

Abstract
The clustered regularly-interspaced short palindromic repeats (CRISPR)—CRISPR-associated (Cas) system from Streptococcus pyogenes (Spy) has been successfully adapted for RNA-guided genome editing in a wide range of organisms. However, numerous reports have indicated that Spy CRISPR-Cas9 systems may have significant off-target cleavage of genomic DNA sequences differing from the intended on-target site. Here, we report the performance of the Neisseria meningitidis (Nme) CRISPR-Cas9 system that requires a longer protospacer-adjacent motif for site-specific cleavage, and present a comparison between the Spy and Nme CRISPR-Cas9 systems targeting the same protospacer sequence. The results with the native crRNA and tracrRNA as well as a chimeric single guide RNA for the Nme CRISPR-Cas9 system were also compared. Our results suggest that, compared with the Spy system, the Nme CRISPR-Cas9 system has similar or lower on-target cleavage activity but a reduced overall off-target effect on a genomic level when sites containing three or fewer mismatches are considered. Thus, the Nme CRISPR-Cas9 system may represent a safer alternative for precision genome engineering applications.
Introduction
Clustered, regularly-interspaced short palindromic repeats (CRISPR) and CRIPSR-associated (Cas) proteins constitute an acquired immune system in bacteria and archaea.1 The type II CRISPR-Cas9 system from Streptococcus pyogenes (Spy) has been adapted to target distinct genomic loci in a wide variety of organisms.2,3,4,5,6,7,8,9 This system is composed of a Cas9 protein, a trans-activating CRISPR RNA (tracrRNA), and a targeting CRISPR RNA (crRNA) that can be programmed to recognize a genomic sequence via Watson-Crick base pairing.10 Furthermore, the tracrRNA and crRNA (2-RNA system) can be fused to create a single guide RNA (sgRNA).10 Unlike other genome-editing tools such as zinc finger nucleases (ZFNs)11 and transcription activator-like effector nucleases (TALENs)12 that require engineered protein domains to target specific DNA sequences, in a CRISPR-Cas9 system the binding of endonuclease (Cas9) to DNA is guided by a short RNA. The targeting region of a crRNA (2-RNA system) or a sgRNA (single guide system) will herein be referred to as the guide RNA (gRNA). DNA-targeting specificity is determined by two key factors: a DNA sequence matching the gRNA variable region and a protospacer adjacent motif (PAM) directly downstream of the target sequence.10 It has been demonstrated that, although Spy CRISPR-Cas9 systems can cleave the intended target sequence with high efficiency, off-target cleavage occurs at DNA sequences that have base mismatches, deletions, or insertions relative to that of the gRNA.13,14,15,16,17,18,19,20 Current strategies to reduce CRISPR-Cas9 off-target effects include the use of partially inactivated Cas9 nickases21,22 and a catalytically dead Cas9 fused to the FokI endonuclease domain,23,24,25 both of which require the design and delivery of two gRNAs. The requirement of a specific PAM sequence is quite strict, with Spy Cas9 recognizing NGG, and to a lesser extent NAG, and in rare instances other noncanonical sequences.17 Although CRISPR-Cas9 systems from other species have also been studied,26 apart from the Staphylococcus aureus (Sau) CRISPR-Cas9 system,27 only very limited investigations have been made for their use to target endogenous loci in mammalian cells.28
The PAM sequence of a CRISPR system has important implications to its target specificity. Although base mismatches may be tolerated between the gRNA and targeted sequences, the PAM sequence has a stricter adherence to the consensus. This led to the hypothesis that an orthogonal CRISPR-Cas9 system with a longer PAM sequence may have fewer off-target cleavage events. Here, we report our studies of genome editing in mammalian cells using the CRISPR-Cas9 system from Neisseria meningitidis (Nme) that has previously been demonstrated to recognize a canonical PAM sequence NNNNGATT.26,29,30 On average, the optimal PAM sequence (NGG) for the Spy CRISPR-Cas9 system is present once every eight bases in a random sequence when both sense and antisense orientations are taken into consideration, rising to once every 4 bp for all recognizable PAM sequences (NRG). In contrast, the optimal Nme Cas9 PAM sequence (NNNNGATT) occurs once every 128 bases in a random sequence, with the degenerate PAM NNNNGHTT occurring once every 96 bases, thus reducing the number of potential off-target sites. We show that compared with the Spy system, Nme systems exhibit a lower off-target effect at endogenous loci, while achieving similar or lower on-target activities. Therefore, the Nme CRISPR-Cas9 system may represent a more specific alternative for precision genome engineering applications.
Results
Determination of optimum guide RNA length
Previous studies of the Nme CRISPR-Cas9 systems used 24 and 20 nucleotides (nt) long gRNAs with 24 nt gRNAs more closely resembling the native length.26,29,30 Since Spy Cas9 gRNAs with truncations were reported to have lower off-target activity,31 we systematically tested Nme gRNAs ranging from 24 to 20 nt to determine the optimum gRNA length for high Nme Cas9 activity for both the native crRNA and tracrRNA system (2-RNA) where the crRNA and tracrRNA are separate and expressed from different U6 promoters, and a chimeric single guide RNA (sgRNA) system with crRNA and tracrRNA fused together and expressed from a single U6 promoter (Figure 1a,b). We found that the Nme CRISPR-Cas9 systems had non-homologous end joining-induced indel (small insertion and/or deletion) rates ranging from 3 to 75%. The levels of cleavage activity observed at each target locus varied based on the length of gRNA used (Supplementary Table S1). The 2-RNA system has a preference for a longer gRNA sequence (Figure 1c,d), whereas the sgRNA system seems to tolerate more variance in the length of the gRNA. Our results agree with a previous report that compared to T, nucleotide A is not favored as the first N base in the Nme PAM sequence (ANNNGATT).29 We also observed lower activity when nucleotide T was the PAM proximal base of the target sequence (Supplementary Figure S1).
Figure 1.

