

Abstract
Programmable nucleases allow defined alterations in the genome with ease-of-use, efficiency, and specificity. Their availability has led to accurate and widespread genome engineering, with multiple applications in basic research, biotechnology, and therapy. With regard to human gene therapy, nuclease-based gene editing has facilitated development of a broad range of therapeutic strategies based on both nonhomologous end joining and homology-dependent repair. This review discusses current progress in nuclease-based therapeutic applications for a subset of inherited monogenic diseases including cystic fibrosis, Duchenne muscular dystrophy, diseases of the bone marrow, and hemophilia and highlights associated challenges and future prospects.
Introduction
There are 5,000–8,000 monogenic diseases, defined as inherited conditions arising from mutations on a single gene.1 These often manifest during childhood and lead to morbidity and sometimes premature death. While each monogenic disease is rare, it has been estimated that together they will affect about 6% of people at some point in their lives.1 Diagnosis and treatment for these diseases remain largely insufficient, and the care is primarily palliative, focusing on disease management without addressing the underlying genetic defects. The realization of the social and economic importance of rare diseases and the acute need for diagnostics and treatments has led to initiatives like the International Rare Disease Research Consortium (IRDiRC, http://www.irdirc.org), the Undiagnosed Diseases Network (UDN, http://www.genome.gov/27562471) and Syndromes Without a Name UK (SWAN UK, http://www.geneticalliance.org.uk/projects/swan.htm).
Gene therapy, which encompasses a range of strategies, aimed from the outset to treat inherited disorders, assuming that monogenic diseases would be the easiest to target. Classical gene therapy approaches have centered on the delivery of DNA to augment endogenous gene expression. Predominantly, these approaches rely on the transfer of functional genes using a variety of viral vectors, due to their intrinsic ability to effectively transduce human cells. Retroviral vectors provided the first clear demonstrations of therapeutic benefit in primary immunodeficiencies, and they also highlighted the risk of adverse events attributable to insertional mutagenesis due to genomic integration of proviruses.2 Among several, other success stories include Glybera,3 the first clinically approved gene therapy in the European Union, which uses an adeno-associated virus (AAV) vector drug for lipoprotein lipase deficiency; and in the case of cystic fibrosis, repeated nebulization of liposomes encoding the cystic fibrosis transmembrane conductance regulator (CFTR) gene has shown some therapeutic benefit.4 Thus, gene augmentation shows great therapeutic promise and has set the stage for the gene-editing approaches reviewed here.
Gene editing is a gene therapy approach that relies on designer nucleases to recognize and cut specific DNA sequences, and subsequently exploits innate cellular DNA repair pathways, namely nonhomologous end joining (NHEJ) and homology directed repair (HDR), to introduce targeted modifications in the genome (Figure 1a). Four nuclease families have been used in this context: meganucleases, zinc-finger nucleases (ZFNs), transcription activator-like effector nucleases (TALENs), and clustered regulatory interspaced short palindromic repeats associated RNA-guided Cas9 (CRISPR-Cas9) nucleases.5 These can be designed to precisely introduce a double stranded break at the target locus of interest. The double stranded break will then be repaired by either NHEJ or HDR. NHEJ involves direct ligation of DNA ends in a highly efficient but error-prone manner, which causes small insertions and/or deletions (Indels) at the break site. In the context of a disease-causing locus, NHEJ can be exploited to excise or disrupt deleterious sequences, or even restore the reading frame of a gene (Figure 1b). In contrast, HDR requires a donor DNA containing sequences homologous to those adjacent to the double stranded break. The donor DNA can be used to repair a mutation or to knock-in a block of exons (“superexon”) or a full cDNA at either the endogenous locus (reconstituting the wild-type sequence) or at a genomic “safe harbor” (a region of DNA where transgenes can integrate and express in a predictable manner without insertional mutagenesis or perturbation of gene function)6 (Figure 1b). Gene editing thus opens up the possibility of permanently modifying a genomic sequence of interest by enabling targeted disruption, insertion, excision, and correction in both ex vivo and in vivo settings (Figure 1c). While these advances are expected to revolutionize the field at large, current gene-editing approaches are limited by efficacy of modification, safety concerns related to the specificity of nucleases, and delivery of gene-editing tools to target cell types. This review aims to outline prominent gene-editing research across a range of monogenic disorders (Table 1) and to highlight recent advancements and current challenges.
Figure 1.

Overview of therapeutic gene editing. (a) DNA repair pathways for the resolution of a double stranded break (DSB). A nuclease is targeted toward a defined genomic locus, introducing a DSB. This may undergo one of two major repair pathways known as nonhomologous end joining (NHEJ) or homology-directed repair (HDR), depending on cell cycle stage and availability of a DNA donor template. NHEJ is an error-prone mechanism, which causes small insertions or deletions (Indels) upon ligating the ends of the DNA break. HDR is a precise mechanism which repairs the break by using a homologous donor template. (b) Functional gene-editing strategies using DNA repair pathways. 1) NHEJ can be used to disrupt genomic sequences as a consequence of Indels. This can cause frameshift mutations leading to an early stop codon (or restoration of the reading frame by splice site disruption). 2) NHEJ can mediate targeted deletions. This requires generation of DSBs on both sides of the target genomic sequence, which then deletes the intervening sequence while NHEJ re-joins the DNA ends. 3) HDR can be used to correct a specific mutation by introducing a nuclease-mediated DSB (in proximity to the target site) in the presence of a homologous donor DNA containing corrective sequence. Upon recombination, the repair template corrects the mutated locus. 4) Likewise, by supplying exogenous DNA on the donor template flanked between regions of homology, HDR can be used to mediate targeted gene insertion or knock-in. (c) Schematic diagram comparing ex vivo and in vivo approaches. In vivo approaches involve direct transfer (denoted by the syringe) of genome-editing reagents such as programmable nucleases and donor templates to the human body. In this instance, two prominent gene transfer agents, viral vectors, and liposomes are shown. Ex vivo is centered on correction of the genetic defect outside of the body. This is a staged-approach whereby: 1) Patient cells are obtained. 2) Gene editing is performed in vitro. This involves delivery of nucleases on their own or concomitantly with repair template. The patient cells can be programmed into induced pluripotent stem cells (iPSCs) before or after gene editing. Once corrected, iPSCs may be differentiated into cell types of interest. 3) The genetically corrected cells are characterized and expanded. 4) The corrected cells are then re-grafted back into the patient through autologous transplantation.
Table 1. Therapeutic gene-editing approaches applied in selected monogenic diseases.
