

Abstract
Plasmid-based Sleeping Beauty (SB) transposon vectors were developed and used to deliver genes for low-density lipoprotein and very-low-density lipoprotein receptors (LDLR and VLDLR, respectively) or lacZ reporter into liver of an LDLR-deficient mouse model of familial hypercholesterolemia (FH). SB transposase, SB100x, was used to integrate the therapeutic transposons into mice livers for evaluating the feasibility of the vectors in reducing high blood cholesterol and the progression of atherosclerosis. Hydrodynamic gene delivery of transposon-VLDLR into the livers of the mice resulted in initial 17–19% reductions in plasma cholesterol, and at the later time points, in a significant stabilization of the cholesterol level for the 6.5-month duration of the study compared to the control mice. Transposon-LDLR-treated animals also demonstrated a trend of stabilization in the cholesterol levels in the long term. Vector-treated mice had slightly less lipid accumulation in the liver and reduced aortic atherosclerosis. Clinical chemistry and histological analyses revealed normal liver function and morphology comparable to that of the controls during the follow-up with no safety issues regarding the vector type, transgenes, or the gene transfer method. The study demonstrates the safety and potential benefits of the SB transposon vectors in the treatment of FH.
Introduction
Low-density lipoprotein (LDL) receptor (LDLR)-associated familial hypercholesterolemia (FH) is an inherited, autosomal-dominant disease of cholesterol metabolism, characterized by a lifelong elevation of circulating LDL-cholesterol (LDL-C). It is the most frequent Mendelian disorder and a classical example of a monogenic condition predominantly caused by mutations affecting the expression or proper function of the gene encoding LDLR, the receptor responsible for clearing LDL-C from the circulation.1,2 In more than 90% of the FH cases in which a gene mutation has been identified, the primary cause of the disease is a defective LDLR gene.3 Patients suffering from the homozygous form of FH (hoFH) suffer from severe hypercholesterolemia with total cholesterol 12–30 mmol/l (460–1,160 mg/dl), and they develop atherosclerosis and coronary artery disease early during their childhood. Patients with heterozygous FH (heFH) have more moderately elevated plasma LDL-C, total cholesterol levels of 8–15 mmol/l (310–580 mg/dl), and also they develop premature coronary artery disease.4,5 The estimated theoretical prevalences for hoFH and heFH are ~1/1,000,000 and 1/500 in most populations worldwide, respectively. However, recent population-based investigations have screened ~1/137 to 1/200 individuals with heFH, suggesting that the disease is more common than previously thought.5,6 The hoFH patients with less than 2% of normal LDLR activity are considered as being receptor-negative and patients with residual 2–25% of normal LDLR activity as receptor-defective. If untreated, the receptor-negative hoFH patients rarely survive beyond adolescence; the prognosis for the receptor-defective hoFH patients is usually more positive; however, they too tend to develop clinically significant atherosclerosis before the age of 30 years.7
The present treatments for FH are based on dietary changes, lifelong LDL-C-lowering medication, and especially in the case of hoFH patients, regular LDL apheresis. Conventional statin medication may help heFH patients, but in particular for the individuals with hoFH, the response is often attenuated and/or inadequate.8 The new lipid-lowering drug therapies, mipomersen and lomitapide, that act via different mechanisms of action may improve the management of FH, especially the hoFH.9 LDL apheresis, the current standard treatment for hoFH or severe heFH, may transiently reduce the plasma LDL-C level by 70–80%, but the effect is not long lasting, and the treatment needs to be repeated regularly as the circulating LDL-C rapidly reaccumulates.8 Presently, liver transplantation is the only curative measure for the most severe hoFH cases as a liver transplant with functional LDLRs greatly improves the lipid metabolism of a patient.10
Gene replacement therapy is thought to offer a curative method for the FH. The liver is the organ mainly responsible for the clearance of LDL-C from the circulation, and the introduction of a functional copy of the LDLR gene into the patient's hepatocytes could ensure long-term expression of the receptor protein.11 Preclinical studies with the LDLR and very-low-density lipoprotein receptor (VLDLR) in animal models of hoFH have demonstrated a proof-of-principle for the therapeutic approach resulting in reduction in LDL-C levels and atherosclerosis.12,13 In addition, clinical investigations for the treatment of FH in the 1990s resulted in a modest LDLR expression in some hoFH patients.14,15 However, preclinical studies in the LDLR null animals, which have used the LDLR as a transgene, have revealed immune-mediated elimination of the liver-associated vector DNA12 or the transgene expressing hepatocytes,16,17 leading to only a transient reduction of LDL-C. Thus, the expression of transgenic VLDLR in the liver has been hypothesized to possibly provide a better route for the uptake of LDL-C into the hepatocytes.17,18
The major obstacle to the further development of the FH gene therapy has been the suboptimal success in developing safe and efficient vectors that could provide long-term expression. The recent successes of gene therapy in preclinical animal models of hoFH using an AAV-based gene therapy support the further development of the FH therapy.19,20,21 In addition, a plasmid-based transposon vector called Sleeping Beauty (SB) has pioneered the use of DNA transposon vectors in mammalian cells.22 In addition to the general safety elements of nonviral DNA vectors,23 the major beneficial feature of SB for gene therapy applications is its ability to integrate its transgene cargo into the genome of its target cells. The integrative cut-and-paste transposition mechanism of SB transposon has been described in detail in ref. 24 and presented in Supplementary Figure S1. The integration efficacy of the latest and most active version of SB, named SB100x, when coupled with the enhanced inverted terminal repeat structure T2 type of the SB transposon,25 is now found to be comparable with the integration rates of viral vectors.26 Interestingly, there are studies suggesting that the SB transposase-mediated integration might be safer than that of other integrating vectors, due to the near-random integration profile of SB: SB transposition is not biased toward actively transcribing genes.27 In contrast to SB, the commonly used retroviral, lentiviral, and AAV-derived vectors as well as two other recent transposon systems, piggyBac and Tol2, exhibit a rather clear integration bias into transcriptionally active units in genome.28,29,30,31,32 The integration bias encountered with these vectors may increase the risk of insertional mutagenesis, in comparison with SB.27,33 SB can also be considered as relatively safe due to its lack of association with genomic recombinations, deletions or translocations at the genomic integration sites.27,34 The potency of SB for gene therapy applications has been proven in several preclinical studies in which the vector system has been used to treat and to correct disease phenotypes of various different animal models of inheritable diseases.35 In addition, clinical gene therapy trials have been initiated using the SB vector.36 To date, there are no reports on the use of the SB vector system in the treatment of FH. The present investigation aimed to develop and characterize novel therapeutic non-viral SB vectors for the treatment of FH and to study their safety and efficacy in an in vivo gene delivery of the LDLR and VLDLR to decrease the abnormally elevated LDL-C levels and atherosclerosis in a FH model.
Results
In vitro studies
To optimize the SB-mediated transposition for liver-directed gene transfer, transposon plasmids carrying reporter genes were constructed and used to transfect cell types relevant to the investigations of treatment of the FH disease, HepG2 cells, and the LDLR-deficient WHHL (Watanabe heritable hyperlipidemic rabbit) fibroblasts, in combination with SB100x transposase (Figure 1a). The optimal transposase to transposon (Ts:Tn) ratio of these cell types was examined, due to the overproduction inhibition phenomenon, a mechanism by which transposition activity is downregulated when the transposase is over concentrated in cells. Titration of Ts:Tn ratio is typically required in order to achieve optimal results with the SB system.24 In the colony-forming assay studies in HepG2 cells, the transposition efficacy of the Ts:Tn ratio 1:1 was surprisingly found to be significantly higher than the ratio 1:10, a very commonly used Ts:Tn ratio for SB24 (Figure 1b) which led to a conclusion of development of a helper plasmid-independent cis transposon vector for ensuring the delivery of both the transposase and transposon into a single cell. The “all-in-one” cis vector outperformed the transposition efficacy of the dual plasmid system at the Ts:Tn ratio 1:10 too, but not at the Ts:Tn ratio 1:1 (Figure 1b). Figure 1c shows the high (left panels, A and C) and low (right panels, B and D) colony yields achieved with and without active transposase, respectively, in HepG2 and WHHL cells. The transfection efficacy of the SB100x transposase in combination with the T2/GFP transposon at 2 or 3 days following the gene transfers was measured as GFP fluorescence using the fluorescence-activated cell sorting (FACS) method, and relatively high transfection efficacies were observed (Figure 1d,e). The long-term follow-up of 7 weeks by fluorescence microscopy and FACS analyses verified the superiority of the Ts:Tn ratio 1:1 over the ratio 1:10 for HepG2 cells (Figure 1d). It also confirmed the functionality and feasibility of the “all-in-one” cis vectors in the hepatocyte cell line, thus adding the cis vectors to the list of vectors to be investigated further. Without the assistance of selection pressure, the GFP reporter gene showed high and steady long-term transgene expression following the transfections with the transposon vectors at all investigated time points, whereas the control transfections led to the transgene expression plummeting down to the autofluorescence level of the untreated cells around days 10 to 15 posttransfection (Figure 1d). Moreover, the GFP expression achieved with SB100x treatment did not show any signs of decline after reaching its steady expression level around day 20 (Figure 1d,e).
Figure 1.

