

ABSTRACT
Congenital cataracts are a significant cause of lifelong visual loss. They may be isolated or associated with microcornea, microphthalmia, anterior segment dysgenesis (ASD) and glaucoma, and there can be syndromic associations. Genetic diagnosis is challenging due to marked genetic heterogeneity. In this study, next‐generation sequencing (NGS) of 32 cataract‐associated genes was undertaken in 46 apparently nonsyndromic congenital cataract probands, around half sporadic and half familial cases. We identified pathogenic variants in 70% of cases, and over 68% of these were novel. In almost two‐thirds (20/33) of these cases, this resulted in new information about the diagnosis and/or inheritance pattern. This included identification of: new syndromic diagnoses due to NHS or BCOR mutations; complex ocular phenotypes due to PAX6 mutations; de novo autosomal‐dominant or X‐linked mutations in sporadic cases; and mutations in two separate cataract genes in one family. Variants were found in the crystallin and gap junction genes, including the first report of severe microphthalmia and sclerocornea associated with a novel GJA8 mutation. Mutations were also found in rarely reported genes including MAF, VIM, MIP, and BFSP1. Targeted NGS in presumed nonsyndromic congenital cataract patients provided significant diagnostic information in both familial and sporadic cases.
Keywords: next‐generation sequencing, congenital cataract, microcornea, microphthalmia, eye
Introduction
Congenital cataracts are responsible for approximately 10% of childhood blindness worldwide [Gilbert et al., 1993; Foster et al., 1997; Gilbert and Foster, 2001; Muhit and Gilbert, 2003]. Management is often difficult due to the risk of amblyopia in the developing visual system and complicated by the presence of glaucoma with attendant further loss of vision [Swamy et al., 2007]. Congenital cataracts occur due to a disruption to the lens microarchitecture or the accumulation of protein aggregates in the lens [Shiels and Hejtmancik, 2007]. There are autosomal‐dominant, autosomal‐recessive, and X‐linked genetic forms of congenital cataracts, which may be isolated (nonsyndromic) or associated with systemic disease or syndromes (syndromic). In only approximately 18% of cases is there a known family history [Wirth et al., 2002], so a significant proportion are sporadic cases where it is not known if there may be an underlying genetic cause for the lens abnormality.
Mutations in over 30 genes are known to cause nonsyndromic forms of congenital cataracts [Hejtmancik, 2008; Huang and He, 2010]. These code for a variety of lens structural proteins including crystallin, gap junction, intermediate filament, and lens membrane proteins, as well as transcription factors, and there are over 150 other cataract loci [Shiels et al., 2010]. Around 18% of patients with congenital cataracts also have microcornea, defined by horizontal corneal diameter (HCD) <10 mm at birth in an eye of normal axial length (AXL) [Hansen et al., 2007]. There can be other associated ophthalmic abnormalities including microphthalmia, anterior chamber abnormalities, and retinal degeneration. Some syndromic forms of congenital cataracts can be subtle with associated systemic features such as learning difficulties and facial features presenting or becoming apparent only in later childhood [Slavotinek, 2011]. Owing to the marked genetic and phenotypic heterogeneity, genetic testing has been a difficult, expensive, and time‐consuming task using conventional Sanger sequencing. When this was undertaken on a research basis, mutations were identified in known cataract genes in 33%–50% of familial nonsyndromic cases [Hansen et al., 2007; Kumar et al., 2011; Sun et al., 2011], but sporadic cases were not examined.
For such a heterogeneous condition, high‐throughput sequencing techniques offer a much more time and cost‐effective method of mutation detection. Using whole‐exome sequencing (WES) with target gene analysis, mutations were identified in approximately 50% of congenital nonsyndromic cataract families with a clear autosomal‐dominant inheritance pattern [Reis et al., 2013; Prokudin et al., 2014]. Application of targeted next‐generation sequencing (NGS) in 36 families with a high proportion of consanguineous and syndromic cases led to a mutation detection rate of 75% [Gillespie et al., 2014].
In this study, to investigate NGS in nonsyndromic cases with a variety of inheritance patterns including a high proportion of sporadic cases in nonconsanguineous families, we applied targeted NGS of cataract genes in a cohort of 46 apparently nonsyndromic congenital cataract families, the largest series of congenital cataract cases studied to date. Half the cases were sporadic, and the remainder had apparent autosomal‐dominant or autosomal‐recessive inheritance patterns. Mutations were identified in 17 genes and we found a high mutation detection rate of approximately 70% in these sporadic and familial apparently nonsyndromic cases. New diagnoses and/or new information about inheritance patterns were identified in almost two‐thirds of the cases where pathogenic variants were identified.