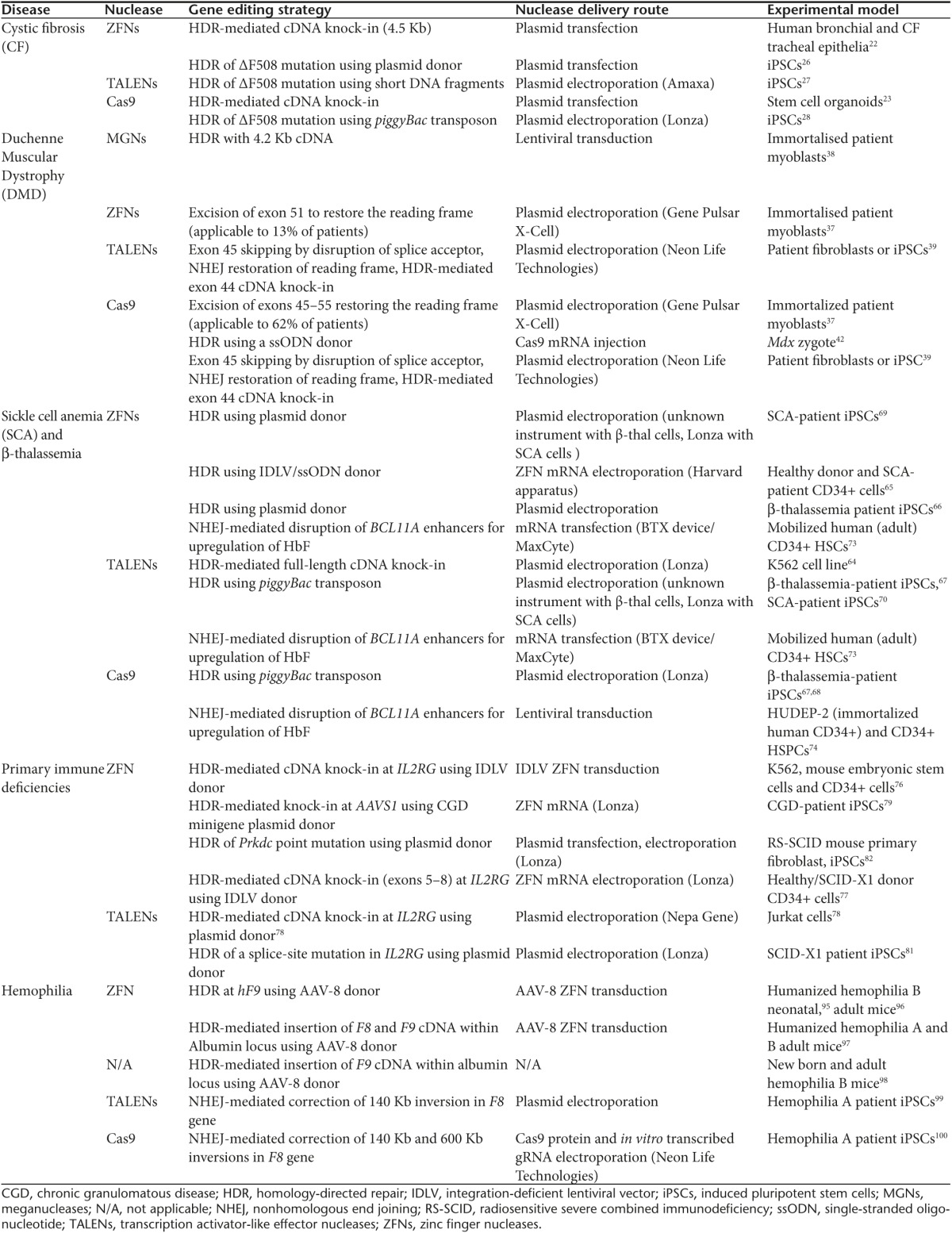
Cystic Fibrosis
Cystic fibrosis (CF) is an autosomal-recessive disease resulting from mutations in the CFTR gene, which encodes an epithelial anion channel. CFTR is distributed across a wide range of organs including pancreas, kidney, liver, lungs, gastrointestinal tracts, and reproductive tracts, making CF a multiorgan disease. Mutations in CFTR lead to suboptimal ion transport and fluid retention, causing the prominent clinical manifestations of abnormal thickening of the mucus in lungs and pancreatic insufficiency.7 In the lung, dysfunctional CFTR hinders mucociliary clearance, rendering the organ susceptible to bacterial infections and inflammation, ultimately leading to airway occlusion, respiratory failure, and premature death.8 CF remains the most common and lethal genetic disease among the Caucasian population with 70,000–100,000 sufferers estimated worldwide, highlighting a real need for the development of better treatments.
One major challenge to the development of a therapeutic strategy for CF is the huge diversity of mutation types. ΔF508 (deletion of phenylalanine at codon 508) mutation, with a prevalence of >80% in CF patients, is by far the most common, but more than 1,990 CFTR-mutations have been described.9 They cause premature stop codons, aberrant splicing, incorrect protein folding or trafficking to the cell surface, and dysfunctional CFTRs with limited channel-opening capacity.9 Pharmacological interventions have been targeted to several of these processes, in the form of: (i) read-through therapeutics, which recognize premature stop codons, thereby allowing full-length protein production and a decline in associated nonsense-mediated decay10; (ii) correctors, which enable slightly misfolded proteins to evade endoplasmic reticulum quality control and insert at the primary epithelia11; and (iii) potentiators, which target gating mutations and increase channel opening.8,12 One prominent drug, the potentiator ivacaftor (Kalydeco), has demonstrated significant improvement of numerous clinical endpoints such as forced expiratory volume, weight gain, reduction of hospital admissions (related to requirement of i.v. antibiotic administration), and increase in lung clearance index in diverse patient subsets bearing the G551D-CFTR missense mutation.13,14,15,16 Ivacaftor also demonstrated clinical benefit in other gating mutation types: G1244E, G1349D, G178R, G551S, G970R, S1251N, S1255P, S549N and S549R, with G970R being one exception.17 In the case of ΔF508, ivacaftor has been tested in a phase 3 clinical trial in combination with lumacaftor.18 The improvements seen in forced expiratory volume were modest compared to ivacaftor monotherapy in G551D studies.13,15 Thus, while drug administration is therapeutic in some gating mutation types, the commonly occurring ΔF508 still requires a more effective treatment. QR-010 is a drug based on modified single-stranded RNA and designed to repair ΔF508 mRNA; research in human cell lines and mice appears promising and is currently progressing to a phase 1b clinical trial.19,20 However, this still would not address mutations resulting from aberrant splicing; it is in these instances gene editing could prove most beneficial.
The premise of permanent correction by gene editing, as opposed to drug and recent nonviral gene therapy treatments (previously reviewed in refs. 9,21) which require repeated administration, is promising. Such CFTR correction at the genome level has been trialled across many of the gene-editing platforms. ZFNs were first used to achieve HDR-mediated knock-in of a 4.5 kb genomic donor harboring CFTR exons 10–24. The efficiency in patient-derived epithelial cells was modest, with DNA cleavage as measured by NHEJ estimated at 7.8% and the subsequent HDR occurring at <1%, respectively.22 The ΔF508 mutation was targeted and functional repair obtained in stem cell organoids, using CRISPR-Cas9-mediated HDR and an exon 11 and puromycin resistance-containing DNA donor. In most instances, this resulted in a heterozygous phenotype or monoallelic correction. Moreover, the forskolin-induced swelling assay, which demonstrates functional correction of the organoids via fluid secretion into the epithelia, showed more improvement than chemical correctors.23,24 Although for stem cell organoids there are colon engraftment data, extension of these studies to lung would be helpful to assess the therapeutic promise of this approach.25 Similar HDR strategies have demonstrated CFTR correction in induced pluripotent stem cells (iPSCs) derived from CF patients, bearing heterozygous ΔF508/ΔI507 mutations in the ZFN study,26 and Δ508 homozygous mutations in the TALENs,27 as well as Cas9 (ref. 28) studies. Genetically modified iPSCs can be selected and amplified at clonal level, allowing the production of pure populations of corrected cells, something unlikely to be achieved with primary cells. Two of these reports demonstrated that corrected clones could be differentiated successfully into epithelial cells, while retaining CFTR expression.26,27 Even with these advances, the multiorgan involvement of CF would limit an ex vivo approach as engraftment of corrected cells would only provide localized correction. In addition, there is a lack of consensus regarding which cells of the sinuses and lungs should be targeted in the amelioration of CF.
Given that drug therapy has demonstrated moderate repair, the focus may now be upon reaching a therapeutic threshold. Current threshold estimates are in the range of 10–24% of normal expression,9 similar to improvements in forced expiratory volume following the use of ivacaftor,9 and perhaps achievable for other mutations by a combination of gene and drug therapies. The development of a catalytically inactive Cas9 in which the DNA cleavage domains harbor point mutations, tethered to a VP64 transcriptional activator, could be used to upregulate CFTR expression.29,30 This transcriptional enhancement of the mutant gene, coupled to corrector and potentiator therapy, could be beneficial as the pool of protein available to be inserted within the epithelial surface would be increased.
