

Abstract
The rapid advancement in targeted genome editing using engineered nucleases such as ZFNs, TALENs, and CRISPR/Cas9 systems has resulted in a suite of powerful methods that allows researchers to target any genomic locus of interest. A complementary set of design tools has been developed to aid researchers with nuclease design, target site selection, and experimental validation. Here, we review the various tools available for target selection in designing engineered nucleases, and for quantifying nuclease activity and specificity, including web-based search tools and experimental methods. We also elucidate challenges in target selection, especially in predicting off-target effects, and discuss future directions in precision genome editing and its applications.
Targeted genome-editing technology continues to create intense excitement with each new technological advance.1,2,3 The development of tools to generate DNA breaks, activate,4 repress or label genomic loci,5,6 and remodel chromatin7 in a controlled, targeted manner will greatly aid the studies of a wide range of biological issues, including gene and genomic functions. The ability to specifically modify the genome also holds great promise for targeted gene therapies. Early work with meganucleases and zinc finger nucleases (ZFNs) showed that targeted site-specific DNA breaks could greatly increase the rate of homology-directed repair (HDR) at the specified locus.8,9 More recent developments include TAL effector nucleases (TALENs)10,11 and CRISPR/Cas9 systems (Figure 1).12,13,14 ZFNs consist of zinc finger motifs, which bind to DNA triplets, and the FokI nuclease domain which cleaves DNA upon dimerization.15,16 TALENs are composed of TAL effectors fused to the FokI nuclease domain and recognize DNA bases via conserved repeats that differ by two residues known as the repeat variable diresidue (RVD), which confers specificity to individual bases.17,18 Unlike ZFNs and TALENs that use protein domains to recognize target DNA sequences, the widely used CRISPR/Cas9 system adapted from Streptococcus pyogenes (Spy) uses both RNA and protein-based DNA recognition. These RNA-guided nucleases (RGENs) use a short guide RNA strand (gRNA), which targets a 20-nucleotide sequence, and the CRISPR associated (Cas) endonuclease Cas9, which binds to the fixed protospacer adjacent motif (PAM) NGG.12,13 Although there is a strict adherence to PAM recognition, due to the short length of the PAM the specificity of RGENs is largely controlled by gRNA-DNA interaction. With these engineered nucleases, we now have efficient molecular scissors that can cut genomic DNA in cells at preselected locations and introduce mutagenic errors via the nonhomologous end joining (NHEJ) DNA repair pathway for targeted gene knockout or targeted deletion of large chromosomal segments. Alternatively, if an exogenous DNA donor template is introduced in concert with the nuclease, DNA cleavage (DNA double strand breaks or nicks) may trigger endogenous HDR with the supplied DNA donor template, resulting in precise DNA modifications (Figure 1). These abilities have led to the emerging field of genome editing, a new field in engineering and life sciences focusing on precisely modifying genomes using engineered nucleases.
Figure 1.

Classes of designer nucleases and gene-editing outcomes. Targeted double-strand breaks can be induced using ZFNs, TALENs, or CRISPR/Cas9. DNA breaks are repaired via endogenous repair pathways such as non-homologous end joining (NHEJ) and homologous recombination (HR). The NHEJ pathway results in short deletions or insertions at the target site that can result in a targeted gene knock-out. The HR pathway is a high fidelity pathway that uses the sister chromatid as a template to correct the DNA break. An exogenous DNA template may be provided for homology directed repair (HDR). This pathway can be exploited to repair mutations or modify DNA at the resolution of a single nucleotide.
With the rapid advancement of genome-editing research, a suite of nuclease design and validation tools has been developed, significantly facilitating nuclease target site selection and experimental validation in terms of on-target and off-target activities. For most of the biological and medical applications of genome editing, high efficiency and high specificity of engineered nucleases are among the most important functional requirements; both are closely related to target site selection. For each endogenous genomic locus, the efficiency of DNA cleavage, both on-target and off-target, depends not only on the intrinsic nuclease activity (such as that of FokI domains and Cas9 protein) but also on target site accessibility and the affinity of DNA binding domain(s) (such as Tal effector domains and gRNA) to the target sequence. The specificity of engineered nucleases is significantly affected by the affinity of nuclease-DNA binding, such as zinc finger—DNA binding (ZFNs), Tal effector—DNA binding (TALENs) and gRNA—DNA hybridization (CRISPR), although the dimerization of FokI domains (ZFNs and TALENs) and the Cas9-PAM interactions may also play important roles. There is a lack of understanding on the behavior and functions of engineered nucleases in living cells, especially the dynamics of their interactions with DNA, and the cell cycle-dependent cleavage activity. Due to the limited biological knowledge and understanding of the structure and dynamics of the cell nucleus, especially chromatin structure, prediction of nuclease target accessibility and cleavage rates in living cells remains difficult. Further, the efficiency of homology directed repair also depends on the design, accessibility, and binding affinity of the donor templates as well. Therefore, experimental validation of target site selection is necessary. Herein, we use “true off-target sites” to indicate the off-target sites that are experimentally confirmed using polymerase chain reaction, sequencing or other methods.
In this article, we review some of the web-based tools available for target selection in designing engineered nucleases, and selected experimental methods for quantifying nuclease activity and specificity. Due to space limitations and the rapid development of the genome-editing field, only a subset of available tools will be discussed, rather than having a comprehensive review. Challenges in target selection, especially in predicting off-target effects, and future directions in precision genome editing will also be discussed.
Web-Based Design Tools for Nuclease Target Selection
A range of bioinformatics and experimental-based nuclease design tools have been developed that aid the target site selection of engineered nucleases. These tools fall into the following three main categories: (i) choice of target sites/design of nucleases, (ii) genomic searches for possible off-target sites, and (iii) determining the level of on- and off-target cleavage rates. A list of the available design tools is given in Table 1, together with a brief description of the functionality for each tool. Most of the tools listed in Table 1 are for the design of CRISPR/Cas9 systems, with a few for ZFNs and TALENs.
Table 1. Nuclease design tools.

ZFN design tools
Zinc finger proteins (ZFPs) can be designed to target many novel sequences based on the 3 bp specificity of individual fingers.19,20 Phage display-based selections and rational design techniques have been used by certain companies and research labs to generate high-affinity ZFPs and ZFNs.21,22,23,24,25,26 However, zinc finger (ZF) design remains difficult due to positional effects and a lack of straightforward ZFP design principles—a number of amino acid sequences in a given finger can specify a given triplet, but the activity of any given zinc finger is strongly dependent on its position in the ZFP and the nature of the neighboring zinc fingers.27,28,29 Tools such as ZiFit were developed to address this issue by taking the context dependence into account. However, designing a highly active and specific ZFN pair remains challenging.30,31 Alternatively, a bacteria two-hybrid screening platform is also available for custom ZFP production.32 However, the substantial amount of work required has limited its use outside of a small number of dedicated labs.
TALEN design tools
For designing TALENs, the DNA-targeting specificity of TAL effector RVDs is more straightforward than that of ZFs, allowing easier design of TALENs. There are four main RVDs, one for each DNA base.17,18 Based on this simple 1 to 1 recognition code and the requirement for a flanking 5' thymine base, first-generation design programs output many potential target sites.10,11,33,34,35,36 Despite the ability of well-designed nucleases to target defined loci with high efficiency, the widespread use of TALENs has been hampered by poor performance of some TALEN pairs designed, thereby necessitating the screening of a large number of candidates to find a validated TALEN pair with a high level of activity. For example, a high-throughput study that looked at the activity of 96 TALEN pairs determined that 12 pairs had no activity and 43 pairs had activities below 20% in a model cell line.37 Some TALEN design tools incorporate ranking of TALEN pairs. The E-TALEN webtool incorporates a scoring algorithm for ranking potential TALENs, but this scoring system was not experimentally validated.38 The second-generation TALEN design tool SAPTA (Scoring Algorithm for Predicting TALEN Activity) uses improved guidelines for TALEN design based on rules derived from experimentally testing 205 individual TALEN monomers.39 The SAPTA algorithm was designed to identify target sites for highly active TALENs34 that use the NK (Asparagine-Lysine) RVD which displays higher specificity for guanines compared to the standard NN (Asparagine-Asparagine) RVD.40 It was clear in constructing SAPTA that affinity plays an important role in having high cleavage activity, especially when the G-C content of the target site is high.39 However, the current version of SAPTA is based on experimental results from TALENs with NK RVD, and issues with target accessibility may render the predicted activity inaccurate. Therefore, further improvements to SAPTA are being conducted to make it a more useful design tool.
