together with a plasmid expressing mCherry. Cell survival is calculated as the fraction of mCherry-positive cells at day 5 as compared to day 2 posttransfection as measured by flow cytometry, relative to a control sample transfected with a plasmid expressing the nontoxic I-SceI meganuclease (n = 3). A toxic ZFN pair harboring a WT FokI cleavage domain was used as control (ZFNtox). Error bars indicate SEM. St1, St3, and Sp refer to RGNs based on Streptococcus thermophilus CRISPR1, CRISPR3, and Streptococcus pyogenes CRISPR-Cas9, respectively. NHEJ, nonhomologous end joining; PAM, protospacer adjacent motif; RGN, RNA-guided nuclease.
The cleavage activity of the six RGNs was initially assessed on an episomal reporter plasmid in which the PRKDC or CARD11 target sites were cloned between the translational start codon ATG and the open reading frame of a destabilized EGFP (dsEGFP) cassette.33 Upon transfection, GFP expression can be monitored by flow cytometry. In the presence of an active nuclease, cleavage of the target site will either induce plasmid degradation or lead to NHEJ-based indel mutations that disrupt the dsEGFP expression cassette. Thereby, the loss of GFP fluorescence is an indirect measure of nuclease activity. Expression of all six RGNs, targeted either to PRKDC or CARD11, led to a significant reduction of GFP-positive cells (Figure 1b). As an internal control, previously published ZFNs targeting GFP (ZFNGFP)34 were used. To confirm that the reduction of GFP-positive cells was a result of RGN cleavage activity, the corresponding catalytically inactive forms were generated and used as controls. To this end, the RuvC and HNH cleavage domains of St1Cas9 and St3Cas9 proteins were mutated based on sequence homology with the previously published catalytically “dead” SpCas9 (dCas9)35 and St3Cas9 nicking mutants36 (Supplementary Figure S3). A weak—but not significant—reduction in the number of GFP-positive cells was observed for all dCas9 proteins in the presence of the corresponding gRNA (Figure 1b). Since dCas9 proteins retain their ability to bind to the target DNA, we hypothesized that the reduction was due to their binding to the target site and subsequent interference with transcription, but not the result of residual cleavage activity. To confirm this hypothesis, plasmid DNA was extracted from cells transfected with plasmids encoding active Cas9 or dCas9 in combination with the corresponding gRNA. The extent of NHEJ-induced indel mutations at the episomal target sites was quantified using the mismatch-sensitive T7 endonuclease I (T7EI) assay (Figure 1c).37 T7EI activity was detected only in samples, in which catalytically active Cas9 proteins were expressed, confirming that St1-dCas9 and St3-dCas9 proteins have lost their DNA cleavage properties. To assess the cytotoxicity associated with overexpression of these RGNs, we employed a well-established cell survival assay.38 All six RGNs showed no significant cytotoxicity when compared to control cells transfected with the homing endonuclease I-SceI (Figure 1d). In contrast, cells overexpressing a previously described cytotoxic ZFN pair (ZFNtox)39 showed a significant decrease in cell survival. Taken together, these results show that the St-based RGNs display similar cleavage activities in an episomal reporter assay as the conventional SpCas9-based RGN and that these StCas9 proteins are well tolerated in human cells.
Since transfected reporter plasmids are present in multiple copies within the nucleus and their cleavage is not affected by the chromatin status, we next investigated the activities of the St-based RGNs in targeting endogenous loci in their native chromatin context. The chosen target sites in the PRKDC or CARD11 loci share a consensus motif that fulfills the PAM requirements of all three Cas9 proteins (Figure 1a). The PRKDC locus was efficiently targeted by both St3Cas9- and SpCas9-based RGNs with ~42% of alleles modified, while the St1Cas9-based RGN showed lower targeting efficiency (~23%; Figure 2a). All three CARD11-specific RGNs showed similar cleavage activities, ranging from 17 to 22% of targeted alleles (Figure 2b). The lower overall efficiency in targeting the CARD11 locus may be due to different chromatin status and distinct transcriptional activity but this aspect was not investigated further. In conclusion, our findings highlight that both St1- and St3-based RGNs are well tolerated in human cells and can be used to efficiently modify the human genome.
Figure 2.

Comparison of RGN cleavage activities at endogenous human loci. The cleavage activity of the six RGNs was assessed at two human loci, PRKDC (a) and CARD11 (b). HEK293T cells were transfected with the indicated RGNs and genomic DNA was extracted 3 days later. Upon PCR amplification of the target regions, the frequency of target disruption was assessed with the mismatch-sensitive T7EI assay. Catalytically inactive Cas9 (dCas9) variants were used as controls. Arrows indicate the positions of the expected cleavage products. Numbers at the bottom show the average percentage of modified alleles (n = 3). RGN, RNA-guided nuclease.
Tolerance of St CRISPR-Cas9 systems for truncated gRNA
In some publications, SpCas9-based RGNs were shown to induce mutations at unwanted off-target sites at high frequencies.22 This deleterious aspect could be ameliorated by using truncated gRNAs in which the protospacer was shortened from 20 to up to 17 nucleotides, leading to an up to 5,000-fold decrease in off-target activity.26 We thereby thought to investigate whether StCas9-based RGNs have similar tolerance for truncated gRNAs as SpCas9. We tested the cleavage efficiencies of the six RGNs in the presence of canonical gRNAs targeting 20 nucleotides long protospacers and truncated gRNA versions targeting 19, 18, and 17 nucleotide long protospacers, respectively (Figure 3a,b). Interestingly, both PRKDC- and CARD11-specific SpCas9-based RGNs showed a significant decrease in cleavage activities when the spacers were shortened to 18 or 17 nucleotides, regardless of the presence of a mismatched 5′-G. This suggests that this approach cannot be universally applied to every target site without impairing the on-target activity. Similarly, for both St1Cas9- and St3Cas9-based RGNs, the protospacer could be shortened to 19 nucleotides with only minor effects on the targeting efficiencies, while shorter gRNAs with protospacers of 18 or 17 nucleotides almost abolished their cleavage activities (Figure 3a,b). In conclusion, St1Cas9- and St3Cas9-based RGNs tolerate truncated gRNA similarly to SpCas9-based RGNs and, depending on the target site, this approach can potentially be used to reduce off-target mutagenesis.