Duchenne Muscular Dystrophy
Duchenne muscular dystrophy (DMD) is an inherited X-linked disease resulting from mutations that disrupt the reading frame in the gene encoding dystrophin. This protein plays a crucial role in stabilization of muscle sarcolemma and signaling. In the absence of dystrophin, progressive muscle wasting, with concomitant declines in respiratory and cardiac function, occur. Over time, this results in the loss of ambulation, necessitates the use of invasive ventilation, and ultimately leads to premature death.31,32
Presently, there is no effective treatment for DMD. Current interventions including the use of prednisolone are inadequate, targeting only secondary characteristics of inflammation and muscle loss.33 Despite being recognized as a prime candidate for gene therapy since the discovery of DMD in 1987, advancement has been hindered because DMD is the largest naturally occurring gene, spanning 2.5 Mb, with a cDNA of 11.2 Kb. A great deal of work has focused upon mutation-specific strategies using pharmacological and gene therapy approaches. Many of the current approaches, such as exon skipping, aim to restore the reading frame by targeting mRNA, masking splice sites or enhancers of exons that shift the reading frame.34,35 The aim of these strategies is to produce a truncated but viable dystrophin protein, resulting in a clinically milder Becker rather than DMD phenotype.36
Genome-editing approaches for DMD include promoting permanent exon removal,37 and HDR-mediated cDNA knock-in.38,39 Notable demonstrations in primary patient myoblasts have been the permanent excision of exon 51 (which would be applicable to 13% of patients) achieved using ZFNs, leading to restoration of the reading frame in an approach akin to exon skipping,40 and permanent removal of exons 45–55 by multiplexed Cas9 (Figure 1b). The latter is a mutational hotspot which if targeted could be therapeutically applicable for 62% of patients.37 Both of these approaches result in a truncated but functional dystrophin protein.37 The mutational hotspot has also been targeted by HDR, with meganuclease-mediated repair of exons 45–52 in immortalized patient cells.38 The benefit of the HDR-mediated approach is that the subsequent correction would enable the restoration of full-length dystrophin (Figure 1b).
DMD has also been restored by genome editing in iPSCs from a patient lacking exon 44 by three different strategies: exon 45 skipping by disruption of its splicing acceptor, Indel-mediated frameshift of exon 45, and exon 44 knock-in, utilizing both TALEN and CRISPR-Cas9 approaches.39 Targeted differentiation of such iPSCs could eventually progress to correction of individual mutation types via engraftment of corrected proliferative cells into muscles. Some concerns with this approach are the modulation of cell proliferation, efficiency of the engraftment process, and localized intramuscular regeneration.41
Model systems are also providing data of relevance to human therapy. Direct germline correction of a murine Dmd mutation has recently been demonstrated in the mdx mouse, in which the exon 23 nonsense mutation was corrected via Cas9 mRNA injection followed by implantation into pseudo pregnant females. This resulted in mosaicism within targeted animals, showing correction ranging from 2 to 100%; this type of work could establish the level of dystrophin expression required to provide therapeutic benefit, which is currently predicted to be between 15 and 20% (ref. 42). Two genotypically distinct rat models have also been produced, one with a C>T nonsense mutation in exon 23 analogous to that of the mdx mouse and another with a large deletion spanning exons 6–13 (refs. 43,44). Additionally, the use of CRISPR-Cas9 in pigs and nonhuman primates for the generation of DMD phenotypes demonstrates that such gene-editing can be easily transitioned into larger animals.45,46,47 This ease of generation of new DMD animal models allows for a range of mutation types to be produced, thereby providing a greater diversity of models for translational research. Moreover, larger animal models such as rats, pigs, and primates tend to exhibit muscle phenotypes such as fibrosis, which is more representative of the clinical manifestations seen in patients than those in the commonly used mdx mouse.48,49 Thus, such models could serve to enrich our understanding of DMD at large and produce more robust and reliable end points to determine efficacy of treatments.
Disorders of The Bone Marrow
Bone marrow diseases comprise a variety of conditions including severe anemic hemoglobinopathies and more than 250 different primary immunodeficiencies. Current treatment modalities include transfusion of blood, or blood-derived products such as erythrocytes, immunoglobulins, and platelets. However, hematopoietic stem cell (HSC) transplantation remains the only curative therapy to achieve permanent reconstitution. Despite the growing number of donor depositories, a human leukocyte antigen-matched donor cannot be found for some patients. In these cases, gene therapy using gene addition in autologous patient cells may offer a potentially safe and efficacious strategy. Successful and durable reconstitution using retroviral and/or lentiviral vectors has been achieved in patients suffering with various bone marrow disorders, including X-linked severe combined immunodeficiency (SCID-X1),2,50,51 adenosine deaminase deficiency,52,53,54,55,56 Wiskott–Aldrich syndrome,57,58 and β-thalassemia.59,60
Hemoglobinopathies: beta-thalassemia and sickle cell anemia
Hemoglobinopathies or “genetic anemias” result from defects of mature hemoglobin. Both β-thalassemia and sickle cell anemia (SCA) are caused by mutations on the HBB gene which encodes the β-globin chain. β-Thalassemias are heterogeneous, with >200 mutation types across the HBB locus affecting different steps of β-globin production (initiation of transcription, splicing, and posttranslational modification). The subsequent excess of α-globin causes apoptosis of red blood cells (RBCs) resulting in severe anemia. In contrast, SCA is caused by a missense mutation (A-to-T transversion) at codon 6 of HBB, which causes RBCs to distort to a “sickle” shape. Sickled RBCs constrict small capillaries causing severe tissue damage, acute painful crises, respiratory insufficiency, and progressive organ damage.61 SCA is one of the most common monogenic diseases with more than 250,000 affected infants born every year and is a major cause of morbidity and mortality worldwide.62
Although nontargeted gene augmentation for hemoglobinopathies has made considerable progress,59,60,63 the high levels of gene expression required and the potential risk of insertional mutagenesis associated with uncontrolled viral integration remain challenging. Editing the HBB locus using programmable nucleases can instead allow for permanent β-globin correction. Recently, TALENs were used to achieve ~20% HDR-mediated knock-in of therapeutic β-globin full-length cDNA to the endogenous β-globin locus in K562 erythroleukemia cells.64 Such a strategy would result in expression of β-globin from the cDNA instead of wild-type genomic sequence and would be therapeutic for both SCA and β-thalassemia. Separately, ZFNs were used to repair the SCA point mutation in CD34+ hematopoietic stem progenitor cells (HSPCs). Co-delivery of ZFN mRNA and a donor template led to 15 and 18% HDR in CD34+ cells derived from healthy donor and patients with SCA, respectively.65 Furthermore, correction of SCA mutation in patient cells led to production of wild-type hemoglobin in vitro. As only 10–30% of corrected donor cells are required to generate sufficient RBCs, gene repair in HSCs obtained from patients can reach a therapeutic threshold.65
Another strategy being explored in the context of hemoglobinopathies is gene editing of disease-causing mutations in iPSCs. These can be differentiated into HSCs, which can then be used for autologous transplantation. Recently, independent groups have demonstrated progress in this area by applying ZFNs,66 TALENs,67 or Cas9 (refs. 67,68) to correct different β-thalassemia mutations in various patient iPSCs. Corrected iPSCs maintained their ability to differentiate into erythroblasts with increased transcription of β-globin.68 Similar progress has also been made for SCA where ZFNs69 and TALENs70 were used to repair the sickle cell point mutation in patient iPSCs. For treatment of sickle cell disease, it is particularly important to ensure that the highly paralogous genes encoding γ-globin and δ-globin are not inadvertently mutagenized. Both studies specifically analyzed these loci and demonstrated no nuclease-associated off-target activity. These studies demonstrate that human stem cells including HSCs and iPSCs, and nuclease-induced gene-editing approaches can be used in combination to create corrected patient-derived cells. Despite the many advantages of iPSC technology, for immune-based therapy, potential concerns of immunogenicity in iPSCs and their derivatives should be thoroughly examined before clinical translation.71
Apart from adult hemoglobin, the level of fetal hemoglobin (HbF) is also a key modifier of clinical severity of hemoglobinopathies. In patients with SCA, high HbF is associated with generally milder disease phenotype. This has been attributed to naturally occurring variants in the enhancer regions of BCL11A, a transcriptional repressor of HbF production in adult erythroid cells.72 Recently, TALENs/ZFNs73 and Cas9-nuclease74 were used to specifically disrupt enhancer regions in BCL11A resulting in substantial HbF induction without the detrimental effects associated with complete loss of BCL11A. Such therapeutic gene-editing approach could be used to elevate HbF to clinically relevant levels thereby ameliorating β-globin disorders.
