

Abstract
Background
ASP3026 is a second-generation anaplastic lymphoma kinase (ALK) inhibitor that has potent in vitro activity against crizotinib-resistant ALK-positive tumors. This open-label, multicenter, first-in-human phase I study (NCT01284192) assessed the safety, pharmacokinetic profile, and antitumor activity of ASP3026.
Methods
Advanced solid tumor patients received oral ASP3026 in 3 + 3 dose-escalation cohorts at doses of 25–800 mg once daily in 28-day cycles. The endpoints were to identify the maximum tolerated dose (MTD), the recommended phase II dose (RP2D), and the pharmacokinetic profile of ASP3026. A phase Ib expansion cohort enrolled patients with metastatic, crizotinib-resistant ALK-positive solid tumors at the RP2D, and response was evaluated by RECIST 1.1.
Results
The dose-escalation cohort enrolled 33 patients, including three crizotinib-resistant, ALK-positive patients, and the dose-expansion cohort enrolled another 13 crizotinib-resistant, ALK-positive non-small cell lung cancer (NSCLC) patients. ASP3026 demonstrated both linear pharmacokinetics and dose-proportional exposure for area under the plasma concentration–time curve and maximum concentration observed with a median terminal half-life of 35 h, supporting the daily dosing. Grade 3 rash and elevated transaminase concentrations were dose-limiting toxicities observed at 800 mg; hence, 525 mg daily was the MTD and RP2D. The most common treatment-related adverse events were nausea (38 %), fatigue (35 %), and vomiting (35 %). Among the 16 patients with crizotinib-resistant ALK-positive tumors (15 NSCLC, 1 neuroblastoma), eight patients achieved partial response (overall response rate 50 %; 95 % confidence interval 25–75 %) and seven patients (44 %) achieved stable disease.
Conclusions
ASP3026 was well tolerated and had therapeutic activity in patients with crizotinib-resistant ALK-positive advanced tumors.
Trial registration
ClinTrials.gov: NCT01284192
Keywords: ASP3026, Neoplasms, ALK inhibitor, Phase I, Pharmacokinetics
Background
In recent years, aberrant expression of anaplastic lymphoma kinase (ALK) tyrosine kinase receptor has emerged as a relevant biomarker and therapeutic target for a number of solid tumors [1, 2]. Different types of alterations in the ALK gene have been implicated in human cancer tumorigenesis, and different tumor types have different structural alterations in the ALK gene [3, 4]. In non-small cell lung cancer (NSCLC), echinoderm microtubule-associated protein-like 4 (EML-4) and kinesin family member 5B (KIF5B) account for the majority of ALK gene rearrangement lung cancer [5]. The presence of ALK gene rearrangement defines ~3–13 % of NSCLC [6–10] that are highly sensitive to the first-generation ALK tyrosine kinase inhibitor (TKI), crizotinib. Crizotinib (250 mg twice daily) has high efficacy in patients with NSCLC harboring this oncogenic kinase, with overall response rates of >50 % [11–13]. Crizotinib has been established as the standard first-line treatment for patients with advanced ALK-positive NSCLC [13]. The current National Comprehensive Cancer Network (NCCN) guideline recommend crizotinib use in patients with advanced NSCLC harboring ALK gene rearrangement [14]. However, almost all patients develop resistance, typically within 10 months [11, 13, 15, 16].
The most common molecular mechanisms of resistance include amplification of the ALK fusion gene, development of resistance mutations, and activation of alternative or bypass signaling pathways or progression in the CNS [17]. One strategy to overcome crizotinib resistance is to develop potent small molecule TKIs for ALK-rearranged genes and/or specifically target the common resistant mutations, such as gatekeeper mutation L1196M [17].
ASP3026 is a selective, ATP-competitive, second-generation ALK TKI that was identified through a medicinal chemistry campaign designed to obtain compounds with a better pharmacologic profile compared with crizotinib. The kinase selectivity of ASP3026 was evaluated and compared with that of crizotinib against a panel of 86 tyrosine kinases [18]. ASP3026 at 1 μmol/L inhibited 11 tyrosine kinases by more than 50 %, with the highest selectivity for ALK, ROS1, and ACK kinases, showing that the kinase selectivity of ASP3026 differed from crizotinib. ASP3026 was more selective for FRK, YES, ACK, TNK1, and EGFR (L858R), whereas crizotinib had higher selectivity for MET, RON, LCK, JAK2, MUSK, TRKs, TYRO3, AXL, MER, and EPHs [18]. ASP3026 fits within the ATP-binding pocket of both wild-type and L1196M ALK kinase domains and inhibits their kinase activities with IC50 values of 10 and 32 nmol/L, respectively. By contrast, crizotinib fits within the ATP-binding pocket of wild-type ALK kinase domain but not the L1196M ALK kinase domain. Thus, crizotinib displays tenfold weaker activity for the mutated EML4-ALK compared with the wild-type ALK gene [18]. In mice bearing subcutaneous and intracranial xenograft tumors, ASP3026 has potent antitumor activity against both wild-type ALK and EML4-ALK L1196M xenograft tumors compared with crizotinib [18]. ASP3026 also has a higher tissue-to-plasma ratio compared with crizotinib, which could translate into a wide therapeutic margin between efficacious and toxic doses [18]. Preclinical data indicated that ASP3026 may have potential therapeutic effects for patients with crizotinib-resistant ALK-positive NSCLC and potentially for patients with other cancer types of ALK-driven tumors.
