

Abstract
Costly coagulation factor VIII (FVIII) replacement therapy is a barrier to optimal clinical management of hemophilia A. Therapy using FVIII-secreting autologous primary cells is potentially efficacious and more affordable. Zinc finger nucleases (ZFN) mediate transgene integration into the AAVS1 locus but comprehensive evaluation of off-target genome effects is currently lacking. In light of serious adverse effects in clinical trials which employed genome-integrating viral vectors, this study evaluated potential genotoxicity of ZFN-mediated transgenesis using different techniques. We employed deep sequencing of predicted off-target sites, copy number analysis, whole-genome sequencing, and RNA-seq in primary human umbilical cord-lining epithelial cells (CLECs) with AAVS1 ZFN-mediated FVIII transgene integration. We combined molecular features to enhance the accuracy and activity of ZFN-mediated transgenesis. Our data showed a low frequency of ZFN-associated indels, no detectable off-target transgene integrations or chromosomal rearrangements. ZFN-modified CLECs had very few dysregulated transcripts and no evidence of activated oncogenic pathways. We also showed AAVS1 ZFN activity and durable FVIII transgene secretion in primary human dermal fibroblasts, bone marrow- and adipose tissue-derived stromal cells. Our study suggests that, with close attention to the molecular design of genome-modifying constructs, AAVS1 ZFN-mediated FVIII integration in several primary human cell types may be safe and efficacious.
The bedrock of hemophilia A treatment is factor VIII (FVIII) protein replacement to restore hemostatic capacity to a level sufficient to enable normal blood coagulation during activities of daily living. Regular prophylaxis with plasma-free recombinant FVIII products is the treatment of choice as it greatly reduces the frequency of acute bleeding episodes, chronic musculoskeletal disability, and improves health-related quality of life.1,2 Of the current global population of about 140,000 people with hemophilia A, 75% receive little or no FVIII replacement.3 Even when FVIII products are affordable, regular prophylaxis is associated with frequent breakthrough bleeding,4 while the need for frequent intravenous access limits acceptance, especially among children for whom effective early intervention is especially important.5
The high cost of FVIII replacement products for more than half the world's population of hemophilia A patients motivates attempts to develop alternative therapies. In vivo gene therapy using viral vectors is appealing for FVIII deficiency. Although it has not yet achieved the same success as gene therapy for hemophilia B,6 improvements in FVIII transgene expression and packaging in AAV vectors appear promising, as are approaches to minimize immune responses to AAV vectors.
An alternative strategy is nonviral delivery of a FVIII transgene into autologous cells ex vivo. Proof of concept was demonstrated in a clinical trial of autologous dermal fibroblasts transfected ex vivo with a plasmid that delivered a B domain-deleted FVIII transgene.7 Since then, several programmable nucleases with the potential to modify genomes with high precision have emerged and can be delivered by nonviral vectors. Among these, zinc finger nuclease (ZFN) technology is currently most advanced towards possible clinical applications. A phase-1 clinical trial of ZFN-mediated CCR5 inactivation in autologous T cells reported no adverse event attributable to ZFN.8 Nonetheless, there is heightened awareness of potential oncogenic complications because clinical trials of transgene integration mediated by gammaretroviral vectors were marred by treatment-induced leukemias and myelodysplasia.9,10,11 The biosafety of all genome-modifying techniques is therefore crucial for clinical approval.
Off-target genome modifications in ZFN-treated cells have not been comprehensively evaluated. They have been identified by screening bioinformatically predicted off-target sites,12,13 in vitro cleavage of biased libraries14 or sequencing the integration sites of integrase-defective lentiviral vectors.15 These studies reported frequencies of off-target events ranging from 1 to 6%. A machine-learning classifier16 has partially resolved the issue of largely nonoverlapping off-target sites generated by different methods but there remain nontrivial method-dependent discrepancies in off-target site identifications.14,15 No single technique for interrogating the genome suffices to reveal off-target modifications comprehensively; neither are there consensus standards for assessing biosafety ex vivo.17,18,19 We have adopted a suite of techniques comprising genomic analysis of bioinformatically predicted off-target sites, whole-genome sequencing (WGS), integration junction and donor vector copy number analysis, and RNA-seq transcriptome profiling to holistically evaluate the biosafety of AAVS1 ZFN-mediated FVIII transgene integration in primary human cells. We assembled codon-optimized AAVS1 ZFN monomers expressed from a single plasmid for stoichiometric expression of each monomer in transfected cells. This study aimed to address potential genotoxicity using AAVS1 ZFN designed for accurate and efficient on-target cleavage activity. In brief, monomers were mutated to suppress homodimerization,20 and additional mutations were introduced to restore nuclease activity which is greatly reduced in obligate heterodimeric FokI catalytic domains.21,22 The fully modified ZFN, named Enhanced Sharkey, could be expected to have substantially lower off-target activity (18.9% homodimer versus 1.7% heterodimer),20 while mutations introduced to restore nuclease activity neither decrease nor increase cleavage specificity of the intended targets.21,22 Thus, Enhanced Sharkey AAVS1 ZFN is novel in incorporating several molecular features that ought to favor accurate and efficient integration. We show that Enhanced Sharkey AAVS1 ZFN integrated a FVIII transgene and induced durable FVIII secretion by primary human umbilical cord-lining epithelial cells (CLECs) with negligible off-target effects.
Results
Evaluation of different AAVS1 ZFN constructs
We tested three AAVS1 ZFN constructs: obligate heterodimer20; Sharkey21 and Enhanced Sharkey22 to quantify integration of pZDonor (50 bp) in K562 cells (Supplementary Figure S1 shows all plasmid constructs used). The obligate heterodimer was mutated as reported (E490K; I538K and Q486E; I499L) to disfavor homodimerization of FokI catalytic domains such that ZFN mainly functions as heterodimers. Two amino acid substitutions (S418P and K441E) in each monomer of the obligate heterodimer were introduced to enhance cleavage activity in the Sharkey variant. Enhanced Sharkey was a further modification to the Sharkey variant (H537R substitution in E490K; I538K; S418P; K441E nuclease and N496D substitution in Q486E; I499L; S418P; K441E nuclease) which was reported to confer superior cleavage activity while suppressing homodimerization. We further compared each construct under conditions of mild hypothermia (30 °C) and at 37 °C. Transient hypothermia increases the efficiency of ZFN-induced changes probably by decreasing global mRNA and protein turnover and enhancing viability of transfected cells.23 Both Sharkey and Enhanced Sharkey ZFNs at 30 °C induced highest efficiencies of donor DNA integration (44.5 and 47.9%, respectively) assessed by restriction fragment length polymorphism (RFLP) (Figure 1). Enhanced Sharkey ZFN was used at 30 °C in all subsequent experiments. Under conditions of efficient transfection (Figure 2a, left panel), integration of 50-bp (Figure 2a, right panel), 3.75-kb (Figure 2b), and 9.1-kb (Figure 2c) donor DNAs into the AAVS1 locus of K562 cells was demonstrated by RFLP and integration junction PCR. Four out of 40 single-cell K562 clones transfected with the 9.1-kb donor were positive on screening by integration junction PCR, yielding an estimated 10% integration efficiency for large donor DNAs. Sequencing of integration junction PCR amplicons confirmed precise integration of donor DNA into intron 1 of PPP1R12C (Figure 2b,c). Cells electroporated with donor DNA alone in the absence of ZFNs did not show evidence of transgene integration by integration junction PCR and RFLP (Figure 2a,c). The 9.1-kb donor DNA delivered a hybrid human-porcine B domain-deleted FVIII cDNA (Supplementary Figure S2).24
Figure 1.

Comparison of site-specific cleavage activities of ZFN constructs. (a) Comparison of AAVS1 ZFN variants and transient hypothermia on cleavage efficiency. Genomic DNA from K562 cells which were coelectroporated with pZDonor and the following AAVS1 ZFN variants: obligate heterodimer (OH), Sharkey or Enhanced Sharkey (see Supplementary Figure S1 legend for details) and cultured at either 37 °C or 30 °C. pEGFP in each electroporation served as an index of transfection efficiency. Site-specific cleavage was evaluated by restriction fragment length polymorphism. Results shown are the mean ± SD of triplicate densitometric measurements of the AAVS1 modified locus expressed as a percentage of the combined unmodified and modified locus. (b) Graphical representation of panel a data. Different percentages of K562 genomic DNA attaining AAVS1-specific integration of the 50-bp donor DNA for each ZFN variant (determined from panel a) (left axis) are shown against comparable efficiencies of pEGFP gene transfer by electroporation (right axis). Data are mean ± SD (n = 3). Integration was significantly greater (P < 0.05) for cells incubated at 30 °C compared to 37 °C for all ZFN variants. At 37 °C, significantly greater integration was achieved with Enhanced Sharkey (P = 0.0024) and Sharkey (P = 0.03) compared to OH. At 30 °C, Enhanced Sharkey and Sharkey had comparable activity and the former was significantly more active than OH (P = 0.02).
Figure 2.