SB transposition efficacy was determined by colony-forming assays and long-term follow-up FACS analyses of the cell cultures transfected with SB100x transposase and T2/NeoR transposon or T2/GFP transposon, respectively. (a) Schematic representation of the vectors in the in vitro transfection and transposition efficacy studies (from top to bottom): pCMV/SB100x (4,756 bp), pT2/CAG-NeoR (6,268 bp), pT2/CAG-GFP (5,611 bp), pT2/LSP-GFP (6,356 bp), pCMV/SB100x-T2/CAG-NeoR(in-one) (7,732 bp), pCMV/SB100x-T2/CAG-GFP(in-one) (7,075 bp), and pCMV/SB100x-T2/LSP-GFP(in-one) (8,336 bp); plasmid size of each construct is given here after the plasmid names. (b) In the colony-forming assay in the HepG2 cell cultures transfected with SB100x transposase and T2/CAG-NeoR transposons, the relative transposition efficacies (± SEM) are presented. The efficacy of the Ts:Tn ratio 1:1 was found significantly higher than the ratio 1:10 (P = 0.0010). The “all-in-one” cis vector performed better than the dual plasmid system at the Ts:Tn ratio 1:10 (P = 0.0002) too; but in comparison with the Ts:Tn ratio 1:1, the difference was not significant (P = 0.1209). In WHHL fibroblasts, the Ts:Tn ratio 1:10 outperformed the ratio 1:1 in the colony-forming assay, but the exact transposition efficacy could not be determined due to the cell type's carpet-like dish surface covering growth manner (see c). When compared to the control transfections conducted with inactive SB, the performance of all the vector combinations with SB100x was statistically significant (P < 0.0001). (c) Representative images of NeoR-positive cell colonies on cell culturing dishes, 21 days after transfection with PEI: HepG2 (A and B) and WHHL cells (C and D) co-transfected with T2/NeoR transposon in combination with (A and C) or without (B and D) an active transposase. (d) Long-term FACS follow-up of the HepG2 cell cultures transfected with SB100x + T2/CAG-GFP vectors using Microporator electroporator. The follow-up confirms the superiority of the Ts:Tn ratio 1:1 over the ratio 1:10 for HepG2 cell line and shows the efficient transposition from the “all-in-one” cis plasmid (the upper row of stars indicates the Ts:Tn 1:1 comparison with the ratio 1:10, and the lower row the comparison between the “all-in-one” vector and the Ts:Tn ratio 1:10). No difference was detected between the two top-performing vectors at any of the investigated time points (P > 0.9999), and in comparison with the control transfections, all the combinations with SB100x showed statistically significant transposition efficacy (P < 0.0001). (e) Long-term comparison of the CAG and LSP promoter driven GFP expression in HepG2 cells following the transfections with Neon electroporator and different Ts:Tn ratios. The investigated time points in d and e are indicated below the graphs. One-way or two-way ANOVA with Bonferroni's posttest was used in the data analyses. *P < 0.05, **P < 0.01, ***P < 0.001. Data are expressed as percentage (mean ± SD) of three independent transfection replicates.
The use of tissue-specific promoters is known to be advantageous as they are less sensitive to promoter inactivation and less likely to activate the host cell defense machinery. This may improve the stability and longevity of transgene expression,37 and thus the potential differences between the abilities of the universal CAG promoter and a liver-specific mouse alpha fetoprotein (AFP) enhancer II/mouse minimal albumin promoter (from here on referred as LSP, liver-specific promoter) to drive transgene expression were investigated. On account of the sequential liver-directed gene transfers, T2/CAG-GFP or T2/LSP-GFP transposons were investigated in combination with SB100x in HepG2 cells. Although the initial transfection efficacy of the dual vector combinations with T2/CAG-GFP were higher than those with T2/LSP-GFP, in the long-term, the difference was not significant (P > 0.9999 at day (d) 25), or it was in favor of the use of the T2/LSP-GFP vector (P = 0.0144 and P < 0.0001 for the Ts:Tn 1:1 and 1:10 vector combinations, respectively, at d31) (Figure 1e). The two “all-in-one” vectors with the different promoters were found to perform equally well at all investigated time points.
The differences between the CAG and LSP promoter-driven expression of the transgenic mouse LDLR, mLDLR, were compared at d20 by FACS analyzing transfected HepG2 cells that were immunolabeled with fluorescein isothiocyanate-conjugated antibody against LDLR. The results revealed no differences in the amount of mLDLR-expressing cells between the respective SB100x + T2/CAG-mLDLR and SB100x + T2/LSP-mLDLR vector combinations (P = 0.9975, P = 0.0538, and P = 0.1705 for the vectors at the Ts:Tn ratios 1:1, 1:10, and the “all-in-one” vectors, respectively; Figure 2a). Moreover, no differences were found in the gene expression levels driven by the two different promoters, as reflected in the total fluorescence intensity (P = 0.4254, P = 0.9976, and P > 0.9999 for the vectors at the Ts:Tn ratios 1:1, 1:10, and the “all-in-one” vectors, respectively; Figure 2b). The comparisons within either CAG or LSP promoter vector combinations (Ts:Tn ratios 1:1 versus 1:10 versus “all-in-one” vectors) demonstrated a trend for the Ts:Tn ratio 1:1 to yield the highest percentage of mLDLR-expressing cells but the difference between the two Ts:Tn ratios was found to be significant only with the LSP vectors (P = 0.8454 and P = 0.0139 for the vectors with CAG and LSP, respectively). In comparison with the “all-in-one” vectors, the dual vector systems were found to perform better (P < 0.0001 for the both CAG vector combinations; and P < 0.0001 and P = 0.0002 for LSP vectors at the ratios 1:1 and 1:10, respectively).
Figure 2.

Comparison of the effect of the CAG and LSP promoters on gene expression of transgenic mLDLR in HepG2 cell cultures and degradation assay in WHHL fibroblasts. The FACS was conducted for the anti-LDLR immunolabelled cells at day 20 following the gene transfers by Neon electroporation using different vector combinations, indicated in the figure. Transgenic mLDLR expression, driven by the two promoters, was determined by measuring FITC fluorescence of the (a) transgene-expressing cells as well as the gene expression levels of the cells, which correspond to the (b) total intensity of fluorescence. The two promoters performed equally well: no differences were found in the percentage of positive cells or promoter-driven gene expression in total FI between the respective SB100x + T2/CAG-mLDLR and SB100x + T2/LSP-mLDLR vector combinations at any Ts:Tn ratios. In comparison with the nontransfected control cells, the performance of all the investigated vector combinations was found to be significantly improved in terms of both the percentage of positive cells (P < 0.001) and total FI (P < 0.01). Data are expressed as percentage (mean ± SD) of three independent transfection replicates. One-way ANOVA with Tukey's posttest was used in analysis. In degradation assay (c), following the gene transfers with the LDLR carrying transposons, the change in transgenic LDLR mediated 125I-LDL degradation was investigated. In WHHL fibroblasts, transfected with mLDLR, degradation of 125I-LDL increased 34.4 ± 5.9% (SEM) when compared to the nontransfected control cells, and 146.3 ± 12.9% in the cells transfected with rLDLR. The data were analyzed with one-way ANOVA and are expressed as percentage (mean ± SEM) of three independent transfection replicates. *P < 0.05, ***P < 0.001. a.u., arbitrary units; FI, fluorescence intensity; FITC, fluorescein isothiocyanate; mLDLR, mouse LDLR; rLDLR, rabbit LDLR.
The functionality and biological activity of the transfected LDLR was investigated by studying the LDLR-mediated degradation of 125I-labeled LDL particles in WHHL fibroblasts, which have been widely used to examine LDL receptor biology due to the naturally occurring LDLR mutation in the WHHL rabbits. The 125I-LDL degradation assay was conducted using mouse and rabbit LDLR transgenes (mLDLR and rLDLR, respectively) cloned into the transposon vectors under the universal CAG promoter. The rLDLR transgene was included into the study because of its already previously verified functionality (data not shown). The results of the 125I-LDL degradation assay showed that the LDLR transgenes, delivered into cell cultures in transposon plasmids, were functional, as they induced an increase in the cells' ability to degrade 125I-LDL particles (Figure 2c) and hence feasible for the in vivo studies.
In vivo studies
Animal well-being after hydrodynamic injections. In general, the mice tolerated very well the gene transfer procedure via hydrodynamic tail vein injection. All animals recovered from the hydrodynamic injections completely in 5 to 10 minutes without any signs of distress or problems. The blood creatinine levels did not raise from the prehydrodynamic injection baseline values, indicating that the putatively harsh gene transfer method was not harmful to the kidney function, or that 1 week was long enough to allow kidney function to fully recover from the hydrodynamic stress (Table 1). No statistically significant differences were detected in the creatinine levels of the hydrodynamically injected mice and the nontreated control mice at the analyzed time points. No statistically significant differences were found in the mean alanine aminotransferase (ALAT) values of gene transfer receiving mice and the nontransfected control either. Generally, the plasma ALAT levels of the mice increased gradually throughout the study. However, as the weights of Western type (WT) diet-consuming mice increased throughout the study (Supplementary Figure S2) and as obesity is a known cause of hepatic steatosis and fatty liver disease, which causes elevation in ALAT levels,38 obesity is the likely explanation behind the continuous increase in the ALAT levels in this study. The presence of hepatic steatosis was confirmed in all groups in the histopathological analyses (see below). No loss of body weight occurred after the initial 1 week time point following the gene transfers. The mean increase in the weights of the male and female mice were 33.1 ± 2.8% and 42.4 ± 4.1% (SEM), respectively, from the beginning of the study until the last measurement (Supplementary Figure S2). In comparison to the pretreatment values of each individual animal, no statistically significant differences were found in the body weight changes between the groups at the investigated time points (P > 0.05 in all the cases when the groups were compared to each other using one-way analysis of variance (ANOVA)).
Table 1. Clinical chemistry values of LDLR knockout mice in several timepoints after the plasmid-mediated gene transfers.