Primary immune deficiencies
Primary immune deficiencies comprise a heterogeneous group of rare diseases in which part of the immune system is missing or functions improperly. On the clinical spectrum, SCID is the most severe form of primary immunodeficiency, resulting in a development block in production of T-cells, with additional defects of B- and natural killer cells. The most common form of SCID, SCID-X1, is caused by mutations in the gene encoding the interleukin 2 receptor common gamma chain (IL2RG). Several groups have successfully used ZFNs to target and induce HDR in the IL2RG locus in human HSCs and embryonic stem cells, albeit with relatively low efficiencies.75,76,77 A notable demonstration was the ZFN-mediated insertion of corrective cDNA (exons 5–8 that would correct all SCID-X1 mutations downstream of exon 4) into the IL2RG mutational hotspot in long-term repopulating HSCs. This led to the correction of defective IL2RG in HSPCs from a subject with SCID-X1 and multilineage differentiation upon transplantation into immunodeficient SCID mice.77 Separately, TALENs were used to specifically target and induce HDR in the IL2RG locus of Jurkat cells.78
While direct genome editing in HSCs is an attractive alternative to viral gene addition therapy, both approaches are dependent on the capability to efficiently culture and expand HSCs ex vivo. In addition, HSC-based transplantation approaches generally involve myeloablative conditioning, which given the young age and immunocompromised state of SCID patients, poses significant risk. An iPSC-based approach could provide an unlimited source of subject-derived, corrected cells from which immune cells could be derived continuously for transfusion, but it would be a cumbersome approach. Issues regarding the efficiency of iPSC differentiation toward the hematopoietic lineage also need addressing. On the other hand, in diseases where the number of HSC is compromised, iPSCs could provide a ready source of corrected cells. ZFNs have been used to correct various types of chronic granulomatous disease by introducing five different functional genes into the AAVS1 “safe harbor” in iPSCs generated from peripheral HSCs. Using in vitro myeloid differentiation, normal granulocytes were generated from the corrected iPSCs.79 Provirus-free iPSCs resulting from methods in which no transgene integration events are required have been generated.80 Such iPSCs overcome concerns related to insertional mutagenesis and spurious transgene expression and are preferred for clinical application. Provirus-free iPSCs have been generated from a SCID-X1 patient, and the genetic defect corrected utilizing TALENs.81 These iPSCs retained their differentiation potential into NK cells, which expressed mature cell markers and had the correctly spliced IL2RG. This provided the first evidence of gene repair of SCID-X1 patient iPSCs resulting in regeneration of mature lymphoid cells in vitro. For radiosensitive SCID, ZFNs were utilized to correct a mutation on Prkdc gene in primary mouse fibroblasts and iPSCs.82 The corrected cells retained their potential to differentiate into functional T-cells in vitro, overcoming the developmental block.
Even though gene editing in the context of primary immunodeficiencies is not yet ready to be applied in a clinical setting, it already offers valuable tools to study and model immune disorders at the cellular level. With CRISPR-Cas9, zygote injections can be done in a 1-day procedure generating gene-modified mice in less than 4 weeks.83 NSG mice have been efficiently manipulated in this way.84 Similarly, a rat model of SCID-X1 has been generated using ZFNs.85 Additionally, endonucleases have been used to generate knockout models in animals previously unamenable to efficient genetic modification. These include: rabbits with IL2RG, IGHM, RAG1, or RAG2 knockout86,87,88,89; hamsters with STAT2 knockout90; and monkeys with RAG1 knockout.91
Hemophilia
Hemophilia is a group of inherited bleeding disorders that affect the blood-clotting process. This deficit is most often caused by mutations in genes coding for either clotting factor VIII (hemophilia A) or factor IX (hemophilia B), two crucial components of the blood coagulation cascade. People with hemophilia often experience internal bleeding into knee, hip, elbow, and ankle and subsequent joint disease. Current care for hemophilia involves lifelong infusions of clotting factors. Although highly effective at disease management, clotting factors are short lived in circulation. This necessitates i.v. delivery at least 2–3 times per week, which is both invasive and expensive.92 Alternatively, gene therapy, via transfer of a normal copy of F8 or F9 gene (encoding FVIII and FIX), may enable permanent correction and stable endogenous expression. Estimates suggest that an increase of only 1% in plasma FVIII or FIX levels would be therapeutic and encouraging results have been obtained by AAV vector-mediated gene transfer.93,94
Genome editing using programmable nucleases has also shown promise by allowing in situ targeting of hemophilia A and B. For hemophilia B, where mutations span across the entire coding region of human F9 (hF9) gene, HDR has been demonstrated by direct injections of AAV8-ZFNs and a corrective cDNA (promoterless exons 2–8 bearing a splice acceptor and a poly A signal flanked by homology arms) into livers of neonatal95 and adult humanized hemophilic mice.96 In neonatal mice, the level of gene repair was sufficient to correct clotting times, and partial hepatectomy showed stable genome modification, as levels of FIX were stable in the genome-edited liver. In sharp contrast, episomal AAV F9 transgene delivery could not overcome partial hepatectomy and FIX levels decreased to almost background levels, highlighting the advantage of gene editing.95 Analyses in adult hF9 mice showed sustained production of human FIX, averaging 23% of normal levels at week 60 (ref. 96).
In a novel strategy, ZFNs were utilized for targeted integration of promoterless F8 and F9 therapeutic transgenes within the highly expressed albumin gene.97 The albumin gene is expressed at high levels in liver cells and loss of its expression from a few percent of cells does not appear to be detrimental. This study demonstrated AAV8-ZFN-mediated long-term expression of both hF8 and hF9 at therapeutic levels in hemophilia A and B mouse models, respectively97 and is progressing toward clinical application. This highlights that certain genomic sequences, such as the albumin locus, are both permissive and amenable for transgene integration. In this context, it is worth mentioning a parallel study, which demonstrated nuclease-free, AAV vector-mediated targeting of the albumin locus to drive expression of hF9 in new born and adult mice. The animals produced levels of clotting factor that were between 7 and 20% of normal.98
Efforts to re-introduce F8 in hemophilia A have been more challenging. Such an approach has primarily been limited by the large size (7 Kb) of its cDNA, inefficient protein production, and complex mutations, comprising mostly of large inversions and duplications. Among the genotypes that result in hemophilia A, two different types of chromosomal inversions involving a portion of F8 gene are most frequent, accounting for 50% of cases. An initial study using TALENs led to correction of an inversion mutation in an iPSC model, establishing proof-of-concept.99 Building up on this, a separate study demonstrated correction of a ~600 Kb inversion using CRISPR-Cas9 system in iPSCs derived from hemophilia A patients.100 This was achieved by using Cas9 nucleases with target sites on either side of the inversion. Corrected iPSC colonies were clonally expanded before differentiating into epithelial cells, which produced FVIII protein in vitro. Transplantation of corrected endothelial cells rescued injury mortality in hemophilic mice in a short-term experiment.100
Hemophilia gene therapy is a forerunner in the field, with promising clinical development using safer viral vectors. Although the genome-editing strategies outlined here are still under development and currently lack safety validation, they can be combined with existing advancements such as AAV vector-mediated delivery to the liver to further clinical application. Furthermore, manipulation of the albumin locus may allow for this condition to be treated in the absence of nucleases, and the locus could also be used as a “safe harbor” for expression of other secreted proteins.98
Conclusion
The past few years have seen notable demonstrations of genome editing being applied across a multitude of diseases. While the application of nucleases holds significant therapeutic promise, optimum progress can only be achieved by examining the advancement of gene editing holistically. A number of ubiquitous challenges need to be considered, mostly relating to efficacy of genome editing at the target sequence, safety concerns related to nuclease-associated off-target effects, and delivery of gene-editing tools.
Editing efficiency is dependent on the DNA repair process being relied upon. In instances where the desired effect can be achieved by NHEJ, the correction will most likely occur at a relatively high frequency as NHEJ is the major repair pathway in mammalian cells, although the usefulness of this approach may be limited by the stochastic nature of the Indels being formed. As discussed earlier, NHEJ has been used to mediate disruption of coding and regulatory sequences, targeted deletions of exons or large intervening sequences, and disruption of splice sites. Methods that can predict and evaluate micro-homology sites can be used to bias the repair toward frameshift mutations in protein coding sequence.101 This would partially address the potential reduction in efficiency caused by micro-homology-mediated end joining, a secondary end-joining pathway with a bias for in-frame deletions.102
Precise HDR-based locus alterations allow targeted insertion or in situ correction of mutated DNA sequence, which are suitable for a large subset of disease-causing mutations. However, they are reliant upon homologous recombination, which is normally limited to S and G2 phases of the cell cycle, requires a DNA template, and inherently occurs at lower frequencies. HDR-based strategies may also require enrichment and expansion of corrected cells, normally restricted to ex vivo approaches. While ex vivo manipulation may be possible in diseases like those affecting bone marrow HSCs, it would limit applications in diseases with multiorgan involvement or those where transplantation is not an option. Further progress into enabling HDR with higher efficacy would therefore be beneficial. In this respect, a recent report has demonstrated that it may be possible to transiently activate HDR in G1 cells by restoring BRCA1–PALB2 interaction,103 possibly facilitating HDR genome editing in quiescent cells.