AAVS1 locus-specific integration of different size donor DNAs. (a) ZFN-dependent integration of donor DNA. K562 cells were coelectroporated with pEGFP (reporter for transfection efficiency) and pZDonor with or without AAVS1 ZFN mRNA. (Left): Brightfield and fluorescence images of transfected cells. Bar = 100 µm (Right): PCR spanning the integration junction (top) and RFLP assay (bottom) performed on genomic DNA from cells 4 days after treatment with pZdonor only; or 4 (Day 4) and 16 days (Day 16) after treatment with pZdonor and AAVS1 ZFN mRNA provided evidence of site-specific integration of 50-bp donor DNA. Control PCR amplified a 900-bp region of the AAVS1 locus. (b) Accuracy of Enhanced Sharkey AAVS1 ZFN-mediated integration of pZdonor EGFP. Left: PCR amplification of the left and right integration junctions performed on genomic DNA of K562 cells coelectroporated with pZDonor EGFP and Enhanced Sharkey ZFN with or without G418 selection. Right: DNA sequence chromatogram of left (top) and right (bottom) junctional PCR amplicons. Vector sequences are underlined in blue; Enhanced Sharkey AAVS1 ZFN recognition half-sites are underlined in red. Control PCR amplified a 900-bp region of the AAVS1 locus. WT K562 denotes untransfected control K562 cells. (c) Accuracy of Enhanced Sharkey AAVS1 ZFN-mediated integration of pZDonor Hybrid FVIII. Left: PCR amplification of the left and right integration junctions performed on genomic DNA of K562 cells electroporated with pZDonor Hybrid FVIII only or coelectroporated with Enhanced Sharkey ZFN followed by G418 selection. Right: DNA sequence chromatogram of left (top) and right (bottom) junctional PCR amplicons. Vector sequences are underlined in blue; Enhanced Sharkey AAVS1 ZFN recognition half-sites are underlined in red. Control PCR amplified a 900-bp region of the AAVS1 locus. WT K562 denotes untransfected control K562 cells. White vertical lines in the gel images demarcate lanes that were merged for clarity.
Enhanced Sharkey AAVS1 ZFN activity in CLECs
RT-PCR showed highest levels of ZFN expression 8–48 hours after electroporation with Enhanced Sharkey AAVS1 ZFN plasmids (Supplementary Figure S3a). ZFN protein expression was also higher in CLECs subjected to transient (1–3 days) mild hypothermia after transfection compared to CLECs which were never exposed to hypothermia (Supplementary Figure S3b). ZFN activity was assessed by the CEL-1 assay (reporting DNA break repair by nonhomologous end joining), integration junction PCR and RFLP (evidence of site-specific donor integration). CEL-1 assay results showed significantly higher ZFN activity when right and left Enhanced Sharkey AAVS1 ZFN monomers were delivered as a single construct (43 ± 1.9%) compared to ZFN monomers delivered as two constructs (35 ± 1.8%) (P = 0.045) (Supplementary Figure S4a). Thus, subsequent experiments were conducted using a single plasmid that delivered both Enhanced Sharkey AAVS1 ZFN monomers and transient hypothermia. Under these conditions, integration junction PCR and RFLP analysis showed no donor DNA integration in CLECs electroporated with pZDonor only (Supplementary Figure S4b). Varying doses of Enhanced Sharkey AAVS1 ZFN and donor DNA were tested to determine conditions that induced least cellular toxicity quantified by cell viability and phosphorylated histone H2AX. The overall results showed that coelectroporation of 5–10 µg Enhanced Sharkey AAVS1 ZFN with 10 µg donor DNA induced least cellular toxicity (Supplementary Figure S5).
Gene trap integration of FVIII transgene
As the size of FVIII transgene donor DNA was relatively large (9 kb) and because accurate integration was paramount, we first assessed the accuracy of genome targeting in CLECs using gene trap donor vectors, AAVS1 SA-2A-puromycin-pA, and AAV-CAGGS-EGFP,12 which delivered smaller sized donor DNAs. PCR of puromycin-resistant CLECs demonstrated integration of 1.3- and 4.2-kb donors, respectively, using genome-specific primers which overlapped the intended integration site (Supplementary Figures S6 and S7). Sequencing of PCR amplicons confirmed complete integration of the 1.3-kb donor. Most of the integrated 4.2-kb donor was sequenced (except for a 1-kb GC-rich region within the CAGGS promoter), and showed no insertions, deletions or rearrangements. PCR of integration junctions and donor DNA were negative in untreated CLECs.
Using the same gene trap strategy, integration junction and long PCR of CLECs coelectroporated with the FVIII transgene donor DNA and Enhanced Sharkey AAVS1 ZFN showed integration of the complete donor in intron 1 of PPP1R12C (Figure 3a,b), confirmed by AAVS1 site-specific PCR and sequencing of the complete FVIII transgene (Figure 3b,c; Supplementary Table S1). Puromycin-resistance selected CLECs with the integrated transgene (puro-CLECs). This selected population secreted substantially higher FVIII levels (2,131 ± 17 mU/106 cells/24 hours) 37 days postelectroporation than unselected CLECs (952 ± 8 mU/106 cells/24 hours) 1 day postelectroporation (Figure 3d).
Figure 3.

AAVS1 locus-specific integration of FVIII donor DNA in CLECs. (a) Schematic of donor DNA integrated in AAVS1 locus by homology-directed integration (not drawn to scale). FVIII transgene was expressed from the human ferritin light chain promoter. Heavy red bars indicate homology arms. Dashed red lines indicate AAVS1 locus genomic DNA flanking the homology arms. Arrows indicate primers for integration junction and overlapping PCR to document integration of the complete FVIII transgene DNA. (b) Accuracy of Enhanced Sharkey AAVS1 ZFN-mediated integration of pSA-2A-Puro Hybrid FVIII. Integration junction PCR (JPCR) showing donor DNA integration in intron 1 of PPP1R12C. Left and right JPCR amplified diagnostic 5′ and 3′ genome junctions, respectively, created by donor integration using primers specific to donor DNA and adjacent genomic regions beyond the homology arms (all primer sequences in Supplementary Table S5). Control positive PCR amplified a 900-bp sequence in the AAVS1 locus 2 kb away from the integration site. DNA size markers are 10, 3, and 1 kb. The right panel shows two different long PCRs encompassing the full-length transgene performed with donor- and locus-specific primers. Only puro-CLEC genomic DNA was positive for the predicted 6.9- and 4.2-kb amplicons which were sequenced to confirm integration of the complete FVIII transgene cDNA (Supplementary Table S1). DNA size markers of 10, 5, and 3 kb are indicated. White vertical line in the gel image demarcates lanes that were merged for clarity. (c) JPCR amplicons were sequenced to confirm site-specific left and right integration junctions. Donor-specific sequences are underlined in green and Enhanced Sharkey AAVS1 ZFN binding sites are underlined in red. (d) FVIII activity in conditioned media of wt- and puro-CLECs 1 day and 37 days postelectroporation. Control CLECs were transfected with FVIII donor plasmid only (C1-CLEC) or cotransfected with FVIII donor plasmid and ZFNs without puromycin selection (C2-CLEC). Puro-CLECs were transfected like C2-CLEC and subjected to puromycin selection (0.5 mg/ml for 7 days beginning 4 days after electroporation). Data are mean ± SEM; n = 3. (e) Copy number of total integrated donor DNA and integration junctions at AAVS1 locus relative to copy number of a control locus on chromosome 19q13.42. Digital droplet PCR of puro-CLEC genomic DNA was performed to determine copies of integrated vector, left integration junction, right integration junction and two control loci in chromosome 19q13.42. Puro-CLEC refers to puromycin-selected cells with stable integration of FVIII transgene 1 month postelectroporation. Reactions performed without template DNA were negative controls. Data are mean ± SD; n = 4. (f) Quantitative RT-PCR of PPP1R12C transcript levels (exons 4–6) in wt-CLECs and puro-CLECs. Data are mean ± SEM; n = 3.
Quantifying on- and off-target FVIII transgene integration
Quantifying on-target versus off-target transgene integrations by digital droplet PCR of integration junction amplicons (on-target) versus vector-specific amplicons (on- and off-target integrations) showed no significant difference in copy number, relative to control 1 diploid locus, of total vector (0.55 ± 0.11 copies) compared with left junction (0.57 ± 0.07 copies; P = 0.85) and right junction (0.65 ± 0.05 copies; P = 0.35) amplicons, suggesting minimal off-target integrations (Figure 3e). RT-PCR showed twofold lower PPP1R12C expression in puro-CLECs compared to untreated CLECs, consistent with monoallelic transgene integration (Figure 3f).