Figure 3.

Tolerance of Cas9 orthologs for truncated gRNAs. The efficiencies of RGN-mediated cleavage using truncated gRNA was assessed at human PRKDC (a) and CARD11 (b) loci. Genomic DNA was extracted 3 days upon transfection of HEK293T cells and extent of cleavage was monitored with T7EI assay. Untreated cells were used as a control (−). The lengths of the truncated gRNAs are indicated above each lane and mismatched 5′-G are highlighted with asterisks (*). Arrows indicate the positions of the expected cleavage products. The average frequencies of modified alleles are indicated at the bottom of each panel (n = 3). RGN, RNA-guided nuclease.
Titration of St CRISPR-Cas9 activity
Alternative methods to reduce nuclease-mediated off-target mutagenesis rely on optimizing the concentration of active nucleases in the target cells. Indeed, protein delivery of TALENs, ZFNs, or RGNs has led to a substantial decrease of off-target effects for all three nuclease platforms, likely by limiting their intranuclear availability both temporally and quantitatively.40,41,42 To determine the minimal effective dose of the three RGNs systems, we transfected HEK293T cells with increasing amount of Cas9 and gRNA expression plasmids. All six tested RGNs showed a correlation between the amount of transfected plasmids and the cleavage activities (Figure 4a–c and Supplementary Figure S2). Of note, the activities of the PRKDC-specific SpCas9 and St3Cas9-based RGNs peaked with the second highest amount of transfected plasmids (Figure 4b), suggesting that higher amounts induce toxicity. In conclusion, fine-tuning the amount of CRISPR-Cas9 in the respective target cells may improve the ratio of on-target activity to nuclease-associated cytotoxicity.
Figure 4.

Titration of RGN activity. (a) Correlation between RGN dose and cleavage activity. HEK293T cells were transfected with decreasing amounts of plasmids expressing the indicated Cas9 and its corresponding gRNA in a fixed ratio of 1:3. The cleavage activities of the RGNs based on the indicated Cas9 orthologs in the different samples was assessed via T7EI assay. Shown are T7EI assays performed at the PRKDC locus. Arrows indicate the positions of the expected cleavage products. Numbers at the bottom show the average percentages of modified alleles (n = 3). (b,c) Quantification of RGN activity. The bar graphs show the percentages of modified alleles (n = 3) in correlation with the indicated plasmid amounts for PRKDC-specific (b) and CARD11-specific RGNs (c). Error bars indicate SEM. RGN, RNA-guided nuclease.
Specificity of St CRISPR-Cas9 systems
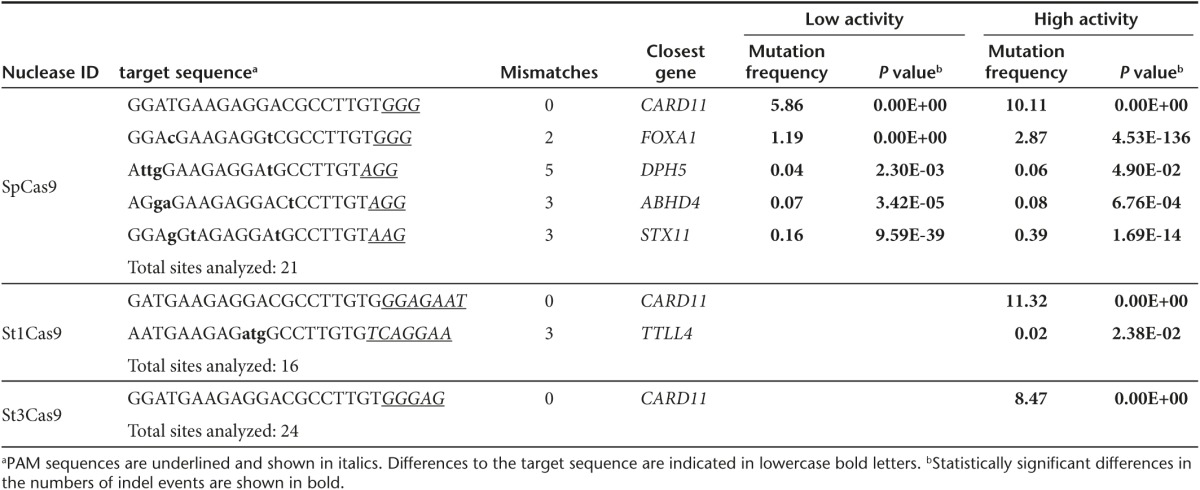
Having defined the requirements for efficient on-target cleavage activity, we sought to test our hypothesis that more stringent PAM requirements reduce the occurrence of off-target mutagenesis. We used COSMID, a web-based tool for identifying CRISPR-Cas9 off-target sites,43 to predict the top 24 off-target sites of all 6 RGNs, including sites with canonical and noncanonical PAMs. High-throughput sequence analysis confirmed comparable on-target activities of St3Cas9- and SpCas9-based RGNs at the PRKDC locus (Table 1 and Supplementary Table S2) and similar activities of all three RGNs at the CARD11 locus (Table 2 and Supplementary Table S3). When comparing off-target events, it became obvious that both the number of off-target sites, as well as the frequencies of off-target cleavage events, were reduced for St1Cas9- and St3Cas9-based systems as compared to the SpCas9-based RGNs. This observation was true for both PRKDC- and CARD11-specific RGNs. Of note, in cells transfected with the St3Cas9-based CRISPR-Cas9 system, we could only detect one off-target site for the PRKDC-specific RGN and no statistically significant signs of off-target mutagenesis for the CARD11-specific RGN (Tables 1 and 2; Supplementary Tables S2 and S3). In conclusion, these results strongly support the notion that implementing Cas9 proteins with more stringent PAM requirements result in RGNs with improved specificity.