Overview of the Nme CRISPR-Cas9 system. Outline of the expression systems and the predicted structures of the of the 2-RNA (a) and sgRNA (b) systems. (c) T7EI assay showing activity of the Nme 2-RNA system and the Nme sgRNA system at the IL2RG locus in HEK 293T cells. (d) Activity of gRNAs with different lengths. Bars represent the average cutting efficiency for each length and the cutting activity for each locus tested is represented as circles. Data for the Nme 2-RNA system (blue bars) is taken from T7EI assay data from 13 endogenous loci and the data for the Nme sgRNA system (red bars) is taken from T7EI assay data from 10 endogenous loci. All experiments were carried out in HEK293T cells. gRNA lengths that had activity levels that differed significantly from the highest active gRNA length at denoted by *. Significance was determined using a two-tailed t-test. P < 0.05 (e) Luciferase single-strand annealing (SSA) assays to investigate protospacer adjacent motif (PAM) recognition of Nme Cas9. All data from biological triplicates. The significance of 2-RNA activity levels (*) and sgRNA activity levels (†) were determined using a two-tailed t-test. P < 0.05 (f) Extrapolated PAM sequence preference of the Nme CRISPR-Cas9 systems with 2-RNA (top) and the Nme sgRNA (bottom) based on results from Luciferase SSA assays.
Characterization of the dependence on protospacer-adjacent motif
The canonical PAM sequence for Nme Cas9 was bioinformatically determined to be NNNNGATT.32 Similar to the Spy Cas9 that can recognize PAM sequences other than the canonical NGG motif, previous reports suggest that Nme Cas9 can also recognize non-canonical PAM sequences. However, these studies tested PAM recognition either in a bacteria cell29 or with a limited number of possible PAMs.30 To investigate the PAM preference of the Nme Cas9 system in mammalian cells, PAM sequences differing by one nucleotide at each position were tested in HEK293T cells to determine the effect on Nme CRISPR-Cas9 induced cleavage using a quantitative luciferase-based single-strand annealing (SSA) assay.33 In this assay, two direct repeats of the firefly luciferase open reading frame are separated by a nuclease target site from HPRT1 and two stop codons. Recognition and cleavage of the target site by a designer nuclease leads to repair of the DNA double-strand break via single-strand annealing between the two direct repeats, rescuing the expression of luciferase protein (Figure 2a). The use of this surrogate reporter system also removes any bias from site accessibility issues that may arise when targeting different endogenous loci. As expected, Nme Cas9 had high activity in the presence of the bioinformatically determined PAM sequence NNNNGATT. Consistent with previously reports, the NNNNGCTT and NNNNGTTT PAM sequences also facilitated high levels of Nme Cas9 activity.26,29 Lower activities were observed with several other PAM sequences (Figure 1e). We found that the PAM motifs that facilitated Nme Cas9 activity differed between the two gRNA conformations, with the 2-RNA Nme system displaying more variation in base recognition than the sgRNA Nme system (Figure 1f). Although the reduced activity of these novel PAM sequences precludes their use for targeted DNA cleavage, they should be considered when searching for potential off-target sites.
Figure 2.

Comparison of mismatch tolerance between Nme and Spy CRISPR-Cas9 systems. (a) Schematic of the luciferase based single-strand annealing (SSA) assay used to determine gRNA specificity. Following successful cleavage of the target site by Cas9, the shaded homologous regions can recombine via SSA resulting in the restoration of the full length luciferase open reading frame and expression of luciferase. The level of luciferase expressed is measured by the amount of D-luciferin which is converted into oxyluciferin and light. The activity of gRNA variants containing mismatches (b), insertions (c), or deletions (d) are displayed as heatmaps based on relative single-strand annealing activity. The activity of all gRNA variants was normalized to the original guide sequence. gRNAs of 20, 21, and 23 nt were used for Spy sgRNA, Nme sgRNA, and Nme 2-RNA systems respectively. All data derived from biological triplicates. The sequences and raw data for all gRNA variants can be found in Supplementary Table S3.
Comparison of targeting specificity between Nme Cas9 and Spy Cas9
Previous studies have shown that Spy CRISPR-Cas9 systems can tolerate genomic target sites containing base mismatches, single-base DNA insertions (DNA bulges) and single-base DNA deletions (RNA bulges),13,14,15,16 which have significant implications to the targeting specificity of CRISPR-Cas9. To study the sequence mismatch tolerance of the Nme CRISPR-Cas9 system, we systematically modified single nucleotides at all positions throughout the HPRT1 targeting gRNA (Figure 2b) and tested the ability of these gRNA variants to direct Cas9-mediated DNA cleavage by determining luciferase expression in an SSA based assay. The optimal gRNA length for each system was used and nucleotides at all positions were modified except the base at the 5′ end. This base must be a guanine as it is the transcription start site for the U6 promoter. Specifically, 23-base gRNAs were used for the 2-RNA Nme system, 21 bp gRNAs for the sgRNA Nme system, and 20bp gRNAs for the Spy system respectively. We found that the off-target cleavage due to single-base mismatches varied significantly among the three systems in a position-dependent fashion, with the Spy Cas9 system having a higher off-target effect at most of the locations when compared with the Nme Cas9 systems (Figure 2b, Supplementary Table S2).
To determine if the Nme CRISPR-Cas9 system could also tolerate target sites containing one-base DNA bulges, we systematically removed single nucleotides at all possible positions throughout the original HPRT1-targeting gRNAs for the three CRISPR-Cas9 systems (spy, 2-RNA Nme, and sgRNA Nme), resulting in gRNA-DNA interfaces containing one-base DNA bulges (Figure 2d, Supplementary Table S2). Luciferase-based SSA assays were used to determine CRISPR-Cas9-induced cleavage activity for each gRNA variant. We found that, similar to what we observed previously,16 the Spy CRISPR-Cas9 system tolerated DNA bugles at the 5′ and 3′ ends, as well as at positions 10–14 nucleotides from the PAM site. The Nme CRISPR-Cas9 systems also showed DNA bulge tolerance, but with lower overall activity compared with the Spy system (Figure 2d). Specifically, the 2-RNA Nme system showed no DNA bulge tolerance in the central region, whereas the sgRNA Nme system displayed a significant level of activity with a DNA bulge at position 6/7/8. Note that this DNA bulge could also be modeled as having 3 mismatches at positions 1, 4, and 6, and a noncanonical PAM that is not recognized by Nme (NNNNTTTA) (Supplementary Figure S2a). Similarly, for the Spy system, certain DNA bugles may be interpreted as base mismatches. For example, removing the 5′-end bases (base 17, 18, or 19) results in a truncated gRNA with one or two base mismatches, and the 3′-end base deletions (base 1/2/3) results in a 19-nt gRNA with a shifted PAM sequence (GGG to AGG) (Supplementary Figure S2b), which may explain why the activity for the Spy Δ3/2/1 gRNA is similar to the original HPRT1 gRNA. However, off-target activities associated with centrally located DNA bulges (position 10–14) cannot be modeled as a result of base-mismatches.