We conducted this phase I dose-escalation trial to evaluate the safety and pharmacokinetics (PKs) of ASP3026 as an oral single agent in patients with advanced solid malignancies. A planned phase Ib dose-expansion cohort at the recommended phase II dose (RP2D) was conducted to evaluate the tumor response of ASP3026 in patients with metastatic ALK-positive NSCLC who progressed on crizotinib.
Methods
Clinical study summary
The clinical trial design was a phase I, multicenter, open-label, dose-escalation and dose-expansion study (NCT01284192) of ASP3026 in patients with advanced malignancies. The study was conducted in accordance with all applicable regulatory requirements and had institutional review board approval prior to study initiation at participating institutions. Written informed consent was obtained from all patients prior to the initiation of any study-specific procedures.
Patient population
Adult patients (≥18 years of age) with histologically or cytologically confirmed diagnosis of relapsed/refractory tumor were included in the dose-escalation study. Patients had to have Eastern Cooperative Oncology Group (ECOG) performance status ≤2, adequate life expectancy >12 weeks, be non-child bearing (or be using protocol-specified contraceptive measures), and be able to swallow oral medications. Additional criteria for patients with ALK abnormalities in the dose-escalation phase included patients to be positive for ALK abnormalities (by any molecular method including, but not limited to, polymerase chain reaction, direct sequencing, in situ hybridization, or be previously confirmed by fluorescence in situ hybridization), to not have symptomatic brain metastases, to not to be taking >5 mg prednisone daily, or to not require hepatic enzyme-inducing anti-seizure medication.
Inclusion into the dose-expansion cohort required patients to have ALK-positive tumors that had progressed on crizotinib. Key exclusion criteria for the dose-escalation cohort included patients with leptomeningeal involvement (as assessed through medical history review or through physical examination), inadequate bone marrow, renal and/or hepatic function, and a known history of long QT syndrome. Brain magnetic resonance imaging (MRI) was carried out for all dose-expansion patients and for all dose-escalation patients known to have brain metastases at screening. For patients with baseline brain metastases, MRI was performed at the end of cycle 2 and then every 2 cycles thereafter. Only ALK-positive subjects were eligible for the dose-expansion cohort; ROS1 was not included.
Study design and treatments
The study was divided into two parts: dose escalation and dose expansion. Dose escalation used a traditional 3 + 3 dose-escalation design. Cycles of treatment were every 28 days with continuous dosing of ASP3026. Patients were followed-up for safety assessments 30 days (±7 days) after the last ASP3026 dose. The starting dose for ASP3026 was 25 mg administered orally once daily. Dose escalation proceeded to the next seven cohorts of 50, 75, 125, 200, 325, 525, and 800 mg. The first patient in each dose-escalation cohort was evaluated for dose-limiting toxicities (DLTs) in cycle 1, day 4. If no DLTs were reported by the investigator, the remaining patients in the cohort were enrolled. Therapeutic concentrations were projected to be reached above 325 mg. The protocol allowed that and subsequent doses to be expanded to enroll an additional three patients who were known to have tumors tested positive for ALK abnormalities once safety was established in the first three subjects for that cohort. To further address the antitumor effects and the safety of ASP3026 in patients who progressed on crizotinib, the dose-expansion part of the study focused on crizotinib-refractory ALK-positive NSCLC patients.
The primary objectives of the study were to determine the safety and tolerability of ASP3026 in patients with advanced malignancies and to determine the maximum tolerated dose (MTD) and RP2D for ASP3026. The MTD was defined as the highest dose of ASP3026 at which <33 % of patients experienced a DLT during cycle 1. Secondary objectives were to determine the PKs and antitumor activity of ASP3026.
Safety/tolerability assessments
The safety and tolerability of ASP3026 were assessed by adverse events (AEs) (graded based on NCI-CTCAE v4.03), laboratory tests, vital signs, electrocardiograms, and clinical observations.
Dose-limiting toxicity criteria were grade 4 neutropenia lasting ≥7 days, febrile neutropenia, grade 3 thrombocytopenia, grade 3 non-hematologic toxicity, except for nausea/vomiting or diarrhea (nausea/vomiting or diarrhea was considered a DLT in patients who had grade 3 toxicity for ≥3 days or grade 4 toxicity of any duration), and any study drug-related toxicity resulting in treatment delay >2 weeks or discontinuation of treatment at the assigned dose level.
Pharmacokinetic assessments
Plasma PKs samples were taken on days 1, 2, 8, 15, 22, and 28 (±2 days) of cycle 1. On day 1, the following PK parameters were assessed for ASP3026: area under the plasma concentration–time curve (AUC24), AUClast, maximum concentration observed (Cmax), and time of maximum concentration observed (tmax). On day 28, AUCtau, Cmax, Ctrough, tmax, CL/F, Vz/F, and t½, calculated based on the ratio of accumulation for AUC (day 28 AUCtau/day 1 AUC24) (Rac [AUC]), were assessed. Cumulative effects on steady-state ASP3026 plasma levels were assessed by comparing Ctrough on day 28 divided by Ctrough on days 8, 15, and 22.
