

Abstract
DNA hypermethylation is extensively explored as therapeutic target for gene expression modulation in cancer. Here, we re-activated hypermethylated candidate tumor suppressor genes (TSGs) (C13ORF18, CCNA1, TFPI2, and Maspin) by TET2-induced demethylation in cervical cancer cell lines. To redirect TET2 to hypermethylated TSGs, we engineered zinc finger proteins (ZFPs), which were first fused to the transcriptional activator VP64 to validate effective gene re-expression and confirm TSG function. ChIP-Seq not only revealed enriched binding of ZFPs to their intended sequence, but also considerable off-target binding, especially at promoter regions. Nevertheless, results obtained by targeted re-expression using ZFP-VP64 constructs were in line with cDNA overexpression; both revealed strong growth inhibition for C13ORF18 and TFPI2, but not for CCNA1 and Maspin. To explore effectivity of locus-targeted demethylation, ZFP–TET2 fusions were constructed which efficiently demethylated genes with subsequent gene re-activation. Moreover, targeting TET2 to TFPI2 and C13ORF18, but not CCNA1, significantly decreased cell growth, viability, and colony formation in cervical cancer cells compared to a catalytically inactive mutant of TET2. These data underline that effective re-activation of hypermethylated genes can be achieved through targeted DNA demethylation by TET2, which can assist in realizing sustained re-expression of genes of interest.
Introduction
Besides genetic mutations, epigenetic silencing of tumor suppressor genes (TSGs) is another important event by which normal cells can transform to cancer cells.1 Especially, the CpG hypermethylation observed in core promoter regions is well associated with gene silencing2 and is the target of a variety of therapeutic interventions aimed to restore TSG expression in cancer. In this regard, genome-wide epigenetic drugs, such as 5-Aza-2′-deoxycytidine (5-aza-dC), have been FDA approved for hematological cancers, but their toxicity and lack of specificity seem to limit their efficacy for solid tumors.3 However, developments in the field of gene editing offer a promising new approach to correct CpG methylation in a targeted fashion by epigenetic/epigenome editing (gene-targeted epigenetic reprogramming).4,5 Indeed, methylcytosine dioxygenases, ten-eleven translocation (TET) proteins, have been targeted to sequences within hypermethylated promoter regions resulting in successful removal of CpG methylation at the site.6,7,8,9
For a long time, targeted DNA demethylation seemed unfeasible in mammalian cells as no enzymes were identified with the capacity to actively demethylate DNA. A breakthrough in the field was the identification of the TET proteins as important players in the active DNA demethylation pathway,10 as they catalyze the oxidation of methylated CpGs (5mC) to 5-hydroxymethylcytosine (5hmC) and other oxidized 5mC derivatives. These intermediates recruit a variety of DNA repair proteins/glycosylases, such as thymine-DNA glycosylase11,12 to trigger the final step of active DNA demethylation by the base excision repair system. The oxidizing properties of TET proteins make them powerful biological tools to demethylate DNA strands in vitro11 and in vivo.13 Gene-targeted demethylation initiated by TET-enzymes has attracted attention as an innovative approach to re-express silenced TSGs, and may provide new avenues to battle cancer.
Interestingly, increasing evidence is revealing that silencing of the TET-enzymes themselves is an important factor responsible for TSG silencing14 and re-introduction of TET enzymes can re-activate hypermethylated TSGs.15,16 To re-activate a chosen TSG by targeted demethylation, TET enzymes have been linked to a variety of DNA targeting tools, such as zinc finger proteins (ZFPs)6,9 and transcription activator-like effector proteins.7,8 Previously, the fusion of ZFPs to various effector domains (such as the strong transcriptional activator VP64, generating an artificial transcription factor (ATF)), has proven to be an effective tool for modulation of gene expression in many disease models and clinical trials have been performed with ZFP fusions, indicating their therapeutic potential.17
Epigenetically silenced TSGs in cancer are attractive targets for therapeutic interventions aiming to decrease DNA methylation, as re-activation of these, as opposed to genetically mutated TSGs, will result in functional proteins. In this study, we focused on four candidate TSGs in cervical cancer (C13ORF18, CCNA1, TFPI2, and Maspin), which all have been reported to be methylated/silenced in this malignancy.1,18,19,20,21 Of these, Maspin (SERPIN5B) has been well studied in cancer, but despite this, its role remains controversial.22 TFPI2 is identified in an increasing number of cancers as DNA methylation marker18,23 with potent tumor suppressive activities,24 but its function as TSG in cervical cancer is unknown. C13ORF18 was previously identified by us as DNA methylation marker18,19 with putative tumor suppressive activities25 in cervical cancer. CCNA1 is specifically methylated in various cancers.18,26
Here, we aim to validate the putative TSG function of these methylated genes in order to select suitable targets for targeted re-expression by TET2-induced DNA demethylation. First, we studied the epigenetic regulation of these genes in cervical cancer cell lines and induced effective ZFP-VP64-based gene regulation. Next, we showed that targeting TET2 could induce DNA demethylation and gene re-activation that translated to decreased cancer growth and induction of apoptosis.
Results
Epigenetic Regulation of Target Genes
First, we confirmed epigenetic dysregulation of CCNA1, TFPI2, C13ORF18, and Maspin in a panel of cervical cancer cell lines. Examination of the mRNA expression revealed that CCNA1, TFPI2, and C13ORF18 were silenced or expressed at very low levels in at least 5/8 cell lines. Interestingly, Maspin was highly expressed in 7/8 cell lines (Figure 1a). This unexpected finding was also found on protein level in a panel of cervical cancer cell lines, and could be confirmed for cervical cancer patient samples (Supplementary Figure S1); we found high Maspin expression in tumor cells as opposed to the normal adjacent cells, while Maspin methylation levels in cervical cancer patients were decreased. In the cell lines, we also confirmed that Maspin was not mutated, indicating that Maspin is functionally active. For HeLa, SiHa, and Caski, the expression of Maspin was associated with the presence of active histone marks (H3K4Me3 and H3K9Ac) and the absence of repressive marks (H3K9Me3 and H3K27Me3). The silencing of CCNA1 and C13ORF18, and to a lesser extend TFPI2, was associated with the presence of H3K9Me3 in these cell lines (Figure 1b). In addition to the histone marks, DNA methylation was closely correlated with the gene expression status (Figure 1c), without a clear association with cellular TET expression (Supplementary Figure S2).
Figure 1.