Specificity of genome editing is one of the major safety concerns for translational research. Owing to sequence similarities within the genome, endonucleases can cleave and modify off-target regions that are distinct from the site of interest. Off-target effects can lead to unwanted genetic modifications causing cellular stress, functional impairment or enhancement, and oncogenicity, all of which could have detrimental effects clinically.104 Considerable work is being undertaken to increase fidelity through better design of nuclease components, which has led to improved variants such as megaTALs,105 dead Cas9-Fok1 fusion nucleases,106 Cas9 nickases,107 and Cas9 nucleases with truncated guide RNAs.108 Furthermore, screens of bacterial strains have led to discovery of several alternative Cas9-nucleases with varying specificities.109,110,111,112 More recently, Cpf1, a prominent CRISPR variant that requires a shorter RNA and generates a staggered cut which could improve HDR, has also been described.113 All of these variants highlight the progress in the field but still require extensive examination prior to their application in a translational research setting. Specificity of modification can also be helped by careful target site selection and use of delivery methods that would allow for efficient but transient expression of nucleases. New methods, such as GUIDE-seq114 and BLESS,115 have also been developed for unbiased evaluation of off-target modifications on a genome-wide scale.
The final challenge pertains to the delivery of gene-editing reagents including nucleases and a donor template in case of HDR. A variety of delivery approaches are being explored depending on cell types to be targeted. Cells that can be cultured and engrafted under ex vivo conditions are amenable to delivery via nucleic acids, proteins, and viral vector systems; mRNA and protein delivery of the nucleases are now well-established procedures. However, for in vivo gene-editing applications, the most promising delivery systems are viral vectors. Both integrating and nonintegrating viral vector systems have been explored in this context, although the latter are favored due to their safety profile. In particular, AAV vectors, with a wide range of serotypes and ability to transduce a variety of tissue types, are promising candidates. However, AAV vectors are restricted by their small packaging capacity, which poses challenges for large nuclease proteins such as TALENs or Cas9.
Despite the outlined challenges, genome editing is advancing at fast pace, with continued focus on pioneering and improving strategies. The successful use of ZFNs targeting CCR5, the co-receptor necessary for HIV to infect T-cells, to control AIDS, remains the single demonstration of gene editing in a therapeutic setting116 but will be quickly followed by others. Our developing understanding of programmable nucleases and DNA repair, coupled to general progress in regenerative medicine and knowledge of inherited disease pathophysiology, should warrant exciting outcomes from therapeutic genome editing, which we could only dream of in the nineties117—we look forward to the next 20 years.
Acknowledgments
We thank Linda Popplewell for critical reading of the manuscript and for helpful discussions. R. J.Y.-M. acknowledges financial support from the 7th EU Framework Programme (PERSIST project, grant agreement no. 222878) and Royal Holloway, University of London; his work has also received research support from Sangamo Biosciences, Inc. Sangamo has had no role in the current study. V.P. and M.M. acknowledge financial support from Royal Holloway, University of London, and Muscular Dystrophy UK.
References
- In: Rodwell, C., Aymé, S. (eds.) (2014). 2014 Report on the State of the Art of Rare Disease Activities in Europe Part Ii: Key Developments in the Field of Rare Diseases in Europe in 2013. 1–90. http://www.eucerd.eu/upload/file/Reports/2014ReportStateofArtRDActivities.pdf.
- Hacein-Bey-Abina, S, Hauer, J, Lim, A, Picard, C, Wang, GP, Berry, CC et al. (2010). Efficacy of gene therapy for X-linked severe combined immunodeficiency. N Engl J Med 363: 355–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ylä-Herttuala, S (2012). Endgame: glybera finally recommended for approval as the first gene therapy drug in the European union. Mol Ther 20: 1831–1832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alton, EW, Armstrong, DK, Ashby, D, Bayfield, KJ, Bilton, D, Bloomfield, EV et al.; UK Cystic Fibrosis Gene Therapy Consortium. (2015). Repeated nebulisation of non-viral CFTR gene therapy in patients with cystic fibrosis: a randomised, double-blind, placebo-controlled, phase 2b trial. Lancet Respir Med 3: 684–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaj, T, Gersbach, CA and Barbas, CF 3rd (2013). ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol 31: 397–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadelain, M, Papapetrou, EP and Bushman, FD (2012). Safe harbours for the integration of new DNA in the human genome. Nat Rev Cancer 12: 51–58. [DOI] [PubMed] [Google Scholar]
- O'Sullivan, BP and Freedman, SD (2009). Cystic fibrosis. Lancet 373: 1891–1904. [DOI] [PubMed] [Google Scholar]
- Fanen, P, Wohlhuter-Haddad, A and Hinzpeter, A (2014). Genetics of cystic fibrosis: CFTR mutation classifications toward genotype-based CF therapies. Int J Biochem Cell Biol 52: 94–102. [DOI] [PubMed] [Google Scholar]
- Griesenbach, U, Pytel, KM and Alton, EW (2015). Cystic fibrosis gene therapy in the UK and elsewhere. Hum Gene Ther 26: 266–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welch, EM, Barton, ER, Zhuo, J, Tomizawa, Y, Friesen, WJ, Trifillis, P et al. (2007). PTC124 targets genetic disorders caused by nonsense mutations. Nature 447: 87–91. [DOI] [PubMed] [Google Scholar]
- Ren, HY, Grove, DE, De La Rosa, O, Houck, SA, Sopha, P, Van Goor, F et al. (2013). VX-809 corrects folding defects in cystic fibrosis transmembrane conductance regulator protein through action on membrane-spanning domain 1. Mol Biol Cell 24: 3016–3024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckford, PD, Li, C, Ramjeesingh, M and Bear, CE (2012). Cystic fibrosis transmembrane conductance regulator (CFTR) potentiator VX-770 (ivacaftor) opens the defective channel gate of mutant CFTR in a phosphorylation-dependent but ATP-independent manner. J Biol Chem 287: 36639–36649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies, JC, Wainwright, CE, Canny, GJ, Chilvers, MA, Howenstine, MS, Munck, A et al.; VX08-770-103 (ENVISION) Study Group. (2013). Efficacy and safety of ivacaftor in patients aged 6 to 11 years with cystic fibrosis with a G551D mutation. Am J Respir Crit Care Med 187: 1219–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies, J, Sheridan, H, Bell, N, Cunningham, S, Davis, SD, Elborn, JS et al. (2013). Assessment of clinical response to ivacaftor with lung clearance index in cystic fibrosis patients with a G551D-CFTR mutation and preserved spirometry: a randomised controlled trial. Lancet Respir Med 1: 630–638. [DOI] [PubMed] [Google Scholar]
- Ramsey, BW, Davies, J, McElvaney, NG, Tullis, E, Bell, SC, Dřevínek, P et al.; VX08-770-102 Study Group. (2011). A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N Engl J Med 365: 1663–1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barry, PJ, Plant, BJ, Nair, A, Bicknell, S, Simmonds, NJ, Bell, NJ et al. (2014). Effects of ivacaftor in patients with cystic fibrosis who carry the G551D mutation and have severe lung disease. Chest 146: 152–158. [DOI] [PubMed] [Google Scholar]
- Yu, H, Burton, B, Huang, CJ, Worley, J, Cao, D, Johnson, JP Jr et al. (2012). Ivacaftor potentiation of multiple CFTR channels with gating mutations. J Cyst Fibros 11: 237–245. [DOI] [PubMed] [Google Scholar]
- Wainwright, CE, Elborn, JS, Ramsey, BW, Marigowda, G, Huang, X, Cipolli, M et al.; TRAFFIC Study Group; TRANSPORT Study Group. (2015). Lumacaftor-ivacaftor in patients with cystic fibrosis homozygous for Phe508del CFTR. N Engl J Med 373: 220–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beumer, W, Swildens, J, Henig, N, Anthonijsz, H, Biasutto, P, Leal, T, et al. (2015). WS01.2 QR-010, an RNA therapy, restores CFTR function using in vitro and in vivo models of ΔF508 CFTR. J Cyst Fibros 14: S1. [Google Scholar]
- Henig, N, Beumer, W, Anthonijsz, H, Beka, M, Panin, N, Leal, T, et al. (2015). QR-010, an RNA therapy, restores CFTR function in the saliva secretion assay. American Thoracic Society International Conference Meetings Abstracts.