RGEN design tools
The ability of the Spy CRISPR/Cas9 system to target any 20 nucleotide sequence that is adjacent to an NGG PAM simplifies the design of gRNAs, since it is easy to locate PAM sequences in a gene or region of interest using a bioinformatics tool (Table 1). Although in general the CRISPR/Cas9 systems may have a much higher DNA cleavage rate when compared to ZFNs and TALENs, it is still desirable to identify optimal target sites in silico. Efforts have been made recently to develop web-based tools to predict high nuclease activity sites in a genomic region of interest. For example, sgRNA Designer41 (Table 1) attempts to predict the optimal sequence composition for high CRISPR/Cas9 activity. However, although the algorithm was validated with a previous CRISPR knockout library screen in human and mouse cells, it was not tested for designing a gRNA for a given input sequence. Similarly, sgRNA Scorer42 (Table 1) ranks gRNAs for high activity based on an algorithm generated using data from gRNAs tested in HEK293T cells. This study noted some correlation between site accessibility and gRNA activity, but it is unknown whether the predicted scores are valid for other cell types. The ranking from sgRNA Designer and sgRNA Scorer were shown to have a weak correlation. The web-based tool CRISPR Scan43 (Table 1) more accurately predicts gRNA activity in zebrafish than sgRNA Designer. Although this is likely due to a more accurate algorithm in CRISPR Scan, it could also be a consequence of using data from zebrafish in constructing the algorithm, resulting in a somewhat biased comparison. However, unlike sgRNA Designer and sgRNA Scorer (both with algorithms based on library screens), to date CRISPR Scan is the only tool with demonstrated ability to correlate gRNA activities with predicted scores. Although CRISPR Scan could also identify highly active gRNAs in Xenopus tropicalis, it remains to be seen if it can predict sgRNA activity in human cells.
Web-Based Tools for Nuclease Off-Target Site Prediction
The advancement of ZFN and TALEN technology sparked a growing concern for potential off-target cleavage that may occur throughout the genome. Nuclease specificity was often measured indirectly by cellular toxicity levels.44,45 More sophisticated techniques aim to directly measure nuclease activity at predefined genomic loci or screen libraries of sequences to identify potential off-target sites.46,47 Large genome size and the large number of potential nuclease cleavage sites have made determining the most likely off-target sites very difficult, especially as genomic context can greatly influence the cleavage of identical sites at different loci.48 A number of tools have been developed that search genomes for possible off-target sites for engineered nucleases, including scripts that systematically scan genomes and web-based bioinformatics tools that aid in the determination of potential off-target sites.31 Some of these tools are well validated using other existing approaches and/or experimental methods, including next-generation sequencing (NGS) of targeted amplicons. One example of using true off-target sites of a well characterized ZFN pair for establishing a bioinformatics tool is PROGNOS (Predicted Report Of Genome-wide Nuclease Off-target Sites) (Figure 2a), which was validated using results from different methods and comparisons of the level of overlap and the number of sites identified by each method are shown in Figure 2a46,47,49,50,51 Interestingly, PROGNOS, an exhaustive search tool, identified a true off-target site that was not found with experimental based methods.46 However, highly active off-target sites may not be ranked highly by PROGNOS, suggesting that there are unknown factors influencing ZFN and TALEN off-target activity but not yet accounted for in PROGNOS. Therefore, further improvements of PROGNOS are needed based on unbiased genome-wide analysis of off-target activity of ZFNs and TALENs.
Figure 2.

Comparison of off-target analysis by different methods. (a) The 38 heterodimeric bona fide off-target sites for CCR5 ZFNs42 found by four different experiment-based prediction methods and the refined “ZFN v2.0” PROGNOS algorithm. The PROGNOS sites are drawn from the top rankings spanning 3× the number of predictions by the Bayesian abstraction of the in vitro cleavage profile. (**) Note that only six of the sites found using ChIP-Seq were described,44 so the full degree of overlap of all ChIP-Seq sites with sites found by other methods remains unknown. Adopted from Fine et al.46. (b) A comparison of the off-target predictions by the MIT CRISPR Design Tool (solely bioinformatics-based) to the bona fide off-target sites found for nine different RGENs by the GUIDE-Seq method (experimental-based). (c) A comparison analogous to (b) but using the E-CRISP bioinformatics-based prediction tool. GUIDE-Seq figures adopted from Tsai et al.55.
Compared with ZFNs and TALENs, the CRISPR/Cas9 systems are easier to use, more efficient, and can readily target multiple genes. The potential drawback of using CRISPR/Cas9 systems to target genomic loci is possible off-target effects, since their target specificity relies on Watson-Crick base pairing, thus a gRNA can hybridize to sequences containing base mismatches, resulting in off-target cleavage.52,53,54 Although many web-based tools have been developed to identify off-target sites (Table 1), none can predict off-target sites with high accuracy, as discussed below. For example, a recent comparison of off-target predictions by the MIT CRISPR Design and E-CRISP tools for nine different gRNA designs demonstrated that in predicting CRISPR/Cas9 off-target sites these tools performed poorly, indicating that off-target activity cannot be accurately identified when predictions are solely based on sequence homology (Figure 2b,c).55 Further, it was revealed that CRISPR/Cas9 systems could tolerate DNA bulges and RNA bulges at the cleavage site, in addition to base mismatches.56 Consequently, a more sophisticated program, COSMID (CRISPR Off-target Sites with Mismatches, Insertions and Deletions) was developed that ranks potential off-target sites by considering base mismatches, insertions and deletions between gRNA and DNA sequences,48 and some other search tools have since incorporated insertions and deletions as an additional search option.57
A comparison of existing web-based tools for predicting CRISPR/Cas9 off-target sites revealed a wide range of agreements and discrepancies (Table 2). The inability of some tools to identify off-target sites containing only mismatches suggests that these tools use a repeat masker (Table 2). With DNA or RNA bulges, tools with the ability to search for bulge-containing sites perform better than those without, although some tools can identify bulge-containing sites that can be modeled as base mismatches (Table 2). However, they failed to identify true off-target sites with bulges that cannot be modeled by base mismatches alone (Table 2). Since there is still a lack of understanding about target site accessibility and RGEN binding to DNA in living cells, the existing CRISPR design tools may not predict off-target effects (sites and cleavage rates) with high accuracy, therefore readers are advised to consider using several tools (Table 1) to compare outputs for initial design of gRNAs and perform experimental validation to determine true off-target sites.
Table 2. Comparison of COSMID with other available tools in predicting off-target sitesa.
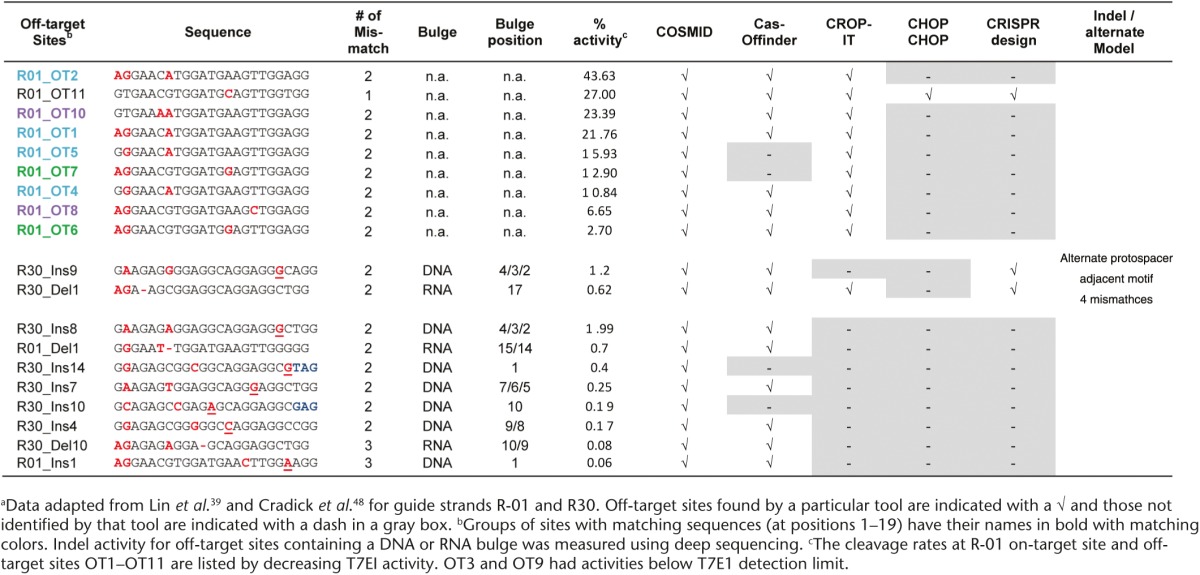
The CROP-IT web tool integrates whole-genome information from existing Cas9 off-target binding and cutting data sets in an effort to improve off-target identification and prediction.58 Even though this tool makes use of experimental data and outperforms some other search algorithms, it still performed poorly when compared to the results obtained using the Guide-Seq method, since only ~60% of the true off-target sites were identified for three gRNAs even when the top 500 predicted sites were considered. This high level of false positive hits demonstrates a major drawback of current in silico algorithms for RGEN off-target identification.