Antitumor assessments
Solid tumor assessment was based on Response Evaluation Criteria in Solid Tumors (RECIST) v1.1. For target lesions, complete response (CR) was defined as disappearance of all target lesions and pathologic lymph nodes with a reduction in short axis to <10 mm. Partial response (PR) was defined as a ≥30 % decrease in the sum of diameters of the target lesions, taking as reference the baseline sum of diameters. Progressive disease (PD) was defined as a ≥20 % increase in the sum of diameters from the smallest sum on the study and the sum of diameters to be ≥5 mm from the smallest sum on study. Stable disease (SD) was defined as neither PR nor PD.
For patients in the dose-expansion cohort with evaluable tumor diameter, the best tumor reduction was calculated as the decrease from baseline in the sum of the target lesions.
Statistical analyses
For continuous variables, descriptive statistics included the number of patients, mean (standard deviation), median, and range. For antitumor assessments, the number and percentage (95 % CI) of patients with CR, PR, SD, and PD were summarized. The overall best tumor response was also summarized. For the objective response rates, the exact confidence interval of response was calculated only for the dose-expansion cohort using the Clopper–Pearson method. For continuous PK parameters, the coefficient of variation was calculated. For Cmax and AUC, geometric mean was calculated. All data processing and analyses were performed using SAS® Version 9.1.3 or higher. The safety analysis set was defined as patients who received at least one ASP3026 dose. The PK analysis set was defined as patients who received at least one dose of ASP3026 and provided the values of drug concentrations for at least one time point.
Results
Baseline patient characteristics
Enrollment began on 11 January 2011 and closed on 28 June 2013. The data cut-off date was 19 February 2014. Forty-six patients (33 patients in the dose-escalation cohort; 13 in the dose-expansion cohort) were included in the current analyses.
Table 1 summarizes patients’ demographics. For all study patients, 22 (48 %) were men and the median (range) age was 61 (19–77) years. In the dose-escalation cohort, the most common primary tumor types were breast and lung adenocarcinoma (both n = 4), leiomyosarcoma and adenocarcinoma (unspecified primary) (both n = 3), and bile duct, colon, and ovarian cancer (each n = 2). The median (range) duration of prior chemotherapy/targeted therapy was 46 (1–171) days. Patients were not mandatorily screened for brain metastases at baseline in the dose-escalation cohort.
Table 1.
Patient demographics and baseline disease characteristics
| Parameter/statistics | Dose-escalation cohort | Dose-expansion cohort (ALK-positive) | Both cohorts |
|---|---|---|---|
| Total (n = 30)a | 525 mg (n = 16)b | All patients (n = 46) | |
| Sex, n (%) | |||
| Male | 14 (47) | 8 (50) | 22 (48) |
| Female | 16 (53) | 8 (50) | 24 (52) |
| Race, n (%) | |||
| White | 25 (83) | 1 (6) | 26 (57) |
| Black or African American | 5 (17) | 14 (88) | 19 (41) |
| Asian | 0 | 1 (6) | 1 (2) |
| Age (years) | |||
| Mean (standard deviation) | 61.6 (9.6) | 51.1 (11.8) | 57.9 (11.5) |
| Median (range) | 64 (44–77) | 51 (19–71) | 61 (19–77) |
| Weight (kg), mean (standard deviation) | 80.3 (20.4) | 75.0 (11.6) | 78.5 (17.9) |
| ECOG performance status, n (%) | |||
| Grade 0 | 6 (20) | 9 (56) | 15 (33) |
| Grade 1 | 19 (63) | 7 (44) | 26 (57) |
| Grade 2 | 5 (17) | 0 | 5 (11) |
| Primary tumor type, n (%) | |||
| Lung adenocarcinoma | 4 (13) | 7 (44) | 11 (24) |
| NSCLC | 0 | 6 (38) | 6 (13) |
| Malignant lung neoplasm | 0 | 2 (13) | 2 (4) |
| Breast | 4 (13) | 0 | 4 (9) |
| Adenocarcinoma (unspecified primary) | 3 (10) | 0 | 3 (7) |
| Leiomyosarcoma | 3 (10) | 0 | 3 (7) |
| Colon | 2 (7) | 0 | 2 (4) |
| Bile duct | 2 (7) | 0 | 2 (4) |
| Ovarian | 2 (7) | 0 | 2 (4) |
| Other | 10 (33) | 1 (6) | 11 (24) |
| Brain metastases history, n (%) | – | 9 (56) | – |
| Prior radiation therapy, n (%) | 18 (60) | 14 (88) | 32 (70) |
ALK anaplastic lymphoma kinase, ECOG Eastern Cooperative Oncology Group, NSCLC non-small cell lung carcinoma, UNK unknown
aExcludes 3 ALK-positive patients
bIncludes 3 ALK-positive patients from the dose-escalation cohort
In the 16 ALK-positive patients (including three ALK-positive patients from the dose-escalation cohort and 13 from the dose-expansion cohort), 15 NSCLC patients had ALK rearrangement and one neuroblastoma patient had an oncogenic ALK gene mutation F1174L [19–21]. Eight patients (50 %) were male, and the median (range) age of the patients was 51 (19–71) years (Table 1); nine patients (56 %) had brain metastases. All ALK-positive patients had progressed on crizotinib.