Epigenetic regulation of putative tumor suppressor genes in a panel of cervical cancer cell lines. (a) mRNA expression relative to GAPDH of CCNA1, TFPI2, C13ORF18 and Maspin in a panel of eight cervical cancer cell lines. (b) Quantitative ChIP for repressive histone marks (H3K9me3 and H3K27me3) and histone marks associated with active gene-transcription (H3K4me3, H3K9Ac, H3Ac, and H4Ac) for the gene promoters of CCNA1, TFPI2, C13ORF18 and Maspin in HeLa, SiHa, and CaSki (H3K9Ac, n = 2). (c) Methylation status of the CCNA1, TFPI2, C13ORF18 and Maspin promoter of CpGs in the indicated region. Methylation levels were analyzed by bisulfite sequencing (each circle represents the average of three or more clones) or bisulfite pyrosequencing (average methylation of cell population). Re-expression/upregulation of CCNA1 (d), TFPI2 (e), and Maspin (f) mRNA after treatment with different concentrations of epigenetic drugs (500 nM (+), 5 µM 5-aza-dC (++) and/or 500 nM TSA (+)). Values represent the mean of at least three independent experiments, unless stated otherwise, measured in triplicate ± SEM.
All silenced genes could be re-expressed by epigenetic drugs, with induction levels reaching up to ~2700-fold and ~6900-fold for CCNA1 and TFPI2, respectively, and 20-fold for Maspin (Figure 1d–f). Compared to C13ORF18 (±50-fold),25 CCNA1 and TFPI2 were much more responsive to epigenetic drug treatment. Methylation-specific PCRs proved that upregulations were at least partly the result of promoter demethylation (Supplementary Figure S1). Together, these results confirmed that CCNA1, TFPI2, and C13ORF18 were epigenetically repressed in cervical cancer, but Maspin seemed to be overexpressed.
Target Validation by ZFPs Fused to VP64
To achieve ZFP-mediated gene targeting for CCNA1 and TFPI2, we engineered in total 17 ZFPs (Figure 2a). ZFPs were subsequently fused to VP64 and screened for their ability to modulate endogenous expression of CCNA1 (6 ZFPs) or TFPI2 (11 ZFPs). From the six ATFs targeted to the CCNA1 promoter, an initial screening in HeLa revealed 12ab-VP64 as a robust inducer of CCNA1 expression (Supplementary Figure S3a). Further validation of 12ab-VP64 in the CCNA1 repressed cell lines SiHa, CaSki, and C33A revealed that 12ab-VP64 consistently induced expression of CCNA1 mRNA up to 66-fold (Supplementary Figure S3a). From the 11 ATFs targeted at the TFPI2 promoter, 43ab-VP64 was the most robust re-activator of TFPI2 in methylated HeLa and SiHa cells, with induction levels up to 23-fold (Supplementary Figure S3b). We also confirmed induction of Maspin expression by published Maspin-targeting ATFs (126-VP64 and 97-VP64)27 in the cervical cancer cell lines CaSki and C33A (Supplementary Figure S3c). Previously, we showed induction of C13ORF18 in these cervical cancer cell lines.25
Figure 2.

Re-expression of candidate TSGs (C13ORF18, CCNA1, TFPI2 and Maspin) in tumor-derived cell lines. (a) Schematic representations of 18-bp ZFPs (not on scale) binding to DNA. Also shown are the promoter region of CCNA1, TFPI2, Maspin and C13ORF18 stretching from −750 to +750 bp relative to the main TSS of the corresponding genes. Indicated are the targeted sites of the ZFPs directed to the CCNA1, TFPI2, Maspin and C13ORF18 promoter, as well as ZFPs which successfully demonstrated gene expression modulation when fused to effector domains. CpGs are indicated as vertical bars. Gene expression levels of C13ORF18 (b), CCNA1 (c), TFPI2 (d) and Maspin (e) mRNA relative to empty vector after retroviral transduction with gene-targeting ATFs (C13ORF18: 3ab-VP64, 5ab-VP64; CCNA1: 12ab-VP64; TFPI2: 43ab-VP64; and Maspin: 126-VP64) in CSCC-7, CSCC-8, CC-10 and CC-11. CC-11 cells were also transduced with cDNA overexpression constructs of C13ORF18, CCNA1, TFPI2 and Maspin. Each bar represents the mean of in general three independent measured in triplicate ± SEM. Quantification of mRNA was performed using qRT-PCR and induction levels were normalized to GAPDH and relative to an empty vector.
We further validated the selected VP64-ATFs in four low passage cervical cancer cell lines.28 Two of these cell lines grow as spheres (CSCC-7 and CC-11), better representing primary tumor characteristics (Supplementary Figure S4a). For comparison, CC-11 cells were also transduced with viruses to directly express the target cDNA (Figure 2b–e, right bars). The ATFs consistently upregulated their target genes C13ORF18, CCNA1 or TFPI2 in all cell lines (Figure 2b–e) with no/very low cross-inductions of these other genes (Supplementary Figure S4b–g). Maspin was highly expressed in all four cell lines, and ATFs could not further upregulate Maspin in these cells.
Binding Profiles of Engineered ZFPs
In order to gain greater insight into the specificity of our ZFPs, ZFP-binding profiles were determined by ChIP-Seq. ZFPs without effector domain were used, as it was observed that effector domains, such as VP64, can shield the HA tag from detection by our antibody. PCR on the sonicated fragments revealed a close association of the ZFPs with their targeted promoter (Figure 3a).
Figure 3.

Genome association of ZFPs. (a) ATF association of C13ORF18-, CCNA1-, and TFPI2-targeting ZFPs with their respective gene promoters (C13ORF18: empty vector, 3ab-NoEf and 5ab-NoEf; CCNA1: 12ab-NoEf; and TFPI2: 43ab-NoEf) as detected by ChIP using an anti-HA antibody. (b) Coverage plots showing the local enrichment for binding of 43ab-NoEf at the targeted TFPI2 promoter in a 20-kb spanning region visualized with RStudio (cells expressing an empty vector were used as control). Local enrichment was determined using the coverage distribution report obtained from NextGENe. See Supplementary Figure S5 for binding of 3ab-, 5ab-, 12ab-, and 43ab-NoEf to all promoters (C13ORF18, CCNA1, TFPI2, and Maspin). (c) Representation of the identified ChIP-Seq peaks with coverage of five or more for the gene-targeting constructs and an empty vector as determined by the peak identification report obtained from NextGENe. (d) Percentage of ChIP-Seq peaks (with coverage of ten or more reads) bound to promoters, distal promoters and gene bodies (thus the region −10,000 until +10,000 bp relative to the TSS with the exception of the region +/− 100 bp relative to the TSS) or other regions (such as non-coding DNA). (e) Graphical representation of the number of peaks and their coverage at the mistargeted promoters from d.