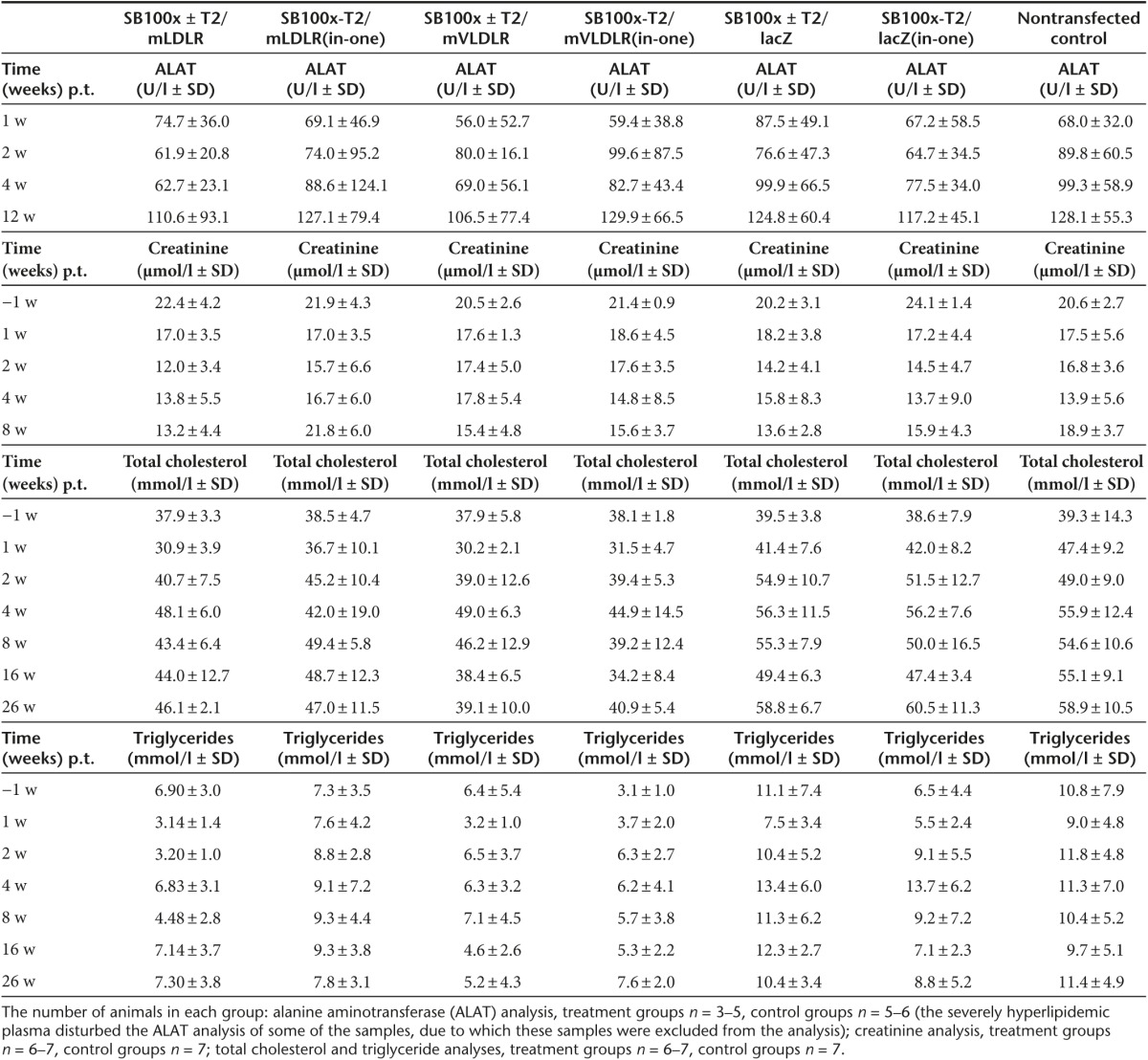
Long-term follow-up of blood lipid levels. Before the WT diet, all mice showed very similar plasma cholesterol levels (mean total cholesterol 5.3 ± 0.7 mmol/l, SD) and the WT diet induced a substantial increase in the pre-gene transfer cholesterol levels (mean cholesterol 38.6 ± 7.4 mmol/l, SD) (Table 1). Gene transfers with the T2/mLDLR and T2/mVLDLR vectors (in combination with SB100x, or as cis plasmids) to LDLR knockout (KO) mice resulted in a trend toward a cholesterol-lowering and stabilizing effect in the treated groups (Figure 3a). One week after the gene transfers, all the T2/mLDLR- and T2/mVLDLR-treated mice displayed lowered cholesterol (change in total cholesterol as compared to their individual pretreatment values (±SEM): −17.4 ± 6.9%, −6.0 ± 7.2%, −19.4 ± 3.5%, and −17.3 ± 5.1% for the T2/mLDLR, SB100x-T2/mLDLR(in-one), T2/mVLDLR, and SB100x-T2/mVLDLR(in-one) treatment groups, respectively), with the exception of two individual mice in the SB100x-T2/mLDLR(in-one) treatment group. In contrast, elevated cholesterol levels were observed in all control animals. After the initial declines at 1-week time point, the cholesterol levels of the T2/mLDLR, T2/mVLDLR, and SB100x-T2/mVLDLR(in-one) groups returned to the pregene transfer level or were even slightly above it and continued to rise from thereafter. However, the increase in the cholesterol levels in the treatment groups was moderate in comparison to that seen in the control animals. Some fluctuation was seen in the cholesterol levels: the levels peaked at the 4-week time point, followed by a gradual declination of the cholesterol for the mVLDLR-treated groups that continued until 16-week time point before stabilization. Similarly as seen herein, a rise in total cholesterol in the treated animals after the initial reduction, followed by second, more gradual decline, has also been observed in the previous studies.12,13 Sixteen weeks after the gene transfers, the T2/mVLDLR and the SB100x-T2/mVLDLR(in-one) treated groups displayed 2.7 ± 9.4% and −10.2 ± 9.3% change in their total cholesterol, and 2.0 ± 6.0% and 7.5 ± 6.6% change at week 26, respectively. These observed cholesterol levels were significantly lower in comparison to the levels of nontransfected controls (P = 0.0068 and P = 0.0005 at 16 weeks; P = 0.0025 and P = 0.0073 at the 26 weeks time points, respectively, in two-way ANOVA analysis). Cholesterol levels of the T2/mLDLR and SB100x-T2/mLDLR(in-one) treated groups did not decline after the 8-week time point but levelled below the cholesterol levels of the control groups; however, statistically significant differences were present. The lacZ-treated groups and the nontransfected control animals showed no treatment effect (P = 0.2754 and P = 0.2469 at 16 weeks; and P = 0.9403 and P = 0.9999 at the 26 weeks for the T2/lacZ and SB100x-T2/lacZ(in-one) groups, respectively) and experienced an elevation in their plasma cholesterol levels after the time point of gene transfers.
Figure 3.

Effect of direct in vivo plasmid vector-mediated mLDLR and mVLDLR gene transfers on plasma lipids in LDLR knockout mice. Changes in plasma (a) cholesterol and (b) triglyceride levels after the gene transfers via hydrodynamic tail vein injections. Data are expressed as percentage (mean ± SEM) change in comparison to the pretreatment values in each individual animal. Comparisons were performed to the levels of nontransfected control animals using two-way ANOVA, followed by Dunnet's posttest. *P < 0.05, **P < 0.01, ***P < 0.001.
Plasma triglyceride (TG) levels of the animals were also analyzed. Prior to the WT diet, all the mice displayed very similar TG levels (mean TG 1.2 ± 0.36 mmol/l, SD). After the gene transfers, the change in TG values did not completely follow the patterns of the cholesterol values (Figure 3b). However, cholesterol and TGs are not metabolized via the same mechanisms. Therefore, the gene therapy approach used herein to increase the LDL-C uptake was not expected to similarly affect the circulating TG levels. No significant differences were found in comparison with the nontransfected controls in the TG levels in a long-term follow-up (at 16 and 26 weeks P values were: P = 0.9805 and P = 0.9666 for the T2/mLDLR group; P = 0.9982 and P = 0.9920 for the SB100x-T2/mLDLR(in-one) group; P = 0.9509 and P = 0.9930 for the T2/mVLDLR group; P = 0.9980 and P = 0.4829 for the SB100x-T2/mVLDLR(in-one) group; P = 0.9196 and P = 0.9835 for the T2/lacZ group; P = 0.9996 and P = 0.9961 for the SB100x-T2/lacZ (in-one) group, respectively).
Development of atherosclerosis from en face stained aortas. Atherosclerosis was measured from LDLR KO mice aortas as a percentage area of lesions in the entire aorta. The en face analysis revealed that all the mice in the study, fed a WT diet for more than 6 months, had developed atherosclerosis with aortic lesions of varying sizes (Figure 4a). At the very least, the aortic arches were found covered with plaques in all mice, even in those individuals that otherwise displayed a more moderate aortic lesion development. The mean lesion areas in the aortas (±SEM) were 39.3 ± 4.2% and 41.7 ± 5.9% for the T2/mLDLR and SB100x-T2/mLDLR(in-one) treated mice; 36.3 ± 9.0% and 45.9 ± 2.5% for the T2/mVLDLR and SB100x-T2/mVLDLR(in-one) treated mice; 54.9 ± 2.0% and 48.8 ± 2.6% for the T2/lacZ and SB100x-T2/lacZ(in-one) treated mice; and 59.3 ± 3.6% for the nontransfected control mice (Figure 4b). The groups treated with T2/mLDLR and T2/mVLDLR transposons had modestly decreased areas of atherosclerotic lesions in en face opened aortas in comparison to the control mice, the difference reaching statistical significance in the SB100x + T2/mLDLR and SB100x + T2/mVLDLR groups (P = 0.0418 and P = 0.0154, respectively). In general, the aortas were found to be extremely fragile due to loss of elasticity caused by atherosclerosis.
Figure 4.

Atherosclerotic lesions in aortas of WT diet-fed LDLR knockout mice. (a) Pictures of en face aortas of the (A) SB100x + T2/mLDLR, (B) SB100x-T2/mLDLR(in-one), (C) SB100x + T2/mVLDLR, (D) SB100x-T2/mVLDLR(in-one), (E) SB100x + T2/lacZ, and (F) SB100x-T2/lacZ(in-one) treated mice, and (G) the nontransfected control mice. In the pictures, atherosclerotic lesions are stained red/pink and the non-atherosclerotic tissue is seen as nearly transparent (showing the black background). (b) Mean lesion areas of the groups as percentages of the total aortic areas, data are expressed as percentage (mean ± SEM). One-way ANOVA, followed by Dunnet's post-test, was used for statistical analyses. *P < 0.05.
Histopathological analyses. The post-mortem examinations of the mice did not reveal any malignancies or abnormalities, except the severe visceral obesity in most of the mice. Histopathological analyses evaluating the safety of liver-targeted gene transfers were performed from randomly selected liver sections. All the microscopically analyzed liver samples were found to display typical features of fatty liver, where vacuoles of fat accumulate in hepatocytes (Figure 5). The extensive macro- and microvesicular steatosis observed in the livers is likely caused by the obesity, due to the mice consuming the WT diet for more than 6 months. Macrovesicular steatosis in particular is a common form of fatty degeneration, which can typically be caused by oversupply of lipids, and it is seen in obesity.39,40 Although lipid vacuoles were present in the liver sections of all groups, a trend toward slightly less pronounced steatosis (smaller and fewer vacuoles) was noted in the liver sections from SB100x + T2/mVLDLR, SB100x-T2/mVLDLR(in-one), and SB100x + T2/mLDLR vector treated groups compared to the rest of the groups. Some clustering of inflammatory cells (monocytes) was seen in the investigated liver sections, indicating apoptotic and/or necrotic cells within the tissue and inflammation. No evidence of extensive fibrotic tissue development was observed, but marked necrotic areas were found in the liver of one mouse (not shown). This necrosis was not gene delivery nor vector related as the individual mouse was one of the nontransfected control animals.
Figure 5.