Discontinuations, dose-escalation, and dose-limiting toxicities
All 30 enrolled patients (excluding three ALK-positive patients) discontinued treatment in the dose-escalation cohort (28 due to PD and two for other reasons [see “Safety/tolerability assessments” section]). At the time of data cut-off, 12 (75 %) discontinued treatment, 11 due to PD and one patient died in the dose-expansion cohort.
In the dose-escalation cohort, two patients receiving 800 mg ASP3026 experienced protocol-defined DLTs, probably related to the study drug, of increased aspartate aminotransferase and drug eruption (maculopapular rash on trunk, lower extremities, face, arm, and back), both grade 3. MTD was determined to be 525 mg, which was subsequently administered in the dose-expansion cohort.
Adverse events
Within the dose-escalation and dose-expansion cohorts, 29 patients (97 %) and 16 patients (100 %) experienced ≥1 AE, respectively. Drug-related AEs were reported by 20 patients (67 %) in the dose-escalation cohort and 15 patients (94 %) in the dose-expansion cohort (Table 2).
Table 2.
Summary of AEs possibly or probably related to study drug occurring in ≥2 patients in either cohort
| Dose-escalation cohort | Dose-expansion cohort (ALK-positive) | Both cohorts | |
|---|---|---|---|
| Total (n = 30)a | 525 mg (n = 16)b | Total (n = 46) | |
| Overall | 20 (67) | 15 (94) | 35 (76) |
| Nausea | 7 (23) | 10 (63) | 17 (37) |
| Vomiting | 6 (20) | 10 (63) | 16 (35) |
| Fatigue | 13 (43) | 3 (19) | 16 (35) |
| Decreased appetite | 1 (3) | 4 (25) | 5 (11) |
| Diarrhea | 3 (10) | 2 (13) | 5 (11) |
| Rash | 0 | 3 (19) | 3 (7) |
| Headache | 1 (33) | 2 (13) | 3 (7) |
| Constipation | 2 (7) | 1 (6) | 3 (7) |
| Peripheral neuropathy | 0 | 2 (13) | 2 (4) |
| Cataract nuclear | 0 | 2 (13) | 2 (4) |
| Periorbital edema | 0 | 2 (13) | 2 (4) |
| Blurred vision | 0 | 2 (13) | 2 (4) |
| Anemia | 2 (7) | 0 | 2 (4) |
| Increased blood creatinine | 2 (7) | 0 | 2 (4) |
ALK anaplastic lymphoma kinase
aExcludes 3 ALK-positive patients
bIncludes 3 ALK-positive patients from the dose-escalation cohort
Serious AEs (SAE) were reported by seven patients (23 %) in the dose-escalation cohort; one SAE was possibly related to the study drug (international normalized ratio [INR] increased that resolved after drug withdrawal). Serious AEs were reported by five patients (31 %) in the dose-expansion cohort; one SAE was possibly related to the study drug (abnormal liver function test that did not resolve after drug withdrawal) and another probably related to the study drug (keratoacanthoma that resolved after drug withdrawal). There were three deaths on study, all of which were due to PD and not related to the study drug.
Adverse events leading to drug discontinuation occurred in two patients in the dose-escalation cohort (one patient experienced a pulmonary embolism [not study drug-related] and an increased INR [possibly study drug-related]); one patient experienced increased aspartate aminotransferase that was probably related to the study drug. AEs leading to drug discontinuation occurred in one patient in the dose-expansion cohort (an abnormal liver function test [possibly study drug-related] and bilateral pleural effusion, constrictive pericarditis, and disease progression [all not study drug-related]).
Laboratory parameters and electrocardiograms
There were no meaningful changes in any clinical laboratory parameters or vital signs over time or any overall shifts from baseline in hematology or laboratory tests.
There was an increase in QTcF values in some patients. Mean (standard deviation) maximal increase of 47.0 (10.7) ms was reported at the 325-mg dose; at the MTD (525 mg), the mean (standard deviation) maximal increase was 25.8 (16.8) ms in the dose-expansion cohort.
ASP3026 pharmacokinetics
Mean ASP3026 plasma concentrations by dose are shown in Fig. 1, and PK parameters are provided in Tables 3 and 4. ASP3026 had rapid oral absorption, with a tmax of approximately 3 h. The mean (standard deviation) accumulation half-life was 25 (37) h (median half-life, 35 h (range, 22–85 h). Steady-state plasma concentrations were reached by day 8 for both the dose-escalation and dose-expansion cohorts. After multiple dosing (day 28), ASP3026 showed both linear and dose-proportional exposure (Cmax and AUClast) over the 25- to 800-mg dose range; slope estimates (95 % CI of slope) comparing dose-normalized exposure to dose were 0.170 (−0.018, 0.358) and 0.090 (−0.097, 0.277) for Cmax and AUClast, respectively.
Fig. 1.