Analysis of in silico predicted off-target sites
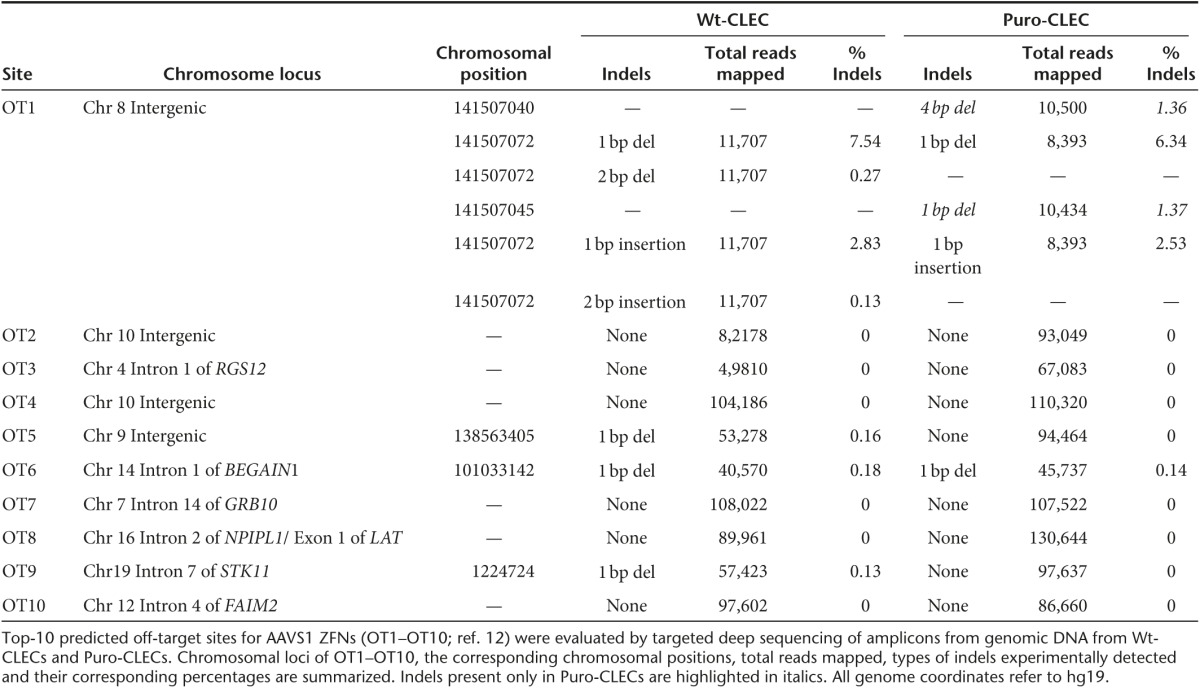
Targeted deep sequencing of the predicted ten most likely off-target sites (OT1–OT10)12 (Supplementary Table S2) showed 4- and 1-bp deletions at low frequency (1.36 and 1.37%, respectively) only in OT1, an 8q24.3 intergenic locus. No other indels in OT2–OT10 specific to puro-CLECs were detected (Table 1). Sensitivity of indel detection was determined by spiking wild-type AAVS1 amplicon with a synthetic amplicon having a 5-bp deletion at the AAVS1 target site at molar ratios of 1:10 to 1:1,000 (mutant:wild type). Deep sequencing detected the spiked deletion at all ratios with a highly linear relationship between spike-in and observed levels (R2 = 0.999), establishing a detection sensitivity of 0.1% (Supplementary Figure S8).
Table 1. Targeted deep sequencing of in silico predicted 10 most likely AAVS1 ZFN off-target sites.
Whole-genome sequencing
VarScan analysis25 of WGS data generated 2,736 high-confidence indels specific to puro-CLECs, but none were in silico predicted off-target sites.26 We considered only loci harboring multiple indels to be true off-target events.27 Genomic coordinates of high-confidence indels revealed 196 loci with 2–5 indels each. All were in repetitive DNA regions. Three off-target loci had the highest combined number of indels and single nucleotide polymorphisms (SNPs) (4–6) and also higher proportions (33–50%) of mapped reads than on-target indels (19–46%). On-target indels were identified by aligning all reads to a 980-base sequence of the reference AAVS1 locus (all genome coordinates refer to hg19). The ZFN cleavage site was between positions 401 and 430 of this sequence. Four of 21 mapped reads (19%) showed a single A deletion at position 400 of this sequence. Twenty-three of 50 mapped reads (46%) showed either a single T deletion or a single C insertion at position 407. The putative off-target events mapped to intergenic regions in chr2_237939600 (indel 1); chr21_15213700 (indel 2); and chr8_1365500 (indel 3). All lacked potential AAVS1 ZFN-binding sites. The nearest protein-coding genes were COPS8 (54 kb from indel 1), C21orf15 (1.7 kb from indel 2), and DLGAP2 (84 kb from indel 3). Indel 1 was a false-positive indel because sequence data showed only two single-base substitutions (Figure 4a). Indel 3 comprised insertions of two different satellite DNAs. The larger insertion (1,489 bp) was a 15-fold expansion of satellite DNA while the smaller insertion (186 bp) was a twofold expansion of a different satellite (Figure 4b). (Indel 2 could not be sequenced continuously owing to highly repetitive sequence motifs.) Tandem repeats are intrinsically unstable because of replication slippage and unequal sister chromatid exchange during mitosis. Off-target cleavage by FokI possibly facilitated repeat expansion by strand invasion. Our data indicate that off-target events caused by FokI dimerization independent of ZFN binding are rare.
Figure 4.

Off-target genomic changes associated with Enhanced Sharkey AAVS1 ZFN-mediated FVIII transgene integration. (a) DNA sequence of indel 1. Two single-base substitutions were present in this position without either insertion or deletion. (b) Sequence of indel 3. Indel 3 comprised expansion of two different satellite DNAs (weblogo.berkeley.edu). Each red and green rectangle represents a single copy of the respective satellite DNA. (c) Putative breakpoints of unbalanced structural variants (SVs) in puro-CLEC from bioinformatic analysis of WGS data. All chromosome positions refer to hg19. (d) Relative genome copy number at putative breakpoints by quantitative PCR. The mean CT value at each breakpoint locus was normalized to its own actin CT value. The ratio of normalized copy number of puro-CLEC:wt-CLEC at each putative breakpoint is shown for SV2 and SV3. Genome copy number could be quantified at only one putative breakpoint of SV1 and SV4. Data are mean ± SD of triplicate reactions.
WGS analysis suggested four chromosomal rearrangements, all having unbalanced genome copy number. Three were interchromosomal and one was an intrachromosomal structural variant (Figure 4c). Genomic PCR repeatedly failed to detect all putative abnormal chromosomal junctions, suggesting that the rearrangements were another false positive finding. However, as it was important to determine if ZFN treatment had induced structural changes in chromosomes, we employed a different validation method based on relative genome copy number analysis. As all breakpoint loci in the four structural variants (SV) were predicted to have unbalanced genome copy number, we used quantitative PCR to determine the copy number of each breakpoint locus in genomic DNA of puro-CLECs relative to the same breakpoint locus in wild-type CLECs (wt-CLECs). Relative copy number at each breakpoint locus was expressed as the ratio of normalized CT values of puro- and wt-CLECs. Normalization was necessary because SV1–SV4 breakpoint loci were amplified at different annealing temperatures to achieve specificity of amplification. The CT value of actin locus amplification was used to normalize the CT value of each breakpoint locus in the same experiment. Breakpoint 2 locus of SV1 could not be amplified. The candidate breakpoint loci of SV4 were 8 bp apart and were amplified as a single locus. The ratios of puro-CLEC to wt-CLEC genome copy numbers of 0.97–1.07 at all candidate breakpoints analyzed were not consistent with a substantive frequency of unbalanced structural alterations (Figure 4d). Based on the absence of abnormal chromosomal junctions by PCR amplification and the absence of abnormal genome copy number at breakpoint loci by experimental validation, it was unlikely that ZFN treatment had induced biologically meaningful chromosomal rearrangements.
RNA-seq
RNA-seq of wt-CLECs and puro-CLECs identified 17,751 transcripts in total, of which only 57 (0.3%) were overexpressed and 33 (0.2%) were underexpressed at least twofold in puro-CLECs (Figure 5a,b; Supplementary Table S3). FVIII was among the overexpressed transcripts. Pathway analysis showed that 10 dysregulated transcripts mapped to cytokine–cytokine receptor interaction by DAVID analysis28 (Benjamini-corrected P = 0.011) (Supplementary Figure S9), consistent with PPP1R12C's role in inflammation.29 Although seven dysregulated genes were potential proto-oncogenes in a consolidated catalogue of more than 1,600 oncogenes (http://www.bushmanlab.org/links/genelists), none mapped to any of the canonical cancer pathways in KEGG.
Figure 5.

Gene Ontology classification of over- and underexpressed genes in puro-CLECs and quantitative RT-PCR of selected transcripts. RNA-seq of wt-CLECs and puro-CLECs identified 17,751 transcripts in total, of which only (a) 57 were overexpressed and (b) 33 were underexpressed at least twofold in puro-CLECs. (c) Real-time RT-PCR was performed on wt-CLEC and puro-CLEC to confirm transcriptome data of selected neighboring genes located within 1Mb of the AAVS1 integration site (left) and interacting partners of PPP1R12C (right). GAPDH-normalized transcript levels (determined by the 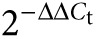 method) in puro-CLEC are shown relative to wt-CLEC. Data are mean ± SEM; n = 2 experiments per group and 3 replicates per sample.
method) in puro-CLEC are shown relative to wt-CLEC. Data are mean ± SEM; n = 2 experiments per group and 3 replicates per sample.