Subsequently, these samples were subjected to genome-wide sequencing and again we demonstrated binding of the ZFPs to their intended promoters (Figure 3b and Supplementary Figure S5). For example, 3ab-targeting C13ORF18 showed substantial enrichment at the methylated C13ORF18 promoter. However, also off-targets were detected (Supplementary Figure S6 and S7). While the empty vector revealed a relatively steady background signal throughout the genome (Supplementary Figure S6a) (many peaks, but with low coverage (Figure 3c), ZFPs seemed to have site preferences (Supplementary Figure S6a). Interestingly, for all analyzed ZFPs targeting closed chromatin (3ab, 5ab, and 12ab), off-target binding events within our four target loci were most pronounced in the promoter region of active (TFPI2, Maspin in CaSki cells) and not repressed loci (C13ORF18, CCNA1 in CaSki cells). Strikingly, such off-target effects can be very pronounced especially for DNA segments with a high sequence similarity with the 18-bp target region. As an example, we analyzed binding sites for the 12ab ZFP and found significant enrichment for DNA segments with a single mismatch within the ZFP target site (Supplementary Figure S6b and S7). Such off-targets by sequence similarity can result in peaks with high coverage, even for 2 bp mismatches, as shown for 12ab-NoEf off-targets with mismatches at the edges of the target site (Supplementary Figure S7).
Then, for each ZFP the total number of off-targets was determined (with a coverage of five or more). We found many peaks for the ZFPs (Figure 3c), although also empty vector had many peaks, albeit mostly with low coverage. Compared to empty vector, ZFP binding was enriched at promoter regions, but not at distal promoter segments and gene bodies, as shown for peaks with ten or more reads (Figure 3d). The total number of mistargeted promoters was several-fold higher compared to an empty vector (between 806 and 5984 for the various constructs, empty vector 204) (Figure 3e). The TFPI2-ZFP showed the least off-targets, although a difference in viral load between the different ZFP constructs may have influenced the number of mistargeted promoters.
Re-expression of Silenced TSGs Results in Suppressive Effects in Tumor-derived Cell Lines
To determine which genes within our panel of hypermethylated genes have a tumor suppressive function, we re-activated gene expression using the VP64-ATFs. In HeLa cells, TFPI2-induced expression by 43ab-VP64 significantly decreased cell viability (−85% (P < 0.01)) 4 days after transduction compared to empty vector (Figure 4a), but this effect was not seen for 12ab-VP64 targeting CCNA1. In CC-11 cells, all silenced genes could be upregulated by our ATFs (Figure 2), and therefore this cell line was chosen to compare the differences in the effect on cell growth between these genes (Figure 4b). We found that C13ORF18- and TFPI2-inducing constructs significantly reduced cell growth compared to empty vector (3ab-VP64 −45% (P < 0.05), 43ab-VP64 −58% (P < 0.01)), while CCNA1-inducing constructs had no effect on cell growth. As controls, we expressed cDNA constructs of the four genes in CC-11 cells and measured the percentage of late apoptotic/necrotic cells (Figure 4c). We found again indications that TFPI2 and C13ORF18 are TSGs; both C13ORF18 and TFPI2 cDNA overexpression significantly induced cell death compared to an empty vector, while the cell death induced by CCNA1 or Maspin overexpression did not reach significance. In all assays, TFPI2 was identified as the strongest growth inhibitor.
Figure 4.

Tumor suppressive effects of candidate TSGs. (a) Cell viability of HeLa cells after re-expression of silenced CCNA1 and TFPI2 by 12ab-VP64 and 43ab-VP64, respectively. (b) Cell viability of CC-11 cells after gene induction/upregulation of C13ORF18, CCNA1, TFPI2, and Maspin by 3ab-VP64, 12ab-VP64, 43ab-VP64, and 126-VP64, respectively. Cell proliferation was assessed using a MTS assay during 5 days and empty vector and ZFP-NoEf were used as controls. (c) Percentage of cell death after treatment with empty vector, C13ORF18-, CCNA1-, TFPI2-, and Maspin-cDNA overexpression constructs in CC-11 cells. Each data point represents the mean of three or more independent experiments measured in triplicate ± SEM.
Epigenetic Editing by TET2
In contrast to transcriptional activators, such as VP64, (a combination of) epigenetic editing approaches could provide us with the tools to ultimately re-express silenced TSGs in a sustained fashion.9 Therefore, to assist in realizing this goal, we fused TET2 and its catalytically inactive mutant to our selected panel of ZFPs targeting silenced genes. TET2 was selected based on our previous observations that TET2 was the most efficient DNA demethylating enzyme when targeted to the ICAM-1 promoter.6 To reach optimal/more effective levels of ZFP–TET2 constructs, cells were GFP sorted and re-transduced before analysis (superinfection) (Figure 5a and Supplementary Figure S8a,b); this procedure increased transgene expression to ~10-fold. Similarly, superinfection compared to regular infections of VP64-ATFs resulted in higher gene inductions for all four genes (Supplementary Figure S8c–e). Subsequently, we showed that the promoter of C13ORF18 could be significantly re-activated by TET2-mediated gene targeting in CaSki cells, whereas the TET2 mutant did not significantly affect gene expression (Figure 5a). Re-expression of C13ORF18 by 5ab fused to the catalytic domain of TET2 (5ab-TET2-CD) reached induction levels up to 59-fold (P < 0.01) (Figure 5a) relative to empty vector. Expression of GFP (as surrogate marker for construct expression) showed only minor differences between TET2 and its mutant (Supplementary Figure S8a). To determine whether the C13ORF18 induction by TET2 was associated with DNA demethylation at the promoter region, an initial screening was performed using bisulfite sequencing upon expression of ATFs and TET2 fusions on regularly infected cells. This screen revealed that the promoter region of C13ORF18 was partially demethylated by the TET fusions, especially around the 5ab-binding site (Supplementary Figure S9a,b). To quantify the reduction in methylation levels on single CpGs, we performed bisulfite pyrosequencing for six CpGs surrounding the 5ab-target site on superinfected cells (Figure 5b,c). We observed effective DNA demethylation of the CpGs by the TET2 constructs both at the three CpGs at the 5′ flanking side of the ZFP-binding site (reduction of 48% (P < 0.001), 50% (P < 0.001) and 44% (P < 0.001)) as well as at the 3′ flanking side of the ZFP-binding site (reduction of 19% (P < 0.01), 37% (P < 0.01) and 11% (P < 0.09)). Also the simultaneous expression of 3ab+5ab-TET2-CD resulted in significant demethylation of the targeted CpGs, although to a lesser extent (up to 21% (P < 0.001), Figure 5c). The control constructs (empty vector, VP64-ATF, and NoEf) did not induce significant demethylation of the analyzed CpGs around the 5ab target site.