To determine if Nme CRISPR-Cas9 systems could tolerate one-base RNA bulges, single nucleotides were added at each position throughout the original HPRT1 gRNA sequences, resulting in one-base RNA bugles at the gRNA-DNA interfaces. Inserted nucleotides differed from both flanking bases to ensure a single-base bulge at each position along the gRNA (Supplementary Table S2). The luciferase-based SSA assay was again used to determine the activity of all gRNA variants. Surprisingly, we found that for both the Spy and Nme sgRNA systems, RNA bulges outside the 5′ end abolished cleavage activity almost completely (Figure 2c). However, the Nme 2-RNA system with separate crRNA and tracrRNAs tolerated RNA bulges at the central region (position 10–15). These findings are similar to previously tested Spy gRNAs with similar GC contents.16
Comparison of Nme and Spy Cas9 on-target activity at overlapping endogenous loci
Direct comparison of the off-target profiles of Nme and Spy CRISPR/Cas9 systems at endogenous loci requires genomic sites with overlapping Nme and Spy PAM sequences and a shared protospacer sequence since it is widely accepted that different gRNAs may have different activity levels and off-target profiles. To identify shared overlapping Spy and Nme protospacers, genomic regions of interest were searched for the sequence NGGNGHTT containing the PAM sequence of Nme Cas9 (NNNNGHTT) and that of Spy Cas9 (NGG). In total, eight sites were selected with one site (HPRT1) containing the PAM sequence NGGNGTTT, and the other seven sites having the PAM sequence NGGNGATT. The activity of Nme and Spy CRISPR/Cas9 systems targeting these sites was assayed in HEK293T cells, with the activity levels determined using the T7EI assay. We found that the Nme systems targeting HPRT1, F8, and DHFR had activity levels as high as 40%, but were generally lower than that of Spy Cas9 (Figure 3). The cleavage activity of the Nme CRISPR/Cas9 systems at the remaining 5 target sites was substantially lower than that observed with the Spy system (Figure 3, Supplementary Figure S3).
Figure 3.

On-target activity of Nme and Spy CRISPR-Cas9 systems targeting endogenous loci in HEK293T cells. Both Nme and Spy gRNAs target overlapping protospacer sequences. Shown are cleavage activities of different CRISPR/Cas9 systems at eight genomic loci as measured using the T7EI assay. Data from biological replicates. For each locus both Nme Cas9 2-RNA and sgRNA gRNAs were compared to Spy Cas9. Nme 2-RNA gRNAs with significantly different values are denoted by † whereas Nme sgRNA gRNAs with significant differences in activity compared to Spy Cas9 are denoted by *. Statistical analysis was carried out for the gRNA length with the highest activity at each locus. P < 0.05.
The off-target activity profiles of Nme and Spy systems at overlapping endogenous loci
After comparing the ability of Spy and Nme CRISPR-Cas9 systems to tolerate single base mismatches, insertions, and deletions in a surrogate reporter system, we further investigated the off-target activity of Spy and Nme systems by determining if partially matched endogenous sequences are cleaved. Specifically, potential off-target sites for Nme and Spy gRNAs targeting HPRT1, DHFR, and F8 were identified and ranked using the web-based tool COSMID.34 For Spy gRNAs, sites with three or less base mismatches containing the NRG PAM were identified whereas for Nme gRNAs, sites with three base mismatches in the first 19 PAM proximal bases containing the NNNNGNTT PAM were identified. Sites with one base insertions and deletions were also identified for both the Spy and Nme systems. Overall, the number of potential off-target sites identified for the Nme CRISPR-Cas9 systems was dramatically lower than that of the Spy system due to the requirement for a longer PAM sequence (Table 1). The top scoring off-target sites were amplified using locus-specific primers and sequenced using the Illumina Miseq platform. The on-target activity was also validated using deep sequencing for both the Spy and Nme systems. For each of the three target sites (HPRT1, DHFR, and F8), differences were observed in the mutation profiles between target loci. This is most likely due to differences in microhomology around the cleavage site35 (Supplementary Figure S4). The measured indel rates at the sites tested for Spy are listed in Table 2 while that for Nme are listed in Table 3. Interestingly, with the Spy CRISPR-Cas9 system, for each of the three target genes (HPRT1, DHFR, and F8), one off-target site was found to have a measurable level of cleavage activity (Table 2, Figure 4). In contrast, at all sites tested for the Nme CRISPR/Cas9 systems, no measurable off-target activities were detected (Table 3).
Table 1. Number of potential off-target (OT) sites identified by COSMID.
Table 2. On- and off-target mutations and indel rates induced by Spy Cas9 designed to cleave endogenous genes.