In view of Enhanced Sharkey AAVS1 ZFN's minimal footprint on the transcriptome, we broadened our analysis to investigate possible indirect effects of haploinsufficient PPP1R12C expression on its interacting protein partners and downstream effectors. RNA-seq data of 74 other protein phosphatases, 50 myosin-related and downstream genes, 29 known protein interacting partners, and 43 neighboring genes of PPP1R12C within 1 Mb centered on the AAVS1 integration site (Supplementary Table S4) showed dysregulation of only DUSP6, a PPP1R12C interacting partner, whose expression was 5.5-fold higher in puro-CLECs. Quantitative RT-PCR confirmed 4.2-fold increase in DUSP6 expression in puro-CLECs (Figure 5c). DUSP6 negatively regulates ERK1/230 and high expression impairs epithelial–mesenchymal transition and tumorigenicity.31 Proliferation of puro-CLECs was slightly but not significantly reduced (Supplementary Figure S10), possibly reflecting the combined effects of high DUSP6 expression and PPP1R12C haploinsufficiency, the latter being required for completion of mitosis and cytokinesis.32
ZFN-mediated cleavage at the AAVS1 locus was also readily induced in primary human dermal fibroblasts, human bone marrow- and adipose tissue-derived stromal cells with overall efficiencies of 20% or higher (Supplementary Figure S11). Using the same conditions which integrated FVIII transgene in CLECs, primary human dermal fibroblasts, bone marrow- and adipose tissue-derived stromal cells could also be induced to integrate and secrete transgenic FVIII (Figure 6; Supplementary Figure S12).
Figure 6.

Enhanced Sharkey AAVS1 ZFN activity and FVIII transgene secretion in other primary human cell types. (a) AAVS1 ZFN-mediated FVIII transgene integration in adult human primary cells. PCR of genomic DNA from puromycin-resistant human dermal fibroblasts (Fibroblast FVIII stables) and bone marrow stromal cells (BMSC FVIII stables) coelectroporated with FVIII donor plasmid and Enhanced Sharkey AAVS1 ZFN showed the presence of integrated vector (521 bp), left (602 bp) and right (551 bp) integration junctions. Control PCR amplified a 510-bp region of the AAVS1 locus. Minus template lanes show negative control reactions performed without genomic DNA. White vertical line in the gel image demarcates lanes that were merged for clarity. (b) FVIII secretion by ZFN-modified cells. FVIII activity in conditioned media of wild type- and puromycin-resistant human dermal fibroblasts (152 ± 14 mU/106 cells/24 hours) and bone marrow stromal cells (253.3 ± 6.4 mU/106 cells/24 hours) 6 weeks postelectroporation. Data are mean ± SEM; n = 3.
Discussion
Leukemias and preleukemia complicating retroviral gene therapy trials have refocused emphasis on biosafety. Genome editing by nonvirally delivered programmable nucleases could be well suited for safe and efficacious therapeutic transgene integration by designing constructs which avoid integration in potentially hazardous genomic regions (i.e., regulatory elements, transcription start sites, and within transcription units). Although five ZFN clinical trials have been completed or are in progress (www.clinicaltrials.gov; accessed on 8 June 2015), assessment of genotoxic risks sufficiently robust to meet clinical standards is a vexing challenge. There is currently no consensus on how modified genomes should be examined and analyzed for off-target alterations. Moreover, each application should be assessed independently as many technical and biological factors affect the accuracy of genome editing.
We selected the AAVS1 locus for integrating the FVIII transgene because other investigators have reported evidence suggesting it is likely to be a safe genomic harbor, although this has not been rigorously demonstrated to date.33,34,35 The locus has endogenous insulator elements which favor durable and reproducible transgene expression in different cell types, while shielding neighboring genes from unintended transcriptional effects.33,36 These features have enabled different integrated transgenes to be effectively expressed from this locus.37,38 Moreover, disruption of the AAVS1 locus has no known pathological or functional consequences.39
We integrated a hybrid human-porcine FVIII transgene in primary human somatic cells as a test bed for autologous FVIII cell therapy. Hybrid human-porcine FVIII proteins are known to be better secreted than human FVIII,24,40 possibly because the hybrid protein activates the unfolded protein response less than recombinant human FVIII protein.41 Nonhematopoietic primary human cells have limited replicative capacity in vitro which makes derivation and in vitro expansion of single-cell clones for clinical therapy impractical. Thus, bulk cell populations of gene-modified CD34+ hematopoietic stem cells were used in clinical trials of SCID-X1, chronic granulomatous disease and Wiskott–Aldrich syndrome.42,43,44 We reasoned that profiling ZFN-modified CLECs by different methods would more likely identify potentially oncogenic alterations than reliance on a single technique. To this end, we profiled ZFN-modified CLECs by targeted deep sequencing of the in silico predicted off-target sites, complemented with WGS. This showed a low frequency of microdeletions in only one predicted off-target site in 8q24.3 which was 38.2-kb distant from TRAPPC9 (on the 5′ side) and 14.6 kb from CHRAC1 (on the 3′ side). Neither gene was transcriptionally dysregulated in puro-CLECs.
WGS is used increasingly for unbiased identification of SNPs, indels, and rearranged chromosomes. Indels have been identified from data obtained at 24× and 28.4× depths of coverage.45,46 SNPs were detected with 95% sensitivity at 14× depth47 but the depth for comparable sensitivity of indel detection is unknown. Our results showing a low incidence of indels mirror the absence of SNPs and indels induced by ZFN correction of SOD1 mutation in human induced pluripotent stem cells.48 Aside from cost, WGS has limitations as a standalone method for identifying off-target events. Even at high depths of coverage, sequencing artifacts and bioinformatic filters applied to WGS data generate false results. Furthermore, to identify off-target events present at frequencies of 10, 1, and 0.1% with 95% sensitivity would require sequencing 15, 150 or 1,500 diploid single-cell clones, respectively.49 Detecting a mutation present in 1% of cells sequenced at 500× depth would, on average, be based on only 2.5 reads.50 These considerations make WGS impractical as the sole or primary technique for biosafety assessment given the current state of technology and costs.
Recognizing these limitations, we broadened genomic profiling with quantitative analysis of integration junction and transgene copy number and RNA-seq. Overall, our results revealed minimal evidence of off-target alterations and none that would be considered potentially oncogenic. PPP1R12C encodes a regulatory subunit of protein phosphatase 1 which is involved in mitotic exit, apoptosis, DNA damage response, signaling and metabolism. Its interacting protein partners (CAMKK1, CDC42BPB, MPRIP, MYL2, MYL5, MYL7, MYL9, MYL10, MYL12A, MYL12B, MYLPF, PHLPP2 (string-db.org)), PRKG1, pro-IL-16, and SRF function in cell proliferation, differentiation, survival, migration, apoptosis, transcription, cytoskeletal organization, and signal transduction. Given these multiple functions, RNA-seq data of puro-CLECs gave very little evidence that haploinsufficiency of PPP1R12C was functionally deleterious. This is concordant with the tolerance of embryonic stem cells and induced pluripotent stem cells for AAVS1 ZFN-induced biallelic disruption of PPP1R12C.12,33,34
We tested three types of readily procured primary human somatic cells, i.e., dermal fibroblasts, bone marrow- and adipose tissue-derived stromal cells. All three cell types were permissive for reasonably high levels of DNA cleavage activity using the same Enhanced Sharkey AAVS1 ZFN construct. Like puro-CLECs, ZFN-modified primary fibroblasts and bone marrow-derived stromal cells also secreted FVIII (Figure 6). Among the cell types tested in this study, CLECs and bone marrow-derived stromal cells are promising choices for autologous cell therapy. CLECs are more abundant in their primary source, a waste product of parturition (about 6 × 109 cells per umbilical cord), than bone marrow-derived stromal cells (about 0.5–3 × 104 cells/ml bone marrow aspirate).51,52 They are readily expanded ex vivo to clinically relevant numbers because they do not undergo spontaneous replicative senescence which occurs with bone marrow-derived stromal cells at comparable population doublings.53 CLECs highly express a nonclassical human leucocyte antigen, HLA-G,54 which has intrinsic tolerogenic and immunosuppressive activity.55,56 CLEC banks have been established in anticipation of their utility for autologous or allogeneic cell therapy applications, including treatment of infants and children with hemophilia. Nonetheless, bone marrow-derived stromal cells have an extensive record of safety in clinical trials. The US National Institutes of Health registry of approved clinical trials (clinicaltrials.gov; accessed on 1 October 2015) lists 92 completed and ongoing “bone marrow stromal cell” trials and 250 “bone marrow mesenchymal stem cell” trials. This depth of clinical experience coupled with the ease of obtaining fresh bone marrow justifies development of bone marrow-derived stromal cells for cell therapy of adult patients. These considerations point to potentially wide application of our approach for autologous cell therapy for pediatric and adult patients using cells whose proficiency for synthesizing and secreting therapeutic proteins is well known.
In summary, our study has shown that Enhanced Sharkey AAVS1 ZFN designed for high biosafety has broad applicability for autologous cell therapy using several primary human cell types and could be developed as potential FVIII-secreting bioimplants with a low risk of unintended oncogenic effects.
Materials and Methods
Cell culture. K562 cells were purchased from the American Type Culture Collection (Manassas, VA). Primary human cells (dermal fibroblasts, adipose-derived stromal cells, bone marrow-derived stromal cells and CLECs) were derived and provided by CellResearch Corporation, Singapore with National University Health System IRB approval.
K562 cells were cultured in Iscove's modified Eagle medium (Sigma-Aldrich, St. Louis, MO) supplemented with 10% fetal bovine serum (Hyclone, Waltham, MA). All primary human cells, except CLECs, were cultured in Dulbecco's modified Eagle medium (DMEM)—25 mmol/l glucose (Sigma-Aldrich) supplemented with 10% fetal bovine serum. CLECs were cultured in Medium 171 (Cascade Biologics, Portland, OR) supplemented with 50 ng/ml IGF-1, 50 ng/ml PDGF-BB, 5 ng/ml TGF-β1, and 5 ng/ml insulin (all from R&D Systems, Minneapolis, MN).