The tools for ZFN, TALEN, and RGEN off-target predictions differ in their input parameters, search features, degree of exhaustive search, accuracy, and the amount of information in output. In some cases, a number of sequence-validated off-target sites could be identified only by a single tool;46 in some other cases, predictions from several tools overlap.48 As shown in Table 3, although not perfect, in silico off-target search tools can be very helpful in quickly establishing a nuclease design, synthesis and testing workflow. For example, the current web-based tools are useful in screening potential gRNA designs for identifying closely matched sites, and tools that do not contain a repeat masker can help identify gRNAs that have perfectly matched off-target sites or that target repetitive elements.
Table 3. gRNA design overview.

Unlike PROGNOS which has algorithms built upon molecular information of protein-DNA interactions for both zinc finger motifs and TAL effector RVDs, existing web-based tools for the prediction of RGEN off-target sites31,35,48,52 rely heavily on sequence homology between the gRNA and potential cleavage sites. This often renders the prediction and ranking of potential off-target sites inaccurate. There is an unmet need to establish broadly applicable “in silico” rules for searching and ranking RGEN off-target sites due to the fundamental challenges, including the lack of detailed molecular information on Cas9, gRNA, and DNA interactions, the quantitative measurements of affinity between gRNA and DNA target, and target accessibility. To improve the first-generation search algorithms, a better understanding of gRNA-DNA interaction, nuclease-DNA binding and cleavage dynamics, as well as target accessibility is required. With newer genome-wide methods for determining nuclease off-target cleavage,55,59,60,61 it is likely that more true off-target sites for engineered nucleases (especially CRISPR/Cas9 systems) will be confirmed and a better understanding of nuclease off-target effects emerge, which will help to improve the bioinformatics based off-target search and prediction tools.
Methods for Experimental Evaluation of Target Site Selection
Many experimental methods have been developed to quantify the activity of engineered nucleases, including enzyme-based assays62,63 and sequencing-based assays.64,65,66 Most of these methods detect small insertions and/or deletions (indels) that arise from imperfect NHEJ-mediated repair of DNA double-strand breaks (DSBs). The most widely used enzyme-based methods rely on mismatch-sensitive enzymes such as CEL-I nuclease and T7 endonuclease I (T7EI).62,63,67 They work by detecting heteroduplexes formed by hybridizing wild-type and mutant DNA sequences or hybridizing two different mutant sequences together, and the relative intensity of cleavage products resolved by agarose gel electrophoresis provides a measure of mutation frequency in a population of cells. Alternatively, if the nuclease cut site is within a unique restriction enzyme motif, a restriction fragment length polymorphism (RFLP)-based assay can be used in place of CEL-I or T7EI. In this assay, nuclease-induced indels destroy the restriction site. When these cleavage products are resolved on a gel, the band corresponding to the uncut DNA represents the mutant population.68 Although these enzyme-based assays are quick and cost effective, they have a detection limit of 1–5% and are sensitive to endogenous mismatches (such as heterozygous SNPs) leading to potential false positive results.
Sanger sequencing of DNA from individual clones has been the gold standard for confirming nuclease induced indels, but this method is time consuming and not cost-effective due to the high number of samples that need to be analyzed.69 Alternatively, Sanger sequencing of a bulk population can be used in conjunction with the recently developed web tool tracking of indels by decomposition (TIDE).64 TIDE deconvolutes the mixed chromatogram signals from nuclease-treated cells to accurately determine the mutation frequency in the population. The TIDE tool also outputs the frequency of each deletion and insertion size in the population and is insensitive to endogenous SNPs. However, as with the enzyme-based methods, TIDE analysis has a lower limit of detection of 1–5%. To accurately detect rare cleavage events, high-throughput sequencing approaches enable accurate measurement of mutation rates as low as 0.1%, although careful consideration should be made to discard false positives due to polymerase chain reaction or sequencing error.70
Single-molecule real-time (SMRT) sequencing is an alternative platform that has been demonstrated to perform as well as sanger sequencing of single cell clones but with higher throughput, and it is possible to use SMRT sequencing to measure HDR and NHEJ events simultaneously due to the longer read length.65 Other less common protocols available for detecting nuclease induced indel rates include fluorescent polymerase chain reaction,71 DNA melting analysis,72 and CRISPR/Cas9 restriction fragment length polymorphism 73 that can distinguish between mono and biallelic mutagenesis in clones. These methods indirectly measure nuclease activity as they depend on the mutagenic susceptibility of the endogenous repair machinery in the cell type employed. One method that directly measures the levels of DNA DSBs is BLESS (direct in situ breaks labeling, enrichment on streptavidin and next-generation sequencing).74 Although this method detects free DNA DSB ends, it cannot detect any alleles that have undergone NHEJ repair. As the price of NGS has dropped markedly, it is now possible to very precisely measure the percentage of alleles that are wild-type, mis-repaired or have correctly undergone HDR.65 Many laboratories use internal pipelines for the analysis of sequencing results; though several web-based tools have been recently developed, including CRISPR-GA75 and CRISPResso (Table 1).
Methods for Determining Off-Target Effects
Although engineered nucleases are designed to cleave at a predefined genomic locus, off-target effects at similar sequences have been observed.45,53,76 ZFNs and TALENs display promiscuity due to the ability of ZFPs and TAL effectors to bind to sites in the genome that have high degrees of homology to on-target sites. RGEN induced DSBs can be caused by binding promiscuity of both the gRNA and the Cas9 endonuclease. The optimal PAM for Spy Cas9 is NGG, although active off-target sites with NAG, NGA, NCG, NGC, NGT, NTG, and NAA PAM sequences have been identified.52,55 Mismatches as well as base insertions or deletions that form bulges between the gRNA and the target DNA strand may also be tolerated.52,53,56 The functional consequence of the off-target activity of engineered nucleases is still largely unclear and the off-target effects (both sites and cleavage rates) are likely to vary within the major classes of nucleases due to the requirement for homology with the on-target site, and between the major classes of nucleases due to the nature of nuclease-DNA binding. However, any active off-target site in an exonic or regulatory sequence in a genome would likely have detrimental effects on gene expression and could possibly lead to aberrant cellular function. In addition to nuclease-induced small indels, there is the possibility of a chromosomal deletion,53,77 inversion,78 or translocation between the on-target and off-target sites (Figure 3).79 Indeed, the potential for chromosomal translocations is a real concern in the use of multiplex gene targeting for therapeutic purposes, although it presents a novel system for modeling oncogenic translocations in vivo.80
Figure 3.

Gross chromosomal rearrangements as a consequence of genome editing. Multiplex gene targeting can result in targeted large deletions, inversions, or translocations. However, these gross chromosomal rearrangements can also occur between nuclease on- and off-target sites. Cut sites represented by red arrows.
Given the potentially dire consequences of nuclease off-target activity, it is pertinent to identify and characterize potential off-target effects when using genome editing for therapeutic applications. Experimental determination of active off-target sites is a laborious task due to the size of the genome and the large number of potential off-target sites. Early studies of nuclease specificity focused on experimental methods, such as in vitro SELEX,49,81,82 integrase-defective lentiviral vector (IDLV) capture,50 in vitro cleavage,47 and bacteria one-hybrid screening83 to determine potential off-target sites and provide a shortlist of candidate sites for testing. All of these methods are laborious, costly and require highly specialized protocols which have prevented their widespread use. It is therefore very beneficial to use bioinformatics-based tools to identify potential nuclease off-target sites, as discussed above. The fact that PROGNOS46 has identified bona fide off-target sites for more ZFNs and TALENs constructed than available experimental based methods such as SELEX and IDLV capture is a clear demonstration of the power of in silico prediction methods.46
As for the CRISPR/Cas9 systems, issues with target sequence accessibility and the tolerance of base mismatches and DNA/RNA bulges make accurate prediction of true off-target sites difficult. For example, existing web-based tools for RGEN off-target prediction may identify hundreds or even thousands of potential off-target sites,52 but the scoring/ranking of these sites is usually inaccurate or even misleading, since typically few of the top-ranked sites are true off-target sites as revealed by experimental evaluation. The most widely used algorithm for scoring potential off-target sites predominantly relies on data from four gRNAs targeting a single gene and determines the likelihood of cleavage at a given site based on the total number of mismatches (up to four), mismatch position, and distance between mismatches.52 However, given the high number of false positive hits and the failure of many tools to identify true off-target sites, it is likely that there are other factors apart from sequence homology that influence off-target cleavage. Neither experimentally testing all the potential off-target sites nor relying on rudimentary ranking of these sites is ideal for confirming the true off-target sites.