Table 1. Analysis of the off-target cleavage activities of PRKDC-specific RGNs.
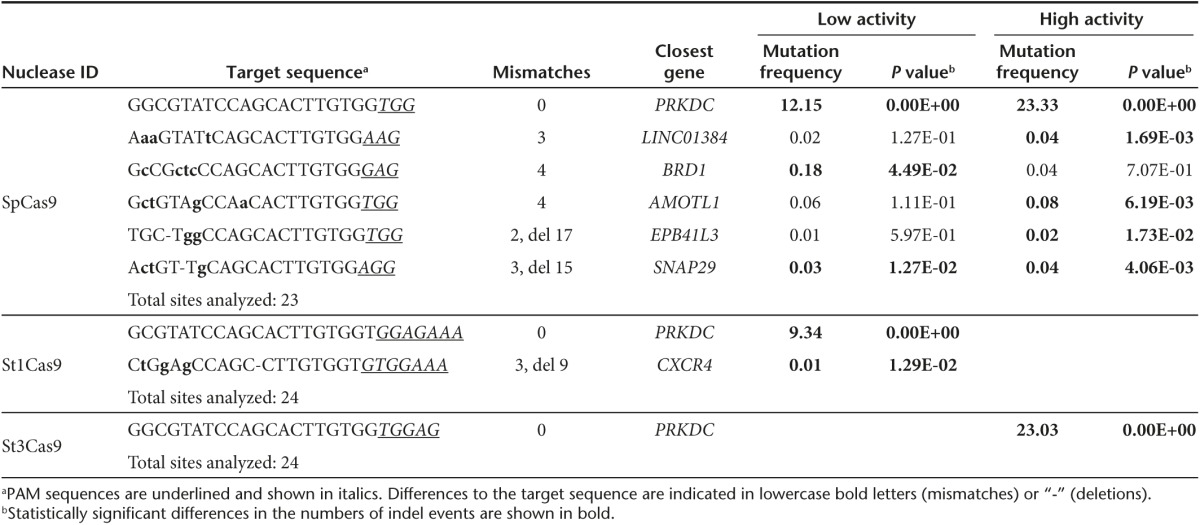
Table 2. Analysis of the off-target cleavage activities of CARD11-specific RGNs.
Discussion
Genome editing with designer nucleases has seen an unprecedented rapid development in the last 3 years.44,45 In particular, the introduction of RGNs has provided a novel and easily customizable tool for genome engineering, which has been adopted by researchers all over the world.46 RGNs are based on type II CRISPR-Cas9 system of bacteria that represent a primordial immune system helping these organisms to defend against invading exogenous DNA, such as bacteriophages.47 Target site recognition of CRISPR-Cas9 relies on Watson/Crick base pairing between the invading DNA target (the protospacer) and 20 nucleotides of an RNA molecule that guides the system (the spacer). Upon target site recognition, the invading DNA is cleaved by the Cas9 protein within the complex.35,36 This DNA–RNA interaction is flexible and tolerates a few mismatches, as well as insertions or deletions that lead to DNA or RNA bulges.48 Such mismatches are generally located within the 5′-proximal region of the target site22 while the 3′-end, the so-called “seed” sequence, is less permissive.24 While this flexibility is of paramount importance for bacteria, since it allows the host to recognize exogenous DNA harboring sequence alterations due to evolutionary pressure, it is a major concern when RGNs are applied for human genome editing. Indeed, DNA cleavage at off-target sites leads to unwanted mutagenesis and genotoxicity that could limit the widespread use of this technology, in particular for those applications aimed at clinical translation. In the last years, many improvements have been adopted to reduce RGN-mediated off-target mutagenesis, including the use of truncated gRNAs or of dimeric RNA-guided FokI nucleases or nickases.26,27,28 While most of these improvements act at the level of the DNA–RNA interaction, another element that plays a major role in target site recognition is the PAM, a short DNA sequence directly recognized by the Cas9 protein. Its presence is instrumental for target site cleavage because RGNs fail to cleave the DNA even if a perfectly matching target site is not followed by a proper PAM.49 Since the PAM has a major role in target (and consequently off-target) recognition, we hypothesized that implementing Cas9 proteins with more stringent PAM requirements may increase RGN specificity. To this end, we thoroughly compared the activities, cytotoxicities and specificities of RGNs based on Cas9 of different species that recognize progressively longer PAM sequences, ranging from the three nucleotide PAM of the commonly used SpCas9 (5′-NGG) to five and seven nucleotides long PAMs recognized by St3Cas9 (5′-NGGNG) and St1Cas9 (5′-NNAGAAW), respectively.