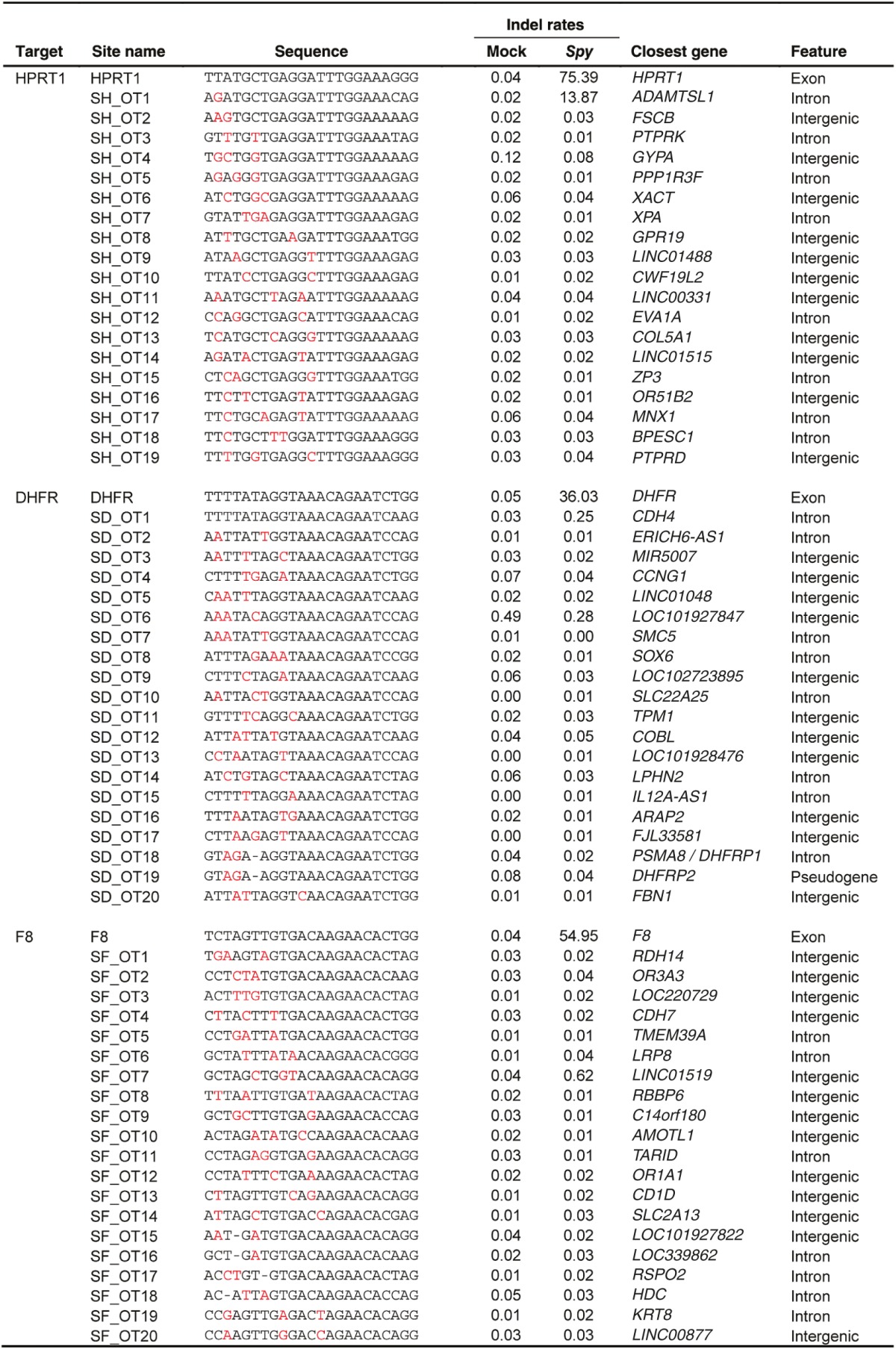
Table 3. On- and off-target mutations and indel rates induced by Nme Cas9 designed to cleave endogenous genes.
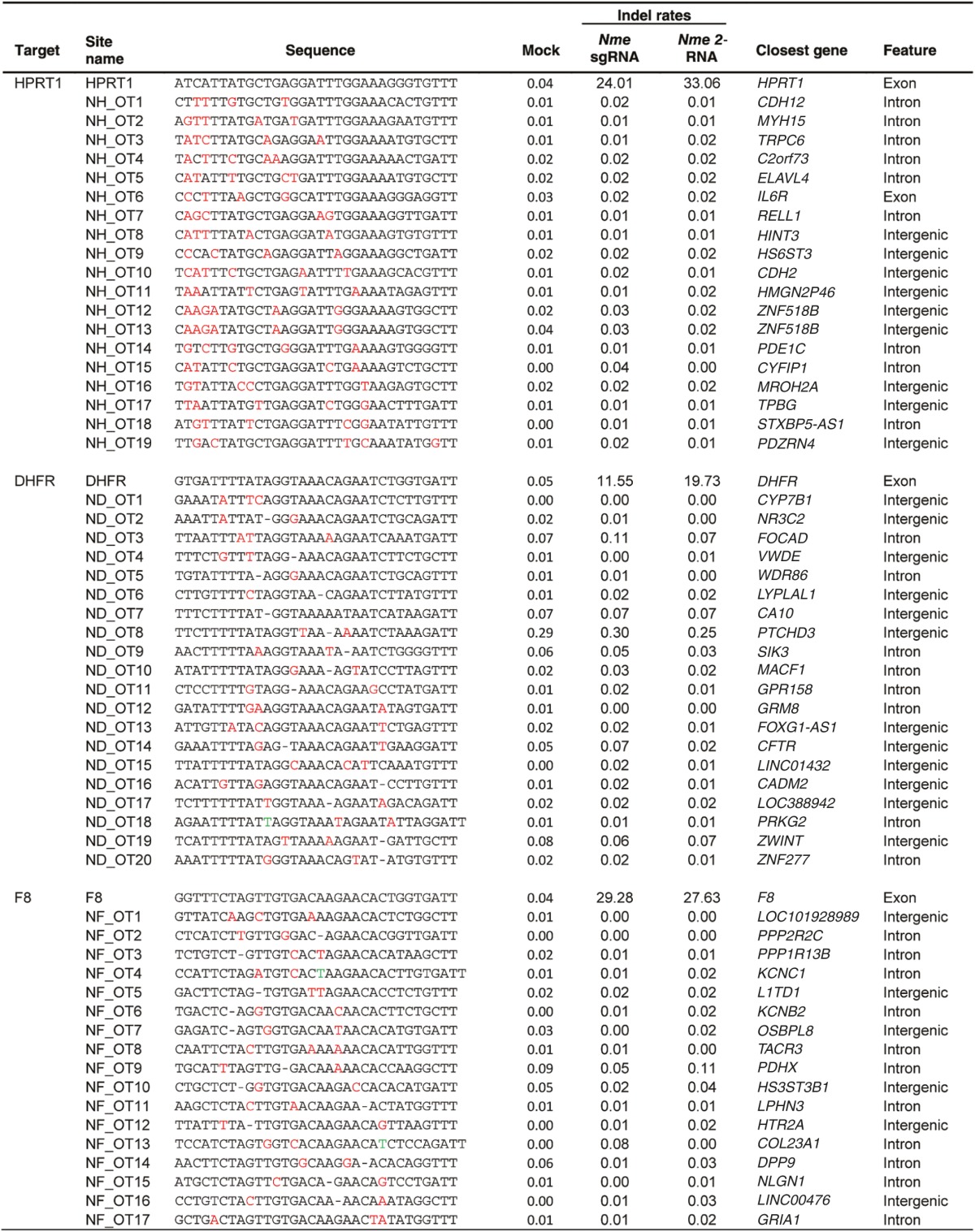
Figure 4.