Representative images of liver sections of WT diet-fed LDLR knockout mice livers showing typical findings in the histopathological analysis. (a–d) Mild and (e, f) marked hepatocellular steatosis and fatty liver with lipid vacuoles ranging from small (microvesicular, some of the vacuoles indicated with arrows) to large (macrovesicular, some of the vacuoles indicated with asterisks), the largest filling and expanding the cell and displacing the nucleus to periphery. In e and f, steatosis is advanced: healthy hepatocytes are fewer, and lipid vacuoles are even more pronounced in size than in a–d. Clustering of monocytes was seen in some of the investigated liver sections. Images a and b represent the typical general appearance of liver sections from the animals treated with SB100x + T2/mVLDLR and SB100x-T2/mVLDLR(in-one), respectively; images c and d are from SB100x + T2/mLDLR and SB100x-T2/mLDLR(in-one) treated animals, respectively; and the most advanced steatosis (images e and f) was mostly observed in the livers of the controls (SB100x-T2/lacZ(in-one) and non-treated control mice, respectively). Light microscopic images of HE-stained liver sections. Bar = 100 µm. HE, hematoxylin and eosin.
Immunohistological analyses of transgene expression. The initial efficiency of the gene transfer method was evaluated by investigating the lacZ expression in the liver 3 days after the hydrodynamical tail vein gene transfers. The anti-β-galactosidase stained liver sections of the mice, treated with SB100x + T2/lacZ trans and SB100x-T2/lacZ(in-one) cis vectors and terminated at day 3 after the gene transfers, displayed evident lacZ expression in the nuclei of the hepatocytes (Figure 6a–c); however, lacZ-positive cells were not detected in every liver section per lacZ-positive mice, indicating only partial transfection of the livers. It is known that following systemic hydrodynamic gene transfer in mice, the transduced hepatocytes are predominantly located in the pericentral region,41 which may be explained by the wider and straighter sinusoids with more fenestrae per unit of surface in the pericentral area than in the periportal area.42,43 Long-term transgene expression could be verified 6.5 months after the gene transfers from the anti-β-galactosidase-stained liver sections of the mice treated with lacZ carrying vectors. Nonetheless, the persistent transgene expression was assessed as being very modest as only a few lacZ-positive cells could be detected per positive section (Figure 6d). The result suggests low in vivo transposition efficacy or inefficient transfer of the dual plasmid system into the same cells. Indeed, more lacZ-positive liver sections could be found from the liver sections of the mice treated with the SB100x-T2/lacZ(in-one) cis vector than from the dual plasmid system-treated livers. No β-galactosidase staining was found in the sections cut from the livers of nontransfected mice (Figure 6e). Long-term expression of the mLDLR transgene was also investigated 6.5 months after the gene transfers using an anti-LDLR antibody. The long-term transgenic mLDLR expression could be seen in some liver sections of a few of the mLDLR-treated mice (Figure 6f–l), and more mLDLR-positive liver sections were detected in the samples of SB100x-T2/mLDLR(in-one) vector-treated animals (Figure 6g,h,k,l) than from the samples of dual plasmid system-treated animals (Figure 6f,j). Limitations in detecting the potentially very low expression levels of the transgene with immunohistochemistry, or a suboptimal choice of anti-LDLR antibody used may have affected the sensitivity of the method. It is also possible that some of the genome invading transposons could have been silenced post-integration, as this phenomenon has been observed previously.44 No fluorescence signal was detected in any of the livers of nontransfected mice (Figure 6i,m).
Figure 6.

Transgene expression after in vivo injection of SB transposon vectors into the tail vein of LDLR knockout mice. Representative light microscope images of paraffin-embedded liver sections, stained with antibody against lacZ, 3 days after gene transfers with (a and b) SB100x + T2/lacZ and (c) SB100x-T2/lacZ(in-one) vectors, and 6.5 months after (d) SB100x-T2/lacZ(in-one) vector gene transfer. Red arrow heads indicate the lacZ-positive nuclei (dark brown), whereas the nuclei of the nontransfected cells are bright blue, seen in the close-up (b) of a lacZ-positive d3 liver section (a). Nontransfected control mouse liver is shown in e. Bar = 200 µm (a, c–e). Representative fluorescent microscope images showing long-term mLDLR transgene expression as green fluorescence in the immunolabelled liver tissue of (f,j) SB100x + T2/mLDLR and (g, h, k, l) SB100x-T2/mLDLR(in-one) vector treated mice 6.5 months after gene transfers. DAPI stain was used to visualize the nuclei (blue). The fluorescence images (j–m) represent close-ups of the images next to them (f–i), respectively. (i) Nontransfected mouse liver section is included for comparison with a (m) close-up. Bar = 200 µm (f–i); 100 µm (j–m).
PCRs verify the presence and the copy numbers of the transposons. With the conventional PCR, the lacZ transgene was detected in all the investigated liver gDNA samples of the mice terminated 3 days after the gene transfers, and the qPCR analysis confirmed the finding (Figure 7a). In the long-term follow-up mice, terminated 6.5 months after the gene transfers, conventional PCR was able to detect the lacZ transgene in most of the investigated liver gDNA samples; however, with qPCR, the T2/lacZ transposon target sequence was detected in only five of the samples, three of which were the samples from the mice co-transfected with SB100x and T2/lacZ transposon, and two from the “all-in-one” vector-treated mice (Figure 7b). These lacZ-transposon copy numbers observed in the long-term follow-up samples were declined in comparison with the early time point (Figure 7a). The higher early copy numbers are thus likely to reflect nonintegrated, episomal transgene copies. No amplification could be detected from the nontransfected control animal samples.
Figure 7.

PCR on the liver gDNA of individual LDLR knockout mice transfected with the SB transposon vectors. In the conventional PCR, amplification of (a) 228 bp DNA fragment (indicated in the figure with arrowheads) from lacZ transgene that was observed in all the liver gDNA samples of the individual mice terminated 3 days after the gene transfers and (b) in most of the long-term (6.5 months) follow-up samples. The samples from individual mice that were found positive also in the qPCR (graphs shown next to the respective gel images), at 3 days or 6.5 months after the gene transfers, respectively, are indicated with white font in the gel images a–d. The transposon copy numbers, unveiled in the copy number assays, are indicated above the columns. The light gray columns indicate the liver samples from dual plasmid co-transfected individual animals, and the dark gray columns are samples from “all-in-one” vector transfected animals. In the conventional PCR, amplification of 152 or 173 bp fragment from the transgenic (c) mLDLR or (d) mVLDLR, respectively, was verified in many of the long-term (6.5 months) follow-up samples. The samples from the individual mice in the T2/mLDLR or T2/mVLDLR vector treated groups that were found positive also in the qPCR are indicated with white font in the gel images, and the transposon copy numbers per diploid genomes are indicated above the respective columns in the graphs. The number of genomes per qPCR was normalized to the genomic copy number of the mTfrc, known to be present in two copies in a diploid mouse genome. The regression curve equations for the generated standard curves in qPCR were y = −3.229 + 36.176, and y = −3.395 + 39.962 for T2/lacZ and Tfrc endogenous control assays, respectively. In c, a copy-number assay designed to detect the T2 transposon IR/DR(L) was used, and the regression curve equations for the generated standard curves were y = −2.7695 + 36.456, and y = −3.1311 + 40.115 for IR/DR(L) and Tfrc endogenous control assays, respectively. In d, the regression curve equations were y = −3.496 + 34.637, and y = −3.3917 + 38.616 for T2/mVLDLR and Tfrc endogenous control assays, respectively. The numbered lanes in gel images represent PCR samples analyzed from the livers of individual transfected mice. In e, biodistribution analysis on spleen, lung, kidney, and heart gDNA of mice treated with SB100x+T2/lacZ or SB100x/lacZ “all-in-one” vectors shows amplification of 228 the lacZ transgene fragment in the tissues at d3. The samples from the individual mice that were found positive also in the qPCR are indicated in black font in the gel image e; the transposon copy numbers in these samples were 0.016, 0.02, 0.03, and 0.037 lacZ copies per diploid genome, respectively. S, spleen; L, lung; K, kidney; and H, heart; cntrl represents a nontransduced control animal sample; + is a positive control for amplification; and – is a negative control for amplification (H2O). Gene Ruler DNA Ladder Mix (Fermentas) was used as a molecular weight marker.
Similarly, the transgenic mLDLR and mVLDLR were investigated from the liver gDNA of the mice followed up for 6.5 months. Conventional PCR resulted in target sequence amplification from the mLDLR transgene in most of the studied liver gDNA samples, although the amplified DNA fragments were faint in most of the cases (Figure 7c). In the qPCR analysis, only three transposon-positive samples could be verified, one of which was a SB100x + T2/mLDLR co-transfected mouse sample and the other two in “all-in-one” vector-treated samples (Figure 7c), with minute transposon copy numbers. The results were similar for the T2/mVLDLR-treated samples: conventional PCR detected more positive samples than qPCR (Figure 7d), although some of the positive amplicons in the conventional PCR were very faint. In addition, more T2/mVLDLR-positive livers were found in the samples of SB100x-T2/mVLDLR(in-one)-treated animals than in the dual vector system-treated animals. Two of the “all-in-one” vector transfected samples harbored surprisingly high mVLDLR-transposon copy numbers (Figure 7d), making the scale of the copy numbers very wide. Nonetheless, these very high copy numbers were in line with the results of the conventional PCR analysis, in which the respective samples have yielded the strongest amplicons (Figure 7d).
A small-scale biodistribution study was conducted to investigate the transposon distribution into the major organs other than liver following the hydrodynamic injections. gDNA extracted from the spleen, lung, heart, and kidney of the SB100x + T2/lacZ and SB100x-T2/lacZ(in-one) transfected mice were PCR analyzed. Initial distribution of the plasmid vectors in the system was detected in all investigated tissue types at d3, but not all investigated individual tissues were positive (Figure 7e). The positive lacZ amplicon was clearer in the d3 spleen and lung samples; kidney and heart samples appeared to be the least positive. In qPCR, only three of the spleens and one lung were assayed as positive. However, no T2/lacZ-positive signal was observed in the PCR conducted on the 6.5-month follow-up mice (data not shown), the livers of which had been previously identified lacZ positive. Thus, it was concluded that no detectable transposition had occurred in tissues.
Gene expression analyses by qPCR. The lacZ expression could be confirmed in the liver samples (Figure 8a) that had been previously verified positive for the lacZ-transposons in the gDNA PCR analyses (Figure 7a,b). Altogether six mLDLR vector-treated animals were found to express the transgene, four of which belong to the SB100x + T2/mLDLR cotransfected and two to the SB100x-T2/mLDLR(in-one) vector-transfected groups (Figure 8b). The gene expression analyses on the liver samples from the SB100x + T2/mVLDLR co-transfected and SB100x-T2/mVLDLR(in-one) vector-transfected animals could confirm the gene expression in four samples, two of which were treated with the dual plasmid system (Figure 8c). The samples from which the transgene expression could be detected with the method had been previously PCR and/or qPCR-verified to carry the transposons within the liver gDNA.