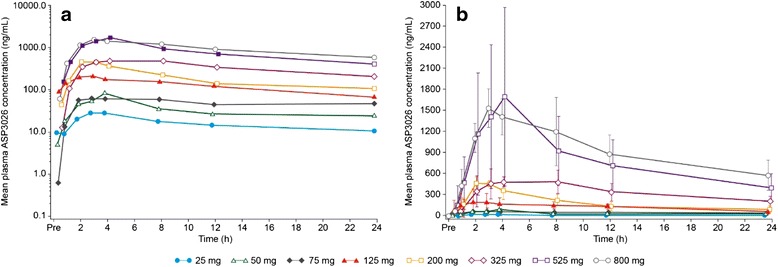
Mean plasma concentration of ASP3026, cycle 1, day 1. a Semi-log plot. b Linear plot. For patient numbers at each dose, refer to Tables 3 and 4
Table 3.
Pharmacokinetic parameters for ASP3026 (cycle 1, day 1)
| Dose (mg; once daily) | Number | C max (ng/mL) | t max (h)a | AUC24 (ng h/mL) |
|---|---|---|---|---|
| Dose-escalation cohort | ||||
| 25 | 4 | 32.0 (10.2) | 3.0 (0.5–4.0) | 378 (104) |
| 50 | 3 | 99.7 (56.6) | 3.0 (2.0–4.0) | 846 (225) |
| 75 | 3 | 87.6 (40.3) | 8.2 (2.0–24.2) | 1155 (442) |
| 125 | 4 | 261.7 (131.1) | 5.0 (1.0–8.0) | 3000 (860) |
| 200 | 4 | 490.5 (68.6) | 2.5 (2.0–3.0) | 4585 (1310) |
| 325 | 3 | 586.2 (109.7) | 3.0 (3.0–8.0) | 7950 (1957) |
| 525 | 6 | 1750 (1279) | 4.0 (3.0–4.2) | 18,543 (10,812) |
| 800 | 3 | 1633 (252.7) | 3.0 (2.0–8.0) | 21,796 (5990) |
| Dose-expansion cohort | ||||
| 525 | 16 | 961.0 (563.8) | 3.1 (2.0–8.0) | 11,746 (9063) |
Calculated accumulation ratio (AUCd28, tau/AUC24h of cycle 1, day 1)
AUC area under the concentration–time curve, C max maximum concentration observed, NA not applicable, t max time of maximum concentration observed
aMedian (range); mean (standard deviation) for other parameters
Table 4.
Pharmacokinetic parameters for ASP3026 (cycle 1, day 28)
| Dose (mg; once daily) | Number | C max (ng/mL) | t max (h)a | AUC24 (ng h/mL) | t ½ (h) |
|---|---|---|---|---|---|
| Dose-escalation cohort | |||||
| 25 | 3 | 68.4 (42.3) | 3.0 (1.0–8.1) | 1038 (335) | 36.6 (15.7) |
| 50 | 3 | 143 (62.4) | 3.0 (2.0–4.0) | 2111 (541) | 35.2 (18.7) |
| 75 | 3 | 352.5 (147.5) | 3.0 (2.0–4.0) | 5627 (1791) | 84.7 (53.9) |
| 125 | 1 | 667.9 (NA) | 2.1 (NA) | 8967 (NA) | 36.7 (NA) |
| 200 | 3 | 681.8 (104.8) | 3.0 (2.1–3.0) | 7620 (1699) | 21.9 (1.4) |
| 325 | 3 | 1159 (856.9) | 4.0 (3.0–4.0) | 18,764 (17,647) | 26.3 (29.1) |
| 525 | 6 | 2819 (1681) | 3.5 (2.0–4.1) | 40,114 (24,479) | 27.3 (5.0) |
| 800 | 1 | 4854 (NA) | 4.0 (NA) | 78,081 (NA) | 37.9 (NA) |
| Dose-expansion cohort | |||||
| 525 | 15 | 1331 (813.7) | 4.0 (0–4.1) | 19,993 (10,552) | 24.9 (12.7) |
Calculated accumulation ratio (AUCd28, tau/AUC24h of cycle 1, day 1)
AUC area under the concentration–time curve, C max maximum concentration observed, NA not applicable, t max time of maximum concentration observed
aMedian (range); mean (standard deviation) for other parameters
ASP3026 antitumor effects
In the dose-expansion cohort (crizotinib-resistant ALK-positive; 15 NSCLC; one neuroblastoma), the best overall response was PR in eight patients (50 %) and SD in seven patients (44 %) (Table 5). The objective response rate (CR + PR) was 50 % (95 % CI, 25–75 %). Of the eight patients experiencing a PR, six had lung target tumors. The other two patients had the following target tumors: patient 1 had liver and adrenal tumors; patient 2 had pancreas tail, soft tissue peritoneum, paraaortic, and uterine tumors.
Table 5.