Materials and Methods
Patients
Forty‐six probands with apparently isolated, nonsyndromic bilateral congenital cataracts were investigated. These patients were seen in the genetic eye clinic of a major pediatric referral hospital over a period of 12 years. The majority of the families were from a Caucasian background (37/46) and the remainder had Middle Eastern (7/46) or Asian (2/46) ethnicity, representing the diverse cultural background of the Western Sydney region. Patients were selected on the basis of DNA sample availability, and preferably with parental and other family member samples also available. Approximately half the cases (22/46) were familial, 19 with a likely autosomal‐dominant mode of inheritance (Families 3–6, 8, 10, 11, 14, 16, 19, 22, 23, 26, 31–33, 38–40), although one of these (Family 14) was in a consanguineous family, and in three families the pedigree was suggestive of autosomal‐recessive inheritance (Families 1, 24, and 46), with consanguinity in two of these (Families 24 and 46). The other 24 of the 46 probands were sporadic cases (Supp. Fig. S1). Most patients had bilateral congenital cataracts diagnosed in the 1st year of life, and some also had microcornea and additional ophthalmological features such as microphthalmia, nystagmus, and glaucoma (Supp. Table S1). Genomic DNA was isolated from leukocytes of peripheral venous blood in all cases, except for the proband from Family 40 where only a saliva sample could be obtained. Two probands were previously investigated using a WES approach and were independently investigated in this study (Families 8 and 11 in this study correspond to Families 4 and 2, respectively, in Prokudin et al. [2014]). Ophthalmological details and samples for genomic DNA extraction were collected from family members when available (Supp. Fig. S1). All experiments were approved by the Human Research Ethics Committee of Sydney Children's Hospital Network, Sydney, Australia.
NGS, Variant Prioritization, and Validation
Thirty‐eight probands (from Families 1–38) were analyzed using the Illumina TruSeq Custom Amplicon (Version 1.5; Illumina Inc., 2011–2013, CA, USA) approach. As part of our work in developmental eye disease, a panel of amplicons targeted the coding exons and 10 bp flanking intronic sequences were designed to include 32 known congenital cataract disease genes (Supp. Table S2 – contains RefSeq [NCBI] accession numbers). Data were based on the GRCh37/hg19 version of the reference genome. Nonsyndromic and some syndromic congenital cataract disease genes were included owing to our clinical observation that some forms of syndromic cataracts may be subtle and missed by referring pediatricians and ophthalmologists, especially in infancy. A capture‐based approach was also used in this study when it became available, using the Illumina TruSight and TruSight One Clinical Exomes (Illumina Inc., 2013–2014). These both used the Nextera capture method (Illumina Inc.) designed to focus on disease‐causing genes including those in HGMD (http://www.hgmd.org/) and OMIM (http://www.omim.org/). Both clinical exome panels included the 32 congenital cataract genes we targeted in our custom amplicon (TruSeq) approach. The Illumina TruSight Clinical Exome was performed on two probands (Families 39 and 40) and one additional family member (father from Family 6). This captured 2,761 OMIM‐identified disease genes. The expanded TruSight One Clinical Exome, targeting 4,813 disease genes, was also used on 23 probands. These included 17 mutation‐negative patients from the TruSeq custom amplicon cohort (Families 1, 3, 4, 5, 7, 9, 11, 15, 17, 19, 21, 26, 29, 30, 34, 36, 38) and six probands (from Families 41 to 46). The custom amplicon samples were sequenced on the Illumina MiSeq (Illumina Inc.) with 2×250 bp paired‐end reads (Ramaciotti Centre, Sydney, Australia). The clinical exome samples were sequenced with the Illumina HiSeq 2500 (Illumina Inc.) using a 2×150 bp paired‐end read protocol (Ramaciotti Centre).
Alignment was undertaken using NextGene software (v2.4.1, 2015, SoftGenetics, PA, http://www.softgenetics.com/NextGENe.html), and this was also used for variant identification. Variants in the 32 cataract genes were annotated using Annovar [Wang et al., 2010] (March 22, 2015 Build, http://annovar.openbioinformatics.org) and Alamut‐Batch (Version 1.4, 2015; Interactive Biosoftware, Rouen, France, http://www.interactive‐biosoftware.com/alamut‐batch/) (Table 1).
Table 1.