We demonstrated that St3Cas9-based RGNs are similarly active as the conventional SpCas9-based system, with NHEJ efficiencies of up to ~40% at PRKDC and ~17% at CARD11 in transfected HEK293T cells. The lower activity of St1Cas9-based RGN at both loci is probably due to different DNA binding affinities or a lower efficiency in assembling an active complex with its corresponding gRNA in human cells. Nevertheless, the cleavage frequencies obtained for the RGNs tested in this study are comparable with previously published reports that either used the same RGNs or RGN based on NmCas9. The minor observed differences could be attributed either to alternative delivery methods, to the use of a different gRNA scaffold10,20,32 or to the differential accessibility of the target loci. As a matter of fact, the CARD11 locus was consistently targeted with lower efficiencies by all three RGN systems, suggesting a more restrictive chromatin architecture. This aspect was not further investigated, as differences in target site accessibility has been observed previously.50
Interestingly, all RGNs showed significant activities at the lowest amount of transfected plasmids, suggesting that the RGN concentration can be fine-tuned in order to retain on-target cleavage activity but reduce off-target mutagenesis. Further experiments are needed to thoroughly address this aspect. While fine-tuning the concentration of RGNs within the target cells is important to decrease genotoxicity, destabilizing the DNA–RNA interface by shortening the gRNA was shown previously to reduce off-target mutagenesis.26 In our experiments, all six RGNs revealed similar tolerance towards truncated gRNAs, although the use of shorter gRNAs led to a considerable reduction in targeting efficiencies when the gRNAs was shortened by more than one nucleotide. These findings are in contrast with previously published data showing that canonical and truncated gRNA led to similar on-target activity.26 We assumed that these discrepancies are mostly due to differences in the impact of the chromatin environment of the investigated loci.
Here, we have compared for the first time the specificities of RGNs based on different Cas9 proteins. We have used COSMID, a publicly available web-based tool, to define the top 24 potential off-target sites of the six different RGNs in silico and analyzed the extent of off-target mutagenesis by high-throughput sequencing. Because the three Cas9 proteins have previously shown a certain tolerance for alternative PAMs,51,52,53 we have extended the in silico analysis to potential off-target sites flanked by noncanonical PAM sequences (see Materials and Methods for details). Our findings demonstrate that the PAM sequence is indeed a major determinant of RGN specificity. Hence, implementing the use of alternative Cas9 proteins that recognize more restrictive PAMs has a direct impact on their specificity. Even at low on-target activity, we identified several off-target sites for the PRKDC- and CARD11-specific RGNs based on the conventional SpCas9. In contrast, RGNs based on St1Cas9 and St3Cas9 showed few to no off-target mutagenesis under conditions for which on-target activity was as high as for the SpCas9-based counterpart. Taken together, our results support the notion that the use of Cas9 orthologs with longer and more restrictive PAMs may represent an easy strategy to improve RGN specificity.
Despite the advantage in terms of reduced genotoxicity, longer PAM sequences will decrease the targeting range, i.e., the overall frequency of potential target sites in a given genome. Given that many researchers express the gRNA from a U6 promoter containing plasmid that requires a 5′-G for efficient transcription, the occurrence of potential SpCas9-based RGN target sites (5′-GN19 NGG) is about 1 in 64 bp of a random DNA sequence. This number will decrease to about 1 in 256 for the St3Cas9-based system (5′-GN19 NGGNG). As the RGN targeting range can be increased by removing the 5′-end restriction using post-transcription enzymatic processing,28 expanding the RGN toolbox with several well-characterized CRISPR-Cas9 systems from different bacteria or archaea may represent a valuable alternative for future clinical translation. Our results extend the targeting range of CRISPR-Cas9 systems by complementing the genome editing toolbox with additional enzymes that have different PAM requirements and are less prone to off-targeting. Depending on the target site and specificity needs, one can choose a particular RGN system that matches these requirements. This will become particularly important when precise targeting is required, as for example for allele-specific targeting in order to discriminate between the disease causing mutation and the wild-type allele. Moreover, having available CRISPR-Cas9 systems of different origins expands the possibility of simultaneous orthogonal targeting.51,54 Additionally, implementing the use of Cas9 protein that are smaller in size than SpCas9, such as the St1Cas9, can be of particular interest for clinical translation. Many viral vector systems currently used in the clinic, such as vectors based on adeno-associated virus, have a low cargo capacity.21 Thus, implementing smaller and more specific CRISPR-Cas9 systems will further expand their use in human gene therapy.