Figure 8.

Relative quantity (RQ) of transgene expression in the livers of the LDLR knockout mice. LacZ expression (a) in the SB100x + T2/lacZ, and SB100x-T2/lacZ(in-one) vector transfected animals 3 days (1–6) and 6.5 months (7–19) after the gene transfers via hydrodynamic injections. The light gray columns indicate the liver samples from SB100x + T2/lacZ co-transfected individual animals, the dark gray columns are samples from SB100x-T2/lacZ(in-one) vector transfected animals. (b) The SB100x + T2/mLDLR (light gray columns) and SB100x-T2/mLDLR(in-one) (dark gray columns) vector transfected animals and (c) the SB100x + T2/mVLDLR (light gray columns) and SB100x-T2/mVLDLR(in-one) (dark gray columns) vector transfected animals 6.5 months after the gene transfers. Gene expression assays detecting the endogenous mouse Hprt or 18S rRNA expression in the samples was used in normalizing the target gene expression levels of the transgenic lacZ, or mLDLR and mVLDLR, respectively.
Discussion
In this study, the development and testing of the novel nonviral transposon system yielded SB transposon vectors carrying therapeutic LDLR and VLDLR genes for restoring the LDL metabolism in the hepatocytes of FH animal models. Because even a minimal amount of LDLR activity is beneficial for receptor defective subjects, it was hypothesized that plasmid-based vectors might be able to provide therapeutic LDLR expression sufficiently efficacious to treat the condition, if expressed stably. The transient gene expression has been a great challenge for the non–viral plasmid-based vectors, as many gene therapy approaches require stable or long-term expression of the therapeutic gene in order to achieve a long-lasting and effectual cure. The development of integrating transposon vector systems in the recent years has eliminated this problem. One of the novel transposon vectors, the plasmid-based SB transposon system, has been shown to correct several genetic disorders in preclinical settings (summarized in ref. 35), and the vector has already entered clinical trials.36
The safety and mutagenic potential of any given integrating vector system is one of the most important basic considerations for the application in clinical gene therapy. The SB transposon system is generally considered as rather safe in its near-random genomic integration pattern when compared to the other integrating vectors, including the commonly used viral vectors.45 The SB transposase element itself is not actively integrated from the dual plasmid system or the “all-in-one” vectors designed herein, as it is not incorporated between the two IR/DR sequences in the ends of transposon element that are substrates of the transposase (Supplementary Figure S1), but the spontaneous integration process could involve an integration of the SB transposase into genome, which in theory could lead to the mobilization of integrated transposon sequences. However, even though a previous study could detect some SB transposase sequences in genomic DNA of blood T cells following a gene transfer conducted with a highly efficient electroporation method, no detectable transposase expression could be found in the subsequent analyses.46 From safety perspective, it is also notable that the human genome does not contain Tc1-like elements related to SB, that theoretically could be cross-mobilized as a result of SB transposase transfer into human cells.24 Furthermore, by using a promoter that is active for only a few days in expressing the SB transposase, no evidence of remobilization of the transposons already inserted in the genome has been obtainable, suggesting genomic stability of the SB transposition.47 No cytotoxicity has been associated with the transient expression of the SB transposase protein in the numerous studies in which it has been used.48,49,50
Although the devastating manifestations of FH are primarily extrahepatic, the liver is the target organ for FH gene therapy. The rapid resolution of severe hypercholesterolemia following liver transplantation in hoFH10 supports the hypothesis that the newly gained hepatic expression of LDLRs lowers markedly the plasma LDL-C levels. Presently, the gene transfer via hydrodynamic injection51 has been assessed the most efficient way of delivering naked DNA into the liver, at least in rodents.52 Following the hydrodynamic injection into the systemic circulation, the injected large volume cannot pass through the heart rapidly enough, and the overload results in a retrograde flow through the vena cava, especially into the hepatic veins. As the intrahepatic pressure increases, the injected DNA solution is forced out of the hepatic sinusoids and into the hepatocytes, typically meaning that more than 90% of the injected DNA ends up in the liver.43
The SB transposon vectors generated herein were found both functional and able to ensure the long-term gene expression in vitro when paired with the hyperactive SB100x transposase. After the liver-directed gene transfers, the transposon-LDLR-treated animals demonstrated a trend of stabilization in the cholesterol levels in the long term. A greater improvement in the hypercholesterolemia and more prolonged therapeutic effect was achieved by using the VLDLR instead of the LDLR in the gene transfers. Gene delivery of mVLDLR into the livers of the mice resulted in initial 17–19% reductions in plasma cholesterol, and at the later time points, in a significant stabilization of the cholesterol level compared to the control mice.
VLDLR is structurally and functionally closely related to LDLR, and it predominantly modulates the extrahepatic metabolism of apoE-rich lipoproteins by their direct uptake into endothelial cells and through upregulating lipoprotein lipase-mediated metabolism of these lipoproteins.53 In contrast to LDLR, the expression of VLDLR is not regulated by intracellular free cholesterol.54 In the hepatocytes, where LDLRs are abundant, VLDLR is normally expressed only in minute amounts; however, its hepatic expression can be upregulated, e.g., by administration of certain molecules.55,56 Importantly, VLDLR was selected in the study in an attempt to circumvent possible immune responses, reported to be mounted against transgenic LDLR in LDLR-deficient subjects.12,16,17 In WHHL rabbit and LDLR−/− mice, adenovirus vector-mediated overexpression of LDLR has been found to promote cytotoxic T-lymphocyte responses against both the viral-encoded proteins and the LDLR transgene, leading to elimination of the transgene-expressing hepatocytes.16,17 Similarly, investigations in LDLR-deficient mice with AAV vector delivered LDLR transgene under the control of a strong ubiquitous promoter resulted in immune responses being mounted against the transgene and in the loss of liver-associated vector DNA, which led to only a transient reduction of plasma cholesterol.12 FH gene therapy using VLDLR is a good option as VLDLR is known to bind the metabolic precursors to LDL (VLDL and IDL lipoproteins that contain apoE) with high affinity.57 In FH therapy, VLDLR does not elicit immune responses to the transgene product because it is expressed in the tissues of LDLR-deficient subjects, and the hepatic VLDLR gene transfers into LDLR KO mice have been found to improve the clearance of the IDL fraction, lower the IDL/LDL fraction notably,17,58 and provide a long-term lowering of plasma cholesterol and prevention of the development of atherosclerosis in mice.18 Prior to this study, there have not been any reports on the usage of the SB vectors in the treatment of LDLR-deficient FH.
To summarize, this study demonstrated high in vitro transfection efficiency with the developed SB vectors, followed by transposition and high and stable long-term transgene expression. Successful target organ transfections were accomplished with the developed vectors, as transgene expression could be detected in the liver 3 days after the gene transfers. In the long-term in vivo follow-up, the numbers of transposon copies and the transgene-expressing cells had declined substantially but were still detectable. Despite the limited transposon copies found 6.5 months after the gene transfers, a modest, yet significant, long-term therapeutic effect could be demonstrated following the liver-directed VLDLR gene transfers. As the LDLR KO mouse model of FH does not express LDLR, the therapeutic effect of gene transfers on the plasma lipoproteins can be attributed to the VLDLR expression. In conclusion, this study demonstrates the safety and potential feasibility of the SB transposon system in the treatment of FH using liver-directed gene therapy. However, in the future, determining the optimal in vivo transposase/transposon vector combination and dose contingent on target species will be important for achieving an optimal efficacy and response to the therapy.
Materials and Methods
Cloning of transposon expression vectors. The pT2/BH-CAG transposon plasmid was constructed from the pT2/BH plasmid25 as described previously.59 PCR amplified NeoR (neomycin resistance), GFP (green fluorescence protein), and lacZ (β-galactosidase) reporter genes, as well as the mouse and rabbit LDL receptors (mLDLR and rLDLR, respectively) and mouse VLDL receptor (mVLDLR) transgenes were inserted into EcoRV site of the pT2/BH-CAG plasmid to create pT2/CAG-NeoR, pT2/CAG-GFP, pT2/CAG-mLDLR, pT2/CAG-mVLDLR, and pT2/CAG-rLDLR plasmids, respectively. Transposon plasmids carrying a liver-specific mouse alpha fetoprotein (AFP) enhancer II/mouse minimal albumin promoter (referred as LSP, liver-specific promoter) were constructed by inserting the PacI/PvuII cut and blunt-ended LSP expression cassette from pLIVE (Mirus Bio, Madison, WI) into EcoRV site of pT2/BH plasmid to create pT2/BH-LSP plasmid and then continued by inserting the PCR-amplified transgenes into SacII site in the expression cassettes MCS to create pT2/LSP-GFP, pT2/LSP-mLDLR, pT2/LSP-mVLDLR, and pT2/LSP-rLDLR plasmids. Both marker and therapeutic gene-carrying transposons were further developed to also bear the SB100x transposase to create “all-in-one” vectors. SB100x transposase expression cassette (with promoter/enhancer region, transposase open-reading frame, and terminal regions) was PCR amplified from pCMV-SB100x plasmid26 and inserted between the SspI sites in pT2/BH-CAG transposons with the previously mentioned transgenes, as well as into a AloI site of pT2/BH-LSP transposons with the previously mentioned transgenes. All the cloned plasmids were first analyzed with restriction enzyme analyses using appropriate restriction endonucleases to confirm the success of the cloning and then sequenced with MegaBACE 750 (Amersham Biosciences, Buckinghamshire, UK) DNA Analysis System in DNA Sequencing Facility of A. I. V. Institute (Kuopio, Finland) to verify the fidelity of the insertions. All the primers used in cloning are listed in Supplementary Table S1.
Cell culturing conditions and seeding of the cells. The characterization and testing of the constructed vectors and the optimization of the SB Ts:Tn ratio were performed in the immortalized HepG2 (human hepatocellular carcinoma, HB-8065, ATCC, Manassas, VA), and in primary WHHL fibroblast cells (obtained from a WHHL rabbit's skin in house). HepG2 cells were cultured as described previously.59 WHHL fibroblasts were cultured in Dulbecco's modified Eagle's medium (Sigma-Aldrich, St. Louis, MO) supplemented with 10% fetal bovine serum and antibiotics (100 units/ml of penicillin and 100 µg/ml of streptomycin; Gibco, Life Technologies, Paisley, UK). The cells were grown at 37 °C in the atmosphere of 95% air and 5% of CO2. For splitting the cultures, the cells were washed with 1× phosphate-buffered saline (PBS) (Sigma-Aldrich) and harvested by trypsinization with TrypLE Express (Invitrogen, Life Technologies, Grand Island, NY) as instructed by the manufacturer. For the experiments, HepG2 cells were seeded on six-well plates at 1 × 105 2 days before transfection or 3 × 105 1 day before transfection or transduction experiments for the cells to reach confluency of 50–75%. WHHL cells were seeded onto six-well plates at 1.6 × 105 or on 12-well plates at 8.0 × 104 a day before transfection experiments for the cells to reach confluency of 50–75%.