Cleavage activity of Spy Cas9 at on- and off-target sites. Activity of Spy gRNAs targeting (a) DHFR, (b) F8, and (c) HPRT1 at both on-target and off-target sites. (d–f) The target sequence at each locus is underlined and mismatches are highlighted in red. Sequence reads contain indels typical of non-homologous end joining repair following Cas9 cleavage. Only sites where activity levels were above 0.1% (as assessed by illumina MiSeq) are shown. Error bars represent 95% confidence intervals and P values were obtained using Fisher's exact test, *P < 0.001.
Discussion
Unlike previous studies which targeted an integrated reporter system28 or used an antibiotic selection system,30 in this work, we directly determined the level of Nme Cas9 activity at endogenous loci in HEK293T cells. However, we found that the Nme Cas9 system has a larger variation in the on-target activity levels between target loci compared to that of the Spy system. This could be due to differences in the Cas9 proteins themselves, which differ in size by 287 amino acids. This difference in size could have an impact on several factors important for cleavage activity such as; site accessibility, binding stability, DNA unwinding and DNA cleavage. A recent report suggests that type II-C Cas9 orthologs, such as Nme Cas9 have poor DNA unwinding ability36 which could account for the lower activity levels observed with Nme Cas9 relative to Spy Cas9. Nme Cas9 also possesses DNase H activity which allows it to efficiently cleave ssDNA.37 A deeper understanding of how the Cas9 nuclease interacts with both the gRNA and the DNA target may also shed some light on these differences. To this end, although the crystal structures of both Spy and Sau Cas9 proteins38,39 have been determined, the crystal structure of Nme Cas9 protein has not yet been reported. We also observed differences in preference of gRNA length between the 2-RNA and sgRNA conformations. The sgRNA conformation used here was described previously.29 This chimeric sgRNA has a shorter hybridization region between the tracrRNA and crRNA than the 2-RNA system and this could affect the stability of the Nme Cas9-gRNA ribonucleoprotein complex. The widely used Spy CRISPR-Cas9 sgRNA backbone sequence has been optimized to generate a highly active gene targeting system, suggesting that further modification of the Nme sgRNA may yield a more robust variant of the Nme CRISPR-Cas9 system with higher cleavage activity.
Interestingly, the effect of base mismatches, insertions (DNA bulges) and deletions (RNA bulges) on cleavage activity differed between Nme and Spy systems, and our results suggest that the Nme system has a lower tolerance to base mismatches and DNA bulges (Figure 2b, Figure 2d), leading to a better specificity. Here again, this difference in specificity could be a result of different Cas9 proteins and how they interact with both the gRNA and the DNA target.
Due to the longer PAM requirement of the Nme Cas9 system, for a specific genomic site to target, there are fewer partially matched sites present in the genome. Specifically, the NNNNGATT motif is expected to occur approximately once every 128 bp whereas the NGG motif recognized by Spy Cas9 occurs once every 8 bp. If all recognizable PAM sequences are considered, the Spy PAM NRG occurs once every 4 bp, whereas the Nme PAM NNNNGHTT occurs once every 96 bp. This lower prevalence of the Nme PAM allowed us to analyze all genomic sites containing at least three mismatches to the target sequence for the degenerate PAM NNNNGNTT. Encouragingly, we observed no activity at any site for three different Nme gRNAs in both the 2-RNA and sgRNA formats. Conversely, for each of three corresponding Spy gRNAs targeting the same endogenous loci we observed detectable levels of off-target activity ranging from 0.25 to 13.87%. This provides clear evidence that the Nme CRISPR-Cas9 system has a better specificity than that of the Spy system. Although having 4 N's in the Nme Cas9 PAM might provide an increased opportunity for non-specific interactions between the gRNA and the PAM, this should not result in increased off-target activity, since any interaction between the gRNA and the NNNN region of the PAM would likely result in low sequence homology between the remaining sequence of gRNA and target DNA, thus preventing Cas9 induced nonspecific cutting. Reduced off-target activity has recently been reported for other Cas9 orthologs with longer PAM sequences. For example, the off-target profile of Sau Cas9 was determined in three different studies.27,40,41 In a direct comparison with Spy Cas9, Sau Cas9 was found to have less off-target effects.27,41 Both the Sth1 and Sth3 orthologs were also shown to have lower off-target activity than Spy Cas9.28
Although all three Spy Cas9 gRNAs tested in this study had off-target activities, the off-target activity levels varied significantly. In general, different Cas9 gRNAs have different numbers of potential off-target sites,17 suggesting that the design of Cas9 gRNAs can be optimized. In particular, we note that all active off-target sites found in this study have a conserved seed region.42,43,44 This region is necessary for DNA binding, and the seed region and PAM alone are sufficient for specific targeting by Spy Cas9.44 Therefore, potential off-target sites containing mismatches in the seed region are less likely to result in Cas9 cleavage activity, and this can be used to select gRNAs with optimal activity and specificity. In silico search tools for off-target site identification may be employed to prescreen gRNAs to identify those with a low propensity for off-target cleavage, i.e., gRNA with a low number of potential off-target sites and most (if not all) of these sites contain mismatches in the seed region.
Unlike Spy Cas9, the smaller size of the Nme Cas9 protein is within the packaging limit of AAV vectors which could be used for more efficient in vivo delivery for genome editing. Indeed, a recent study demonstrated that another small Cas9 ortholog, Sau Cas9 can be delivered efficiently in vivo using AAV.27 The Sau Cas9 system also has a longer PAM sequence which differs slightly with that of Nme (NNGRRT). We believe that the characterization of the Nme system conducted in this study helps add a highly specific Cas9 ortholog to the CRISPR-Cas9 toolbox for a wide range of biological and medical applications.