AAVS1 ZFNs. DNA encoding the wild-type catalytic domain of FokI (pST1374; Addgene; http://www.addgene.org) was mutagenized for heterodimerization.20 The OH (obligate heterodimer) ZFN had two amino acid changes in the FokI monomer fused to the right AAVS1 homology arm (E490K and I538K) and in the monomer fused to the left homology arm (Q468E and I499L). We made further variants of OH ZFN to enhance cleavage activity:
1. The Sharkey variant had S418P and K441E substitutions in both right and left monomers.21
2. The Enhanced Sharkey variant had additional amino acid substitutions: S418P, K441E, and H537R (right FokI monomer) and S418P, K441E, and N496D (left FokI monomer).22
The final ZFN constructs combined both right and left AAVS1-FokI expression cassettes in single plasmids (Supplementary Figure S1a).
Codon-optimized DNA encoding a pair of zinc finger peptides for the AAVS1 locus12 was commercially synthesized (DNA2.0, Menlo Park, CA) and ligated to their corresponding mutagenized FokI monomers in separate plasmid constructs.
Donor DNA. Three plasmids having a neomycin resistance marker were used to integrate donor DNAs of increasing sizes into intron 1 of PPP1R12C (Supplementary Figure S1b):
pZDonor (contains 1.5-kb homology to the AAVS1 locus bisected by a 50-bp multiple cloning site; Sigma-Aldrich)
pZDonor EGFP (3.75-kb CMV-promoter-GFP excised from pEGFP-C1 (Clontech, Mountain View, CA) cloned in pZDonor)
pZDonor Hybrid FVIII (9.1-kb donor encoding human ferritin light chain promoter-hybrid FVIII cDNA cloned in pZDonor; described below).
For integration in primary human cells, donor vectors with a promoterless puromycin resistance selection gene were assembled on pAAVS1 SA-2A-puromycin-pA (Addgene plasmid #22075) and pAAV-CAGGS-EGFP (Addgene plasmid #22212) (Supplementary Figures S1c, S6 and S7).
Hybrid FVIII cDNA. B domain-truncated human-porcine FVIII cDNA consisted of porcine A1 and A3 domains, human signal peptide, A2, residual B (comprising the first 266 amino acids and 8 glycosylation sites), C1 and C2 domains.24 Overlap PCR was used in domain assembly (Supplementary Figure S2). A1 and A3 domains were obtained by RT-PCR of total pig liver RNA based on the reference porcine cDNA sequence (NM_214167.1). Human domains were amplified from complete human FVIII cDNA in pSP64-VIII (American Type Culture Collection, Manassas, VA).
Restriction fragment length polymorphism. RFLP was used to quantify site-specific integration of pZDonor-AAVS1. Two hundred nanograms of genomic DNA was extracted from cells 4 days after electroporation of 10 µg pZDonor-AAVS1 in the absence or presence of AAVS1 ZFN. PCR primers amplified a 1.9-kb region spanning the AAVSI integration site. Amplicons digested with HindIII were resolved by electrophoresis in 8% polyacrylamide gels, poststained with ethidium bromide and imaged (BioRadGel Doc 2000 transilluminator). Two bands (1 and 0.9 kb) indicated donor integration, while a single 1.9-kb band indicated no integration. The intensity and volume of DNA bands were quantified by Quantity One software (Bio-Rad, Hercules, CA).
CEL-1 assay. ZFN activity was assayed by the presence of nonhomologous end-joining repair detected by CEL-1 nuclease using reagents and instructions of the Surveyor mutation detection kit (Transgenomic, Omaha, NE).
Flow cytometry. Transfection efficiencies were evaluated 24 hours after electroporation by fluorescence-activated cell analysis of GFP-expressing cells (BD FACSCalibur flow cytometer; 488 nm argon laser; 530/30 bandpass filter).
DNA damage response was assessed by histone H2AX phosphorylation. CLECs were fixed in 3.7% formaldehyde in phosphate-buffered saline/90% methanol 2 days after electroporation, permeabilized (0.5% Triton-X100, 2% bovine serum albumin) for 10 minutes and incubated with Alexa Fluor 647-conjugated-anti-phosphohistone H2AX antibody (Ser139) (1:40 dilution; Cell Signaling Technology, Danvers, MA) for 1 hour at 25 °C. Cells were washed twice with phosphate-buffered saline, resuspended in 500-µl phosphate-buffered saline and filtered through a 40-µm nylon mesh. Flow cytometry was performed with a 633-nm He-Ne laser and 661/16 bandpass filter.
Data were analyzed using FlowJo v7.22 (FLOWJO, Ashland, OR).
Gene transfer. Two million K562 cells suspended in 100-µl Amaxa Cell Line Nucleofactor Kit V solution containing 10-µg donor DNA plasmid and 5-µg ZFN plasmid or 5-µl AAVS1 ZFN mRNA (Sigma-Aldrich) were electroporated using the T016 setting in Amaxa Nucleofector I (Lonza, Basel, Switzerland). Where indicated, cells were cotransfected with pEGFP to assess efficiency of gene transfer. Stably integrated cells were selected by culture in G418 (0.8 mg/ml) for 14 days.
Two to 10 million primary human cells were electroporated (Amaxa 4D Nucleofector; setting CM113) with 10-µg donor DNA plasmid and 5-µg ZFN plasmid, as indicated in the figure legends. Stably integrated CLECs were resistant to puromycin (0.5 µg/ml for 7 days) (puro-CLECs). Wt-CLECs were untreated with plasmids.
FVIII assay. FVIII activity in overnight conditioned media of wt- and puro-CLECs was assayed using the Coatest SP FVIII kit (Chromogenix, Molndal, Sweden) and recommended protocol. Data are expressed as mUnits FVIII/million cells/24 hours (mean ± SD of triplicates).
Integration junction PCR. Integration junction PCR was performed on 200-ng genomic DNA using DyNAzyme EXT DNA polymerase (Thermo Scientific, Rockford, IL) and primers specific to the integrated vector and genomic sequences immediately adjacent to the integration site. Positive control PCR amplified a separate region within the AAVS1 locus 2 kb away from the integration site. (All primer sequences are in Supplementary Table S5). Integration junction PCR products were sequenced to verify their identity. Overlapping long PCR and sequencing confirmed integration of the complete hybrid FVIII transgene.
Digital droplet PCR. Fifty nanograms genomic DNA from wt- and puro-CLECs, 1 µmol/l of each primer and 0.25 µmol/l BHQ1-FAM probes (Sigma-Aldrich) were added to QX200 dd PCR supermix (Bio-Rad) in a final reaction volume of 20 μl and transferred to QX100 Droplet Generator (Bio-Rad) followed by 40 cycles of PCR amplification (annealing temperature 61 °C and extension time 1 minute per cycle) using C1000 Touch Thermal Cycler. Droplet PCR products were read on a QX100 Droplet Reader (Bio-Rad) and data were analyzed using QuantaSoft software. Control 1 (forward: cctgccttaaacccagccag; reverse: atgacctcatgctcttggccctcgta; probe: aaccaccccagcagatactct) and control 2 (forward: tcccctcccagaaagacctgc; reverse: tcccctcccagaaagacctgc; probe:tacctaacgcactcctgggtga) amplified a genomic locus in human chromosome 19q13.42. Reactions designed for the left (genome specific forward: ttcgggtcacctctcactcc; vector specific reverse: gacgcgcgtgaggaagagttc; probe: aggcgcaccgtgggcttgt) and right (vector specific forward: ctgtggaatgtgtgtcagttag; genome specific reverse: ggctccatcgtaagcaaacc; probe: cgcctctgcctctgagctat) integration junctions amplified AAVS1 site-specific-integrated vector while all integrated vectors regardless of genomic location were detected by vector specific PCR (forward: cgagatgaccgagtacaag; reverse: gctcgtagaaggggaggttg; probe: tcaccgagctgcaagaact). Each PCR reaction was performed in quadruplicate. Data are mean ± SD.
Amplicon deep sequencing. The predicted 10 most likely potential off-target sites previously published for AAVS1 ZFNs12 (OT1–OT10; Supplementary Table S2) were investigated by massively parallel paired-end sequencing (MiSeq, Illumina, San Diego, CA) of amplicons from the AAVS1 integration site and OT1–OT10 of wt- and puro-CLECS (primer sequences in Supplementary Table S5). A commercially synthesized DNA fragment (GenScript, Piscataway, NJ) similar to the AAVS1 locus sequence except for a 5-bp deletion between the ZFN binding half-sites was spiked into the wild-type AAVS1 locus amplicon at molar ratios of 1:10, 1:100, 1:500 and 1:1,000 to determine the sensitivity of indel detection (Supplementary Figure S8). Library construction (Nextera XT DNA Sample Preparation Kit, Illumina) and sequencing were performed by AITbiotech, Singapore.
Whole-genome sequencing. WGS was performed by BGI (Shenzhen, China). Libraries with 500-bp inserts were prepared from 5 μg randomly fragmented genomic DNA from wt- and puro-CLECs and sequenced on HiSeq 2000 (Illumina). Adapter sequences, duplicate reads (marked using Picard tools; picard.sourceforge.net), low-quality reads (more than half the bases in a read having a quality ≤5) and reads with >10% unknown bases were removed from the raw data. Paired-end clean reads (90 bases) from both samples were aligned to the reference human genome (hg19) using Burrows–Wheeler Aligner (BWA)57 and stored in BAM format files. Both samples were sequenced to 23× depth. WGS quality metrics are in Supplementary Table S6.