Figure 5.

TET2-mediated gene activation and demethylation. (a) Relative C13ORF18 mRNA expression (left) after retrovirally induced expression of ZFPs carrying GFP and either TET2-CD, TET2-mutant or NoEf. On the right, the GFP positivity of transduced cells as exemplified for untreated cells or cells expressing an empty vector, 5ab-VP64 or 5ab-TET2 measured by FACS. (b) Schematic representation of the C13ORF18 promoter region carrying the target site of 5ab-TET2-CD and six CpGs surrounding this site. (c) The DNA methylation status of the six CpGs in the C13ORF18 promoter after expressing the gene-targeting constructs or treatment with 5-aza-dC (5 µM for 3 days (n = 3 samples from Figure 1d–f)) as quantified by bisulfite pyrosequencing. Values represent the mean of at least two independent experiments ± SEM. (d) Relative CCNA1 expression after retrovirally induced expression of ZFPs carrying either TET2-CD, TET2-mutant or NoEf in CaSki cells (left). On the right, the DNA methylation status of 10 CpGs in the CCNA1 promoter after expressing the gene-targeting constructs 12ab-TET2-CD and 12ab-VP64 (or treatment with 5-aza-dC (5 µM for 3 days (n = 3)) as quantified by bisulfite pyrosequencing. Statistical differences were determined between empty vector and 12ab-TET2-CD. (e) Relative TFPI2 mRNA expression after retrovirally induced expression of ZFPs carrying either TET2-CD or NoEf in HeLa cells. All cells were GFP sorted (except NoEf) after transduction and re-transduced before analysis. Expression of C13ORF18, CCNA1, and TFPI2 is normalized to GAPDH and relative to an empty vector. mRNA was quantified by qPCR and each bar represents the mean of at least three independent experiments measured in triplicate ± SEM. (f) The off-target effect of the C13ORF18-targeting 5ab-TET2 ZFP and the NoEf control on the methylation status of the CCNA1 promoter. (g) The off-target effect of the CCNA1-targeting 12ab-TET2 ZFP and the NoEf control on the methylation status of the C13ORF18 promoter. Methylation levels were quantified by bisulfite pyrosequencing and each data point represents the mean of three independent experiments ± SEM
Then, we determined robustness of the approach by targeting TET2 to the CCNA1, TFPI2, and Maspin promoters. When fused to 12ab, TET2, but not the mutant, induced upregulation of CCNA1 up to ~58-fold (P < 0.01) (Figure 5d), which was associated with demethylation of several CpGs in the vicinity of the 12ab target site and in the direction of the TET-enzyme (10 CpGs analyzed) (Figure 5d); CpGs were demethylated up to 43% compared to controls. Re-expression of TFPI2 by 43ab-TET2-CD reached induction levels up to ~7-fold (P < 0.05) (Figure 5e), while the targeting of TET2 in Maspin-methylated C33A cells did not result in significant activation of Maspin (data not shown). For the 5-aza-dC-treated cells (Figure 1d), only a limited effect on DNA methylation was observed for most of the analyzed CpGs (Figure 5c,d).
To gain a first insight into whether targeted TET2-constructs only affect methylation of ZFP-bound regions (as determined by ChIP-seq), we investigated the effect of the targeted TET2-constructs on the methylation level of a small selection of non-ZFP bound promoters. Based on the ChIP-Seq analysis, the C13ORF18 targeting ZFP did not bind the CCNA1 promoter and the CCNA1-ZFP did not bind to the C13ORF18 promoter (Supplementary Figure S5). Therefore, these regions were selected for analysis. Indeed, most of the CpGs remained unaffected, although within the C13ORF18 promoter the methylation level of one CpG was significantly reduced (−10%) by the 12ab-TET2-CD targeted at the CCNA1 promoter. Similarly, analysis of a third methylated promoter (EPB41L3, Supplementary Figure S10) showed significant demethylation (−10%) of only 1 out of 30 off-target CpGs analyzed.
Until now, it has not been determined whether re-activation by targeted TET2 to hypermethylated genes can decrease tumor growth. To investigate the feasibility of this approach, we targeted TET2 to the TFPI2 promoter, as cDNA and VP64-ATFs identified TFPI2 as the strongest growth inhibitor (Figure 4). Indeed, in CC-11 cells transduced with TFPI2-targeting constructs, a strong and significant reduction in CC-11 colony size was revealed compared to empty vector (Figure 6a,b); expression of 43ab-TET2 reduced colony size with 68% (P < 0.01). Moreover, targeting TET2 to TFPI2 and C13ORF18, but not CCNA1, decreased cell proliferation of HeLa and Caski cells compared to a catalytically inactive mutant of TET2 (Figure 6c,d), and this was accompanied with an increase in the fraction of late apoptotic/necrotic cells (Figure 6e,f). These data underline that effective re-activation of hypermethylated genes can be achieved through targeted DNA demethylation.
Figure 6.

TET2-mediated reduction of cell growth by targeting TFPI2. (a) Visualization of CC-11 colonies after expressing the TFPI2-targeting ZFPs (43ab) carrying TET2 and controls constructs (empty vector, 43ab-VP64, and TFPI2-cDNA). Pictures are a representative of two independent experiments. (b) Quantification of the colony size of CC-11 cells from a using ImageJ. Each bar represents the average colony size per condition from two independent experiments. For each condition, four pictures were randomly taken. Cell viability of HeLa (c) and CaSki (d) cells 5 days after transduction with the gene-targeting constructs measured with a MTS assay. The fraction of late apoptotic and necrotic cells 5 days after transduction with the gene-targeting zinc fingers (43ab targeting TFPI2, 5ab targeting C13ORF18, and 12ab targeting CCNA1) fused to VP64, TET2 or TET2 mutants measured with a PI assay in HeLa (e) and CaSki cells (f). Transductions were performed on superinfected cells and each data point represents the mean of three independent experiments measured in triplicate ± SEM.
Discussion
Potentially new approaches for cancer treatment are appearing using the TET-methylcytosine dioxygenases as a tool to directly mediate CpG demethylation and restore gene expression.6,7,15,16 In this study, we showed that by targeting TET2 to hypermethylated TSGs, efficient DNA demethylation can be achieved, resulting in significant gene re-activation. We found that the induced demethylation by ZFP–TET2 fusions can result in significantly less tumor growth. Analysis of TET1-3 expression revealed that TET1 was significantly silenced in cervical cancer, while TET2 was highly expressed compared to normal cells, without a clear association with TSG hypermethylation. The most promising therapeutic target we identified from the four putative TSGs (C13ORF18, CCNA1, TFPI2, and Maspin) was TFPI2, and this is the first study describing its role in this malignancy. Moreover, we demonstrate the binding of the engineered ZFPs to their aimed methylated target site, and off-target DNA demethylation events were rather limited within our panel of genes. However, the ChIP-Seq revealed that the ZFPs exhibit off-target binding, especially to promoter areas (several fold more enrichment of the ZFPs at other gene promoters compared to a control). In addition, we found that the well-described TSG Maspin was unexpectedly highly expressed in cervical cancer.