Materials and Methods
Construction of Nme CRISPR-Cas9 vector systems. The Nme Cas9 expressing plasmid pSimple-U6-tracr-U6-BsmBI-NLS-NMCas9-HA-NLS was a gift from Erik Sontheimer and James Thomson (Addgene plasmid #47868).30 The region downstream of the U6 promoter driving crRNA expression was removed using BsmBI. Oligonucleotides containing partial crRNA sequence and two BsmBI sites for gRNA cloning were introduced to create pCL101. This modified cloning strategy requires only the 20–24 nucleotide (nt) guide sequence with appropriate BsmBI cloning overhangs to generate new gRNA constructs. Both U6 promoter regions were removed using AatII and BglII and a G-Block (IDT, Coralville, IA) containing a U6 promoter and a single gRNA (sgRNA) cassette with two BsmBI sites was cloned to create the Nme sgRNA vector pCL103. Both the 2-RNA system (pCL101) and the sgRNA system (pCL103) contain identical BsmBI overhang sequences facilitating simple cloning of new gRNA sequences into both systems using identical oligonucleotides.
CRISPR-Cas9 plasmid assembly. For Nme gRNAs, DNA oligonucleotides containing a G followed by a 19 to 23-nt guide sequence (Supplementary Table S1) were kinased, annealed, and ligated into both pCL101 and pCL103 plasmids. The annealed oligonucleotides have 4-bp overhangs that are compatible with the ends of BsmBI-digested pCL101 and pCL103 plasmids. For Spy gRNAs, DNA oligos containing a G followed by a 19-nt guide sequence were similarly kinased, annealed and ligated into BbsI digested pX330 (gift from Feng Zhang, Addgene plasmid #42230).42 Constructed plasmids were sequenced to confirm the guide strand region using the primer CRISPR_Seq (5′-CGATACAAGGCTGTTAGAGAGATAATTGG-3′) for Spy and Nme sgRNA constructs or Nme2-RNA_Seq (5′-GGCGACACGGAAATGTTGAATACTC-3′) for Nme 2-RNA constructs.
T7 endonuclease I (T7EI) mutation detection assay for measuring endogenous gene modification rates. The cleavage activity of RNA-guided Cas9 at endogenous loci was quantified based on the mutation rates resulting from the imperfect repair of double-stranded breaks by non-homologous end joining. HEK293T cells were seeded 24 hours prior to transfection in 24-well plates at a density of 80,000 cells per well and cultured in Dulbecco's Modified Eagle Medium media supplemented with 10% fetal bovine serum and 2 mmol/l fresh L-glutamine. Cells were transfected with 1,000 ng of Cas9_sgRNA plasmid using 2 µl of Lipofectamine 2000 (ThermoFisher Scientific, Grand Island, NY), according to manufacturer's instructions. Genomic DNA was harvested after 3 days using QuickExtract DNA extraction solution (Epicentre, Madison, WI). T7EI mutation detection assays were performed, as described previously45 and the digestions separated on 2% agarose gels. The cleavage bands were quantified using ImageJ. The percentage of gene modification was determined using the formula, % non-homologous end joining = 100× (1 – (1 – fraction cleaved)0.5), as described.46 All polymerase chain reaction (PCR) reactions were performed using AccuPrime Taq DNA Polymerase High Fidelity (ThermoFisher Scientific, Grand Island, NY) following manufacturer's instructions for 40 cycles (94 °C, 30 seconds; 60 °C, 30 seconds; 68 °C, 45 seconds) in a 50 µl reaction containing 1 µl of the cell lysate, and 1 µl of each 10 µmol/l target region amplification primer (Supplementary Table S1).
Identification of off-target sites. Potential off-target sites in the human genome (hg19) were identified and ranked using the recently developed bioinformatics program COSMID,34 allowing up to three base mismatches without insertions or deletions and two base mismatches with either an inserted or deleted base (bulge). The top ranked sites were further investigated. Primers used for target region amplification were designed by COSMID (Supplementary Table S3).
Luciferase-based single-strand annealing assay. For SSA assays, 10,000 cells were seeded per well in 96-well plates 24 hours prior to transfection. Each transfection contained 100 ng of DNA delivered using 0.4 μl of Lipofectamine 2000. Typically, transfections consisted of 25 ng of SSA target plasmid, 50 ng of CRISPR/Cas9 plasmid, 5 ng of Renilla luciferase plasmid and 20 ng of pUC18. Forty-eight hours post-transfection, luciferase expression was quantified using the Dual-Glo luciferase assay system (Promega, Madison, WI) and a Tecan (Mannedorf, Switzerland) Safire2 microplate reader. All SSA assays were carried out in triplicate.
Deep sequencing to quantify CRISPR-Cas9 activity at genomic loci. Genomic DNA from mock and nuclease-treated cells was amplified via PCR using locus-specific primers that contained adapter sequences for a second round of PCR (Supplementary Table S3). PCR reactions for each locus were performed independently for eight touchdown cycles in which annealing temperature was lowered by 1 °C each cycle from 65 to 57 °C, followed by 35 cycles with annealing temperature at 57 °C. PCR products were purified using Agencourt AmPure XP (Beckman Coulter) according to manufacturer's protocol. A second round of PCR amplification was performed for each individual amplicon using primers containing the adapter sequences from the first PCR, P5 and P7 adapters, and sample barcodes in the reverse primers. PCR products were purified as for first PCR, pooled in an equimolar ratio, and subjected to 2 × 250 paired-end sequencing on the Illumina MiSeq platform.
Paired-end reads from MiSeq were filtered by an average Phred quality (Qscore) greater than 20 and merged into a longer single read from each pair with a minimum overlap of 30 nucleotides using Fast Length Adjustment of SHort reads. Alignments to reference sequences were performed using Burrows-Wheeler Aligner for each barcode and percentage of insertions and deletions containing bases within a ±5-bp window of the predicted cut sites were quantified. Error bounds for indel percentages are Wilson score intervals calculated using binom package for R statistical software (version 3.0.3) with a confidence level of 95%. To determine if each off-target indel percentage from a CRISPR-treated sample is significant compared to a mock-treated sample, a two-tailed P value was calculated using Fisher's exact test.