RNA-seq. Total RNA from wt- and puro-CLECs was qualitatively assessed by Bioanalyzer (Agilent Technologies, Santa Clara, CA) and quantified by Qubit fluorometer (Life Technologies, Beverly, MA). Two micrograms of high quality RNA (RIN > 9) was used for library preparation following the standard protocol (Illumina TruSeq RNA Sample Prep v2 kit). Briefly, poly-A mRNA was purified on oligo-dT magnetic beads. After purification, mRNA was fragmented (150–250 bp) and converted into first-strand cDNA using reverse transcriptase and random primers. Second-strand cDNA synthesis was performed with DNA polymerase I and RNaseH. cDNA fragments were next blunt ended, a single “A” base added for ligation to indexed adapters with complementary T-overhangs. The indexed products were purified and enriched with PCR to create the final cDNA library. Indexed libraries were validated for size and purity by Bioanalyzer, and quantified using Quant-iT PicoGreen dsDNA assay kit (Life Technologies). Libraries were normalized to 10 nmol/l by real-time PCR (iTaq Universal SYBR Green Supermix; BioRad) and equal volumes were pooled. Pooled libraries were denatured and diluted to 20 pmol/l for clustering on the cBot before loading on the HiSeq 2000 to generate paired-end reads of 76bp.
Bioinformatic data analyses
Detection of on-target indels. All reads of the puro-CLEC sample were aligned against a 980-bp sequence containing the intended ZFN cleavage site with e-value cutoff 0.00001. All aligned reads were mapped back to the reference human genome (hg19) using SMALT (www.sanger.ac.uk/resources/software/smalt/) to generate a sequence alignment file. VarScan v2.3.6 was used to detect indels using command “pileup2indel”.25
Detection of off-target indels. All wt- and puro-CLEC reads aligned to hg19 were analyzed for somatic variants using VarScan v2.3.6. The sequence alignments in binary alignment format were transformed to SAMtools pileup format using SAMtools.58 The resulting pileup files were submitted for somatic variant calling by VarScan using the command “somatic” taking wt-CLEC as “normal” and puro-CLEC as “tumor” (java –jar VarScan.jar somatic <pileup file of WT sample> <pileup file of PURO sample> output). Somatic variants identified by VarScan were further classified as high-confidence (.hc) or low-confidence (.lc) using the command “processSomatic” (java –jar VarScan.jar processSomatic output.indel). The high-confidence somatic variants were analyzed further to generate the final list of candidate indels for experimental validation.
Putative off-target sites in hg19 were identified in silico using ZFN-Site.26 The left and right ZFN half-sites were ATCCTGTCCCTA and ACCCCACAGTGG, respectively. The allowed spacing was 5 or 6 bp and the maximum number of mismatches per half-site was set to 2. There was no overlap between high-confidence somatic indels and in silico predicted AAVS1-ZFN off-target sites.
We next adopted another method to identify possible off-target effects. Using an in-house Perl script, we scanned the reads for hot spot genomic regions in which multiple overlapping high-confidence somatic indel variants mapped. These genomic loci were subjected to experimental validation by PCR-Sanger sequencing.
Identification of AAVS1 ZFN-mediated insertion of FVIII donor DNA. All puro-CLEC reads were aligned to AAVS1 sequences modified by integration of FVIII donor DNA (Supplementary Table S1) and the reference human genome (hg19). This identified prokaryotic sequences in the vector that were specific to FVIII donor DNA integration. The absence of these prokaryotic sequences in wt-CLEC sample reads and their presence only in puro-CLEC sample confirmed integration of FVIII donor DNA. To identify off-target integrations, we identified reads from puro-CLEC that were combinations of both human and nonhuman sequences (hereafter called mixed reads). Mixed reads were mapped to the reference human genome. This defined the locations of putative off-target integrations and simultaneously confirmed the absence of nonhuman sequences in hg19. Only reads that aligned to the modified AAVS1 region were considered to be on-target ZFN-mediated insertions of FVIII donor DNA in the AAVS1 locus.
Detection of SV. As SVDetect (version r0.08; http://svdetect.sourceforge.net)59 and BreakDancer (version 1.4.4; http://breakdancer.sourceforge.net/)60 require only abnormally mapped reads, the raw reads were first preprocessed with SAMtools (version 0.1.19; http://samtools.sourceforge.net/) and Picard. The command line used for SAMtools was: samtools view -b -h -F 10 -q 22 input.bam > output.bam.
This command retained in output.bam only reads which were abnormally mapped (mapped to different chromosomes and on different strands) and which had a minimum mapping quality of 22. The resulting bam file was further filtered to discard duplicate reads using the following command in Picard tools: java -jar /opt/picard-1.111/MarkDuplicates.jar INPUT=output.bam OUTPUT=output.nodup.bam REMOVE_DUPLICATES=true ASSUME_SORTED=true METRICS_FILE=metrics.output.txt.
The resulting bam files were submitted to SVDetect and BreakDancer for calling SV. SVDetect and BreakDancer were both configured to detect rearrangements with two or more supporting read pairs using eight times the standard deviation as threshold. After the first step of SVDetect, the resulting ‘.links' file containing all the called SVs was filtered for “imperfect duplicates”61 with in-house Perl script. Links supported solely by clusters of imperfect duplicates were removed. However, links which had only some imperfect duplicates were preserved after removing the supporting imperfect duplicates. Following this, the files were further filtered by SVDetect's own filtering process. The next step was to compare the filtered SVs called for both wt- and puro-CLECs. The option for comparing only the same SV type was turned off. No filtering of imperfect duplicates was done for BreakDancer output as this is not necessary if the anchoring region is set to 3 and the default value in BreakDancer is 7.
The results of BreakDancer and SVDetect were subjected to a final filter to identify overlaps with repetitive DNA and low mappability regions.61 SVs that were only supported by reads that overlapped into any of these regions were removed. Filtering was done with in-house Perl script and BEDtools (version 2.17.0; http://bedtools.readthedocs.org/en/latest/).62 BED files of these regions needed for filtering were extracted as described61 from the UCSC genome browser (http://genome.ucsc.edu/). In the final step, outputs from BreakDancer and SVDetect were inspected for genomic loci that had clusters of reads consistent with inter- or intra-chromosomal SVs specific to puro-CLECs. This was done with in-house Perl script and different degrees of freedom of clustering. The maximum degrees of freedom was set to 3, i.e., genomic coordinates that differed by up to 999 bases were still considered as evidence of clustering.
RNA-seq. Sequence reads were mapped to the reference human genome (hg19) using TopHat (tophat.cbcb.umd.edu). Differential expression was calculated using Cufflinks (cufflinks.cbcb.umd.edu). Transcripts whose expression levels differed by ≥twofold in puro-CLECs compared to wt-CLECs were considered significantly altered. DAVID (Database for Annotation, Visualization and Integrated Discovery) 2.1 Functional Annotation Tool (http://david.abcc.ncifcrf.gov)28 was used to annotate significantly altered transcripts and for pathway mapping. Altered transcripts were also referenced to an aggregated compilation of oncogenes and tumor suppressor genes (http://www.bushmanlab.org/links/genelists).
Validation of potential indels and SV. Genomic DNAs from wt- and puro-CLECs were amplified with phi29 polymerase (REPLI-g kit; Qiagen, Hilden, Germany). High confidence indels were investigated by PCR-Sanger sequencing. Predicted unbalanced SV were investigated by quantitative PCR of genomic breakpoint regions identified by SVDetect and BreakDancer. Each 15 μl reaction, performed in triplicate, comprised 30 ng genomic DNA and 0.3 μmol/l of each primer in iTaq Universal SYBR Green Supermix (Bio-Rad). β-Actin amplification served as the internal control in each experiment (primer sequences in Supplementary Table S5). Fifty amplification cycles were run on CFX96 Touch Real-Time Detection System (Bio-Rad), after which melt curves confirmed product specificity and threshold cycle (CT) values were determined (CFX Manager software, Bio-Rad). The mean CT value of each test locus was normalized to its own actin CT value. Results are expressed as the ratio of normalized CT value of puro-CLEC genomic DNA:normalized CT value of wt-CLEC genomic DNA at each putative breakpoint locus.
Sanger sequencing of indels. PCR amplicons of indels were sequenced using BigDye chemistry in a 3730xI sequencer (Life Technologies). Betaine was added to the sequencing reaction because of highly repetitive DNA sequences in indels 1–3. Indel 2 amplicon could not be obtained as a continuous sequence.