In vitro experiments. Total amount of plasmid DNA used in transfections with PEI transfection reagent (ExGen500, Fermentas, Vilnius, Lithuania) was 1 µg (on a six-well format) and 2 µg in electroporation experiments (Microporator or Neon transfection system by Invitrogen, Life Technologies) (on a 12-well format). The electroporations were carried out with the cells suspended in “Buffer R” (Invitrogen, Life Technologies) with the following device settings: pulse voltage 1,230 V, pulse width 20 ms, and pulse number 3. Twenty to twenty-four hours after the transfections, the cell culturing medium was replaced with fresh medium.
For the NeoR colony-forming assays, the vectors carrying SB100x and T2/NeoR were used with PEI to transfect HepG2 and WHHL cells. For the Ts:Tn ratio investigations, the focus was set to the Ts:Tn ratios 1:1 and 1:10. As a transposition control for the assays, an inactive SB was used instead of SB100x. The transfected cells were selected, starting at 5–7 days posttransfection, by feeding them fresh G418 (1 mg/ml) (InvivoGen, San Diego, CA) containing medium every 2–3 days, and after 21–30 days, the NeoR carrying cell colonies were stained with 0.05% crystal violet solution (Sigma-Aldrich) and counted. The relative transposition efficacy was calculated by dividing the number cell colonies achieved with SB by the number of colonies achieved without an active SB. For FACS analyses of GFP reporter gene, HepG2 cells were transfected with SB100x and T2/GFP vectors using Microporator or Neon electroporator. To determine the percentage and the intensity of fluorescence of the transgene-positive cells during the long-term follow-up, the GFP expression was analyzed by the FACS method (FACSCantoTM II; BD Biosciences, San Jose, CA) as described previously59 and using fluorescence microscope (Olympus IX 81; Olympus UK, UK). For studying the expression of the LDLR cloned in transposon vectors using FACS analysis, HepG2 cells were co-transfected with the SB100x and T2/mLDLR vectors using Neon electroporator. Prior to the FACS analyses, the transfected cells were incubated for 12–24 hours in medium containing 20% fetal bovine serum to downregulate expression of endogenic LDLR. For the FACS analysis, the co-transfected cells were harvested using PBS-based Cell Dissociation Buffer (Gibco, Life Technologies, Paisley, UK) according to the manufacturer's instructions, and then incubated with human FcγR-binding inhibitor (16–9161; eBioscience, San Diego, CA). After the incubation, the cells were immunolabelled with fluorescein isothiocyanate-conjugated rat monoclonal anti-mouse LDLR antibody (25 µg/ml) (FAB2255F; R&D Systems, Minneapolis, MN) according to the manufacturers' instructions. The immunolabelled cells were analyzed using FACS: the percentage of fluorescence-emitting cells and mean fluorescence intensity of cells were measured by counting 10,000 cells from each sample, analyzed with FACSDiva software (BD Biosciences), and total fluorescence intensity was calculated as described in ref. 59. For the LDL degradation assay, LDL was isolated in-house from fresh human plasma by sequential density gradient ultracentrifugation, as described in ref. 60 and labeled with iodine-125 (125I) as described in ref. 61. WHHL fibroblasts were co-transfected with T2/mLDLR or T2/rLDLR transposons using PEI, cultured for 2 days after transfection, and preincubated in medium containing excess serum lipoproteins (20% fetal bovine serum) for 24 hours to downregulate any residual endogenous LDLR activity. The medium was then replaced with medium containing LDL-deficient serum (10% LPDS, Sigma-Aldrich) supplemented with 125I-LDL (10 µg/ml). The cells were incubated with the 125I-LDL-medium for 12 hours to allow the uptake of 125I-LDL by transgenic LDL receptors. The uptake and specific degradation of the 125I-LDL (ng/µg cell protein) were measured as 125I radioactivity in the cell-free medium using a gamma counter (LKB-Wallac, Turku, Finland) as described in ref. 62. The radioactivity measured under identical conditions from control dishes with no cells was subtracted from total degradation to give a measure of cellular degradation.
In vivo experiments
LDLR KO mice. For therapeutic LDLR and VLDLR liver gene transfers, 50 LDLR KO mice were fed a WT high-fat diet (TD.88137; Harlan Laboratories, Indianapolis, IN) ad libitum for 2 weeks for them to develop hyperlipidemic state, induce severe hypercholesterolemia, and initiate the progression of atherosclerosis. The study design had four parallel therapeutic gene transfer treatments versus controls. The SB elements were either co-administrated into mice livers in combination of SB100x and the T2/LSP-mLDLR or T2/LSP-mVLDLR transposon, or using the single “all-in-one” plasmids. The T2/CAG-lacZ transposon vectors were used as gene transfer controls as the two promoters performed equally well in the in vitro testing when the respective T2/CAG-mLDLR and T2/LSP-mLDLR plasmid combinations were compared with each other and as the use of CAG promoter allowed performing a biodistribution study. Also, nontreated mice were included in the study. The mice were randomly divided into different treatment or control groups, and equal numbers of both sexes were selected for all groups. The littermates were divided into individual cages at the start of the high-fat diet. To assess the normal blood lipid profiles, the first blood samples were collected from randomly selected mice before they were put on a high-fat diet. The second blood samples were collected 2 weeks later from all the mice to assess the “null blood samples on a WT diet” before the gene transfers. The gene transfers were conducted using hydrodynamic tail vein injection method after the mice were analyzed to be in a highly hypercholesterolemic state (total cholesterol >30 mmol/l). The general health and appearance of the mice were monitored daily, and they were weighed prior to each blood sample collection. All the animal experiments were approved and authorized by the national Animal Experiment Board and conducted according to The Finnish Act on Animal Experimentation (62/2006) in the experimental animal facilities of University of Eastern Finland.
Gene transfers via hydrodynamic tail vein injection. For the gene transfers via hydrodynamic tail vein injection, total amount of 125 μg of plasmid vectors were diluted in 0.9% saline (total volume of 2.1 ml), equivalent to around 10% of the mouse body weight, and injected into the tail vein within a period of 4 to 7 seconds, essentially as previously described.63 During the injections, the mice were lightly anesthetized with isoflurane inhalation (2% isoflurane–air mixture). Injections taking longer than 4 to 7 seconds were excluded from the study, as were the injections failing to deliver the intended 2 ml volume of the plasmid solution into the tail vein. Following the injections, the mice were kept warm and monitored until a complete recovery to ensure their well-being, and when the recovered mice started moving around normally they were returned to their individual cages.
Clinical chemistry analyses. ALAT, creatinine, and the blood lipid levels were followed-up by regular blood sample collection (once a week during the first month, then once a month until the end of the study). Blood samples (100–150 µl) were collected from the tail vein of the mice into lithium heparin tubes (365971, BD Microtainer; BD, Franklin Lakes, NJ) after >2 hours fasting. Plasma was separated by centrifugation and analyzed for total cholesterol, TGs, ALAT, and creatinine by Movet Oy (Kuopio, Finland).
Histological and immunohistological analyses. After the follow-up period of 6.5 months, the mice were sacrificed with carbon dioxide and perfused with PBS through the left ventricle, and tissue samples were collected in post-mortem examinations for histological, immunohistological, and DNA/RNA level analyses. Some of the mice receiving the T2/lacZ vectors were sacrificed at the day 3 time point post gene transfers and were used for the characterization of gene transfer efficiency. Mice aortas were carefully dissected from aortic arch to bifurcation and fixed in 4% PFA-PBS overnight. The fixed aortas were opened longitudinally, attached to a black surface, and stained with 0.5% Sudan IV dye (Sigma-Aldrich) and photographed for an en face evaluation of atherosclerotic plaques relative to the non-atherosclerotic area, done using AnalySIS software (Olympus Soft Imaging Systems, Munster, Germany). For the hematoxylin–eosin and immunohistochemical staining, the tissue samples were collected in 4% PFA–PBS for overnight fixation, processed to paraffin and cut as 5- or 7-µm sections. Transgene expression in the whole liver samples was studied immunohistochemically and general histological analyses were done for in search for potential tissue abnormalities related to gene transfer. Randomly selected paraffin-embedded tissue sections from the livers were HE stained for general morphology examination. Histopathological examination was conducted in as a blinded analysis. Immunohistochemical stainings were done using primary antibody against LDLR (ab 30532; Abcam, Cambridge, UK) for randomly selected liver sections of the mLDLR-treated mice. To determine the efficacy of the gene transfers and transgene expression, lacZ-treated control mice liver sections were stained with primary antibody against lacZ (AB1211 Chemicon; Merck Millipore, Darmstadt, Germany). For the lacZ stainings, a biotinylated anti-rabbit IgG (H&L) secondary antibody made in goat (BA-100; Vector Laboratories, Burlingame, CA) was used, followed by an avidin–biotin–HRP step (Vector Laboratories) and DAB (Invitrogen, Life Technologies) as a chromogen. In the mLDLR staining, fluorescein isothiocyanate-conjugated goat polyclonal anti-rabbit IgG (H&L) (ab6717; Abcam) was used; Vectashield HardSet Mounting Medium with DAPI (Vector Laboratories) was used to visualize the nuclei.