SUPPLEMENTARY MATERIAL Table S1. Activity of Nm gRNAs at different genomic loci. Table S2. Luciferase SSA assay data. Table S3. Activity of Sp and Nm CRISPR-Cas9 at predicted off-target loci. Figure S1. Effect of sequence on Nm Cas9 activity. Figure S2. Alternative modeling of DNA bulges as shorter gRNAs with alternate PAMs. Figure S3. RepresentativeT7EI assay gel of CRISPR-Cas9 activity with gRNA lengths of 20 to 24 nt. Figure S4. Indel profiles of different CRISPR-Cas9 systems.
Acknowledgments
The authors thank Erik Sontheimer and James Thomson for providing the Nme Cas9 expressing plasmid pSimple-U6-tracr-U6-BsmBI-NLS-NmCas9-HA-NLS. This study was supported by National Institutes of Health (Nanomedicine Development Center Award; grant number (PN2EY018244 to G.B.). Funding for open access charge: National Institutes of Health.
Supplementary Material
Activity of Nm gRNAs at different genomic loci.
Activity of Sp and Nm CRISPR-Cas9 at predicted off-target loci.
References
- Barrangou, R, Fremaux, C, Deveau, H, Richards, M, Boyaval, P, Moineau, S et al. (2007). CRISPR provides acquired resistance against viruses in prokaryotes. Science 315: 1709–1712. [DOI] [PubMed] [Google Scholar]
- Cho, SW, Kim, S, Kim, JM and Kim, JS (2013). Targeted genome engineering in human cells with the Cas9 RNA-guided endonuclease. Nat Biotechnol 31: 230–232. [DOI] [PubMed] [Google Scholar]
- DiCarlo, JE, Norville, JE, Mali, P, Rios, X, Aach, J and Church, GM (2013). Genome engineering in Saccharomyces cerevisiae using CRISPR-Cas systems. Nucleic Acids Res 41: 4336–4343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedland, AE, Tzur, YB, Esvelt, KM, Colaiácovo, MP, Church, GM and Calarco, JA (2013). Heritable genome editing in C. elegans via a CRISPR-Cas9 system. Nat Methods 10: 741–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang, WY, Fu, Y, Reyon, D, Maeder, ML, Tsai, SQ, Sander, JD et al. (2013). Efficient genome editing in zebrafish using a CRISPR-Cas system. Nat Biotechnol 31: 227–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mali, P, Yang, L, Esvelt, KM, Aach, J, Guell, M, DiCarlo, JE et al. (2013). RNA-guided human genome engineering via Cas9. Science 339: 823–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen, B, Zhang, J, Wu, H, Wang, J, Ma, K, Li, Z et al. (2013). Generation of gene-modified mice via Cas9/RNA-mediated gene targeting. Cell Res 23: 720–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, H, Yang, H, Shivalila, CS, Dawlaty, MM, Cheng, AW, Zhang, F et al. (2013). One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell 153: 910–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nekrasov, V, Staskawicz, B, Weigel, D, Jones, JD and Kamoun, S (2013). Targeted mutagenesis in the model plant Nicotiana benthamiana using Cas9 RNA-guided endonuclease. Nat Biotechnol 31: 691–693. [DOI] [PubMed] [Google Scholar]
- Jinek, M, Chylinski, K, Fonfara, I, Hauer, M, Doudna, JA and Charpentier, E (2012). A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337: 816–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urnov, FD, Miller, JC, Lee, YL, Beausejour, CM, Rock, JM, Augustus, S et al. (2005). Highly efficient endogenous human gene correction using designed zinc-finger nucleases. Nature 435: 646–651. [DOI] [PubMed] [Google Scholar]
- Christian, M, Cermak, T, Doyle, EL, Schmidt, C, Zhang, F, Hummel, A et al. (2010). Targeting DNA double-strand breaks with TAL effector nucleases. Genetics 186: 757–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu, Y, Foden, JA, Khayter, C, Maeder, ML, Reyon, D, Joung, JK et al. (2013). High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat Biotechnol 31: 822–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu, PD, Scott, DA, Weinstein, JA, Ran, FA, Konermann, S, Agarwala, V et al. (2013). DNA targeting specificity of RNA-guided Cas9 nucleases. Nat Biotechnol 31: 827–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cradick, TJ, Fine, EJ, Antico, CJ and Bao, G (2013). CRISPR/Cas9 systems targeting β-globin and CCR5 genes have substantial off-target activity. Nucleic Acids Res 41: 9584–9592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, Y, Cradick, TJ, Brown, MT, Deshmukh, H, Ranjan, P, Sarode, N et al. (2014). CRISPR/Cas9 systems have off-target activity with insertions or deletions between target DNA and guide RNA sequences. Nucleic Acids Res 42: 7473–7485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai, SQ, Zheng, Z, Nguyen, NT, Liebers, M, Topkar, VV, Thapar, V et al. (2015). GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nat Biotechnol 33: 187–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frock, RL, Hu, J, Meyers, RM, Ho, YJ, Kii, E and Alt, FW (2015). Genome-wide detection of DNA double-stranded breaks induced by engineered nucleases. Nat Biotechnol 33: 179–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, X, Wang, Y, Wu, X, Wang, J, Wang, Y, Qiu, Z et al. (2015). Unbiased detection of off-target cleavage by CRISPR-Cas9 and TALENs using integrase-defective lentiviral vectors. Nat Biotechnol 33: 175–178. [DOI] [PubMed] [Google Scholar]
- Kim, D, Bae, S, Park, J, Kim, E, Kim, S, Yu, HR et al. (2015). Digenome-seq: genome-wide profiling of CRISPR-Cas9 off-target effects in human cells. Nat Methods 12: 237–43, 1 p following 243. [DOI] [PubMed] [Google Scholar]