Materials and Methods
Plasmids. The expressing plasmid pJDS246 containing a mammalian codon-optimized SpCas9 under the control of a Cytomegalovirus (CMV) promoter was a gift from J. Keith Joung (Addgene plasmid #43861). The genes encoding for St1Cas9 and St3Cas9 were optimized for mammalian codon usage with the online tool available at Integrated DNA Technologies (IDT, Coralville, Iowa) website and ordered as gBlocks from the same provider. To generate the plasmids p-CMV-St1Cas9 and p-CMV-St3Cas9 containing the St1Cas9 and St3Cas9 respectively, the gBlocks were assembled via Gibson Assembly into the pJDS246 digested with NotI and BamHI following standard procedures.55 The plasmid pMLM3636 containing the SpCas9-specific gRNA under the control of U6 promoter was a gift from J. Keith Joung (Addgene plasmid #43860). The catalytically inactive variant of SpCas9 (Sp-dCas9) was a gift from J. Keith Joung (Addgene plasmid #47754) and it was cloned into the pJDS246 backbone digested with NotI and BamHI. The catalytically inactive variants of St1Cas951 and St3Cas949 were created by cloning gBlocks containing inactivating mutations D9A and H599A for St1Cas9 and D10A and N870A for St3Cas9, respectively, in the plasmids p-CMV-St1Cas9 and p-CMV-St3Cas9 digested with NotI and BamHI. To substitute the 3× Flag-tag with a HA-Tag, two complimentary oligonucleotides were cloned into the plasmids containing St1Cas9 and St3Cas9 digested with XhoI and PstI. The resulting plasmids were named p-CMV-Sp-dCas9-HA, p-CMV-St1-dCas9-HA, and p-CMV-St3-dCas9-HA. Single gRNAs for St CRISPR151 and CRISPR331 were designed as previously published.35 The final St1Cas9- and St3Cas9-specific gRNAs contained a target recognition sequence (spacer) at the 5′-end, followed by 20 nucleotides of the crRNA repeat, 4 nucleotides of a self-folding hairpin loop (GTTA), an anti-repeat sequence complementary to the repeat region of the crRNA, and the remaining 3′ sequence of the tracrRNA with Rho-independent terminator (Supplementary Figure S4). The gRNA scaffolds for St1Cas9 and St3Cas9 were ordered as gBlocks (IDT) and cloned into the pMLM3636 vector digested with BsaAI and HindIII via Gibson Assembly. The resulting plasmids were named p-U6-gRNA_St1Cas9 and p-U6-RNA_St1Cas9, respectively. The 20 nucleotides spacers, as well as the truncated spacers, targeting PRKDC and CARD11 loci were cloned into the pMLM 3636 vectors previously described digested with BsmBI via cloning of two complementary oligonucleotides. The sequences of the oligonucleotides used for gRNA cloning are listed in Supplementary Table S4. The dsEGFP reporter constructs used in the episomal gene disruption assay were created by cloning two complementary oligonucleotides containing the protospacer sequences between the ATG and the 5′-end of a destabilized enhanced green fluorescent protein (dsEGFP) into the plasmid pLV.CMV.SceI.dsEGFP digested with PacI and AgeI. The resulting plasmids were named pLV.CMV.CARD11.dsEGFP and pLV.CMV.PRKDC.dsEGFP. All the plasmids were sequenced confirmed by Sanger sequencing (GATC Biotech, Constance, Germany) and can be obtained upon request.
Gene disruption and quantitative cell toxicity assay. HEK293T cells were cultured in Dulbecco's modified Eagle's medium (PAA, GE Healthcare, Munich, Germany) supplemented with 10% fetal calf serum (PAA), 100 U/ml penicillin (PAA), and 100 g/ml streptomycin (PAA). The cells were transfected using polyethylenimine (PEI) as previously described.33 For the episomal dsEGFP knockout assays, 120,000 HEK293T cells per well were seeded on 24-well plates. When ~80% confluent (~24 hours later), cells were PEI transfected with a DNA mixture containing 50 ng of pLV.CMV.CARD11.dsEGFP reporter plasmid, or with pLV.CMV.PRKDC.dsEGFP, respectively, 600 ng of Cas9-expressing plasmid, 200 ng of corresponding gRNA-expressing plasmid, 50 ng of an mCherry-expressing plasmid to normalize for transfection efficiency, and pUC118 to keep the total amount of transfected DNA constant at 1,250 ng. As negative controls, cells were transfected with 600 ng of the plasmid containing the corresponding catalytically inactive Cas9 (dCas9) and 200 ng of gRNA-expressing plasmid or with 600 ng of a Cas9-expressing plasmid without a gRNA-expressing plasmid. For the positive controls, the cells were transfected with 400 ng of a plasmid expressing previously characterized ZFNs targeted to EGFP.34 EGFP disruption was calculated by measuring the fractions of mCherry-positive and EGFP-negative cells by flow cytometry (BD Accuri C6; BD Biosciences, Heidelberg, Germany) 48 hours posttransfection. The survival rate of transfected cells was calculated by measuring the fractions of mCherry-positive cells by flow cytometry 5 and 2 days posttransfection, as previously described.38 As a control, cells were transfected with a pair of ZFNs that were previously shown to be toxic.39 For endogenous gene disruption assays using canonical or truncated gRNAs, HEK293T cells were seeded on a 24-well plates and 24 hours later PEI transfected with a DNA mix containing 600 ng of Cas9 and 200 ng of corresponding gRNA-expressing plasmids, respectively. To titrate the activity of SpCas9, St1Cas9, and St3Cas9, HEK293T cells, seeded as above, were PEI transfected with a DNA mix containing a fixed ratio of Cas9:gRNA encoding plasmids at a 3:1 ratio as indicated in Figure 4a.
T7 Endonuclease I assay. Gene disruption was assayed by T7 Endonuclease I (T7EI; New England Biolabs, Ipswich, MA), as previously described.33 Briefly, genomic DNA was extracted from transfected cells using the QIAamp DNA Mini Kit (Qiagen, Hilden, Germany) 3 days posttransfection. The genomic regions encompassing the RGN target sites in the human PRKDC and CARD11 loci were amplified via PCR using primers #2187 and #2188 for the PRKDC locus and primers #2397 and #2398 for the CARD11 locus, respectively (Supplementary Table S4). The products were cleaned up using the QIAquick PCR Purification Kit (Qiagen) and, upon denaturation and cool down, subjected to T7EI assay. For the T7EI assay on the episomal EGFP reporters shown in Figure 1c, the region encompassing the PRKDC and CARD11 target sites on the corresponding reporter plasmids were amplified via PCR using primers #13 and #77. PCR purification and T7EI assay were performed as described above. To determine the extent of cleavage the band intensities were estimated using Quantity One v.4.6.9 software (Bio-Rad, Hercules, CA) and the background subtracted; the amount of cleavage was then computed as percentage of the total intensities of the cleavage bands, divided by the sum of the intensity of cut and uncut bands. Values below the 5% detection limit of the assay56 were omitted.