Best overall response to ASP3026 in the dose-expansion cohort
| Parameter | Expansion cohort ASP3026 525 mg (n = 16) |
|---|---|
| Best overall responsea, n (%) | |
| Complete response | 0 |
| Partial response | 8 (50) |
| Stable disease | 7 (44) |
| Progressive disease | 0 |
| Unable to evaluate | 1 (6) |
| Objective response (complete response + partial response) | |
| n (%) | 8 (50) |
| 95 % CIb | 25–75 % |
CI confidence interval
aBased on RECIST guidelines (v1.1) and International Working Group revised response criteria
bExact CI obtained using Clopper–Pearson method
For patients in the dose-expansion cohort with evaluable tumor diameter, the best tumor reduction is shown in Fig. 2, along with the duration of response that ranged from 27 to 338 days. For those patients with brain metastasis detected at study entry, brain MRI was performed at the end of cycle 2 and then every 2 cycles thereafter per protocol. Progressive or new brain metastasis was considered as PD in addition to RECIST evaluation for extracranial disease. The median progression-free survival in ALK-positive patients was 6 (95 % CI, 4–9) months.
Fig. 2.
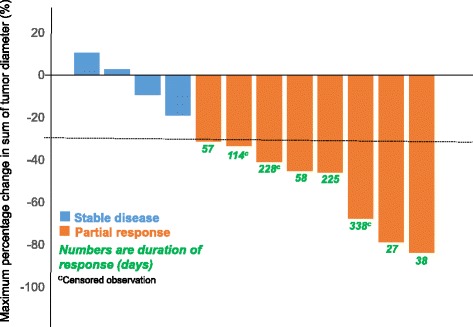
Best tumor response for target lesions in patients treated with ASP3026 in the dose-expansion cohort. Each bar represents one patient. The dotted line at 30 % indicates partial response. Maximum tumor response for the sum of target lesions is shown. The graph shows 12 patients with evaluable primary tumor diameter changes (one patient was non-evaluable, and for three stable disease patients, the overall response was not based on target lesion)
Discussion
Gain-of-function ALK gene alterations have been detected in several types of solid tumors, B cell lymphomas and pediatric tumors [2, 3, 22], for which crizotinib has either established or promising clinical efficacy [17]. Several second- and third-generation ALK inhibitors have also shown either established or promising clinical activity for ALK-positive NSCLC patients who progressed on crizotinib, supporting the development of potent ALK inhibitors as an effective strategy to overcome resistance to crizotinib [17].
We conducted this first-in-human trial to determine the safety, pharmacokinetic, and antitumor effects of a novel second-generation ALK inhibitor ASP3026. Overall, ASP3026 was well tolerated with no treatment-related deaths. The AE profile in the dose-expansion cohort was similar to that reported for crizotinib [11], with gastrointestinal complaints (nausea and vomiting) being some of the most frequently reported. The constellation of AEs in this small sample set was also similar to other agents in this class, such as ceritinib and alectinib, with gastrointestinal AEs commonly reported [16, 23, 24].
ASP3026 had linear PK parameters and demonstrated dose proportionality over the dose range of 25–800 mg once daily. ASP3026 had good oral absorption, with a tmax of approximately 3 h; median half-life was 35 h (range, 22–85 h), confirming that once-daily dosing was suitable.
Of the 16 crizotinib-resistant ALK+ subjects (15 NSCLC and one neuroblastoma) who received 525 mg ASP3026, eight (50 %) achieved PR and seven (44 %) achieved SD at 8 weeks. When restricted to ALK-positive NSCLC patients, the PR and SD rates were 8/15 (53 %) and 6/15 (40 %), respectively. Although caution is warranted due to a lack of head-to-head comparison, this tumor response rate is comparable with those observed in other second-generation ALK inhibitors, such as ceritinib and alectinib, in crizotinib-resistant NSCLC patients [16, 24].
Our study has several other limitations. First, although our study was designed to allow the enrollment of patients with ALK-driven advanced tumors other than NSCLC, only one patient with advanced neuroblastoma, who had a commonly detected ALK F1174L mutation [19–21] and who progressed on prior crizotinib, was identified during the enrollment period. This is because routine molecular testing for other types of tumors did not, and has not, become standard clinical practice. Secondly, multiplexed genomic testing, such as a targeted resistance mutation panel of the ALK kinase domain and targeted next-generation sequencing, was not required at study entry for determining molecular mechanisms of resistance at disease progression to crizotinib. This is unlikely to affect our result. Although different resistance mutations may confer variable responses to subsequent ALK inhibitor therapy [15, 17, 25], most second-generation ALK inhibitors, such as ceritinib and alectinib, as well as ASP3026, have strong efficacy against both secondary mutations in the ALK tyrosine kinase domain and wild-type ALK gene amplification [16, 24]. Nevertheless, with the increasing use of clinical molecular profiling tests at treatment resistance in patients with advanced malignancies, individualized treatment beyond a second-generation ALK inhibitor should be based on the assessment of molecular mechanism of resistance.
Conclusions
The second-generation ALK inhibitor, ASP3026, showed clinical activity in patients with ALK-positive solid tumors, especially NSCLC, with half of the patients achieving partial response and a favorable safety profile with a MTD and R2PD of 525 mg daily.
Ethics approval and consent to participate
The study was conducted in accordance with all applicable regulatory requirements and had institutional review board approval prior to study initiation at participating institutions. Written informed consent was obtained from all patients prior to the initiation of any study-specific procedures.
Consent for publication
Not applicable.