Previously, the targeting of TET1 resulted in substantial DNA demethylation and upregulation of the targeted HOXF2- and beta-globin genes.7 Moreover, targeted demethylation by TET1 of the metastasis suppressor gene CRMP4 abolished metastasis in prostate cancer cells.8 Here, we showed that also the targeting of TET2 to hypermethylated genes results in substantial CpG demethylation and gene re-expression, which was effective enough to translate to reduced cancer cell growth. Demethylation of single CpGs was observed with reductions up to 50% for C13ORF18 and 59% for CCNA1, resulting in significant gene inductions (up to ~59- and ~58-fold, respectively). Compared to our initial report, in which we screened for efficiency of ZFP-targeted TET1, TET2, and TET3, this study describes a more effective DNA demethylation and higher gene induction for TET2 (2-fold induction and ~6% demethylation for targeting the ICAM-1 promoter).6 The improved efficiency may be explained using the superinfection procedure, as we observed more demethylation of target CpGs after re-infecting the cells, thereby increasing the expression of and exposure time to the TET fusions. Also, the addition of epigenetic drugs has shown to facilitate re-expression by DNA demethylating reagents, possibly through inhibition of DNMT3B.9
TET2-induced DNA demethylation was very pronounced at both sides of the binding site (within 50 bp), although the bisulfite sequencing data for C13ORF18 indicates a more widespread effect. These results are in line with the DNA demethylating effects of TET1, which were greatest within 30 bp of either end of the transcription activator-like effector target-binding site, but could reach up to 150–200 bp away of the target site.7 The observed CpG demethylation after targeting TET enzymes could even be an underestimation, as TET enzymes will first convert 5mC to 5hmC, and this modification cannot be differentiated from methylated Cs by bisulfite pyrosequencing. In addition, the targeting of TDG has shown to mediate expressional changes by reducing methylation levels, although to a smaller extend.29 Such rewriting of the methylation code by enzymes that initiate CpG demethylation is of key importance to achieve sustained re-expression of target genes; Indeed, Li et al.8 demonstrated that TET1-induced DNA demethylation of the tumor metastasis suppressor gene CRMP4 resulted in a reduction of metastasis formation up to 60 days after induction of targeted DNA demethylation. VP64-ATF-mediated expressional changes have shown to be only transient, as targeted promoters are incompletely reprogrammed and genes tend to return to the ‘normal' state after the VP64-ATF is removed.9 Previously, it was observed that VP64-induced gene activation of ICAM-1 results in DNA demethylation.6 However, this response seems dependent on the local chromatin environment, as here, we did not observe such an effect on DNA demethylation induced by VP64 for either C13ORF18 or CCNA1. This finding highlights the importance to look into the local chromatin environment to achieve a sustainable response.
Interestingly, 5-aza-dC induced high gene expression levels of our target genes, but this was not associated with much DNA demethylation within the analyzed regions, as demonstrated by bisulfite pyrosequencing of the C13ORF18 and CCNA1-promoter. The last decade, treatment with DNA demethylating agents has been used extensively to demonstrate epigenetic regulation of silenced genes, but indirect 5-aza-dC effects are commonly observed.30 Indeed, lack of gene DNA demethylation after 5-aza-dC-induced expression has been reported many times in literature, for example, for Maspin31, and may also partly explain the commonly observed lack of sustainability on gene expression after such treatment.30 The epigenetic editing approach described here to demethylate genes is believed to be a more specific approach compared to 5-aza-dC. Despite this, as shown by the ChIP-Seq analysis, also the epigenetic editing approach still requires further optimization to make it more specific.
With the development of the transcription activator-like effectors and the recent clustered regularly interspaced short palindromic repeat (CRISPR)/Cas9 system, gene targeting for (epi)genome editing purposes has made a real boost.32 The advantage of ZFPs is their small size, making them efficient in accessing epigenetically silenced regions. With regard to off-target effects, some reports indicated that ZFP-ATFs can manipulate gene expression with single gene resolution29,33 and we previously found that of all protein-coding loci, the E2C-engineered ZFP preferentially binds to its aimed target site.34 All three targeting systems are currently improved to yield highly specific genome editing tools when fused to endonucleases.35 Here, we could demonstrate that our ZFPs bind to their endogenous methylated target site using ChIP-Seq, while no binding to off-target methylated sites was detected for our gene panel. This was in line with little or no off-target DNA demethylation events at these methylated promoters. However, when looking at the genome-wide scale, we did observe that the ZFPs exhibit off-target binding, as the ChIP-Seq revealed that several-fold more off-target promoters showed enrichment for the ZFPs compared to a control. In line with this, Grimmer et al.36 reported thousands of off-target sites, mainly at promoter regions, for ZFPs targeted to the SOX2 promoter. Within our small panel of target genes, we observed preferential off-target binding to the promoters of the actively transcribed genes (TFPI2 or Maspin). For example, ZFP 3ab targeted to C13ORF18 was also enriched at the Maspin promoter. On the other hand, only ZFPs targeted to repressed promoters (12ab to CCNA1, 3ab or 5ab to C13ORF18) were specifically enriched at those difficult to access regions (for our gene panel). High sequence similarity with intended target sites could further enhance off-target effects, as shown here for a 1–2 bp mismatch for the CCNA1-ZFP. Based on ongoing ZFP clinical trials, which did not result in adverse effects, we reason that the off-target issue can be solved/specificity of the approach is likely to be increased by, e.g., the targeting of multiple constructs to a single promoter, as well as the use of split enzyme approaches.37 Alternatively, other DNA binding approaches might provide more specific agents in the future, as is demonstrated by ongoing CRISPR/Cas efforts.32
After the realization that TSG silencing by epigenetic mechanisms is associated with carcinogenesis, enormous efforts have been undertaken to identify those genes for diagnostic or therapeutic strategies, and such studies have contributed to the discovery of novel TSGs. The diagnostic value or observed gene silencing of our chosen panel of genes has been described in several studies,1,18,20,21,38 but so far, only a few studies have been performed to explore the functional consequences of silencing and the potential therapeutic effects of re-expression within this malignancy. In this study, we included low passage cell lines to study functional effects, which may be more representative for primary cervical tumors compared to the long established cell lines. The use of such cell lines may help to bridge the gap with real tumors.