- Mali, P, Aach, J, Stranges, PB, Esvelt, KM, Moosburner, M, Kosuri, S et al. (2013). CAS9 transcriptional activators for target specificity screening and paired nickases for cooperative genome engineering. Nat Biotechnol 31: 833–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran, FA, Hsu, PD, Lin, CY, Gootenberg, JS, Konermann, S, Trevino, AE et al. (2013). Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell 154: 1380–1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guilinger, JP, Thompson, DB and Liu, DR (2014). Fusion of catalytically inactive Cas9 to FokI nuclease improves the specificity of genome modification. Nat Biotechnol 32: 577–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai, SQ, Wyvekens, N, Khayter, C, Foden, JA, Thapar, V, Reyon, D et al. (2014). Dimeric CRISPR RNA-guided FokI nucleases for highly specific genome editing. Nat Biotechnol 32: 569–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aouida, M, Eid, A, Ali, Z, Cradick, TJ, Lee, CM, Deshmukh, H et al. (2015). Efficient fdCas9 synthetic endonuclease with improved specificity for precise genome engineering. PLoS One 10: e0133373 (doi: 10.1371/journal.pone.0133373). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fonfara, I, Le Rhun, A, Chylinski, K, Makarova, KS, Lécrivain, AL, Bzdrenga, J et al. (2014). Phylogeny of Cas9 determines functional exchangeability of dual-RNA and Cas9 among orthologous type II CRISPR-Cas systems. Nucleic Acids Res 42: 2577–2590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran, FA, Cong, L, Yan, WX, Scott, DA, Gootenberg, JS, Kriz, AJ et al. (2015). In vivo genome editing using Staphylococcus aureus Cas9. Nature 520: 186–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller, M, Lee, CM, Gasiunas, G, Davis, TH, Cradick, TJ, Siksnys, V et al. (2015). Streptococcus thermophilus CRISPR-Cas9 systems enable specific editing of the human genome. Mol Ther (epub ahead of print). doi: 10.1038/mt.2015.218. [DOI] [PMC free article] [PubMed]
- Esvelt, KM, Mali, P, Braff, JL, Moosburner, M, Yaung, SJ and Church, GM (2013). Orthogonal Cas9 proteins for RNA-guided gene regulation and editing. Nat Methods 10: 1116–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou, Z, Zhang, Y, Propson, NE, Howden, SE, Chu, LF, Sontheimer, EJ et al. (2013). Efficient genome engineering in human pluripotent stem cells using Cas9 from Neisseria meningitidis. Proc Natl Acad Sci USA 110: 15644–15649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu, Y, Sander, JD, Reyon, D, Cascio, VM and Joung, JK (2014). Improving CRISPR-Cas nuclease specificity using truncated guide RNAs. Nat Biotechnol 32: 279–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Y, Heidrich, N, Ampattu, BJ, Gunderson, CW, Seifert, HS, Schoen, C et al. (2013). Processing-independent CRISPR RNAs limit natural transformation in Neisseria meningitidis. Mol Cell 50: 488–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cradick, TJ, Antico, CJ and Bao, G (2014). High-throughput cellular screening of engineered nuclease activity using the single-strand annealing assay and luciferase reporter. Methods Mol Biol 1114: 339–352. [DOI] [PubMed] [Google Scholar]
- Cradick, TJ, Qiu, P, Lee, CM, Fine, EJ and Bao, G (2014). COSMID: A Web-based Tool for Identifying and Validating CRISPR/Cas Off-target Sites. Mol Ther Nucleic Acids 3: e214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bae, S, Kweon, J, Kim, HS and Kim, JS (2014). Microhomology-based choice of Cas9 nuclease target sites. Nat Methods 11: 705–706. [DOI] [PubMed] [Google Scholar]
- Ma, E, Harrington, LB, O'Connell, MR, Zhou, K and Doudna, JA (2015). Single-Stranded DNA Cleavage by Divergent CRISPR-Cas9 Enzymes. Mol Cell 60: 398–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Y, Rajan, R, Seifert, HS, Mondragón, A and Sontheimer, EJ (2015). DNase H Activity of Neisseria meningitidis Cas9. Mol Cell 60: 242–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimasu, H, Ran, FA, Hsu, PD, Konermann, S, Shehata, SI, Dohmae, N et al. (2014). Crystal structure of Cas9 in complex with guide RNA and target DNA. Cell 156: 935–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimasu, H, Cong, L, Yan, WX, Ran, FA, Zetsche, B, Li, Y et al. (2015). Crystal Structure of Staphylococcus aureus Cas9. Cell 162: 1113–1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinstiver, BP, Prew, MS, Tsai, SQ, Nguyen, NT, Topkar, VV, Zheng, Z et al. (2015). Broadening the targeting range of Staphylococcus aureus CRISPR-Cas9 by modifying PAM recognition. Nat Biotechnol 33: 1293–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedland, AE, Baral, R, Singhal, P, Loveluck, K, Shen, S, Sanchez, M et al. (2015). Characterization of Staphylococcus aureus Cas9: a smaller Cas9 for all-in-one adeno-associated virus delivery and paired nickase applications. Genome Biol 16: 257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong, L, Ran, FA, Cox, D, Lin, S, Barretto, R, Habib, N et al. (2013). Multiplex genome engineering using CRISPR/Cas systems. Science 339: 819–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang, W, Bikard, D, Cox, D, Zhang, F and Marraffini, LA (2013). RNA-guided editing of bacterial genomes using CRISPR-Cas systems. Nat Biotechnol 31: 233–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, X, Scott, DA, Kriz, AJ, Chiu, AC, Hsu, PD, Dadon, DB et al. (2014). Genome-wide binding of the CRISPR endonuclease Cas9 in mammalian cells. Nat Biotechnol 32: 670–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyon, D, Tsai, SQ, Khayter, C, Foden, JA, Sander, JD and Joung, JK (2012). FLASH assembly of TALENs for high-throughput genome editing. Nat Biotechnol 30: 460–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guschin, DY, Waite, AJ, Katibah, GE, Miller, JC, Holmes, MC and Rebar, EJ (2010). A rapid and general assay for monitoring endogenous gene modification. Methods Mol Biol 649: 247–256. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Activity of Nm gRNAs at different genomic loci.
Activity of Sp and Nm CRISPR-Cas9 at predicted off-target loci.