Acknowledgements
We would like to thank Nicola Ray, PhD, Regina Switzer, PhD, and Michelle Utton-Mishra, PhD, of Choice Healthcare Solutions for the medical writing assistance, funded by Astellas Pharma Inc. We would like to thank all the patients and the A3026-CL-0101 study team at Astellas and participating institutions.
Funding
The trial was sponsored by Astellas Pharma Inc.
Abbreviations
- AE
adverse event
- ALK
anaplastic lymphoma kinase
- AUC
area under the concentration–time curve
- Cmax
maximum concentration
- CR
complete response
- ECOG
Eastern Cooperative Oncology Group
- MTD
maximum tolerated dose
- NSCLC
non-small cell lung cancer
- PD
progressive disease
- PKs
pharmacokinetics
- PR
partial response
- RP2D
recommended phase 2 dose
- SD
stable disease
- TKI
tyrosine kinase inhibitor
- tmax
time to maximum concentration
Footnotes
Competing interests
TL: Served on an advisory board for Astellas prior to the conduct of this trial.
PLR: Served on an advisory board for Astellas prior to the conduct of this trial and previous employee received grant funding to conduct this trial. Received consultancy/advisory fees from Pfizer, Genetech and Astex.
SHIO: Received funding from Astellas for this trial.
AT: Serve as a Symphogen board member, received consultancy fees from Nanobiotix, Celator, Janssen, Genmab, Pierre Fabre, Asana, Blend Therapeutics, Merus, Akebia, Johnson and Johnson, Astex, Bayer, Dicema, ArQule, Nektar and Abbvie.
MLM: Received consulting fees from Astellas prior to the conduct of this trial.
EB, HAB, JWP, GY are employees of Astellas Pharma Global Development.
Authors’ contributions
TL, PLR, MLM, EB, HAB, JWP, GY, and AT contributed to the conception and design of the study. TL, PLR, MLM, EB, HAB, JWP, GY, and AT were involved in the development of the methodology. TL, PLR, MLM, S-HIO, EB, HAB, JWP, GY, and AT participated in the analysis and interpretation of the data. All authors contributed in the writing, review, and revision of the article and final approval for submission.
References
- 1.Dirks WG, Fahnrich S, Lis Y, Becker E, MacLeod RA, Drexler HG. Expression and functional analysis of the anaplastic lymphoma kinase (ALK) gene in tumor cell lines. Int J Cancer. 2002;100(1):49–56. doi: 10.1002/ijc.10435. [DOI] [PubMed] [Google Scholar]
- 2.Palmer RH, Vernersson E, Grabbe C, Hallberg B. Anaplastic lymphoma kinase: signalling in development and disease. Biochem J. 2009;420(3):345–361. doi: 10.1042/BJ20090387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Grande E, Bolos MV, Arriola E. Targeting oncogenic ALK: a promising strategy for cancer treatment. Mol Cancer Ther. 2011;10(4):569–579. doi: 10.1158/1535-7163.MCT-10-0615. [DOI] [PubMed] [Google Scholar]
- 4.Iragavarapu C, Mustafa M, Akinleye A, Furqan M, Mittal V, Cang S, Liu D. Novel ALK inhibitors in clinical use and development. J Hematol Oncol. 2015;8:17. doi: 10.1186/s13045-015-0122-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Takeuchi K, Choi YL, Soda M, Inamura K, Togashi Y, Hatano S, et al. Multiplex reverse transcription-PCR screening for EML4-ALK fusion transcripts. Clin Cancer Res. 2008;14(20):6618–6624. doi: 10.1158/1078-0432.CCR-08-1018. [DOI] [PubMed] [Google Scholar]
- 6.Koivunen JP, Mermel C, Zejnullahu K, Murphy C, Lifshits E, Holmes AJ, Choi HG, et al. EML4-ALK fusion gene and efficacy of an ALK kinase inhibitor in lung cancer. Clin Cancer Res. 2008;14(13):4275–4283. doi: 10.1158/1078-0432.CCR-08-0168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li T, Maus MK, Desai SJ, Beckett LA, Stephens C, Huang E, et al. Large-scale screening and molecular characterization of EML4-ALK fusion variants in archival non-small-cell lung cancer tumor specimens using quantitative reverse transcription polymerase chain reaction assays. J Thorac Oncol. 2014;9(1):18–25. doi: 10.1097/JTO.0000000000000030. [DOI] [PubMed] [Google Scholar]
- 8.Gou LY, Niu FY, Wu YL, Zhong WZ. Differences in driver genes between smoking-related and non-smoking-related lung cancer in the Chinese population. Cancer. 2015;121(Suppl 17):3069–3079. doi: 10.1002/cncr.29531. [DOI] [PubMed] [Google Scholar]
- 9.Barlesi F, Mazieres J, Merlio JP, Debieuvre D, Mosser J, Lena H, et al. Routine molecular profiling of patients with advanced non-small-cell lung cancer: results of a 1-year nationwide programme of the French Cooperative Thoracic Intergroup (IFCT). Lancet. 2016. epub ahead of print. doi:http://dx.doi.org/10.1016/S0140-6736(16)00004-0. [DOI] [PubMed]