RT-PCR. Quantitative RT-PCR was performed (primer sequences in Supplementary Table S5) to verify changes in the levels of PPP1R12C and selected transcripts in puro-CLECs. CLECs electroporated without plasmid DNA and of the same number of population doublings served as controls. Intron-spanning exonic primers were used to amplify the endogenous PPP1R12C transcript (exons 4–6), neighboring genes within a 1-Mb interval centered on the AAVS1 integration site (LILRB4, ISOC2, PPP6R1, NAT14, ZNF579, FIZ1, and RDH13), potential interacting partners of PPP1R12C predicted by Gene Network Central (http://www.sabiosciences.com) and Human Protein–Protein Interaction Prediction (http://www.compbio.dundee.ac.uk) that were significantly altered by RNA-seq analysis (DUSP1, DUSP6, CDC6, and DUSP16), and a housekeeping gene, GAPDH. Transcript levels were normalized to GAPDH expression and the fold-change in transcript levels in puro-CLECs was expressed relative to wt-CLECs using the ‘delta-delta C(T) method'.63
Cell proliferation assay. One hundred wt- or puro-CLECs in 100-µl culture medium were seeded into each of quadruplicate wells of a flat bottom 96-well tissue culture plate. MTS assay (CellTiter 96 AQueous One Solution Cell Proliferation Assay; Promega, Madison, WI) was performed after 7 days according to the recommended protocol. Absorbance at 490 nm was quantified using MRX II 96-well plate reader (Dynex, Chantilly, VA).
Statistical analysis. Statistical analysis was performed using GraphPad Prism (GraphPad Software). ANOVA and Tukey–Kramer tests were used to compare the means of three or more groups. Student's unpaired t test was used for comparison between two groups with equal variance and the Mann–Whitney test was used when variances were not assumed to be equal. P values < 0.05 were considered significant.
SUPPLEMENTARY MATERIAL Figure S1. Plasmid DNA constructs used in this study. Figure S2. Hybrid human-porcine B domain-truncated FVIII cDNA and assembly steps. Figure S3. Time-course of ZFN transcription and effect of mild hypothermia on ZFN protein levels in transiently electroporated CLECs. Figure S4. Site-specific double-strand DNA cleavage and homology-directed repair in primary human CLECs. Figure S5. Cellular toxicity and genotoxicity induced by donor DNA or AAVS1 ZFN. Figure S6. AAVS1 site-specific integration of 1.3-kb donor DNA in CLECs. Figure S7. AAVS1 site-specific integration of 4.2-kb donor DNA in CLECs. Figure S8. Sensitivity of indel detection by targeted deep sequencing. Figure S9. Pathway analysis of 90 dysregulated transcripts in puro-CLECs. Figure S10. Comparison of wt-CLEC and puro-CLEC cell proliferation. Figure S11. Site-specific genome modification in different adult primary human cells. Figure S12. FVIII transgene secretion by different primary human cell types. Table S1. Sequence of the complete 9-kb FVIII donor DNA integrated in AAVS1 locus of CLECs. Table S2. In silico predicted ten most likely AAVS1 ZFN off-target sites. Table S3. Gene Ontology classification of 90 transcripts whose levels were ≥twofold different in puro-CLECs compared to wt-CLECs. RNA-Seq was performed on wt-CLECs and puro-CLECs 6 weeks after electroporation of donor DNA and AAVS1 ZFN. Potential oncogenes and tumour suppressor genes (underlined and italicized) did not map to any pathway in puro-CLEC transcriptome analyzed by DAVID. Table S4. Relative transcript levels of 196 selected genes from RNA-Seq data showing their expressions in puro-CLEC compared to wt-CLEC. Altered expression was defined as ≥twofold difference. Table S5. List of PCR primers used in this study. Table S6. Quality metrics of whole-genome sequence data.
Acknowledgments
This work was supported by the National Medical Council, Singapore (EDG/0075/2009 and CIRG/1326/2012). J.S. and O.L.K. planned the research, analyzed data and wrote the paper. J.S., A.J.N., and W.H.N. performed the experiments. T.T.P. and J.M. supplied CLECs. H.H., D.K., and S.M.S. analyzed Ref-Seq and WGS data, reviewed, and edited the manuscript. T.T.P. is founder and shareholder of CellResearch Corporation which owns patents for processing umbilical cord-lining epithelial cells.
Supplementary Material
References
- Manco-Johnson, MJ, Abshire, TC, Shapiro, AD, Riske, B, Hacker, MR, Kilcoyne, R et al. (2007). Prophylaxis versus episodic treatment to prevent joint disease in boys with severe hemophilia. N Engl J Med 357: 535–544. [DOI] [PubMed] [Google Scholar]
- Collins, P, Faradji, A, Morfini, M, Enriquez, MM and Schwartz, L (2010). Efficacy and safety of secondary prophylactic vs. on-demand sucrose-formulated recombinant factor VIII treatment in adults with severe hemophilia A: results from a 13-month crossover study. J Thromb Haemost 8: 83–89. [DOI] [PubMed] [Google Scholar]
- World Federation of Hemophilia (2013). Report on the Annual Global Survey 2012. World Federation of Hemophilia: Montreal, Canada. [Google Scholar]
- Collins, PW, Blanchette, VS, Fischer, K, Björkman, S, Oh, M, Fritsch, S et al.; rAHF-PFM Study Group. (2009). Break-through bleeding in relation to predicted factor VIII levels in patients receiving prophylactic treatment for severe hemophilia A. J Thromb Haemost 7: 413–420. [DOI] [PubMed] [Google Scholar]
- Santagostino, E and Mancuso, ME (2008). Barriers to primary prophylaxis in haemophilic children: the issue of the venous access. Blood Transfus 6:Suppl 2: s12–s16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathwani, AC, Reiss, UM, Tuddenham, EG, Rosales, C, Chowdary, P, McIntosh, J et al. (2014). Long-term safety and efficacy of factor IX gene therapy in hemophilia B. N Engl J Med 371: 1994–2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth, DA, Tawa, NE Jr, O'Brien, JM, Treco, DA and Selden, RF; Factor VIII Transkaryotic Therapy Study Group (2001). Nonviral transfer of the gene encoding coagulation factor VIII in patients with severe hemophilia A. N Engl J Med 344: 1735–1742. [DOI] [PubMed] [Google Scholar]
- Tebas, P, Stein, D, Tang, WW, Frank, I, Wang, SQ, Lee, G et al. (2014). Gene editing of CCR5 in autologous CD4 T cells of persons infected with HIV. N Engl J Med 370: 901–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hacein-Bey-Abina, S, Garrigue, A, Wang, GP, Soulier, J, Lim, A, Morillon, E et al. (2008). Insertional oncogenesis in 4 patients after retrovirus-mediated gene therapy of SCID-X1. J Clin Invest 118: 3132–3142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein, S, Ott, MG, Schultze-Strasser, S, Jauch, A, Burwinkel, B, Kinner, A et al. (2010). Genomic instability and myelodysplasia with monosomy 7 consequent to EVI1 activation after gene therapy for chronic granulomatous disease. Nat Med 16: 198–204. [DOI] [PubMed] [Google Scholar]
- Avedillo Díez, I, Zychlinski, D, Coci, EG, Galla, M, Modlich, U, Dewey, RA et al. (2011). Development of novel efficient SIN vectors with improved safety features for Wiskott–Aldrich syndrome stem cell based gene therapy. Mol Pharm 8: 1525–1537. [DOI] [PubMed] [Google Scholar]
- Hockemeyer, D, Soldner, F, Beard, C, Gao, Q, Mitalipova, M, DeKelver, RC et al. (2009). Efficient targeting of expressed and silent genes in human ESCs and iPSCs using zinc-finger nucleases. Nat Biotechnol 27: 851–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez, EE, Wang, J, Miller, JC, Jouvenot, Y, Kim, KA, Liu, O et al. (2008). Establishment of HIV-1 resistance in CD4+ T cells by genome editing using zinc-finger nucleases. Nat Biotechnol 26: 808–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pattanayak, V, Ramirez, CL, Joung, JK and Liu, DR (2011). Revealing off-target cleavage specificities of zinc-finger nucleases by in vitro selection. Nat Methods 8: 765–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabriel, R, Lombardo, A, Arens, A, Miller, JC, Genovese, P, Kaeppel, C et al. (2011). An unbiased genome-wide analysis of zinc-finger nuclease specificity. Nat Biotechnol 29: 816–823. [DOI] [PubMed] [Google Scholar]
- Sander, JD, Ramirez, CL, Linder, SJ, Pattanayak, V, Shoresh, N, Ku, M et al. (2013). In silico abstraction of zinc finger nuclease cleavage profiles reveals an expanded landscape of off-target sites. Nucleic Acids Res 41: e181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendel, A, Fine, EJ, Bao, G and Porteus, MH (2015). Quantifying on- and off-target genome editing. Trends Biotechnol 33: 132–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pattanayak, V, Guilinger, JP and Liu, DR (2014). Determining the specificities of TALENs, Cas9, and other genome-editing enzymes. Methods Enzymol 546: 47–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koo, T, Lee, J and Kim, JS (2015). Measuring and reducing off-target activities of programmable nucleases including CRISPR-Cas9. Mol Cells 38: 475–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller, JC, Holmes, MC, Wang, J, Guschin, DY, Lee, YL, Rupniewski, I et al. (2007). An improved zinc-finger nuclease architecture for highly specific genome editing. Nat Biotechnol 25: 778–785. [DOI] [PubMed] [Google Scholar]
- Guo, J, Gaj, T and Barbas, CF III (2010). Directed evolution of an enhanced and highly efficient FokI cleavage domain for zinc finger nucleases. J Mol Biol 400: 96–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyon, Y, Vo, TD, Mendel, MC, Greenberg, SG, Wang, J, Xia, DF et al. (2011). Enhancing zinc-finger-nuclease activity with improved obligate heterodimeric architectures. Nat Methods 8: 74–79. [DOI] [PubMed] [Google Scholar]