Immunoblotting. Western blots were performed as described before.37 In brief, HEK293T cell were transfected as described above and protein lysates were prepared 48 hours posttransfections. The three different Cas9 proteins or β-actin were detected using anti-FLAG tag (1:1,000; Cell Signaling Technology, Danvers, MA) or anti-β-actin (1:1,000; Cell Signaling Technology) antibodies, respectively, and visualized with HRP-conjugated anti-rabbit secondary antibody (1:20,000; Dianova, Hamburg, Germany) using West Pico Chemiluminescence substrate (Thermo Fisher Scientific, Waltham, MA).
Off-target site identification and deep sequencing. Potential off-target sites for all Cas9 orthologs were identified using COSMID.43 The query input PAM sequences for SpCas9, St1Cas9, and St3Cas9 were NRG, NNRGRAN, and NGRNK, respectively. The sites were ranked using the weighed mismatch scoring output by COSMID with additional weight matrixes for insertions (+0.7), deletions (+0.51), and PAM sequences (+0.3) added to the scores of the genomic sites containing these features.
Deep sequencing to quantify RGNs activity at genomic loci. The identified genomic loci from mock and nuclease-treated cells were amplified via PCR using locus-specific primers that contained adapter sequences for priming a second round of PCR (Supplementary Tables S5 and S6). PCR reactions for each locus were performed independently for 40 cycles with annealing temperature at 60 °C. A second round of PCR amplification was performed for each individual amplicon using primers containing the adapter sequences from the first PCR, P5 and P7 adapters, and indexes. PCR products were purified using Agencourt AmPure XP (Beckman Coulter) according to manufacturer's protocol. All PCR products were pooled in an equimolar ratio, and subjected to 2 × 250 paired-end sequencing on the Illumina MiSeq platform. De-multiplexed paired-end reads from MiSeq reactions were filtered by an average Phred quality (Qscore) greater than 20 and merged into a longer single read from each pair with a minimum overlap of 30 nucleotides using Fast Length Adjustment of SHort reads (FLASH). Alignments to reference sequences were performed using Burrows-Wheeler Aligner for each barcode and percentage of insertions and deletions containing bases within a ±5 bp window of the predicted cut sites were quantified.
Statistical analysis. All data sets shown as bar graphs represent the average of at least three independent experiments in which each sample is transfected in triplicate. Error bars indicate SEM. Statistical significance was determined using a two-tailed, homoscedastic Student's t-test. To determine if the indel percentage at an off-target site from a RGN-treated sample is significant compared to a sample transfected with an empty vector, two-tailed P values were calculated using Fisher's exact test.
SUPPLEMENTARY MATERIAL Figure S1. Expression levels of Cas9 orthologs. Figure S2. Titration of CARD11-specific RGN activity. Figure S3. Amino acid sequences of Cas9 proteins used in this study. Figure S4. DNA sequences of gRNAs used in this study. Table S1. Cas9 orthologs used in this study. Table S2. Sequencing results for PRKDC-specific RGNs. Table S3. Sequencing results for CARD11-specific RGNs. Table S4. Oligonucleotides used in this study. Table S5. Sequencing primers for PRKDC-specific RGNs. Table S6. Sequencing primers for CARD11-specific RGNs.
Acknowledgments
The authors would like to thank Melina El Gaz, Nicola Bundschuh, Michael Rudnick, Nicolas Wyvekens, and Tautvydas Karvelis for technical assistance and experimental help, Eli J. Fine for bioinformatics support, and J. Keith Joung for plasmids pJDS246 and pMLM3636. This study was supported by the German Federal Ministry of Education and Research (BMBF 01EO0803 to C.M. and T.C.) and the National Institutes of Health (Nanomedicine Development Center Award PN2EY018244 to G.B.). C.M. and T.C. conceived and designed the study, interpreted the data, and wrote the manuscript; M.M., C.M.L., and T.J.C. collected and analyzed the data; G.G. and V.S. provided material and interpreted the data; C.M.L., T.J.C., T.H.D., and G.B. performed and interpreted next-generation sequencing experiments. T.J.C. is a full-time employee of CRISPR Therapeutics. T.C. is a consultant for TRACR Hematology.