PCR and qPCR analyses. Whole liver samples and samples of other major tissues were collected in liquid nitrogen and stored deep frozen for DNA and RNA level investigations. The genomic DNA (gDNA) and total RNA were extracted using ChargeSwitch gDNA Mini Tissue Kit and PureLink RNA Mini Kit (Invitrogen, Life Technologies) or RNeasy Mini Kit (Qiagen, Hilden, Germany), respectively, in accordance with the manufacturers' instructions. For total RNA isolation, Precellys24 lyser/homogenizer with Soft Tissue homogenizing beads (1.4 mm ceramic zirconium oxide beads) (Bertin Technologies, Montigny-le-Bretonneux, France) were used to homogenize the tissues. Resulting gDNA and total RNA was quantified using NanoDrop ND-1000 (NanoDrop Technologies, Wilmington, DE). Conventional PCRs were performed using specific primers for each transgene (listed in Supplementary Table S2). The conventional PCRs contained 200–250 ng of gDNA as a template, specific primers, and Phusion High Fidelity DNA Polymerase (Thermo Fisher Scientific, Waltham, MA) in a final volume of 50 µl, according to the enzyme manufacturer's instructions. As positive controls for the PCRs, plasmids pT2/CAG-lacZ, pT2/BH-CAG-mLDLR, and pT2/BH-CAG-mVLDLR were used as templates. The quantitative PCR (qPCR) for assaying the transposon copy numbers were conducted as described in ref. 59 using TaqMan Custom Copy Number Assays (Applied Biosystems, Life Technologies, Carlsbad, CA) (listed in Supplementary Table S3). To avoid interference of the endogenous mouse genes, the custom assays were designed to span the gene exon–exon boundary, taking advantage of the introns absent in the transgenic genes. Mouse TaqMan Copy Number Reference Assay (Applied Biosystems) for detecting the endogenous transferrin receptor gene (Tfrc), present in two copies in a diploid mouse genome, was used as a control assay for the quantitation. For quantitation, standard curves were generated as described in ref. 59 of the plasmids pT2/BH-CAG-mLDLR, pT2/BH-CAG-mVLDLR, and pT2/BH-CAG-lacZ, and of the gDNA extracted from mice that were not treated with the vectors. For gene expression assays, complementary DNA (cDNA) from each total RNA sample was prepared from 1 µg of DNase I-treated total RNA by reverse transcription with random hexamer primers and Maxima Reverse Transcriptase (Thermo Fisher Scientific). cDNA samples were analyzed on a StepOnePlus Real-Time PCR System (Applied Biosystems, Life Technologies, Carlsbad, CA) using TaqMan Gene Expression Assays (Applied Biosystems, Life Technologies) for detecting the mLDLR, mVLDLR, and lacZ expression (listed in Supplementary Table S3). TaqMan Gene Expression Assays (Applied Biosystems, Life Technologies) detecting endogenous mouse Hprt (hypoxanthine guanine phosphoribosyl transferase) or 18S (eukaryotic 18S subunit ribosomal RNA) were used to normalize the target gene expression levels of the transgenic lacZ, or mLDLR and mVLDLR, respectively. The assays were conducted according to the manufacturer's instructions with TaqMan Gene Expression Master Mix (Applied Biosystems, Life Technologies), and the samples were run in three technical replicates using 50 ng of the cDNA template in each sample. The data were collected simultaneously for both the target gene and the endogenous control gene from the singleplex reactions and analyzed with the StepOne Software, version 2.1 (Applied Biosystems, Life Technologies) using relative quantification based on comparative Ct method (ΔΔCt).64 Liver samples from nontransfected control animals were used as calibrator samples, the relative gene expression ratio of which was set to 1 in the analyses, denoting the baseline. The results are given in log10 graphs (log10 of 1 = 0) to present only the relative quantity of gene expression of the positive livers.
Statistical analyses. Two-way ANOVA or one-way ANOVA (depending on the number of factors to be compared), followed by Bonferroni's, Dunnet's, or Tukey posttest, were performed with GraphPad Prism program, version 5.01 (GraphPad Software, La Jolla, CA) to determine the statistical significance of the results. The tests used in each case are indicated in context. P values < 0.05 were considered statistically significant.
SUPPLEMENTARY MATERIAL Figure S1. Transposition of the SB system, presented as a dual plasmid and a single plasmid system (in trans or in cis, respectively). Figure S2. Development of the LDLR KO mice weight on a Western type diet. Table S1. PCR primers used in cloning of the vectors. Table S2. PCR primers used in standard PCR gDNA analyses. Table S3. TaqMan assays containing the primers and the probes used in qPCR studies.
Acknowledgments
This work was financially supported by Academy of Finland, European Research Council Futuregenes grant (ERC-2009-AdG_20090506), EU INTHER grant (LSHB-CT-2005–018961), Ark Therapeutics, Kuopio University Hospital, and grants from the Finnish Foundation for Cardiovascular Research, the Finnish Cultural Foundation North Savo Regional fund, the Paavo Nurmi Foundation, the Ida Montin Foundation, the Sigrid Juselius Foundation, and the Helena Vuorenmies Foundation. The authors thank Joonas Malinen, Jaana Siponen, Anneli Miettinen, and Seija Sahrio for excellent technical support. The authors declare no conflicts of interest.
Supplementary Material
Transposition of the SB system, presented as a dual plasmid and a single plasmid system (in trans or in cis, respectively).
Development of the LDLR KO mice weight on a Western type diet.
References
- Brown, MS and Goldstein, JL (1974). Familial hypercholesterolemia: defective binding of lipoproteins to cultured fibroblasts associated with impaired regulation of 3-hydroxy-3-methylglutaryl coenzyme A reductase activity. Proc Natl Acad Sci USA 71: 788–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown, MS and Goldstein, JL (1979). Receptor-mediated endocytosis: insights from the lipoprotein receptor system. Proc Natl Acad Sci USA 76: 3330–3337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usifo, E, Leigh, SE, Whittall, RA, Lench, N, Taylor, A, Yeats, C et al. (2012). Low-density lipoprotein receptor gene familial hypercholesterolemia variant database: update and pathological assessment. Ann Hum Genet 76: 387–401. [DOI] [PubMed] [Google Scholar]
- Brown, MS and Goldstein, JL (1986). A receptor-mediated pathway for cholesterol homeostasis. Science 232: 34–47. [DOI] [PubMed] [Google Scholar]
- Nordestgaard, BG, Chapman, MJ, Humphries, SE, Ginsberg, HN, Masana, L, Descamps, OS et al.; European Atherosclerosis Society Consensus Panel. (2013). Familial hypercholesterolaemia is underdiagnosed and undertreated in the general population: guidance for clinicians to prevent coronary heart disease: consensus statement of the European Atherosclerosis Society. Eur Heart J 34: 3478–3490a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benn, M, Watts, GF, Tybjaerg-Hansen, A and Nordestgaard, BG (2012). Familial hypercholesterolemia in the danish general population: prevalence, coronary artery disease, and cholesterol-lowering medication. J Clin Endocrinol Metab 97: 3956–3964. [DOI] [PubMed] [Google Scholar]
- Rader, DJ, Cohen, J and Hobbs, HH (2003). Monogenic hypercholesterolemia: new insights in pathogenesis and treatment. J Clin Invest 111: 1795–1803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemphill, LC (2010). Familial hypercholesterolemia: current treatment options and patient selection for low-density lipoprotein apheresis. J Clin Lipidol 4: 346–349. [DOI] [PubMed] [Google Scholar]
- Robinson, JG (2013). Management of familial hypercholesterolemia: a review of the recommendations from the National Lipid Association Expert Panel on Familial Hypercholesterolemia. J Manag Care Pharm 19: 139–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maiorana, A, Nobili, V, Calandra, S, Francalanci, P, Bernabei, S, El Hachem, M et al. (2011). Preemptive liver transplantation in a child with familial hypercholesterolemia. Pediatr Transplant 15: E25–E29. [DOI] [PubMed] [Google Scholar]
- Rader, DJ (2001). Gene therapy for familial hypercholesterolemia. Nutr Metab Cardiovasc Dis 11 (suppl. 5): 40–44. [PubMed] [Google Scholar]
- Chen, SJ, Rader, DJ, Tazelaar, J, Kawashiri, M, Gao, G and Wilson, JM (2000). Prolonged correction of hyperlipidemia in mice with familial hypercholesterolemia using an adeno-associated viral vector expressing very-low-density lipoprotein receptor. Mol Ther 2: 256–261. [DOI] [PubMed] [Google Scholar]
- Kankkonen, HM, Vähäkangas, E, Marr, RA, Pakkanen, T, Laurema, A, Leppänen, P et al. (2004). Long-term lowering of plasma cholesterol levels in LDL-receptor-deficient WHHL rabbits by gene therapy. Mol Ther 9: 548–556. [DOI] [PubMed] [Google Scholar]
- Grossman, M, Raper, SE, Kozarsky, K, Stein, EA, Engelhardt, JF, Muller, D et al. (1994). Successful ex vivo gene therapy directed to liver in a patient with familial hypercholesterolaemia. Nat Genet 6: 335–341. [DOI] [PubMed] [Google Scholar]
- Grossman, M, Rader, DJ, Muller, DW, Kolansky, DM, Kozarsky, K, Clark, BJ 3rd et al. (1995). A pilot study of ex vivo gene therapy for homozygous familial hypercholesterolaemia. Nat Med 1: 1148–1154. [DOI] [PubMed] [Google Scholar]
- Kozarsky, KF, McKinley, DR, Austin, LL, Raper, SE, Stratford-Perricaudet, LD and Wilson, JM (1994). In vivo correction of low density lipoprotein receptor deficiency in the Watanabe heritable hyperlipidemic rabbit with recombinant adenoviruses. J Biol Chem 269: 13695–13702. [PubMed] [Google Scholar]
- Kozarsky, KF, Jooss, K, Donahee, M, Strauss, JF 3rd and Wilson, JM (1996). Effective treatment of familial hypercholesterolaemia in the mouse model using adenovirus-mediated transfer of the VLDL receptor gene. Nat Genet 13: 54–62. [DOI] [PubMed] [Google Scholar]
- Oka, K, Pastore, L, Kim, IH, Merched, A, Nomura, S, Lee, HJ et al. (2001). Long-term stable correction of low-density lipoprotein receptor-deficient mice with a helper-dependent adenoviral vector expressing the very low-density lipoprotein receptor. Circulation 103: 1274–1281. [DOI] [PubMed] [Google Scholar]