- Doyon, Y, Choi, VM, Xia, DF, Vo, TD, Gregory, PD and Holmes, MC (2010). Transient cold shock enhances zinc-finger nuclease-mediated gene disruption. Nat Methods 7: 459–460. [DOI] [PubMed] [Google Scholar]
- Sivalingam, J, Phan, TT and Kon, OL (2014). Intragenic integration in DLC1 sustains factor VIII expression in primary human cells without insertional oncogenicity. Gene Ther 21: 402–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koboldt, DC, Zhang, Q, Larson, DE, Shen, D, McLellan, MD, Lin, L et al. (2012). VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res 22: 568–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cradick, TJ, Ambrosini, G, Iseli, C, Bucher, P and McCaffrey, AP (2011). ZFN-site searches genomes for zinc finger nuclease target sites and off-target sites. BMC Bioinformatics 12: 152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta, A, Meng, X, Zhu, LJ, Lawson, ND and Wolfe, SA (2011). Zinc finger protein-dependent and -independent contributions to the in vivo off-target activity of zinc finger nucleases. Nucleic Acids Res 39: 381–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang da, W, Sherman, BT, and Lempicki, RA (2009). Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res 37: 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bannert, N, Vollhardt, K, Asomuddinov, B, Haag, M, König, H, Norley, S et al. (2003). PDZ domain-mediated interaction of interleukin-16 precursor proteins with myosin phosphatase targeting subunits. J Biol Chem 278: 42190–42199. [DOI] [PubMed] [Google Scholar]
- Zhang, Z, Kobayashi, S, Borczuk, AC, Leidner, RS, Laframboise, T, Levine, AD et al. (2010). Dual specificity phosphatase 6 (DUSP6) is an ETS-regulated negative feedback mediator of oncogenic ERK signaling in lung cancer cells. Carcinogenesis 31: 577–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong, VC, Chen, H, Ko, JM, Chan, KW, Chan, YP, Law, S et al. (2012). Tumor suppressor dual-specificity phosphatase 6 (DUSP6) impairs cell invasion and epithelial-mesenchymal transition (EMT)-associated phenotype. Int J Cancer 130: 83–95. [DOI] [PubMed] [Google Scholar]
- Banko, MR, Allen, JJ, Schaffer, BE, Wilker, EW, Tsou, P, White, JL et al. (2011). Chemical genetic screen for AMPKα2 substrates uncovers a network of proteins involved in mitosis. Mol Cell 44: 878–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith, JR, Maguire, S, Davis, LA, Alexander, M, Yang, F, Chandran, S et al. (2008). Robust, persistent transgene expression in human embryonic stem cells is achieved with AAVS1-targeted integration. Stem Cells 26: 496–504. [DOI] [PubMed] [Google Scholar]
- DeKelver, RC, Choi, VM, Moehle, EA, Paschon, DE, Hockemeyer, D, Meijsing, SH et al. (2010). Functional genomics, proteomics, and regulatory DNA analysis in isogenic settings using zinc finger nuclease-driven transgenesis into a safe harbor locus in the human genome. Genome Res 20: 1133–1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rio, P, Baños, R, Lombardo, A, Quintana-Bustamante, O, Alvarez, L, Garate, Z et al. (2014). Targeted gene therapy and cell reprogramming in Fanconi anemia. EMBO Mol Med 6: 835–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogata, T, Kozuka, T and Kanda, T (2003). Identification of an insulator in AAVS1, a preferred region for integration of adeno-associated virus DNA. J Virol 77: 9000–9007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang, CJ and Bouhassira, EE (2012). Zinc-finger nuclease-mediated correction of α-thalassemia in iPS cells. Blood 120: 3906–3914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou, C, Chou, BK, Dowey, SN, Tsang, K, Huang, X, Liu, CF et al. (2012). Efficient derivation and genetic modifications of human pluripotent stem cells on engineered human feeder cell lines. Stem Cells Dev 21: 2298–2311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith, RH (2008). Adeno-associated virus integration: virus versus vector. Gene Ther 15: 817–822. [DOI] [PubMed] [Google Scholar]
- Doering, CB, Healey, JF, Parker, ET, Barrow, RT and Lollar, P (2004). Identification of porcine coagulation factor VIII domains responsible for high level expression via enhanced secretion. J Biol Chem 279: 6546–6552. [DOI] [PubMed] [Google Scholar]
- Brown, HC, Gangadharan, B and Doering, CB (2011). Enhanced biosynthesis of coagulation factor VIII through diminished engagement of the unfolded protein response. J Biol Chem 286: 24451–24457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hacein-Bey-Abina, S, Pai, SY, Gaspar, HB, Armant, M, Berry, CC, Blanche, S et al. (2014). A modified γ-retrovirus vector for X-linked severe combined immunodeficiency. N Engl J Med 371: 1407–1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ott, MG, Schmidt, M, Schwarzwaelder, K, Stein, S, Siler, U, Koehl, U et al. (2006). Correction of X-linked chronic granulomatous disease by gene therapy, augmented by insertional activation of MDS1-EVI1, PRDM16 or SETBP1. Nat Med 12: 401–409. [DOI] [PubMed] [Google Scholar]
- Hacein-Bey Abina, S, Gaspar, HB, Blondeau, J, Caccavelli, L, Charrier, S, Buckland, K et al. (2015). Outcomes following gene therapy in patients with severe Wiskott-Aldrich syndrome. JAMA 313: 1550–1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shigemizu, D, Fujimoto, A, Akiyama, S, Abe, T, Nakano, K, Boroevich, KA et al. (2013). A practical method to detect SNVs and indels from whole genome and exome sequencing data. Sci Rep 3: 2161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghoneim, DH, Myers, JR, Tuttle, E and Paciorkowski, AR (2014). Comparison of insertion/deletion calling algorithms on human next-generation sequencing data. BMC Res Notes 7: 864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meynert, AM, Ansari, M, FitzPatrick, DR and Taylor, MS (2014). Variant detection sensitivity and biases in whole genome and exome sequencing. BMC Bioinformatics 15: 247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiskinis, E, Sandoe, J, Williams, LA, Boulting, GL, Moccia, R, Wainger, BJ et al. (2014). Pathways disrupted in human ALS motor neurons identified through genetic correction of mutant SOD1. Cell Stem Cell 14: 781–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai, SQ and Joung, JK (2014). What's changed with genome editing? Cell Stem Cell 15: 3–4. [DOI] [PubMed] [Google Scholar]
- Brash, DE (2015). Cancer. Preprocancer. Science 348: 867–868. [DOI] [PubMed] [Google Scholar]
- Sivalingam, J, Krishnan, S, Ng, WH, Lee, SS, Phan, TT and Kon, OL (2010). Biosafety assessment of site-directed transgene integration in human umbilical cord-lining cells. Mol Ther 18: 1346–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuthbert, R, Boxall, SA, Tan, HB, Giannoudis, PV, McGonagle, D and Jones, E (2012). Single-platform quality control assay to quantify multipotential stromal cells in bone marrow aspirates prior to bulk manufacture or direct therapeutic use. Cytotherapy 14: 431–440. [DOI] [PubMed] [Google Scholar]
- Wagner, W, Horn, P, Castoldi, M, Diehlmann, A, Bork, S, Saffrich, R et al. (2008). Replicative senescence of mesenchymal stem cells: a continuous and organized process. PLoS One 3: e2213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, Y, Gan, SU, Lin, G, Lim, YT, Masilamani, J, Mustafa, FB et al. (2011). Characterization of human umbilical cord lining-derived epithelial cells and transplantation potential. Cell Transplant 20: 1827–1841. [DOI] [PubMed] [Google Scholar]
- Naji, A, Rouas-Freiss, N, Durrbach, A, Carosella, ED, Sensébé, L and Deschaseaux, F (2013). Concise review: combining human leukocyte antigen G and mesenchymal stem cells for immunosuppressant biotherapy. Stem Cells 31: 2296–2303. [DOI] [PubMed] [Google Scholar]
- Amodio, G, Sales de Albuquerque, R and Gregori, S (2014). New insights into HLA-G mediated tolerance. Tissue Antigens 84: 255–263. [DOI] [PubMed] [Google Scholar]
- Li, H and Durbin, R (2009). Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25: 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H, Handsaker, B, Wysoker, A, Fennell, T, Ruan, J, Homer, N et al.; 1000 Genome Project Data Processing Subgroup (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25: 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeitouni, B, Boeva, V, Janoueix-Lerosey, I, Loeillet, S, Legoix-né, P, Nicolas, A et al. (2010). SVDetect: a tool to identify genomic structural variations from paired-end and mate-pair sequencing data. Bioinformatics 26: 1895–1896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, K, Wallis, JW, McLellan, MD, Larson, DE, Kalicki, JM, Pohl, CS et al. (2009). BreakDancer: an algorithm for high-resolution mapping of genomic structural variation. Nat Methods 6: 677–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mijušković, M, Brown, SM, Tang, Z, Lindsay, CR, Efstathiadis, E, Deriano, L et al. (2012). A streamlined method for detecting structural variants in cancer genomes by short read paired-end sequencing. PLoS One 7: e48314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan, AR and Hall, IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26: 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak, KJ and Schmittgen, TD (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25: 402–408. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.