- Griesenbach, U and Alton, EW (2013). Moving forward: cystic fibrosis gene therapy. Hum Mol Genet 22: R52–R58. [DOI] [PubMed] [Google Scholar]
- Lee, CM, Flynn, R, Hollywood, JA, Scallan, MF and Harrison, PT (2012). Correction of the ΔF508 mutation in the cystic fibrosis transmembrane conductance regulator gene by zinc-finger nuclease homology-directed repair. Biores Open Access 1: 99–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwank, G, Koo, BK, Sasselli, V, Dekkers, JF, Heo, I, Demircan, T et al. (2013). Functional repair of CFTR by CRISPR/Cas9 in intestinal stem cell organoids of cystic fibrosis patients. Cell Stem Cell 13: 653–658. [DOI] [PubMed] [Google Scholar]
- Dekkers, JF, Wiegerinck, CL, de Jonge, HR, Bronsveld, I, Janssens, HM, de Winter-de Groot, KM et al. (2013). A functional CFTR assay using primary cystic fibrosis intestinal organoids. Nat Med 19: 939–945. [DOI] [PubMed] [Google Scholar]
- Yui, S, Nakamura, T, Sato, T, Nemoto, Y, Mizutani, T, Zheng, X et al. (2012). Functional engraftment of colon epithelium expanded in vitro from a single adult Lgr5+ stem cell. Nat Med 18: 618–623. [DOI] [PubMed] [Google Scholar]
- Crane, AM, Kramer, P, Bui, JH, Chung, WJ, Li, XS, Gonzalez-Garay, ML et al. (2015). Targeted correction and restored function of the CFTR gene in cystic fibrosis induced pluripotent stem cells. Stem Cell Reports 4: 569–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sargent, RG, Suzuki, S and Gruenert, DC (2014). Nuclease-mediated double-strand break (DSB) enhancement of small fragment homologous recombination (SFHR) gene modification in human-induced pluripotent stem cells (hiPSCs). Methods Mol Biol 1114: 279–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Firth, AL, Menon, T, Parker, GS, Qualls, SJ, Lewis, BM, Ke, E et al. (2015). Functional gene correction for cystic fibrosis in lung epithelial cells generated from patient iPSCs. Cell Rep 12: 1385–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng, AW, Wang, H, Yang, H, Shi, L, Katz, Y, Theunissen, TW et al. (2013). Multiplexed activation of endogenous genes by CRISPR-on, an RNA-guided transcriptional activator system. Cell Res 23: 1163–1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Pinera, P, Kocak, DD, Vockley, CM, Adler, AF, Kabadi, AM, Polstein, LR et al. (2013). RNA-guided gene activation by CRISPR-Cas9-based transcription factors. Nat Methods 10: 973–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bushby, K, Finkel, R, Birnkrant, DJ, Case, LE, Clemens, PR, Cripe, L et al.; DMD Care Considerations Working Group. (2010). Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and pharmacological and psychosocial management. Lancet Neurol 9: 77–93. [DOI] [PubMed] [Google Scholar]
- Bushby, K, Finkel, R, Birnkrant, DJ, Case, LE, Clemens, PR, Cripe, L et al.; DMD Care Considerations Working Group. (2010). Diagnosis and management of Duchenne muscular dystrophy, part 2: implementation of multidisciplinary care. Lancet Neurol 9: 177–189. [DOI] [PubMed] [Google Scholar]
- Ricotti, V, Ridout, DA, Scott, E, Quinlivan, R, Robb, SA, Manzur, AY et al.; NorthStar Clinical Network. (2013). Long-term benefits and adverse effects of intermittent versus daily glucocorticoids in boys with Duchenne muscular dystrophy. J Neurol Neurosurg Psychiatry 84: 698–705. [DOI] [PubMed] [Google Scholar]
- Aartsma-Rus, A, Fokkema, I, Verschuuren, J, Ginjaar, I, van Deutekom, J, van Ommen, GJ et al. (2009). Theoretic applicability of antisense-mediated exon skipping for Duchenne muscular dystrophy mutations. Hum Mutat 30: 293–299. [DOI] [PubMed] [Google Scholar]
- Jarmin, S, Kymalainen, H, Popplewell, L and Dickson, G (2014). New developments in the use of gene therapy to treat Duchenne muscular dystrophy. Expert Opin Biol Ther 14: 209–230. [DOI] [PubMed] [Google Scholar]
- England, SB, Nicholson, LV, Johnson, MA, Forrest, SM, Love, DR, Zubrzycka-Gaarn, EE et al. (1990). Very mild muscular dystrophy associated with the deletion of 46% of dystrophin. Nature 343: 180–182. [DOI] [PubMed] [Google Scholar]
- Ousterout, DG, Kabadi, AM, Thakore, PI, Majoros, WH, Reddy, TE and Gersbach, CA (2015). Multiplex CRISPR/Cas9-based genome editing for correction of dystrophin mutations that cause Duchenne muscular dystrophy. Nat Commun 6: 6244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popplewell, L, Koo, T, Leclerc, X, Duclert, A, Mamchaoui, K, Gouble, A et al. (2013). Gene correction of a Duchenne muscular dystrophy mutation by meganuclease-enhanced exon knock-in. Hum Gene Ther 24: 692–701. [DOI] [PubMed] [Google Scholar]
- Li, HL, Fujimoto, N, Sasakawa, N, Shirai, S, Ohkame, T, Sakuma, T et al. (2015). Precise correction of the dystrophin gene in duchenne muscular dystrophy patient induced pluripotent stem cells by TALEN and CRISPR-Cas9. Stem Cell Reports 4: 143–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ousterout, DG, Kabadi, AM, Thakore, PI, Perez-Pinera, P, Brown, MT, Majoros, WH et al. (2015). Correction of dystrophin expression in cells from Duchenne muscular dystrophy patients through genomic excision of exon 51 by zinc finger nucleases. Mol Ther 23: 523–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briggs, D and Morgan, JE (2013). Recent progress in satellite cell/myoblast engraftment – relevance for therapy. FEBS J 280: 4281–4293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long, C, McAnally, JR, Shelton, JM, Mireault, A, Bassel-Duby, R and Olson, EN (2014). Prevention of muscular dystrophy in mice by CRISPR/Cas9-mediated editing of germline DNA. Science 1184: 1–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura, K, Fujii, W, Tsuboi, M, Tanihata, J, Teramoto, N, Takeuchi, S et al. (2014). Generation of muscular dystrophy model rats with a CRISPR/Cas system. Sci Rep 4: 5635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larcher, T, Lafoux, A, Tesson, L, Remy, S, Thepenier, V, François, V et al. (2014). Characterization of dystrophin deficient rats: a new model for Duchenne muscular dystrophy. PLoS One 9: e110371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, Y, Zheng, Y, Kang, Y, Yang, W, Niu, Y, Guo, X et al. (2015). Functional disruption of the dystrophin gene in rhesus monkey using CRISPR/Cas9. Hum Mol Genet 24: 3764–3774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klymiuk, N, Blutke, A, Graf, A, Krause, S, Burkhardt, K, Wuensch, A et al. (2013). Dystrophin-deficient pigs provide new insights into the hierarchy of physiological derangements of dystrophic muscle. Hum Mol Genet 22: 4368–4382. [DOI] [PubMed] [Google Scholar]
- Hollinger, K, Yang, CX, Montz, RE, Nonneman, D, Ross, JW and Selsby, JT (2014). Dystrophin insufficiency causes selective muscle histopathology and loss of dystrophin-glycoprotein complex assembly in pig skeletal muscle. FASEB J 28: 1600–1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitmore, C and Morgan, J (2014). What do mouse models of muscular dystrophy tell us about the DAPC and its components? Int J Exp Pathol 95: 365–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGreevy, JW, Hakim, CH, McIntosh, MA and Duan, D (2015). Animal models of Duchenne muscular dystrophy: from basic mechanisms to gene therapy. Dis Model Mech 8: 195–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavazzana-Calvo, M, Hacein-Bey, S, de Saint Basile, G, Gross, F, Yvon, E, Nusbaum, P et al. (2000). Gene therapy of human severe combined immunodeficiency (SCID)-X1 disease. Science 288: 669–672. [DOI] [PubMed] [Google Scholar]
- Gaspar, HB, Cooray, S, Gilmour, KC, Parsley, KL, Adams, S, Howe, SJ et al. (2011). Long-term persistence of a polyclonal T cell repertoire after gene therapy for X-linked severe combined immunodeficiency. Sci Transl Med 3: 97ra79. [DOI] [PubMed] [Google Scholar]