Recently, several new experimental methods have been described that attempt to capture the genome-wide activity of RGENs in an “unbiased” manner (Figure 4). These methods use different strategies to detect DNA DSBs with the ultimate goal of identifying RGEN induced DSBs. Cas9 ChIP assays use a catalytically dead version of Cas9 (dCas9) to determine the genome-wide binding profile of dCas9 when combined with a specific gRNA. For all gRNAs tested, ChIP-seq identified the on-target site and hundreds of genome-wide Cas9 binding sites.61,84,85 However, Wu et al.84 reported that only 1 out of 295 ChIP-seq identified sites had off-target activity as confirmed by deep sequencing, whereas Kuscu et al. reported Cas9 cleavage activity at 7 ChIP-seq predicted sites for a single gRNA.61 Independent reanalysis of these seven sites found no evidence for RGEN activity and suggested that the indels observed were due to Illumina sequencing errors in processing homopolymer stretches close to the expected cut sites.55 The lack of overlap between dCas9 binding and Cas9 cleavage activities from these ChIP-seq studies demonstrates that Cas9 binding does not necessarily serve as a marker for RGEN activity. In the absence of gRNA molecules, dCas9 favored DNA regions with open chromatin, raising the possibility that RGEN activity or site preference could be influenced by site accessibility. An alternative approach, Digenome-seq has been developed in which potential off-target sites are identified via in vitro digestion of intact genomic DNA-RGEN complexes coupled with whole genome sequencing.59 This method identifies RGEN off-target sites based on the ability of the nuclease to recognize and cleave genomic off-target sites in vitro.59 When gRNAs targeting HBB and VEGFA were tested using this method, only 4 out of 37 and 8 out of 34 off-target loci identified respectively for the two genes were found to have detectable levels of activity when interrogated by deep sequencing. It is possible that the rest of the sites were false positives or had activity levels below the limit of detection. This discrepancy suggests that cellular or genomic context plays an important role in off-target cleavage.
Figure 4.

Outline of various methods for off-target site identification and validation. In silico prediction tools identify potential off-target (OT) sites that can be analyzed by next-generation sequencing (NGS). There are various experimental methods designed to identify OT sites in an unbiased manner. After OT site identification, a second round of NGS at these sites is required to verify if they are bona fide OT sites.
Genome-wide RGEN off-target sites can be determined by break capture methods, including IDLV capture,60 translocation capture HTGTS (high-throughput, genome-wide translocation sequencing),86 and dsODN capture.55 These methods use different strategies making it difficult to directly compare them. However, there are some striking differences in the results. In a study using IDLV capture,60 six true off-target sites were not found. Each of these sites had activity <1% when assayed by deep sequencing, suggesting that this may be the detection limit of IDLV capture. HTGTS identifies off-target DSBs that have translocated to the on-target site.86 In using HTGTS for identifying the off-target activity of different gRNAs, it was demonstrated that some gRNAs are more specific than others; however, the translocation loci were not analyzed by deep sequencing to determine the activity at identified off-target sites.86 This method is limited by the requirement for DSBs at the on- and off-target sites to occur within the same cell simultaneously. Both breaks must also escape local NHEJ repair which may affect the sensitivity of the assay. The GUIDE-seq method uses a short double stranded oligonucleotide (dsODN) instead of a lentiviral construct to tag DSBs.55 This study found a large number of previously unknown off-target sites for 3 gRNAs and identified off-target sites for 10 additional gRNAs. The GUIDE-seq method is a powerful tool to identify true genome-wide RGEN off-target sites without the restrictions of in silico prediction algorithms. This method makes the assumption that all the sites with RGEN-induced DSBs should take up the blunt ended dsODNs by an NHEJ-dependent pathway. Although this scenario is possible, repair by NHEJ without dsODN insertion is more likely, and sequence homology may influence the integration of dsODNs into certain DSBs. It would be interesting to see if genome-wide GUIDE-seq profiles are consistent using dsODNs of varying sequence. Further, the ability to integrate dsODNs into DSBs by NHEJ may be dependent on the cell type and the nature of the DSB, for example 5' overhangs induced by FokI cleavage and 5' or 3' overhangs induced by Cas9 nickase pairs. The initial study used two cell lines and it remains to be seen if this method can be successfully applied to other cell lines and adapted for use in clinically relevant cell types such as hematopoietic stem cells (HSCs). The only method to directly detect DNA DSBs is BLESS.74,87 The drawback of directly detecting DSBs is that alleles that have undergone NHEJ repair cannot be detected, which makes the assay time sensitive. However, this time sensitivity could allow genome-wide mapping of the chronological order of the activity of engineered nucleases at on- and off-target sites. The BLESS method also outperformed both ChIP-seq and in silico prediction when directly compared with results using two gRNAs with two different Cas9 orthologs (four scenarios).87
A comprehensive comparison of the methods for genome-wide RGEN off-target detection is difficult since there is little overlap in the gRNAs used in these studies. However, the small amount of data that permits direct comparisons shows that GUIDE-seq identifies more off-target sites than any other method, although differences in cell types used in different studies should be taken into account. The establishment of a unified database of all true off-target sites of RGENs would facilitate the design of improved algorithms for in silico prediction of potential off-target sites, which would provide a quick, cost effective means to prescreen candidate gRNAs and greatly enhance the analysis of RGEN genotoxicity.
Methods for Minimizing Off-Target Effects
Several approaches have been developed to reduce off-target activity of engineered nucleases (Figure 5). Early attempts to block off-target activity of ZFNs used mutagenesis of the FokI domain to create heterodimeric versions to reduce homodimerization of ZFNs.44,88,89,90 These modifications are also applicable to other engineered nucleases, such as TALENs and RGENs. However, these heterodimeric modifications can also reduce on-target activity of nucleases, presumably by reducing the binding energy of FokI dimerization. FokI mutagenesis has also been used to generate FokI nickases.91,92,93 For example, ZFNickases can induce HDR at a lower rate than ZFNs, but have a higher HDR to NHEJ ratio. FokI nickases have also been successfully used with TAL effectors.94,95 The Cas9 endonuclease generates DSBs by cleaving DNA strands via conserved RuvC and HNH nuclease domains. The RuvC domain cleaves the non-target DNA strand and the HNH domain cleaves the target DNA strand. Inactivation of one domain results in a partially inactivated Cas9 that can generate DNA single strand breaks.96,97 It has been demonstrated that the Cas9 nickases may have reduced off-target activity while having high on-target activity. It can also be paired to generate a staggered DSB at the on-target locus.86,98 However, if two adjacent 20-base off-target sites with appropriate spacing have sufficient sequence homology to the intended on-target sequence, the Cas9 nickases can bind and become active, resulting in off-target cleavage.56 This suggests that the Cas9 nickase system reduces off-target activity largely by increasing the overall target length from 20 to 40 bases. Further, Cas9 nickases may not be fully inactivated and can still induce DSBs even with a single gRNA.99 The specificity of CRISPR/Cas9 system could be further increased if both of the Cas9 nuclease domains in a Cas9 nickase pair are mutated to create catalytically inactive or dead Cas9s (dCas9s) which are then fused to the FokI nuclease domains respectively, forming a dCas9-FokI pair. In this case, the targeting of DNA sequence is achieved by two gRNAs and dCas9s, and the DNA cleavage is generated by the dimerized FokI domains. Although off-target activity is reduced to a greater degree compared to Cas9 nickases, lower on-target activity is also observed.99,100,101 dCas9-FokI pairs also have a more strict spacer length due to the requirement for FokI dimerization, which limits the number of potential targets in a genome.
Figure 5.