Supplementary Material
References
- Carroll, D (2014). Genome engineering with targetable nucleases. Annu Rev Biochem 83: 409–439. [DOI] [PubMed] [Google Scholar]
- Lieber, MR (2010). The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu Rev Biochem 79: 181–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- San Filippo, J, Sung, P and Klein, H (2008). Mechanism of eukaryotic homologous recombination. Annu Rev Biochem 77: 229–257. [DOI] [PubMed] [Google Scholar]
- Liu, H, Chen, Y, Niu, Y, Zhang, K, Kang, Y, Ge, W et al. (2014). TALEN-mediated gene mutagenesis in rhesus and cynomolgus monkeys. Cell Stem Cell 14: 323–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung, YH, Baek, IJ, Kim, DH, Jeon, J, Lee, J, Lee, K et al. (2013). Knockout mice created by TALEN-mediated gene targeting. Nat Biotechnol 31: 23–24. [DOI] [PubMed] [Google Scholar]
- Tesson, L, Usal, C, Ménoret, S, Leung, E, Niles, BJ, Remy, S et al. (2011). Knockout rats generated by embryo microinjection of TALENs. Nat Biotechnol 29: 695–696. [DOI] [PubMed] [Google Scholar]
- Zu, Y, Tong, X, Wang, Z, Liu, D, Pan, R, Li, Z et al. (2013). TALEN-mediated precise genome modification by homologous recombination in zebrafish. Nat Methods 10: 329–331. [DOI] [PubMed] [Google Scholar]
- Mussolino, C and Cathomen, T (2013). RNA guides genome engineering. Nat Biotechnol 31: 208–209. [DOI] [PubMed] [Google Scholar]
- Deltcheva, E, Chylinski, K, Sharma, CM, Gonzales, K, Chao, Y, Pirzada, ZA et al. (2011). CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature 471: 602–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong, L, Ran, FA, Cox, D, Lin, S, Barretto, R, Habib, N et al. (2013). Multiplex genome engineering using CRISPR/Cas systems. Science 339: 819–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang, W, Bikard, D, Cox, D, Zhang, F and Marraffini, LA (2013). RNA-guided editing of bacterial genomes using CRISPR-Cas systems. Nat Biotechnol 31: 233–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mali, P, Yang, L, Esvelt, KM, Aach, J, Guell, M, DiCarlo, JE et al. (2013). RNA-guided human genome engineering via Cas9. Science 339: 823–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deveau, H, Barrangou, R, Garneau, JE, Labonté, J, Fremaux, C, Boyaval, P et al. (2008). Phage response to CRISPR-encoded resistance in Streptococcus thermophilus. J Bacteriol 190: 1390–1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvath, P, Romero, DA, Coûté-Monvoisin, AC, Richards, M, Deveau, H, Moineau, S et al. (2008). Diversity, activity, and evolution of CRISPR loci in Streptococcus thermophilus. J Bacteriol 190: 1401–1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mojica, FJ, Díez-Villaseñor, C, García-Martínez, J and Almendros, C (2009). Short motif sequences determine the targets of the prokaryotic CRISPR defence system. Microbiology 155 (Pt 3): 733–740. [DOI] [PubMed] [Google Scholar]
- Sternberg, SH, Redding, S, Jinek, M, Greene, EC and Doudna, JA (2014). DNA interrogation by the CRISPR RNA-guided endonuclease Cas9. Nature 507: 62–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders, C, Niewoehner, O, Duerst, A and Jinek, M (2014). Structural basis of PAM-dependent target DNA recognition by the Cas9 endonuclease. Nature 513: 569–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasiunas, G, Sinkunas, T and Siksnys, V (2014). Molecular mechanisms of CRISPR-mediated microbial immunity. Cell Mol Life Sci 71: 449–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mussolino, C, Mlambo, T and Cathomen, T (2015). Proven and novel strategies for efficient editing of the human genome. Curr Opinion Pharmacol 24: 105–112. [DOI] [PubMed] [Google Scholar]
- Hou, Z, Zhang, Y, Propson, NE, Howden, SE, Chu, LF, Sontheimer, EJ et al. (2013). Efficient genome engineering in human pluripotent stem cells using Cas9 from Neisseria meningitidis. Proc Natl Acad Sci USA 110: 15644–15649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran, FA, Cong, L, Yan, WX, Scott, DA, Gootenberg, JS, Kriz, AJ et al. (2015). In vivo genome editing using Staphylococcus aureus Cas9. Nature 520: 186–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu, Y, Foden, JA, Khayter, C, Maeder, ML, Reyon, D, Joung, JK et al. (2013). High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat Biotechnol 31: 822–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koo, T, Lee, J and Kim, JS (2015). Measuring and reducing off-target activities of programmable nucleases including CRISPR-Cas9. Mol Cells 38: 475–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semenova, E, Jore, MM, Datsenko, KA, Semenova, A, Westra, ER, Wanner, B et al. (2011). Interference by clustered regularly interspaced short palindromic repeat (CRISPR) RNA is governed by a seed sequence. Proc Natl Acad Sci USA 108: 10098–10103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiedenheft, B, Lander, GC, Zhou, K, Jore, MM, Brouns, SJ, van der Oost, J et al. (2011). Structures of the RNA-guided surveillance complex from a bacterial immune system. Nature 477: 486–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu, Y, Sander, JD, Reyon, D, Cascio, VM and Joung, JK (2014). Improving CRISPR-Cas nuclease specificity using truncated guide RNAs. Nat Biotechnol 32: 279–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen, B, Zhang, W, Zhang, J, Zhou, J, Wang, J, Chen, L et al. (2014). Efficient genome modification by CRISPR-Cas9 nickase with minimal off-target effects. Nat Methods 11: 399–402. [DOI] [PubMed] [Google Scholar]
- Tsai, SQ, Wyvekens, N, Khayter, C, Foden, JA, Thapar, V, Reyon, D et al. (2014). Dimeric CRISPR RNA-guided FokI nucleases for highly specific genome editing. Nat Biotechnol 32: 569–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garneau, JE, Dupuis, MÈ, Villion, M, Romero, DA, Barrangou, R, Boyaval, P et al. (2010). The CRISPR/Cas bacterial immune system cleaves bacteriophage and plasmid DNA. Nature 468: 67–71. [DOI] [PubMed] [Google Scholar]