For C13ORF18 and TFPI2, we found growth inhibitory effects, but not for CCNA1 and Maspin. To our knowledge, this is the first report that demonstrates that TFPI2 is a TSG in cervical cancer cell lines using either cDNA overexpression or ATF-induced re-expression. Interestingly, Maspin seems to be less important as TSG in cervical cancer. We found that Maspin is unmethylated and highly expressed in cervical cancer, and our constructs did not decrease tumor growth. Although regarded as important therapeutic target in cancer for a long time, the latest insights suggest that the role of Maspin in cancer may need revision and does not have a tumor suppressive function.22
In conclusion, in this study we employed three different approaches to re-activate dormant TSGs and that may affect DNA methylation levels: epigenetic drugs, VP64-ATFs, and ZFP–TET2 constructs. In contrast to ZFP–TET2 constructs, epigenetic drugs and VP64-ATFs did not efficiently initiate demethylation at targeted CpGs, despite resulting in more efficient gene re-activation. Therefore, these two latter approaches seem not to be the most suitable approach for sustained gene re-activation, as this requires changes in the local epigenetic landscape. Our TET2-mediated approach delineates an efficient way to demethylate targeted CpGs, resulting in TSG re-expression and growth inhibition, and such approach can be extended to other genes as well. Such approach may be more sustainable than re-activation by VP64 and more specific compared to epigenetic drugs. Furthermore, our ZFPs may also serve as flexible tools to screen other putative demethylases or to compare demethylating efficiencies between demethylases (e.g., TDG versus TET). We also showed that TFPI2 and C13ORF18 are stronger tumor suppressors compared to CCNA1 and Maspin. For permanent re-expression of silenced genes, future efforts to further unravel the elusive DNA demethylation pathway39 may give clues to increase the efficiency of targeted demethylation to gain permanent control over the transcriptome.
Materials and Methods
Cell Culturing. The human cervical cancer cell lines HeLa, SiHa, CaSki, C33A and the human embryonic kidney cells HEK293T were obtained from the ATCC (Manassas, Virginia) and authenticity of the cell lines was verified by DNA short tandem repeat analysis (Baseclear, Leiden, The Netherlands). The CSCC-7, CSCC-8, CC-10, and CC-11 were derived from cervical squamous cell carcinoma as described previously28 and were kindly provided by Prof. GJ Fleuren (Leiden University Medical Center, Leiden, The Netherlands). The normal healthy human control cells (conditionally immortalized ovarian epithelial cells (OSE-C2) and skin fibroblasts (adult donor)) were, respectively, kindly provided by Dr. Richard Edmondson (Newcastle University, UK) or obtained from ATCC (CCD-1093SK). All cell lines were cultured in DMEM (BioWhittaker, Walkersville, Maryland), supplemented with 10% fetal calf serum (BioWhittaker), 1% l-glutamine and 0.6% gentamycin, at 37 °C under 5% CO2. For treatment with epigenetic drugs, cells were treated with 500 nM or 5 µM 5-aza-dC (Sigma, St Louis, Missouri) for three consecutive days. 500-nM TSA (Sigma) was added on the third day. Cells were harvested on the fourth day for analysis.
Cloning and Delivery. Target sites of engineered ZFPs were selected based on ranking according to Zincfingertools.org40 and the proximity to the transcription start site of targeted genes (−500 bp to +250 bp) (Figure 2a). The coding sequences of their DNA-specific binding domains are shown in Supplementary Table S1 and were established using the amino acid-DNA binding code.41 Sequences were ordered as double stranded DNA oligos (BIO BASIC, Markham, Canada) flanked with SfiI (Thermo Scientific, Leon-Rot, Germany) restriction sites at the N-terminus 5′-GGATCCGAGGCCCAGGCGGCC-3′ and C-terminus 5′-GGCCAGGCCGGCCGAAGATCTGAGGAG-3′. ZFPs were subsequently subcloned in the pMX-IRES-GFP carrying either the transcriptional activator VP64, no effector domain (NoEf), TET2-CD or TET2-mutant6 using SfiI restriction. For ectopic expression of C13ORF18, CCNA1, TFPI2, and Maspin cDNA, DNA fragments of the genes were generated by PCR (Phusion Hot Start II High-Fidelity DNA polymerase, Thermo Scientific) using gene-specific PCR primers (Supplementary Table S2) flanked with BamHI and NotI (C13ORF18, TFPI2) or BglII and EcoRI (CCNA1, Maspin) restriction sites on pre-ordered cDNA carrying plasmids (C13ORF18 isoform a (Invitrogen, Life Technologies, Carlsbad, California), CCNA1 isoform a (Invitrogen) and TFPI2 isoform 1 precursor (BIO-BASIC)) or cDNA obtained from HeLa cells (Maspin) and ligated into the pMX-IRES-GFP.
Retroviral transduction was performed as previously described.25 For superinfection, harvested cells were GFP sorted using the MoFlo-XDP sorter (Beckman Coulter, Woerden, The Netherlands), re-seeded and propagated. Subsequently, cells were re-transduced and harvested using the same procedure as normal transduction, or frozen in liquid nitrogen for later re-transduction.
ChIP(-Seq). ChIP(-Seq) was performed as previously described9 using the following antibodies: normal rabbit IgG (ab46540), H3K36Me3 (ab9050) (Abcam, Cambridge, UK), acH3 (06-599), acH4 (06-598), H3K4Me3 (07-473), H3K9me3 (07-442) and H3K27me3 (07-449, Millipore, Billerica, Massachusetts), H3K9Ac (39137, Active Motif, La Hulpe, Belgium) or anti-HA-tag (101P-200, Covance, Uden, The Netherlands). The coverage for the ChIP-Seq peaks was calculated by NextGENe by dividing the number of bases aligned to the region by the length of the peak. See the Supplementary Materials and Methods for a more extensive description.
Bisulfite Sequencing/Methylation-Specific PCR/Bisulfite Pyrosequencing. Methylation-specific PCR and bisulfite sequencing were performed as previously described25 using gene-specific primers (Supplementary Table S2). Bisulfite pyrosequencing was performed on the Pyromark Q24 MD pyrosequencer (Qiagen) according to the manufacturer's instructions. Methylated levels of single CpGs were determined using Pyromark Q24 Software (Qiagen).
Quantitative Real-time PCR (qRT-PCR). RNA was isolated using the RNeasy Plus Mini Kit (Qiagen, Hilden, Germany) and converted into 1 µg of cDNA (Thermo Scientific). qRT-PCR was performed as previously described9 using gene-specific primers (Supplementary Table S2). Gene expression levels relative to GAPDH were determined with the formula 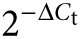 . Fold increase in gene-expression compared to controls was calculated with the formula
. Fold increase in gene-expression compared to controls was calculated with the formula 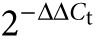 . Samples for which no amplification could be detected were assigned a Ct value of 40, resembling the total number of PCR cycles.