Strategies to reduce off-target events. (a) Modification of the FokI domain to prevent homodimerization of ZFN or TALEN monomers. (b) Modification of the Cas9 nuclease to generate a nicking version of Cas9 (Cas9N). Cas9N can generate single-stranded DNA breaks. (c) Inactivation of the Cas9 endonuclease to create a dead Cas9. Fusion of the FokI domain creates a dCas9-FokI enzyme that requires a pair of dCas9-FokI to achieve dimerization of the FokI domain for DNA cleavage. (d) Cas9 orthologs with longer protospacer adjacent motif sequences can result in less potential off-target sites in the genome.
RGEN off-target effects can also be mitigated by modifying the gRNA, although there is conflicting evidence as to how best to achieve reduced mutagenic potential. Both gRNA truncation102 and gRNA elongation59 have been shown to reduce the off-target activity of certain gRNAs and result in better on- to off-target ratios. More widespread use of these strategies could reveal if they are broadly applicable to all gRNAs, or to which gRNAs they are best suited. Cas9 orthologs with different PAM requirements have been adopted recently for genome editing in mammalian cells.87,103,104,105 Three Cas9 orthologs with longer PAM sequences, Staphylococcus aureus Cas9, Streptococcus thermophilus Cas9 and Neisseria meningitidis Cas987,106,107 have reduced off-target activity. Orthologs with longer PAM sequences are expected to have fewer potential off-target sites genome-wide although the probability of finding a PAM sequence in a gene of interest is also reduced. These orthogonal systems could also be altered to form nickases and dCas9-FokI fusions to further increase the specificity of RGENs.
Challenges and Path Forward
Over the last few years, a new field of precision genome editing has emerged, thanks to the recent advent of engineered nucleases, especially TALENs and CRISPR/Cas9 systems. Although precision genome editing has the potential to revolutionize biology and medicine, and holds great promise for many applications, including disease modeling, molecular pathway dissection, synthetic biology, and therapeutics, many challenges remain. For example, engineered nucleases often generate off-target cleavage, causing mutations, insertions, deletions, inversions, or translocations in the genetic sequence, which may result in aberrant gene expression, cell death, or oncogenesis. Therefore, it is often necessary to maximize the cleavage efficiency of engineered nucleases and minimize genomic risk by reducing or eliminating off-target effects; both are closely related to target site selection. Further, in repairing nuclease-induced DSBs, cells typically favor error-prone pathways such as NHEJ and micro-homology mediated end joining. For therapeutic applications of genome editing where HDR is required, significantly increasing the HDR rate in both dividing and nondividing cells is a major challenge. Another important challenge in further advancing genome editing is efficient delivery of engineered nucleases, activators, repressors and donor molecules into clinically relevant cell types in vitro and in vivo, and developing methods for in vivo tissue-specific delivery.
Although many design tools have been developed for engineered nucleases (Table 1), better tools for target selection are still needed. Since each target locus in a genome requires that a pair of TALENs needs to be constructed and tested, it becomes quite laborious to screen for highly active TALEN pairs. Further, despite the ease in designing and testing CRISPR/Cas9 systems, there is a large variability in their cleavage activity. Although attempts have been made to determine if rational design of highly active gRNAs is possible,41,42,43 when the output of these tools is compared, there is only a modest or no correlation between them, indicating that the broad applicability of the scoring algorithms depends on the experimental results employed in constructing the scoring functions. It remains to be seen if these tools are fully predictive or if over training of the data or selection bias may have skewed the parameters.
Off-target activity of engineered nucleases remains a major concern, especially in therapeutic applications. Off-target DSBs may induce indels that activate oncogenes, and chromosomal rearrangements resulting from on- and off-target DSBs may lead to a cancerous phenotype in nuclease-treated cells. Although great advances have been made in recent years in developing methods for identifying off-target sites, none of the in silico off-target search tools can accurately predict all possible off-target sites, and a better understanding of nuclease-DNA interaction dynamics and target accessibility is required in order to significantly improve these in silico off-target search tools. Also, despite the ability of NGS platforms to identify off-target sites with activity as low as 0.1%, there may be other off-target sites below this limit that go undetected. Another major concern is the variability in sequencing data analysis pipelines implemented by different labs when analyzing NGS data, which makes comparisons between data sets very difficult. It is certainly desirable to have a small number (e.g., 1–3) of “standardized” pipelines that are available to, and acceptable by, the general laboratories in genome editing. Further, the long-term effects of off-target activity are largely unknown. It is estimated that on average, each cell has an estimated steady state of 50,000 endogenous DNA lesions,108 while whole-genome sequencing of 12 individuals revealed over 500,000 indels in each individual with 230–390 occurring in exonic regions.109 Other studies estimate the mutation rate of radiotherapy is at around 20–40 DSBs/cell/Gy and up to 1,000 single strand breaks/cell/Gy.110 Although it is likely that the number of DSBs induced by engineered nucleases is relatively small compared with the endogenous levels of DSB formation and the accumulation of exonic indels in the cell population, significant efforts need to be made to analyze genome-wide off-target effects, develop a database for off-target activities in different cell types, establish consensus guidelines for selecting optimal target sites, and define benchmark assays, best practices and unified standards for determining genotoxicity due to engineered nucleases.
References
- Segal, DJ and Meckler, JF (2013). Genome engineering at the dawn of the golden age. Annu Rev Genomics Hum Genet 14: 135–158. [DOI] [PubMed] [Google Scholar]
- Cai, M and Yang, Y (2014). Targeted genome editing tools for disease modeling and gene therapy. Curr Gene Ther 14: 2–9. [DOI] [PubMed] [Google Scholar]
- Cox, DB, Platt, RJ and Zhang, F (2015). Therapeutic genome editing: prospects and challenges. Nat Med 21: 121–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilber, A, Tschulena, U, Hargrove, PW, Kim, YS, Persons, DA, Barbas, CF 3rd et al. (2010). A zinc-finger transcriptional activator designed to interact with the gamma-globin gene promoters enhances fetal hemoglobin production in primary human adult erythroblasts. Blood 115: 3033–3041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong, L, Zhou, R, Kuo, YC, Cunniff, M and Zhang, F (2012). Comprehensive interrogation of natural TALE DNA-binding modules and transcriptional repressor domains. Nat Commun 3: 968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma, H, Naseri, A, Reyes-Gutierrez, P, Wolfe, SA, Zhang, S and Pederson, T (2015). Multicolor CRISPR labeling of chromosomal loci in human cells. Proc Natl Acad Sci USA 112: 3002–3007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilton, IB, D'Ippolito, AM, Vockley, CM, Thakore, PI, Crawford, GE, Reddy, TE et al. (2015). Epigenome editing by a CRISPR-Cas9-based acetyltransferase activates genes from promoters and enhancers. Nat Biotechnol 33: 510–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouet, P, Smih, F and Jasin, M (1994). Expression of a site-specific endonuclease stimulates homologous recombination in mammalian cells. Proc Natl Acad Sci USA 91: 6064–6068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bibikova, M, Carroll, D, Segal, DJ, Trautman, JK, Smith, J, Kim, YG et al. (2001). Stimulation of homologous recombination through targeted cleavage by chimeric nucleases. Mol Cell Biol 21: 289–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cermak, T, Doyle, EL, Christian, M, Wang, L, Zhang, Y, Schmidt, C et al. (2011). Efficient design and assembly of custom TALEN and other TAL effector-based constructs for DNA targeting. Nucleic Acids Res 39: e82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller, JC, Tan, S, Qiao, G, Barlow, KA, Wang, J, Xia, DF et al. (2011). A TALE nuclease architecture for efficient genome editing. Nat Biotechnol 29: 143–148. [DOI] [PubMed] [Google Scholar]