- Blaese, RM, Culver, KW, Miller, AD, Carter, CS, Fleisher, T, Clerici, M et al. (1995). T lymphocyte-directed gene therapy for ADA- SCID: initial trial results after 4 years. Science 270: 475–480. [DOI] [PubMed] [Google Scholar]
- Kohn, DB, Weinberg, KI, Nolta, JA, Heiss, LN, Lenarsky, C, Crooks, GM et al. (1995). Engraftment of gene-modified umbilical cord blood cells in neonates with adenosine deaminase deficiency. Nat Med 1: 1017–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aiuti, A, Slavin, S, Aker, M, Ficara, F, Deola, S, Mortellaro, A et al. (2002). Correction of ADA-SCID by stem cell gene therapy combined with nonmyeloablative conditioning. Science 296: 2410–2413. [DOI] [PubMed] [Google Scholar]
- Aiuti, A, Cattaneo, F, Galimberti, S, Benninghoff, U, Cassani, B, Callegaro, L et al. (2009). Gene therapy for immunodeficiency due to adenosine deaminase deficiency. N Engl J Med 360: 447–458. [DOI] [PubMed] [Google Scholar]
- Gaspar, HB, Cooray, S, Gilmour, KC, Parsley, KL, Zhang, F, Adams, S et al. (2011). Hematopoietic stem cell gene therapy for adenosine deaminase-deficient severe combined immunodeficiency leads to long-term immunological recovery and metabolic correction. Sci Transl Med 3: 97ra80. [DOI] [PubMed] [Google Scholar]
- Boztug, K, Schmidt, M, Schwarzer, A, Banerjee, PP, Díez, IA, Dewey, RA et al. (2010). Stem-cell gene therapy for the Wiskott-Aldrich syndrome. N Engl J Med 363: 1918–1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aiuti, A, Biasco, L, Scaramuzza, S, Ferrua, F, Cicalese, MP, Baricordi, C et al. (2013). Lentiviral hematopoietic stem cell gene therapy in patients with Wiskott-Aldrich syndrome. Science 341: 1233151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bank, A, Dorazio, R and Leboulch, P (2005). A phase I/II clinical trial of beta-globin gene therapy for beta-thalassemia. Ann NY Acad Sci 1054: 308–316. [DOI] [PubMed] [Google Scholar]
- Cavazzana-Calvo, M, Payen, E, Negre, O, Wang, G, Hehir, K, Fusil, F et al. (2010). Transfusion independence and HMGA2 activation after gene therapy of human β-thalassaemia. Nature 467: 318–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sankaran, VG and Weiss, MJ (2015). Anemia: progress in molecular mechanisms and therapies. Nat Med 21: 221–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modell, B and Darlison, M (2008). Global epidemiology of haemoglobin disorders and derived service indicators. Bull World Health Organ 86: 480–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levasseur, DN, Ryan, TM, Pawlik, KM and Townes, TM (2003). Correction of a mouse model of sickle cell disease: lentiviral/antisickling beta-globin gene transduction of unmobilized, purified hematopoietic stem cells. Blood 102: 4312–4319. [DOI] [PubMed] [Google Scholar]
- Voit, RA, Hendel, A, Pruett-Miller, SM and Porteus, MH (2014). Nuclease-mediated gene editing by homologous recombination of the human globin locus. Nucleic Acids Res 42: 1365–1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoban, MD, Cost, GJ, Mendel, MC, Romero, Z, Kaufman, ML, Joglekar, AV et al. (2015). Correction of the sickle cell disease mutation in human hematopoietic stem/progenitor cells. Blood 125: 2597–2604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma, N, Shan, Y, Liao, B, Kong, G, Wang, C, Huang, K et al. (2015). Factor-induced reprogramming and zinc finger nuclease-aided gene targeting cause different genome instability in β-thalassemia induced pluripotent stem cells (iPSCs). J Biol Chem 290: 12079–12089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, P, Tong, Y, Liu, XZ, Wang, TT, Cheng, L, Wang, BY et al. (2015). Both TALENs and CRISPR/Cas9 directly target the HBB IVS2-654 (C > T) mutation in β-thalassemia-derived iPSCs. Sci Rep 5: 12065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie, F, Ye, L, Chang, JC, Beyer, AI, Wang, J, Muench, MO et al. (2014). Seamless gene correction of β-thalassemia mutations in patient-specific iPSCs using CRISPR/Cas9 and piggyBac. Genome Res 24: 1526–1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebastiano, V, Maeder, ML, Angstman, JF, Haddad, B, Khayter, C, Yeo, DT et al. (2011). In situ genetic correction of the sickle cell anemia mutation in human induced pluripotent stem cells using engineered zinc finger nucleases. Stem Cells 29: 1717–1726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun, N and Zhao, H (2014). Seamless correction of the sickle cell disease mutation of the HBB gene in human induced pluripotent stem cells using TALENs. Biotechnol Bioeng 111: 1048–1053. [DOI] [PubMed] [Google Scholar]
- Jiang, Z, Han, Y and Cao, X (2014). Induced pluripotent stem cell (iPSCs) and their application in immunotherapy. Cell Mol Immunol 11: 17–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer, DE, Kamran, SC, Lessard, S, Xu, J, Fujiwara, Y, Lin, C et al. (2013). An erythroid enhancer of BCL11A subject to genetic variation determines fetal hemoglobin level. Science 342: 253–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vierstra, J, Reik, A, Chang, KH, Stehling-Sun, S, Zhou, Y, Hinkley, SJ et al. (2015). Functional footprinting of regulatory DNA. Nat Methods 12: 927–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canver, MC, Smith, EC, Sher, F, Pinello, L, Sanjana, NE, Shalem, O et al. (2015). BCL11A enhancer dissection by Cas9-mediated in situ saturating mutagenesis. Nature 527: 192–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urnov, FD, Miller, JC, Lee, YL, Beausejour, CM, Rock, JM, Augustus, S et al. (2005). Highly efficient endogenous human gene correction using designed zinc-finger nucleases. Nature 435: 646–651. [DOI] [PubMed] [Google Scholar]
- Lombardo, A, Genovese, P, Beausejour, CM, Colleoni, S, Lee, YL, Kim, KA et al. (2007). Gene editing in human stem cells using zinc finger nucleases and integrase-defective lentiviral vector delivery. Nat Biotechnol 25: 1298–1306. [DOI] [PubMed] [Google Scholar]
- Genovese, P, Schiroli, G, Escobar, G, Di Tomaso, T, Firrito, C, Calabria, A et al. (2014). Targeted genome editing in human repopulating haematopoietic stem cells. Nature 510: 235–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsubara, Y, Chiba, T, Kashimada, K, Morio, T, Takada, S, Mizutani, S et al. (2014). Transcription activator-like effector nuclease-mediated transduction of exogenous gene into IL2RG locus. Sci Rep 4: 5043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merling, RK, Sweeney, CL, Chu, J, Bodansky, A, Choi, U, Priel, DL et al. (2015). An AAVS1-targeted minigene platform for correction of iPSCs from all five types of chronic granulomatous disease. Mol Ther 23: 147–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, M and Izpisua Belmonte, JC (2012). No factor left behind: generation of transgene-free induced pluripotent stem cells. Am J Stem Cells 1: 75–80. [PMC free article] [PubMed] [Google Scholar]
- Menon, T, Firth, AL, Scripture-Adams, DD, Galic, Z, Qualls, SJ, Gilmore, WB et al. (2015). Lymphoid regeneration from gene-corrected SCID-X1 subject-derived iPSCs. Cell Stem Cell 16: 367–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman, SH, Kuehle, J, Reimann, C, Mlambo, T, Alzubi, J, Maeder, ML et al. (2015). Rescue of DNA-PK signaling and T-cell differentiation by targeted genome editing in a prkdc deficient iPSC disease model. PLoS Genet 11: e1005239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, H, Wang, H and Jaenisch, R (2014). Generating genetically modified mice using CRISPR/Cas-mediated genome engineering. Nat Protoc 9: 1956–1968. [DOI] [PubMed] [Google Scholar]