. Samples for which no amplification could be detected were assigned a Ct value of 40, resembling the total number of PCR cycles.
Growth Assay/Propidium Iodide Assay. Cells were seeded in 96-wells plates (2,000 cells per well), transduced and incubated for 1 to 5 days at 37 °C. To measure the fraction of viable cells, 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxy-phenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) (5 mg/ml) was added, followed by 3 hours of incubation at 37 °C. Then, the absorbance was detected at 490 nm with a Versamax microplate reader (Molecular Devices, Sunnyvale, California).
Four days after transduction, the fraction of cell death (late apoptotic and necrotic cells) was determined using propidium iodide as previously described.42 The colony size of CC-11 cells was measured 5 days after viral delivery using ImageJ (version 1.49m).
Statistical Analysis. Statistical significance was determined with the Student's t-test (single group comparison) or one-way ANOVA (multiple group comparison) followed by Dunnett's post hoc test using GraphPad Prism 5 software. A P-value of 0.05 or less was considered statistical significant (*P ≤ 0.05, **P < 0.01, and ***P < 0.001).
SUPPLEMENTARY MATERIAL Figure S1. Methylation and expression status of Maspin and C13ORF18. Figure S2. TET 1-3 expression in healthy control cells and cervical cancer cell lines. Figure S3. Endogenous re-expression of CCNA1, TFPI2 and Maspin mRNA by ATFs. Figure S4. Visualization of the low passage cell lines CSCC-7, CSCC-8, CC-10, and CC-11. Figure S5. ZFP association with their aimed target site. Figure S6. Genome-wide binding of the various gene-targeting constructs. Figure S7. Off-targets caused by sequence similarity. Figure S8. Relative GFP mRNA expression after retroviral treatment with C13ORF18- and CCNA1-targeting constructs in CaSki. Figure S9. Demethylation of the C13ORF18 promoter after ZFP-mediated targeting of TET2 or VP64. Figure S10. Off-target effects of TET-ZFPs on EPB41L3 promoter methylation Table S1. Sequence of ZFPs targeting C13ORF18, CCNA1, TFPI2 and Maspin. Table S2. Primer sequences. Materials and Methods.
Acknowledgments
We acknowledge Marcel Ruiters for helpful discussion and thank Haukeline Volders and Harry Hollema for helping with the Maspin staining. This work was financially supported by the Netherlands Organization for Scientific Research (NWO) through a VIDI grant (number 91786373) and a CHEMTHEM grant (number 728011101) to MGR.
The authors of this manuscript have nothing to declare.
Supplementary Material
References
- Hoque, MO, Kim, MS, Ostrow, KL, Liu, J, Wisman, GB, Park, HL et al. (2008). Genome-wide promoter analysis uncovers portions of the cancer methylome. Cancer Res 68: 2661–2670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esteller, M (2007). Epigenetic gene silencing in cancer: the DNA hypermethylome. Hum Mol Genet 16 Spec No 1: R50–R59. [DOI] [PubMed] [Google Scholar]
- Azad, N, Zahnow, CA, Rudin, CM and Baylin, SB (2013). The future of epigenetic therapy in solid tumours—lessons from the past. Nat Rev Clin Oncol 10: 256–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Groote, ML, Verschure, PJ and Rots, MG (2012). Epigenetic editing: targeted rewriting of epigenetic marks to modulate expression of selected target genes. Nucleic Acids Res 40: 10596–10613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voigt, P and Reinberg, D (2013). Epigenome editing. Nat Biotechnol 31: 1097–1099. [DOI] [PubMed] [Google Scholar]
- Chen, H, Kazemier, HG, de Groote, ML, Ruiters, MH, Xu, GL and Rots, MG (2014). Induced DNA demethylation by targeting Ten-Eleven Translocation 2 to the human ICAM-1 promoter. Nucleic Acids Res 42: 1563–1574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeder, ML, Angstman, JF, Richardson, ME, Linder, SJ, Cascio, VM, Tsai, SQ et al. (2013). Targeted DNA demethylation and activation of endogenous genes using programmable TALE-TET1 fusion proteins. Nat Biotechnol 31: 1137–1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, K, Pang, J, Cheng, H, Liu, WP, Di, JM, Xiao, HJ et al. (2015). Manipulation of prostate cancer metastasis by locus-specific modification of the CRMP4 promoter region using chimeric TALE DNA methyltransferase and demethylase. Oncotarget 6: 10030–10044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huisman, C, van der Wijst, MG, Falahi, F, Overkamp, J, Karsten, G, Terpstra, MM et al. (2015). Prolonged re-expression of the hypermethylated gene EPB41L3 using artificial transcription factors and epigenetic drugs. Epigenetics 10: 384–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tahiliani, M, Koh, KP, Shen, Y, Pastor, WA, Bandukwala, H, Brudno, Y et al. (2009). Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 324: 930–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He, YF, Li, BZ, Li, Z, Liu, P, Wang, Y, Tang, Q et al. (2011). Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science 333: 1303–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller, U, Bauer, C, Siegl, M, Rottach, A and Leonhardt, H (2014). TET-mediated oxidation of methylcytosine causes TDG or NEIL glycosylase dependent gene reactivation. Nucleic Acids Res 42: 8592–8604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin, R, Mao, SQ, Zhao, B, Chong, Z, Yang, Y, Zhao, C et al. (2013). Ascorbic acid enhances Tet-mediated 5-methylcytosine oxidation and promotes DNA demethylation in mammals. J Am Chem Soc 135: 10396–10403. [DOI] [PubMed] [Google Scholar]
- Kroeze, LI, van der Reijden, BA and Jansen, JH (2015). 5-Hydroxymethylcytosine: an epigenetic mark frequently deregulated in cancer. Biochim Biophys Acta 1855: 144–154. [DOI] [PubMed] [Google Scholar]
- Neri, F, Dettori, D, Incarnato, D, Krepelova, A, Rapelli, S, Maldotti, M et al. (2015). TET1 is a tumour suppressor that inhibits colon cancer growth by derepressing inhibitors of the WNT pathway. Oncogene 34: 4168–4176. [DOI] [PubMed] [Google Scholar]
- Hsu, CH, Peng, KL, Kang, ML, Chen, YR, Yang, YC, Tsai, CH et al. (2012). TET1 suppresses cancer invasion by activating the tissue inhibitors of metalloproteinases. Cell Rep 2: 568–579. [DOI] [PubMed] [Google Scholar]