- Jinek, M, Chylinski, K, Fonfara, I, Hauer, M, Doudna, JA and Charpentier, E (2012). A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337: 816–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran, FA, Hsu, PD, Wright, J, Agarwala, V, Scott, DA and Zhang, F (2013). Genome engineering using the CRISPR-Cas9 system. Nat Protoc 8: 2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong, L, Ran, FA, Cox, D, Lin, S, Barretto, R, Habib, N et al. (2013). Multiplex genome engineering using CRISPR/Cas systems. Science 339: 819–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, YG, Cha, J and Chandrasegaran, S (1996). Hybrid restriction enzymes: zinc finger fusions to Fok I cleavage domain. Proc Natl Acad Sci USA 93: 1156–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith, J, Bibikova, M, Whitby, FG, Reddy, AR, Chandrasegaran, S and Carroll, D (2000). Requirements for double-strand cleavage by chimeric restriction enzymes with zinc finger DNA-recognition domains. Nucleic Acids Res 28: 3361–3369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boch, J, Scholze, H, Schornack, S, Landgraf, A, Hahn, S, Kay, S et al. (2009). Breaking the code of DNA binding specificity of TAL-type III effectors. Science 326: 1509–1512. [DOI] [PubMed] [Google Scholar]
- Bogdanove, AJ and Voytas, DF (2011). TAL effectors: customizable proteins for DNA targeting. Science 333: 1843–1846. [DOI] [PubMed] [Google Scholar]
- Pavletich, NP and Pabo, CO (1991). Zinc finger-DNA recognition: crystal structure of a Zif268-DNA complex at 2.1 A. Science 252: 809–817. [DOI] [PubMed] [Google Scholar]
- Pabo, CO, Peisach, E and Grant, RA (2001). Design and selection of novel Cys2His2 zinc finger proteins. Annu Rev Biochem 70: 313–340. [DOI] [PubMed] [Google Scholar]
- Rebar, EJ and Pabo, CO (1994). Zinc finger phage: affinity selection of fingers with new DNA-binding specificities. Science 263: 671–673. [DOI] [PubMed] [Google Scholar]
- Jamieson, AC, Wang, H and Kim, SH (1996). A zinc finger directory for high-affinity DNA recognition. Proc Natl Acad Sci USA 93: 12834–12839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segal, DJ, Dreier, B, Beerli, RR and Barbas, CF 3rd (1999). Toward controlling gene expression at will: selection and design of zinc finger domains recognizing each of the 5'-GNN-3' DNA target sequences. Proc Natl Acad Sci USA 96: 2758–2763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choo, Y and Klug, A (1994). Toward a code for the interactions of zinc fingers with DNA: selection of randomized fingers displayed on phage. Proc Natl Acad Sci USA 91: 11163–11167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porteus, MH and Baltimore, D (2003). Chimeric nucleases stimulate gene targeting in human cells. Science 300: 763. [DOI] [PubMed] [Google Scholar]
- Urnov, FD, Miller, JC, Lee, YL, Beausejour, CM, Rock, JM, Augustus, S et al. (2005). Highly efficient endogenous human gene correction using designed zinc-finger nucleases. Nature 435: 646–651. [DOI] [PubMed] [Google Scholar]
- Isalan, M, Choo, Y and Klug, A (1997). Synergy between adjacent zinc fingers in sequence-specific DNA recognition. Proc Natl Acad Sci USA 94: 5617–5621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez, CL, Foley, JE, Wright, DA, Müller-Lerch, F, Rahman, SH, Cornu, TI et al. (2008). Unexpected failure rates for modular assembly of engineered zinc fingers. Nat Methods 5: 374–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sander, JD, Dahlborg, EJ, Goodwin, MJ, Cade, L, Zhang, F, Cifuentes, D et al. (2011). Selection-free zinc-finger-nuclease engineering by context-dependent assembly (CoDA). Nat Methods 8: 67–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandell, JG and Barbas, CF 3rd (2006). Zinc Finger Tools: custom DNA-binding domains for transcription factors and nucleases. Nucleic Acids Res 34(Web Server issue): W516–W523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sander, JD, Zaback, P, Joung, JK, Voytas, DF and Dobbs, D (2007). Zinc Finger Targeter (ZiFiT): an engineered zinc finger/target site design tool. Nucleic Acids Res 35(Web Server issue): W599–W605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeder, ML, Thibodeau-Beganny, S, Osiak, A, Wright, DA, Anthony, RM, Eichtinger, M et al. (2008). Rapid “open-source” engineering of customized zinc-finger nucleases for highly efficient gene modification. Mol Cell 31: 294–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle, EL, Booher, NJ, Standage, DS, Voytas, DF, Brendel, VP, Vandyk, JK et al. (2012). TAL Effector-Nucleotide Targeter (TALE-NT) 2.0: tools for TAL effector design and target prediction. Nucleic Acids Res 40(Web Server issue): W117–W122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, Y, Cradick, TJ and Bao, G (2014). Designing and testing the activities of TAL effector nucleases. Methods Mol Biol 1114: 203–219. [DOI] [PubMed] [Google Scholar]
- Montague, TG, Cruz, JM, Gagnon, JA, Church, GM and Valen, E (2014). CHOPCHOP: a CRISPR/Cas9 and TALEN web tool for genome editing. Nucleic Acids Res 42(Web Server issue): W401–W407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neff, KL, Argue, DP, Ma, AC, Lee, HB, Clark, KJ and Ekker, SC (2013). Mojo Hand, a TALEN design tool for genome editing applications. BMC Bioinformatics 14: 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyon, D, Tsai, SQ, Khayter, C, Foden, JA, Sander, JD and Joung, JK (2012). FLASH assembly of TALENs for high-throughput genome editing. Nat Biotechnol 30: 460–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heigwer, F, Kerr, G, Walther, N, Glaeser, K, Pelz, O, Breinig, M et al. (2013). E-TALEN: a web tool to design TALENs for genome engineering. Nucleic Acids Res 41: e190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, Y, Fine, EJ, Zheng, Z, Antico, CJ, Voit, RA, Porteus, MH et al. (2014). SAPTA: a new design tool for improving TALE nuclease activity. Nucleic Acid Res 42: e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christian, ML, Demorest, ZL, Starker, CG, Osborn, MJ, Nyquist, MD, Zhang, Y et al. (2012). Targeting G with TAL effectors: a comparison of activities of TALENs constructed with NN and NK repeat variable di-residues. PLoS One 7: e45383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doench, JG, Hartenian, E, Graham, DB, Tothova, Z, Hegde, M, Smith, I et al. (2014). Rational design of highly active sgRNAs for CRISPR-Cas9-mediated gene inactivation. Nat Biotechnol 32: 1262–1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chari, R, Mali, P, Moosburner, M and Church, GM (2015). Unraveling CRISPR-Cas9 genome engineering parameters via a library-on-library approach. Nat Methods 12: 823–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno-Mateos, MA, Vejnar, CE, Beaudoin, JD, Fernandez, JP, Mis, EK, Khokha, MK et al. (2015). CRISPRscan: designing highly efficient sgRNAs for CRISPR-Cas9 targeting in vivo. Nat Methods 12: 982–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szczepek, M, Brondani, V, Büchel, J, Serrano, L, Segal, DJ and Cathomen, T (2007). Structure-based redesign of the dimerization interface reduces the toxicity of zinc-finger nucleases. Nat Biotechnol 25: 786–793. [DOI] [PubMed] [Google Scholar]
- Mussolino, C, Morbitzer, R, Lütge, F, Dannemann, N, Lahaye, T and Cathomen, T (2011). A novel TALE nuclease scaffold enables high genome editing activity in combination with low toxicity. Nucleic Acids Res 39: 9283–9293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fine, EJ, Cradick, TJ, Zhao, CL, Lin, Y and Bao, G (2014). An online bioinformatics tool predicts zinc finger and TALE nuclease off-target cleavage. Nucleic Acids Res 42: e42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pattanayak, V, Ramirez, CL, Joung, JK and Liu, DR (2011). Revealing off-target cleavage specificities of zinc-finger nucleases by in vitro selection. Nat Methods 8: 765–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cradick, TJ, Qiu, P, Lee, CM, Fine, EJ and Bao, G (2014). COSMID: A Web-based Tool for Identifying and Validating CRISPR/Cas Off-target Sites. Mol Ther Nucleic Acids 3: e214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez, EE, Wang, J, Miller, JC, Jouvenot, Y, Kim, KA, Liu, O et al. (2008). Establishment of HIV-1 resistance in CD4+ T cells by genome editing using zinc-finger nucleases. Nat Biotechnol 26: 808–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabriel, R, Lombardo, A, Arens, A, Miller, JC, Genovese, P, Kaeppel, C et al. (2011). An unbiased genome-wide analysis of zinc-finger nuclease specificity. Nat Biotechnol 29: 816–823. [DOI] [PubMed] [Google Scholar]