- Magadán, AH, Dupuis, MÈ, Villion, M and Moineau, S (2012). Cleavage of phage DNA by the Streptococcus thermophilus CRISPR3-Cas system. PLoS One 7: e40913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glemzaite, M, Balciunaite, E, Karvelis, T, Gasiunas, G, Grusyte, MM, Alzbutas, G et al. (2015). Targeted gene editing by transfection of in vitro reconstituted Streptococcus thermophilus Cas9 nuclease complex. RNA Biol 12: 1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, K, Ren, C, Liu, Z, Zhang, T, Zhang, T, Li, D et al. (2015). Efficient genome engineering in eukaryotes using Cas9 from Streptococcus thermophilus. Cell Mol Life Sci 72: 383–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mussolino, C, Morbitzer, R, Lütge, F, Dannemann, N, Lahaye, T and Cathomen, T (2011). A novel TALE nuclease scaffold enables high genome editing activity in combination with low toxicity. Nucleic Acids Res 39: 9283–9293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeder, ML, Thibodeau-Beganny, S, Osiak, A, Wright, DA, Anthony, RM, Eichtinger, M et al. (2008). Rapid “open-source” engineering of customized zinc-finger nucleases for highly efficient gene modification. Mol Cell 31: 294–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinek, M, Chylinski, K, Fonfara, I, Hauer, M, Doudna, JA and Charpentier, E (2012). A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337: 816–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasiunas, G, Barrangou, R, Horvath, P and Siksnys, V (2012). Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc Natl Acad Sci USA 109: E2579–E2586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mussolino, C, Alzubi, J, Fine, EJ, Morbitzer, R, Cradick, TJ, Lahaye, T et al. (2014). TALENs facilitate targeted genome editing in human cells with high specificity and low cytotoxicity. Nucleic Acids Res 42: 6762–6773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornu, TI, Thibodeau-Beganny, S, Guhl, E, Alwin, S, Eichtinger, M, Joung, JK et al. (2008). DNA-binding specificity is a major determinant of the activity and toxicity of zinc-finger nucleases. Mol Ther 16: 352–358. [DOI] [PubMed] [Google Scholar]
- Alwin, S, Gere, MB, Guhl, E, Effertz, K, Barbas, CF 3rd, Segal, DJ et al. (2005). Custom zinc-finger nucleases for use in human cells. Mol Ther 12: 610–617. [DOI] [PubMed] [Google Scholar]
- Liu, J, Gaj, T, Patterson, JT, Sirk, SJ and Barbas, CF 3rd (2014). Cell-penetrating peptide-mediated delivery of TALEN proteins via bioconjugation for genome engineering. PLoS One 9: e85755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaj, T, Guo, J, Kato, Y, Sirk, SJ and Barbas, CF 3rd (2012). Targeted gene knockout by direct delivery of zinc-finger nuclease proteins. Nat Methods 9: 805–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, S, Kim, D, Cho, SW, Kim, J and Kim, JS (2014). Highly efficient RNA-guided genome editing in human cells via delivery of purified Cas9 ribonucleoproteins. Genome Res 24: 1012–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cradick, TJ, Qiu, P, Lee, CM, Fine, EJ and Bao, G (2014). COSMID: a web-based tool for identifying and validating CRISPR/Cas off-target sites. Mol Ther Nucleic Acids 3: e214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cathomen, T and Ehl, S (2014). Translating the genomic revolution - targeted genome editing in primates. N Engl J Med 370: 2342–2345. [DOI] [PubMed] [Google Scholar]
- Hsu, PD, Lander, ES and Zhang, F (2014). Development and applications of CRISPR-Cas9 for genome engineering. Cell 157: 1262–1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doudna, JA and Charpentier, E (2014). Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science 346: 1258096. [DOI] [PubMed] [Google Scholar]
- Barrangou, R, Fremaux, C, Deveau, H, Richards, M, Boyaval, P, Moineau, S et al. (2007). CRISPR provides acquired resistance against viruses in prokaryotes. Science 315: 1709–1712. [DOI] [PubMed] [Google Scholar]
- Lin, Y, Cradick, TJ, Brown, MT, Deshmukh, H, Ranjan, P, Sarode, N et al. (2014). CRISPR/Cas9 systems have off-target activity with insertions or deletions between target DNA and guide RNA sequences. Nucleic Acids Res 42: 7473–7485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sapranauskas, R, Gasiunas, G, Fremaux, C, Barrangou, R, Horvath, P and Siksnys, V (2011). The Streptococcus thermophilus CRISPR/Cas system provides immunity in Escherichia coli. Nucleic Acids Res 39: 9275–9282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, X, Scott, DA, Kriz, AJ, Chiu, AC, Hsu, PD, Dadon, DB et al. (2014). Genome-wide binding of the CRISPR endonuclease Cas9 in mammalian cells. Nat Biotechnol 32: 670–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esvelt, KM, Mali, P, Braff, JL, Moosburner, M, Yaung, SJ and Church, GM (2013). Orthogonal Cas9 proteins for RNA-guided gene regulation and editing. Nat Methods 10: 1116–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fonfara, I, Le Rhun, A, Chylinski, K, Makarova, KS, Lécrivain, AL, Bzdrenga, J et al. (2014). Phylogeny of Cas9 determines functional exchangeability of dual-RNA and Cas9 among orthologous type II CRISPR-Cas systems. Nucleic Acids Res 42: 2577–2590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szczelkun, MD, Tikhomirova, MS, Sinkunas, T, Gasiunas, G, Karvelis, T, Pschera, P et al. (2014). Direct observation of R-loop formation by single RNA-guided Cas9 and Cascade effector complexes. Proc Natl Acad Sci USA 111: 9798–9803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briner, AE, Donohoue, PD, Gomaa, AA, Selle, K, Slorach, EM, Nye, CH et al. (2014). Guide RNA functional modules direct Cas9 activity and orthogonality. Mol Cell 56: 333–339. [DOI] [PubMed] [Google Scholar]
- Gibson, DG, Young, L, Chuang, RY, Venter, JC, Hutchison, CA 3rd and Smith, HO (2009). Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods 6: 343–345. [DOI] [PubMed] [Google Scholar]
- Vouillot, L, Thélie, A and Pollet, N (2015). Comparison of T7E1 and surveyor mismatch cleavage assays to detect mutations triggered by engineered nucleases. G3 (Bethesda) 5: 407–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.