- Tebas, P, Stein, D, Tang, WW, Frank, I, Wang, SQ, Lee, G et al. (2014). Gene editing of CCR5 in autologous CD4 T cells of persons infected with HIV. N Engl J Med 370: 901–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, N, Eijsink, JJ, Lendvai, A, Volders, HH, Klip, H, Buikema, HJ et al. (2009). Methylation markers for CCNA1 and C13ORF18 are strongly associated with high-grade cervical intraepithelial neoplasia and cervical cancer in cervical scrapings. Cancer Epidemiol Biomarkers Prev 18: 3000–3007. [DOI] [PubMed] [Google Scholar]
- Eijsink, JJ, Lendvai, Á, Deregowski, V, Klip, HG, Verpooten, G, Dehaspe, L et al. (2012). A four-gene methylation marker panel as triage test in high-risk human papillomavirus positive patients. Int J Cancer 130: 1861–1869. [DOI] [PubMed] [Google Scholar]
- Xu, C, Quddus, MR, Sung, CJ, Steinhoff, MM, Zhang, C and Lawrence, WD (2005). Maspin expression in CIN 3, microinvasive squamous cell carcinoma, and invasive squamous cell carcinoma of the uterine cervix. Mod Pathol 18: 1102–1106. [DOI] [PubMed] [Google Scholar]
- Zhang, Q, Zhang, Y, Wang, SZ, Wang, N, Jiang, WG, Ji, YH et al. (2012). Reduced expression of tissue factor pathway inhibitor-2 contributes to apoptosis and angiogenesis in cervical cancer. J Exp Clin Cancer Res 31: 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teoh, SS, Vieusseux, J, Prakash, M, Berkowicz, S, Luu, J, Bird, CH et al. (2014). Maspin is not required for embryonic development or tumour suppression. Nat Commun 5: 3164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glöckner, SC, Dhir, M, Yi, JM, McGarvey, KE, Van Neste, L, Louwagie, J et al. (2009). Methylation of TFPI2 in stool DNA: a potential novel biomarker for the detection of colorectal cancer. Cancer Res 69: 4691–4699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin, YY, Gong, W, Weng, MZ, Li, JY and Quan, ZW (2012). The role of tissue factor pathway inhibitor-2 gene in gallbladder cancer. Zhonghua Wai Ke Za Zhi 50: 1099–1103. [PubMed] [Google Scholar]
- Huisman, C, Wisman, GB, Kazemier, HG, van Vugt, MA, van der Zee, AG, Schuuring, E et al. (2013). Functional validation of putative tumor suppressor gene C13ORF18 in cervical cancer by artificial transcription factors. Mol Oncol 7: 669–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivera, A, Mavila, A, Bayless, KJ, Davis, GE and Maxwell, SA (2006). Cyclin A1 is a p53-induced gene that mediates apoptosis, G2/M arrest, and mitotic catastrophe in renal, ovarian, and lung carcinoma cells. Cell Mol Life Sci 63: 1425–1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beltran, AS and Blancafort, P (2011). Reactivation of MASPIN in non-small cell lung carcinoma (NSCLC) cells by artificial transcription factors (ATFs). Epigenetics 6: 224–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koopman, LA, Szuhai, K, van Eendenburg, JD, Bezrookove, V, Kenter, GG, Schuuring, E et al. (1999). Recurrent integration of human papilloma viruses 16, 45, and 67 near translocation breakpoints in new cervical cancer cell lines. Cancer Res 59: 5615–5624. [PubMed] [Google Scholar]
- Gregory, DJ, Zhang, Y, Kobzik, L and Fedulov, AV (2013). Specific transcriptional enhancement of inducible nitric oxide synthase by targeted promoter demethylation. Epigenetics 8: 1205–1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mossman, D, Kim, KT and Scott, RJ (2010). Demethylation by 5-aza-2′-deoxycytidine in colorectal cancer cells targets genomic DNA whilst promoter CpG island methylation persists. BMC Cancer 10: 366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami, J, Asaumi, J, Maki, Y, Tsujigiwa, H, Kuroda, M, Nagai, N et al. (2004). Effects of demethylating agent 5-aza-2(′)-deoxycytidine and histone deacetylase inhibitor FR901228 on maspin gene expression in oral cancer cell lines. Oral Oncol 40: 597–603. [DOI] [PubMed] [Google Scholar]
- Jurkowski, TP, Ravichandran, M and Stepper, P (2015) Synthetic epigenetics-towards intelligent control of epigenetic states and cell identity. Clin Epigenet 7: 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, HS, Liu, D, Huang, Y, Schmidt, S, Hickey, R, Guschin, D et al. (2012). A designed zinc-finger transcriptional repressor of phospholamban improves function of the failing heart. Mol Ther 20: 1508–1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falahi, F, Huisman, C, Kazemier, HG, van der Vlies, P, Kok, K, Hospers, GA et al. (2013). Towards sustained silencing of HER2/neu in cancer by epigenetic editing. Mol Cancer Res 11: 1029–1039. [DOI] [PubMed] [Google Scholar]
- Gaj, T, Gersbach, CA and Barbas, CF III (2013). ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol 31: 397–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimmer, MR, Stolzenburg, S, Ford, E, Lister, R, Blancafort, P and Farnham, PJ (2014). Analysis of an artificial zinc finger epigenetic modulator: widespread binding but limited regulation. Nucleic Acids Res 42: 10856–10868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaikind, B and Ostermeier, M (2014). Directed evolution of improved zinc finger methyltransferases. PLoS One 9: e96931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, Z, Shi, Y, Meng, W, Liu, Y, Yang, K, Wu, S et al. (2014). Expression and localization of maspin in cervical cancer and its role in tumor progression and lymphangiogenesis. Arch Gynecol Obstet 289: 373–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Wijst, MG, Venkiteswaran, M, Chen, H, Xu, GL, Plösch, T and Rots, MG (2015). Local chromatin microenvironment determines DNMT activity: from DNA methyltransferase to DNA demethylase or DNA dehydroxymethylase. Epigenetics 10: 671–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandell, JG and Barbas, CF III (2006). Zinc Finger Tools: custom DNA-binding domains for transcription factors and nucleases. Nucleic Acids Res 34: W516–W523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll, D, Morton, JJ, Beumer, KJ and Segal, DJ (2006). Design, construction and in vitro testing of zinc finger nucleases. Nat Protoc 1: 1329–1341. [DOI] [PubMed] [Google Scholar]
- Li, Q, van der Wijst, MG, Kazemier, HG, Rots, MG and Roelfes, G (2014). Efficient nuclear DNA cleavage in human cancer cells by synthetic bleomycin mimics. ACS Chem Biol 9: 1044–1051. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.