- Sander, JD, Ramirez, CL, Linder, SJ, Pattanayak, V, Shoresh, N, Ku, M et al. (2013). In silico abstraction of zinc finger nuclease cleavage profiles reveals an expanded landscape of off-target sites. Nucleic Acids Res 41: e181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu, PD, Scott, DA, Weinstein, JA, Ran, FA, Konermann, S, Agarwala, V et al. (2013). DNA targeting specificity of RNA-guided Cas9 nucleases. Nat Biotechnol 31: 827–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cradick, TJ, Fine, EJ, Antico, CJ and Bao, G (2013). CRISPR/Cas9 systems targeting β-globin and CCR5 genes have substantial off-target activity. Nucleic Acids Res 41: 9584–9592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu, Y, Foden, JA, Khayter, C, Maeder, ML, Reyon, D, Joung, JK et al. (2013). High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat Biotechnol 31: 822–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai, SQ, Zheng, Z, Nguyen, NT, Liebers, M, Topkar, VV, Thapar, V et al. (2015). GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nat Biotechnol 33: 187–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, Y, Cradick, TJ, Brown, MT, Deshmukh, H, Ranjan, P, Sarode, N et al. (2014). CRISPR/Cas9 systems have off-target activity with insertions or deletions between target DNA and guide RNA sequences. Nucleic Acids Res 42: 7473–7485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bae, S, Park, J and Kim, JS (2014). Cas-OFFinder: a fast and versatile algorithm that searches for potential off-target sites of Cas9 RNA-guided endonucleases. Bioinformatics 30: 1473–1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh, R, Kuscu, C, Quinlan, A, Qi, Y and Adli, M (2015). Cas9-chromatin binding information enables more accurate CRISPR off-target prediction. Nucleic Acids Res 43: e118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, D, Bae, S, Park, J, Kim, E, Kim, S, Yu, HR et al. (2015). Digenome-seq: genome-wide profiling of CRISPR-Cas9 off-target effects in human cells. Nat Methods 12: 237–43, 1 p following 243. [DOI] [PubMed] [Google Scholar]
- Wang, X, Wang, Y, Wu, X, Wang, J, Wang, Y, Qiu, Z et al. (2015). Unbiased detection of off-target cleavage by CRISPR-Cas9 and TALENs using integrase-defective lentiviral vectors. Nat Biotechnol 33: 175–178. [DOI] [PubMed] [Google Scholar]
- Kuscu, C, Arslan, S, Singh, R, Thorpe, J and Adli, M (2014). Genome-wide analysis reveals characteristics of off-target sites bound by the Cas9 endonuclease. Nat Biotechnol 32: 677–683. [DOI] [PubMed] [Google Scholar]
- Qiu, P, Shandilya, H, D'Alessio, JM, O'Connor, K, Durocher, J and Gerard, GF (2004). Mutation detection using Surveyor nuclease. Biotechniques 36: 702–707. [DOI] [PubMed] [Google Scholar]
- Guschin, DY, Waite, AJ, Katibah, GE, Miller, JC, Holmes, MC and Rebar, EJ (2010). A rapid and general assay for monitoring endogenous gene modification. Methods Mol Biol 649: 247–256. [DOI] [PubMed] [Google Scholar]
- Brinkman, EK, Chen, T, Amendola, M and van Steensel, B (2014). Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Res 42: e168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendel, A, Kildebeck, EJ, Fine, EJ, Clark, JT, Punjya, N, Sebastiano, V et al. (2014). Quantifying genome-editing outcomes at endogenous loci with SMRT sequencing. Cell Rep 7: 293–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill, JT, Demarest, BL, Bisgrove, BW, Su, YC, Smith, M and Yost, HJ (2014). Poly peak parser: Method and software for identification of unknown indels using sanger sequencing of polymerase chain reaction products. Dev Dyn 243: 1632–1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, HJ, Lee, HJ, Kim, H, Cho, SW and Kim, JS (2009). Targeted genome editing in human cells with zinc finger nucleases constructed via modular assembly. Genome Res 19: 1279–1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, P, Xiao, A, Zhou, M, Zhu, Z, Lin, S and Zhang, B (2011). Heritable gene targeting in zebrafish using customized TALENs. Nat Biotechnol 29: 699–700. [DOI] [PubMed] [Google Scholar]
- Kim, Y, Kweon, J and Kim, JS (2013). TALENs and ZFNs are associated with different mutation signatures. Nat Methods 10: 185. [DOI] [PubMed] [Google Scholar]
- Chen, S, Oikonomou, G, Chiu, CN, Niles, BJ, Liu, J, Lee, DA et al. (2013). A large-scale in vivo analysis reveals that TALENs are significantly more mutagenic than ZFNs generated using context-dependent assembly. Nucleic Acids Res 41: 2769–2778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, H, Um, E, Cho, SR, Jung, C, Kim, H and Kim, JS (2011). Surrogate reporters for enrichment of cells with nuclease-induced mutations. Nat Methods 8: 941–943. [DOI] [PubMed] [Google Scholar]
- Dahlem, TJ, Hoshijima, K, Jurynec, MJ, Gunther, D, Starker, CG, Locke, AS et al. (2012). Simple methods for generating and detecting locus-specific mutations induced with TALENs in the zebrafish genome. PLoS Genet 8: e1002861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, JM, Kim, D, Kim, S and Kim, JS (2014). Genotyping with CRISPR-Cas-derived RNA-guided endonucleases. Nat Commun 5: 3157. [DOI] [PubMed] [Google Scholar]
- Crosetto, N, Mitra, A, Silva, MJ, Bienko, M, Dojer, N, Wang, Q et al. (2013). Nucleotide-resolution DNA double-strand break mapping by next-generation sequencing. Nat Methods 10: 361–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Güell, M, Yang, L and Church, GM (2014). Genome editing assessment using CRISPR Genome Analyzer (CRISPR-GA). Bioinformatics 30: 2968–2970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mussolino, C, Alzubi, J, Fine, EJ, Morbitzer, R, Cradick, TJ, Lahaye, T et al. (2014). TALENs facilitate targeted genome editing in human cells with high specificity and low cytotoxicity. Nucleic Acids Res 42: 6762–6773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, HJ, Kim, E and Kim, JS (2010). Targeted chromosomal deletions in human cells using zinc finger nucleases. Genome Res 20: 81–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, HJ, Kweon, J, Kim, E, Kim, S and Kim, JS (2012). Targeted chromosomal duplications and inversions in the human genome using zinc finger nucleases. Genome Res 22: 539–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunet, E, Simsek, D, Tomishima, M, DeKelver, R, Choi, VM, Gregory, P et al. (2009). Chromosomal translocations induced at specified loci in human stem cells. Proc Natl Acad Sci USA 106: 10620–10625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maddalo, D, Manchado, E, Concepcion, CP, Bonetti, C, Vidigal, JA, Han, YC et al. (2014). In vivo engineering of oncogenic chromosomal rearrangements with the CRISPR/Cas9 system. Nature 516: 423–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tesson, L, Usal, C, Ménoret, S, Leung, E, Niles, BJ, Remy, S et al. (2011). Knockout rats generated by embryo microinjection of TALENs. Nat Biotechnol 29: 695–696. [DOI] [PubMed] [Google Scholar]
- Hockemeyer, D, Wang, H, Kiani, S, Lai, CS, Gao, Q, Cassady, JP et al. (2011). Genetic engineering of human pluripotent cells using TALE nucleases. Nat Biotechnol 29: 731–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta, A, Meng, X, Zhu, LJ, Lawson, ND and Wolfe, SA (2011). Zinc finger protein-dependent and -independent contributions to the in vivo off-target activity of zinc finger nucleases. Nucleic Acids Res 39: 381–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, X, Scott, DA, Kriz, AJ, Chiu, AC, Hsu, PD, Dadon, DB et al. (2014). Genome-wide binding of the CRISPR endonuclease Cas9 in mammalian cells. Nat Biotechnol 32: 670–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Geen, H, Henry, IM, Bhakta, MS, Meckler, JF and Segal, DJ (2015). A genome-wide analysis of Cas9 binding specificity using ChIP-seq and targeted sequence capture. Nucleic Acids Res 43: 3389–3404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frock, RL, Hu, J, Meyers, RM, Ho, YJ, Kii, E and Alt, FW (2015). Genome-wide detection of DNA double-stranded breaks induced by engineered nucleases. Nat Biotechnol 33: 179–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran, FA, Cong, L, Yan, WX, Scott, DA, Gootenberg, JS, Kriz, AJ et al. (2015). In vivo genome editing using Staphylococcus aureus Cas9. Nature 520: 186–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller, JC, Holmes, MC, Wang, J, Guschin, DY, Lee, YL, Rupniewski, I et al. (2007). An improved zinc-finger nuclease architecture for highly specific genome editing. Nat Biotechnol 25: 778–785. [DOI] [PubMed] [Google Scholar]