- Li, F, Cowley, DO, Banner, D, Holle, E, Zhang, L and Su, L (2014). Efficient genetic manipulation of the NOD-Rag1-/-IL2RgammaC-null mouse by combining in vitro fertilization and CRISPR/Cas9 technology. Sci Rep 4: 5290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mashimo, T, Takizawa, A, Voigt, B, Yoshimi, K, Hiai, H, Kuramoto, T et al. (2010). Generation of knockout rats with X-linked severe combined immunodeficiency (X-SCID) using zinc-finger nucleases. PLoS One 5: e8870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Y, Fan, N, Song, J, Zhong, J, Guo, X, Tian, W et al. (2014). Generation of knockout rabbits using transcription activator-like effector nucleases. Cell Regen (Lond) 3: 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan, Q, Zhang, Q, Yang, H, Zou, Q, Tang, C, Fan, N et al. (2014). Generation of multi-gene knockout rabbits using the Cas9/gRNA system. Cell Regen (Lond) 3: 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flisikowska, T, Thorey, IS, Offner, S, Ros, F, Lifke, V, Zeitler, B et al. (2011). Efficient immunoglobulin gene disruption and targeted replacement in rabbit using zinc finger nucleases. PLoS One 6: e21045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song, J, Zhong, J, Guo, X, Chen, Y, Zou, Q, Huang, J et al. (2013). Generation of RAG 1- and 2-deficient rabbits by embryo microinjection of TALENs. Cell Res 23: 1059–1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan, Z, Li, W, Lee, SR, Meng, Q, Shi, B, Bunch, TD et al. (2014). Efficient gene targeting in golden Syrian hamsters by the CRISPR/Cas9 system. PLoS One 9: e109755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niu, Y, Shen, B, Cui, Y, Chen, Y, Wang, J, Wang, L et al. (2014). Generation of gene-modified cynomolgus monkey via Cas9/RNA-mediated gene targeting in one-cell embryos. Cell 156: 836–843. [DOI] [PubMed] [Google Scholar]
- High, KH, Nathwani, A, Spencer, T and Lillicrap, D (2014). Current status of haemophilia gene therapy. Haemophilia 20 (suppl. 4): 43–49. [DOI] [PubMed] [Google Scholar]
- Nathwani, AC, Tuddenham, EG, Rangarajan, S, Rosales, C, McIntosh, J, Linch, DC et al. (2011). Adenovirus-associated virus vector-mediated gene transfer in hemophilia B. N Engl J Med 365: 2357–2365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathwani, AC, Reiss, UM, Tuddenham, EG, Rosales, C, Chowdary, P, McIntosh, J et al. (2014). Long-term safety and efficacy of factor IX gene therapy in hemophilia B. N Engl J Med 371: 1994–2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H, Haurigot, V, Doyon, Y, Li, T, Wong, SY, Bhagwat, AS et al. (2011). In vivo genome editing restores haemostasis in a mouse model of haemophilia. Nature 475: 217–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anguela, XM, Sharma, R, Doyon, Y, Miller, JC, Li, H, Haurigot, V et al. (2013). Robust ZFN-mediated genome editing in adult hemophilic mice. Blood 122: 3283–3287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma, R, Anguela, XM, Doyon, Y, Wechsler, T, DeKelver, RC, Sproul, S et al. (2015). In vivo genome editing of the albumin locus as a platform for protein replacement therapy. Blood 126: 1777–1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barzel, A, Paulk, NK, Shi, Y, Huang, Y, Chu, K, Zhang, F et al. (2015). Promoterless gene targeting without nucleases ameliorates haemophilia B in mice. Nature 517: 360–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park, CY, Kim, J, Kweon, J, Son, JS, Lee, JS, Yoo, JE et al. (2014). Targeted inversion and reversion of the blood coagulation factor 8 gene in human iPS cells using TALENs. Proc Natl Acad Sci USA 111: 9253–9258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park, CY, Kim, DH, Son, JS, Sung, JJ, Lee, J, Bae, S et al. (2015). Functional correction of large factor VIII gene chromosomal inversions in hemophilia a patient-derived iPSCs using CRISPR-Cas9. Cell Stem Cell 17: 213–220. [DOI] [PubMed] [Google Scholar]
- Bae, S, Kweon, J, Kim, HS and Kim, JS (2014). Microhomology-based choice of Cas9 nuclease target sites. Nat Methods 11: 705–706. [DOI] [PubMed] [Google Scholar]
- Morton, J, Davis, MW, Jorgensen, EM and Carroll, D (2006). Induction and repair of zinc-finger nuclease-targeted double-strand breaks in Caenorhabditis elegans somatic cells. Proc Natl Acad Sci USA 103: 16370–16375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orthwein, A, Noordermeer, SM, Wilson, MD, Landry, S, Enchev, RI, Sherker, A et al. (2015). A mechanism for the suppression of homologous recombination in G1 cells. Nature 528: 422–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maggio, I and Gonçalves, MA (2015). Genome editing at the crossroads of delivery, specificity, and fidelity. Trends Biotechnol 33: 280–291. [DOI] [PubMed] [Google Scholar]
- Boissel, S, Jarjour, J, Astrakhan, A, Adey, A, Gouble, A, Duchateau, P et al. (2014). megaTALs: a rare-cleaving nuclease architecture for therapeutic genome engineering. Nucleic Acids Res 42: 2591–2601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai, SQ, Wyvekens, N, Khayter, C, Foden, JA, Thapar, V, Reyon, D et al. (2014). Dimeric CRISPR RNA-guided FokI nucleases for highly specific genome editing. Nat Biotechnol 32: 569–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran, FA, Hsu, PD, Lin, CY, Gootenberg, JS, Konermann, S, Trevino, AE et al. (2013). Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell 154: 1380–1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu, Y, Sander, JD, Reyon, D, Cascio, VM and Joung, JK (2014). Improving CRISPR-Cas nuclease specificity using truncated guide RNAs. Nat Biotechnol 32: 279–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sapranauskas, R, Gasiunas, G, Fremaux, C, Barrangou, R, Horvath, P and Siksnys, V (2011). The Streptococcus thermophilus CRISPR/Cas system provides immunity in Escherichia coli. Nucleic Acids Res 39: 9275–9282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou, Z, Zhang, Y, Propson, NE, Howden, SE, Chu, LF, Sontheimer, EJ et al. (2013). Efficient genome engineering in human pluripotent stem cells using Cas9 from Neisseria meningitidis. Proc Natl Acad Sci USA 110: 15644–15649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran, FA, Cong, L, Yan, WX, Scott, DA, Gootenberg, JS, Kriz, AJ et al. (2015). In vivo genome editing using Staphylococcus aureus Cas9. Nature 520: 186–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esvelt, KM, Mali, P, Braff, JL, Moosburner, M, Yaung, SJ and Church, GM (2013). Orthogonal Cas9 proteins for RNA-guided gene regulation and editing. Nat Methods 10: 1116–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zetsche, B, Gootenberg, JS, Abudayyeh, OO, Slaymaker, IM, Makarova, KS, Essletzbichler, P et al. (2015). Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell 163: 759–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai, SQ, Zheng, Z, Nguyen, NT, Liebers, M, Topkar, VV, Thapar, V et al. (2015). GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nat Biotechnol 33: 187–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crosetto, N, Mitra, A, Silva, MJ, Bienko, M, Dojer, N, Wang, Q et al. (2013). Nucleotide-resolution DNA double-strand break mapping by next-generation sequencing. Nat Methods 10: 361–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tebas, P, Stein, D, Tang, WW, Frank, I, Wang, SQ, Lee, G et al. (2014). Gene editing of CCR5 in autologous CD4 T cells of persons infected with HIV. N Engl J Med 370: 901–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yáñez, RJ and Porter, AC (1998). Therapeutic gene targeting. Gene Ther 5: 149–159. [DOI] [PubMed] [Google Scholar]