- 10.Sasaki T, Rodig SJ, Chirieac LR, Janne PA. The biology and treatment of EML4-ALK non-small cell lung cancer. Eur J Cancer. 2010;46(10):1773–1780. doi: 10.1016/j.ejca.2010.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Camidge DR, Bang YJ, Kwak EL, Lafrate AJ, Varella-Garcia M, Fox SB, et al. Activity and safety of crizotinib in patients with ALK-positive non-small-cell lung cancer: updated results from a phase 1 study. Lancet Oncol. 2012;13(10):1011–1019. doi: 10.1016/S1470-2045(12)70344-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shaw AT, Kim DW, Nakagawa K, Seto T, Crino L, Ahn MJ, et al. Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. N Engl J Med. 2013;368(25):2385–2394. doi: 10.1056/NEJMoa1214886. [DOI] [PubMed] [Google Scholar]
- 13.Solomon BJ, Mok T, Kim DW, Wu YL, Nakagawa K, Mekhail T, et al. First-line crizotinib versus chemotherapy in ALK-positive lung cancer. N Engl J Med. 2014;371(23):2167–2177. doi: 10.1056/NEJMoa1408440. [DOI] [PubMed] [Google Scholar]
- 14.National Comprehensive Cancer Network (NCCN) Guidelines . Non-small cell lung cancer. Version 4. 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Katayama R, Shaw AT, Khan TM, Mino-Kenudson M, Solomon BJ, Halmos B, et al. Mechanisms of acquired crizotinib resistance in ALK-rearranged lung cancers. Sci Transl Med. 2012;4(120):120ra117. doi: 10.1126/scitranslmed.3003316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shaw AT, Engelman JA. Ceritinib in ALK-rearranged non-small-cell lung cancer. N Engl J Med. 2014;370(26):2537–2539. doi: 10.1056/NEJMc1404894. [DOI] [PubMed] [Google Scholar]
- 17.Awad MM, Shaw AT. ALK inhibitors in non-small cell lung cancer: crizotinib and beyond. Clin Adv Hematol Oncol. 2014;12(7):429–439. [PMC free article] [PubMed] [Google Scholar]
- 18.Mori M, Ueno Y, Konagai S, Fushiki H, Shimada I, Kondoh Y, et al. The selective anaplastic lymphoma receptor tyrosine kinase inhibitor ASP3026 induces tumor regression and prolongs survival in non-small cell lung cancer model mice. Mol Cancer Ther. 2014;13(2):329–340. doi: 10.1158/1535-7163.MCT-13-0395. [DOI] [PubMed] [Google Scholar]
- 19.Bresler SC, Wood AC, Haglund EA, Courtright J, Belcastro LT, Plegaria JS, et al. Differential inhibitor sensitivity of anaplastic lymphoma kinase variants found in neuroblastoma. Sci Transl Med. 2011;3(108):108ra114. doi: 10.1126/scitranslmed.3002950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mazot P, Cazes A, Boutterin MC, Figueiredo A, Raynal V, Combaret V, et al. The constitutive activity of the ALK mutated at positions F1174 or R1275 impairs receptor trafficking. Oncogene. 2011;30(17):2017–2025. doi: 10.1038/onc.2010.595. [DOI] [PubMed] [Google Scholar]
- 21.Schonherr C, Ruuth K, Yamazaki Y, Eriksson T, Christensen J, Palmer RH, et al. Activating ALK mutations found in neuroblastoma are inhibited by crizotinib and NVP-TAE684. Biochem J. 2011;440(3):405–413. doi: 10.1042/BJ20101796. [DOI] [PubMed] [Google Scholar]
- 22.Moran T, Quiroga V, Gil Mde L, Vila L, Pardo N, Carcereny E, et al. Targeting EML4-ALK driven non-small cell lung cancer (NSCLC) Transl Lung Cancer Res. 2013;2(2):128–141. doi: 10.3978/j.issn.2218-6751.2013.03.04. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Seto T, Kiura K, Nishio M, Nakagawa K, Maemondo M, et al. CH5424802 (RO5424802) for patients with ALK-rearranged advanced non-small-cell lung cancer (AF-001JP study): a single-arm, open-label, phase 1-2 study. Lancet Oncol. 2013;14(7):590–598. doi: 10.1016/S1470-2045(13)70142-6. [DOI] [PubMed] [Google Scholar]
- 24.Gadgeel SM, Gandhi L, Riely GJ, Chiappori AA, West HL, Azada MC, et al. Safety and activity of alectinib against systemic disease and brain metastases in patients with crizotinib-resistant ALK-rearranged non-small-cell lung cancer (AF-002JG): results from the dose-finding portion of a phase 1/2 study. Lancet Oncol. 2014;15(10):1119–1128. doi: 10.1016/S1470-2045(14)70362-6. [DOI] [PubMed] [Google Scholar]
- 25.Friboulet L, Li N, Katayama R, Lee CC, Gainor JF, Crystal AS, et al. The ALK inhibitor ceritinib overcomes crizotinib resistance in non-small cell lung cancer. Cancer Discov. 2014;4(6):662–673. doi: 10.1158/2159-8290.CD-13-0846. [DOI] [PMC free article] [PubMed] [Google Scholar]