- Doyon, Y, Vo, TD, Mendel, MC, Greenberg, SG, Wang, J, Xia, DF et al. (2011). Enhancing zinc-finger-nuclease activity with improved obligate heterodimeric architectures. Nat Methods 8: 74–79. [DOI] [PubMed] [Google Scholar]
- Lee, CM, Flynn, R, Hollywood, JA, Scallan, MF and Harrison, PT (2012). Correction of the ΔF508 Mutation in the Cystic Fibrosis Transmembrane Conductance Regulator Gene by Zinc-Finger Nuclease Homology-Directed Repair. Biores Open Access 1: 99–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez, CL, Certo, MT, Mussolino, C, Goodwin, MJ, Cradick, TJ, McCaffrey, AP et al. (2012). Engineered zinc finger nickases induce homology-directed repair with reduced mutagenic effects. Nucleic Acids Res 40: 5560–5568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, X, Wang, Y, Guo, W, Chang, B, Liu, J, Guo, Z et al. (2013). Zinc-finger nickase-mediated insertion of the lysostaphin gene into the beta-casein locus in cloned cows. Nat Commun 4: 2565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, J, Friedman, G, Doyon, Y, Wang, NS, Li, CJ, Miller, JC et al. (2012). Targeted gene addition to a predetermined site in the human genome using a ZFN-based nicking enzyme. Genome Res 22: 1316–1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, Y, Gao, T, Wang, X, Hu, Y, Hu, X, Hu, Z et al. (2014). TALE nickase mediates high efficient targeted transgene integration at the human multi-copy ribosomal DNA locus. Biochem Biophys Res Commun 446: 261–266. [DOI] [PubMed] [Google Scholar]
- Wu, H, Wang, Y, Zhang, Y, Yang, M, Lv, J, Liu, J et al. (2015). TALE nickase-mediated SP110 knockin endows cattle with increased resistance to tuberculosis. Proc Natl Acad Sci USA 112: E1530–E1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mali, P, Aach, J, Stranges, PB, Esvelt, KM, Moosburner, M, Kosuri, S et al. (2013). CAS9 transcriptional activators for target specificity screening and paired nickases for cooperative genome engineering. Nat Biotechnol 31: 833–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran, FA, Hsu, PD, Lin, CY, Gootenberg, JS, Konermann, S, Trevino, AE et al. (2013). Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell 154: 1380–1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duda, K, Lonowski, LA, Kofoed-Nielsen, M, Ibarra, A, Delay, CM, Kang, Q et al. (2014). High-efficiency genome editing via 2A-coupled co-expression of fluorescent proteins and zinc finger nucleases or CRISPR/Cas9 nickase pairs. Nucleic Acids Res 42: e84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai, SQ, Wyvekens, N, Khayter, C, Foden, JA, Thapar, V, Reyon, D et al. (2014). Dimeric CRISPR RNA-guided FokI nucleases for highly specific genome editing. Nat Biotechnol 32: 569–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guilinger, JP, Thompson, DB and Liu, DR (2014). Fusion of catalytically inactive Cas9 to FokI nuclease improves the specificity of genome modification. Nat Biotechnol 32: 577–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aouida, M, Eid, A, Ali, Z, Cradick, T, Lee, C, Deshmukh, H et al. (2015). Efficient fdCas9 Synthetic Endonuclease with Improved Specificity for Precise Genome Engineering. PLoS One 10: e0133373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arribere, JA, Bell, RT, Fu, BX, Artiles, KL, Hartman, PS and Fire, AZ (2014). Efficient marker-free recovery of custom genetic modifications with CRISPR/Cas9 in Caenorhabditis elegans. Genetics 198: 837–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fonfara, I, Le Rhun, A, Chylinski, K, Makarova, KS, Lécrivain, AL, Bzdrenga, J et al. (2014). Phylogeny of Cas9 determines functional exchangeability of dual-RNA and Cas9 among orthologous type II CRISPR-Cas systems. Nucleic Acids Res 42: 2577–2590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esvelt, KM, Mali, P, Braff, JL, Moosburner, M, Yaung, SJ and Church, GM (2013). Orthogonal Cas9 proteins for RNA-guided gene regulation and editing. Nat Methods 10: 1116–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou, Z, Zhang, Y, Propson, NE, Howden, SE, Chu, LF, Sontheimer, EJ et al. (2013). Efficient genome engineering in human pluripotent stem cells using Cas9 from Neisseria meningitidis. Proc Natl Acad Sci USA 110: 15644–15649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller, M, Lee, CM, Gasiunas, G, Davis, T, Cradick, TJ, Siksnys, V et al. (2015). Streptococcus thermophilus CRISPR-Cas9 systems enable specific editing of the human genome. Mol Ther (epub ahead of print). doi: 10.1038/mt.2015.218. [DOI] [PMC free article] [PubMed]
- Lee, CM, Cradick, TJ and Bao, G (2016). The Neisseria meningitidis CRISPR/Cas9 system enables specific genome editing in mammalian cells. Mol Ther (epub ahead of print). doi: 10.1038/mt.2016.8. [DOI] [PMC free article] [PubMed]
- Swenberg, JA, Lu, K, Moeller, BC, Gao, L, Upton, PB, Nakamura, J et al. (2011). Endogenous versus exogenous DNA adducts: their role in carcinogenesis, epidemiology, and risk assessment. Toxicol Sci 120 Suppl 1: S130–S145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dewey, FE, Grove, ME, Pan, C, Goldstein, BA, Bernstein, JA, Chaib, H et al. (2014). Clinical interpretation and implications of whole-genome sequencing. JAMA 311: 1035–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lomax, ME, Folkes, LK and O'Neill, P (2013). Biological consequences of radiation-induced DNA damage: relevance to radiotherapy. Clin Oncol (R Coll Radiol) 25: 578–585. [DOI] [PubMed] [Google Scholar]
- Stemmer, M, Thumberger, T, Del Sol Keyer, M, Wittbrodt, J and Mateo, JL (2015). CCTop: An Intuitive, Flexible and Reliable CRISPR/Cas9 Target Prediction Tool. PLoS One 10: e0124633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prykhozhij, SV, Rajan, V, Gaston, D and Berman, JN (2015). Correction: CRISPR MultiTargeter: a web tool to find common and unique CRISPR single guide RNA targets in a set of similar sequences. PLoS One 10: e0138634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naito, Y, Hino, K, Bono, H and Ui-Tei, K (2015). CRISPRdirect: software for designing CRISPR/Cas guide RNA with reduced off-target sites. Bioinformatics 31: 1120–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grissa, I, Vergnaud, G and Pourcel, C (2007). CRISPRFinder: a web tool to identify clustered regularly interspaced short palindromic repeats. Nucleic Acids Res 35(Web Server issue): W52–W57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu, LJ, Holmes, BR, Aronin, N and Brodsky, MH (2014). CRISPRseek: a bioconductor package to identify target-specific guide RNAs for CRISPR-Cas9 genome-editing systems. PLoS One 9: e108424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heigwer, F, Kerr, G and Boutros, M (2014). E-CRISP: fast CRISPR target site identification. Nat Methods 11: 122–123. [DOI] [PubMed] [Google Scholar]
- Gratz, SJ, Ukken, FP, Rubinstein, CD, Thiede, G, Donohue, LK, Cummings, AM et al. (2014). Highly specific and efficient CRISPR/Cas9-catalyzed homology-directed repair in Drosophila. Genetics 196: 961–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Brien, A and Bailey, TL (2014). GT-Scan: identifying unique genomic targets. Bioinformatics 30: 2673–2675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bae, S, Kweon, J, Kim, HS and Kim, JS (2014). Microhomology-based choice of Cas9 nuclease target sites. Nat Methods 11: 705–706. [DOI] [PubMed] [Google Scholar]
- Xie, S, Shen, B, Zhang, C, Huang, X and Zhang, Y (2014). sgRNAcas9: a software package for designing CRISPR sgRNA and evaluating potential off-target cleavage sites. PloS One 9: e100448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodgkins, A, Farne, A, Perera, S, Grego, T, Parry-Smith, DJ, Skarnes, WC et al. (2015). WGE: a CRISPR database for genome engineering. Bioinformatics 31: 3078–3080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grau, J, Boch, J and Posch, S (2013). TALENoffer: genome-wide TALEN off-target prediction. Bioinformatics 29: 2931–2932. [DOI] [PubMed] [Google Scholar]
- Xiao, A, Wu, Y, Yang, Z, Hu, Y, Wang, W, Zhang, Y et al. (2013). EENdb: a database and knowledge base of ZFNs and TALENs for endonuclease engineering. Nucleic Acids Res 41(Database issue): D415–D422. [DOI] [PMC free article] [PubMed] [Google Scholar]