- Kassim, SH, Li, H, Vandenberghe, LH, Hinderer, C, Bell, P, Marchadier, D et al. (2010). Gene therapy in a humanized mouse model of familial hypercholesterolemia leads to marked regression of atherosclerosis. PLoS One 5: e13424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kassim, SH, Li, H, Bell, P, Somanathan, S, Lagor, W, Jacobs, F et al. (2013). Adeno-associated virus serotype 8 gene therapy leads to significant lowering of plasma cholesterol levels in humanized mouse models of homozygous and heterozygous familial hypercholesterolemia. Hum Gene Ther 24: 19–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somanathan, S, Jacobs, F, Wang, Q, Hanlon, AL, Wilson, JM and Rader, DJ (2014). AAV vectors expressing LDLR gain-of-function variants demonstrate increased efficacy in mouse models of familial hypercholesterolemia. Circ Res 115: 591–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivics, Z, Hackett, PB, Plasterk, RH and Izsvák, Z (1997). Molecular reconstruction of Sleeping Beauty, a Tc1-like transposon from fish, and its transposition in human cells. Cell 91: 501–510. [DOI] [PubMed] [Google Scholar]
- Schmidt-Wolf, GD and Schmidt-Wolf, IG (2003). Non-viral and hybrid vectors in human gene therapy: an update. Trends Mol Med 9: 67–72. [DOI] [PubMed] [Google Scholar]
- Izsvák, Z and Ivics, Z (2004). Sleeping beauty transposition: biology and applications for molecular therapy. Mol Ther 9: 147–156. [DOI] [PubMed] [Google Scholar]
- Cui, Z, Geurts, AM, Liu, G, Kaufman, CD and Hackett, PB (2002). Structure-function analysis of the inverted terminal repeats of the sleeping beauty transposon. J Mol Biol 318: 1221–1235. [DOI] [PubMed] [Google Scholar]
- Mátés, L, Chuah, MK, Belay, E, Jerchow, B, Manoj, N, Acosta-Sanchez, A et al. (2009). Molecular evolution of a novel hyperactive Sleeping Beauty transposase enables robust stable gene transfer in vertebrates. Nat Genet 41: 753–761. [DOI] [PubMed] [Google Scholar]
- Yant, SR, Wu, X, Huang, Y, Garrison, B, Burgess, SM and Kay, MA (2005). High-resolution genome-wide mapping of transposon integration in mammals. Mol Cell Biol 25: 2085–2094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, X, Li, Y, Crise, B and Burgess, SM (2003). Transcription start regions in the human genome are favored targets for MLV integration. Science 300: 1749–1751. [DOI] [PubMed] [Google Scholar]
- Schröder, AR, Shinn, P, Chen, H, Berry, C, Ecker, JR and Bushman, F (2002). HIV-1 integration in the human genome favors active genes and local hotspots. Cell 110: 521–529. [DOI] [PubMed] [Google Scholar]
- Hüser, D, Gogol-Döring, A, Lutter, T, Weger, S, Winter, K, Hammer, EM et al. (2010). Integration preferences of wildtype AAV-2 for consensus rep-binding sites at numerous loci in the human genome. PLoS Pathog 6: e1000985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson, MH, Coates, CJ and George, AL Jr (2007). PiggyBac transposon-mediated gene transfer in human cells. Mol Ther 15: 139–145. [DOI] [PubMed] [Google Scholar]
- Grabundzija, I, Irgang, M, Mátés, L, Belay, E, Matrai, J, Gogol-Döring, A et al. (2010). Comparative analysis of transposable element vector systems in human cells. Mol Ther 18: 1200–1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, G, Geurts, AM, Yae, K, Srinivasan, AR, Fahrenkrug, SC, Largaespada, DA et al. (2005). Target-site preferences of Sleeping Beauty transposons. J Mol Biol 346: 161–173. [DOI] [PubMed] [Google Scholar]
- Vigdal, TJ, Kaufman, CD, Izsvák, Z, Voytas, DF and Ivics, Z (2002). Common physical properties of DNA affecting target site selection of sleeping beauty and other Tc1/mariner transposable elements. J Mol Biol 323: 441–452. [DOI] [PubMed] [Google Scholar]
- Aronovich, EL, McIvor, RS and Hackett, PB (2011). The Sleeping Beauty transposon system: a non-viral vector for gene therapy. Hum Mol Genet 20: R14–R20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiley Online Library. (2013). Gene therapy trials worldwide. J Gene Med. <http://www.wiley.com/legacy/wileychi/genmed/clinical/>.
- Liu, BH, Wang, X, Ma, YX and Wang, S (2004). CMV enhancer/human PDGF-beta promoter for neuron-specific transgene expression. Gene Ther 11: 52–60. [DOI] [PubMed] [Google Scholar]
- Adams, LA and Lindor, KD (2007). Nonalcoholic fatty liver disease. Ann Epidemiol 17: 863–869. [DOI] [PubMed] [Google Scholar]
- Cave, M, Deaciuc, I, Mendez, C, Song, Z, Joshi-Barve, S, Barve, S et al. (2007). Nonalcoholic fatty liver disease: predisposing factors and the role of nutrition. J Nutr Biochem 18: 184–195. [DOI] [PubMed] [Google Scholar]
- Yeh, MM and Brunt, EM (2014). Pathological features of fatty liver disease. Gastroenterology 147: 754–764. [DOI] [PubMed] [Google Scholar]
- Suda, T, Gao, X, Stolz, DB and Liu, D (2007). Structural impact of hydrodynamic injection on mouse liver. Gene Ther 14: 129–137. [DOI] [PubMed] [Google Scholar]
- Wisse, E, De Zanger, RB, Jacobs, R and McCuskey, RS (1983). Scanning electron microscope observations on the structure of portal veins, sinusoids and central veins in rat liver. Scan Electron Microsc(Pt 3): 1441–1452. [PubMed]
- Herweijer, H and Wolff, JA (2007). Gene therapy progress and prospects: hydrodynamic gene delivery. Gene Ther 14: 99–107. [DOI] [PubMed] [Google Scholar]
- Garrison, BS, Yant, SR, Mikkelsen, JG and Kay, MA (2007). Postintegrative gene silencing within the Sleeping Beauty transposition system. Mol Cell Biol 27: 8824–8833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hackett, CS, Geurts, AM and Hackett, PB (2007). Predicting preferential DNA vector insertion sites: implications for functional genomics and gene therapy. Genome Biol 8 (suppl. 1): S12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, X, Haley, K, Wong, M, Guo, H, Lu, C, Wilber, A et al. (2010). Unexpectedly high copy number of random integration but low frequency of persistent expression of the Sleeping Beauty transposase after trans delivery in primary human T cells. Hum Gene Ther 21: 1577–1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell, JB, Aronovich, EL, Schreifels, JM, Beadnell, TC and Hackett, PB (2010). Duration of expression and activity of Sleeping Beauty transposase in mouse liver following hydrodynamic DNA delivery. Mol Ther 18: 1796–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yant, SR, Meuse, L, Chiu, W, Ivics, Z, Izsvak, Z and Kay, MA (2000). Somatic integration and long-term transgene expression in normal and haemophilic mice using a DNA transposon system. Nat Genet 25: 35–41. [DOI] [PubMed] [Google Scholar]
- Ivics, Z and Izsvák, Z (2006). Transposons for gene therapy! Curr Gene Ther 6: 593–607. [DOI] [PubMed] [Google Scholar]
- Galla, M, Schambach, A, Falk, CS, Maetzig, T, Kuehle, J, Lange, K et al. (2011). Avoiding cytotoxicity of transposases by dose-controlled mRNA delivery. Nucleic Acids Res 39: 7147–7160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, G, Budker, V and Wolff, JA (1999). High levels of foreign gene expression in hepatocytes after tail vein injections of naked plasmid DNA. Hum Gene Ther 10: 1735–1737. [DOI] [PubMed] [Google Scholar]
- Jacobs, F, Gordts, SC, Muthuramu, I and De Geest, B (2012). The liver as a target organ for gene therapy: state of the art, challenges, and future perspectives. Pharmaceuticals (Basel) 5: 1372–1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Go, GW and Mani, A (2012). Low-density lipoprotein receptor (LDLR) family orchestrates cholesterol homeostasis. Yale J Biol Med 85: 19–28. [PMC free article] [PubMed] [Google Scholar]
- Takahashi, S, Sakai, J, Fujino, T, Hattori, H, Zenimaru, Y, Suzuki, J et al. (2004). The very low-density lipoprotein (VLDL) receptor: characterization and functions as a peripheral lipoprotein receptor. J Atheroscler Thromb 11: 200–208. [DOI] [PubMed] [Google Scholar]
- Hashizume, M, Yoshida, H, Koike, N, Suzuki, M and Mihara, M (2010). Overproduced interleukin 6 decreases blood lipid levels via upregulation of very-low-density lipoprotein receptor. Ann Rheum Dis 69: 741–746. [DOI] [PubMed] [Google Scholar]
- Gao, Y, Shen, W, Lu, B, Zhang, Q, Hu, Y, Chen, Y et al. (2014). Upregulation of hepatic VLDLR via PPARα is required for the triglyceride-lowering effect of fenofibrate. J Lipid Res 55: 1622–1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi, S, Kawarabayasi, Y, Nakai, T, Sakai, J and Yamamoto, T (1992). Rabbit very low density lipoprotein receptor: a low density lipoprotein receptor-like protein with distinct ligand specificity. Proc Natl Acad Sci USA 89: 9252–9256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi, K, Oka, K, Forte, T, Ishida, B, Teng, B, Ishimura-Oka, K et al. (1996). Reversal of hypercholesterolemia in low density lipoprotein receptor knockout mice by adenovirus-mediated gene transfer of the very low density lipoprotein receptor. J Biol Chem 271: 6852–6860. [DOI] [PubMed] [Google Scholar]
- Turunen, TA, Laakkonen, JP, Alasaarela, L, Airenne, KJ and Ylä-Herttuala, S (2014). Sleeping Beauty-baculovirus hybrid vectors for long-term gene expression in the eye. J Gene Med 16: 40–53. [DOI] [PubMed] [Google Scholar]
- Havel, RJ, Eder, HA and Bragdon, JH (1955). The distribution and chemical composition of ultracentrifugally separated lipoproteins in human serum. J Clin Invest 34: 1345–1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salacinski, PR, McLean, C, Sykes, JE, Clement-Jones, VV and Lowry, PJ (1981). Iodination of proteins, glycoproteins, and peptides using a solid-phase oxidizing agent, 1,3,4,6-tetrachloro-3 alpha,6 alpha-diphenyl glycoluril (iodogen). Anal Biochem 117: 136–146. [DOI] [PubMed] [Google Scholar]
- Weinstein, DB, Carew, TE and Steinberg, D (1976). Uptake and degradation of low density lipoprotein by swine arterial smooth muscle cells with inhibition of cholesterol biosynthesis. Biochim Biophys Acta 424: 404–421. [DOI] [PubMed] [Google Scholar]
- Bell, JB, Podetz-Pedersen, KM, Aronovich, EL, Belur, LR, McIvor, RS and Hackett, PB (2007). Preferential delivery of the Sleeping Beauty transposon system to livers of mice by hydrodynamic injection. Nat Protoc 2: 3153–3165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmittgen, TD and Livak, KJ (2008). Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc 3: 1101–1108. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Transposition of the SB system, presented as a dual plasmid and a single plasmid system (in trans or in cis, respectively).
Development of the LDLR KO mice weight on a Western type diet.