Sporadic and Familial Cases with Likely Causative Variants
| Family (proband) | Inheritance, before/after testing | Gene Refseq ID | Nucleotide change | Predicted amino acid change | Protein domain | In silico: SIFT, MutTaster, PolyPhen, PhyloP, ExAC | Segregation | Novel | ACMG classification |
|---|---|---|---|---|---|---|---|---|---|
| Family 2 (II:1) | #Sporadic/new AD | GJA8 NM_005267.4 | c.119C>T | p.Ala40Val | TM1 | D, D, P, high, nil | Yes: de novo in proband | Yes | LP |
| Family 6 (II:1) | #AD/X‐linked | NHS NM_198270.3 | c.2707delG, hemizygous | p.Glu903Asnfs*4 | Nil | Nil | Yes: hemizygous in affected brothers, heterozygous in mother | Yes | P |
| Family 6 (I:1) | #AD/likely new AD | MAF NM_001031804.2 | c.819G>C | p.Glu273Asp | b‐Zipper | D, D, P, mod, nil | N/K: parental samples unavailable, not present in sons | Yes | LP |
| Family 7 (II:3) | #Sporadic/X‐linked | BCOR NM_001123385.1 | c.4390_4393del | p.Glu1464Profs*19 | Nil | Nil | Yes: de novo in proband. Not found in son. | Yes | P |
| Family 8 (I:2) | AD/AD | CRYGCNM_020989.3 | c.497C>T | p.Ser166Phe | 4th Greek Key | D, D, P, high, nil | Yes: present in affected sons | Prokudin et al. (2014) | P |
| Family 10 (III:3) | AD/AD | PAX6 NM_0001604.4 | c.1119del | p.Thr374Profs*5 | PST rich | Nil | Yes: present in affected sibling and mother | Peter et al. (2013) | P |
| Family 12 (II:1) | #Sporadic/likely new AD | CRYBB1 NM_001887.3 | c.368G>A | p.Arg123His | 3rd Greek key | D, D, P, mod, nil | N/K: Not present in mother, father's sample unavailable | Yes | LP |
| Family 13 (II:2) | #Sporadic/new AD | CRYBB2 NM_000496.2 | c.556T>C | p.Ser186Pro | 4th Greek key | D, D, P, mod, nil | De novo; not found in parents or two unaffected siblings | Yes | LP |
| Family 14 (II:2) | #AD?/AR | GJA8 NM_005267.4 | c.89dupT, homozygous | p.Ile31Hisfs*18 | TM1 | Nil | Yes: consanguineous family, mother heterozygous, father unavailable | Yes | P |
| Family 16 (I:2) | AD/AD | CRYAA NM_000394.3 | c.142T>G | p.Tyr48Asp | N‐terminal | T, D, P, mod, nil | Yes: present in affected son | Yes | LP |
| Family 18 (II:1) | #Sporadic/new AD | PAX6 NM_0001604.4 | c.239T>A | p.Ile80Asn | Paired | D, D, P, high, nil | Yes: de novo in proband | Yes | LP |
| Family 19 (II:2) | AD/AD | GJA3 NM_021954.3 | c.260C>T | p.Thr87Met | TM2 | D, D, P, high, nil | Found in all affected family members | Guleria et al. (2007) | P |
| Family 20 (II:2) | #Sporadic/new AD | GJA8 NM_005267.4 | c.134G>C | p.Trp45Ser | TM1 | D, D, P, high, nil | Yes: de novo in proband | Ma et al. (2005) | P |
| Family 22 (II:3) | AD/AD | CRYGC NM_020989.3 | c.328_329delinsT | p.Pro110Serfs*37 | 3rd Greek key | Nil | Yes: present in affected children | Yes | P |
| Family 23 (I:1) | AD/AD | CRYAA NM_000394.3 | c.61C>T | p.Arg21Trp | N‐terminal | D, D, B, high, nil | Yes: present in affected sons, absent in unaffected son | Devi et al. (2008) | P |
| Family 24 (II:8) | AR/AR | CRYBB3 NM_004076.4 | c.493G>C, homozygous | p.Gly165Arg | 4th Greek key | D, D, P, high, nil | Yes: homozygous in three affected siblings, heterozygous in both parents | Riazuddin et al. (2005) | P |
| Family 25 (II:1) | #Sporadic/likely new AD | VIM NM_003380.3 | c.15del | p.Val6Cysfs*26 | Head | Nil | N/K: parental samples unavailable | Yes | LP |
| Family 26 (II:4) | AD/AD | GJA3 NM_021954.3 | c.176C>T | p.Pro59Leu | EC1 | D, D, P, high, nil | N/K: parental samples unavailable | Bennett et al. (2004) | P |
| Family 27 (II:1) | #Sporadic/likely new AD | MAF NM_001031804.2 | c.915C>T | p.Cys305Trp | b‐Zipper | D, D, P, high, nil | N/K: parental samples unavailable | Yes | LP |
| Family 28 (II:2) | #Sporadic/new AD | MIP NM_012064.3 | c.97C>T | p.Arg33Cys | EC1 | D, D, P, high, nil | Yes: de novo in proband | Gu et al. (2007) | P |
| Family 29 (II:1) | #Sporadic/?new AD | GJA3 NM_021954.3 | c.7G>C | p. Asp3His | SP | T, D, P, high, nil | N/K: parental samples unavailable | Yes | LP |
| Family 30 (II:1) | #Sporadic/?new AD | MAF NM_001031804.2 | c.880C>T | p.Arg294Trp | b‐Zipper | D, D, P, high, nil | N/K: parental samples unavailable | Yes | LP |
| Family 31 (III:1) | AD/AD | CRYGD NM_006891.3 | c.70C>A | p.Pro24Thr | 1st Greek key | T, B, B, weak, nil | Yes: present in affected grandmother | Burdon et al. (2004) | P |
| Family 32 (I:2) | AD/AD | MIP NM_012064.3 | c.597_598ins GGGAACATTCCACT | p.Asn200Glyfs*12 | EC3 | Nil | Yes: present in affected daughter | Yes | P |
| Family 33 (II:2) | AD/AD | CRYAB NM_001885.2 | c.320G>T | p.Arg107Leu | Small heat shock protein | T, D, P, high, nil | Yes: present in affected son and father | Yes | LP |
| Family 35 (II:1) | #Sporadic/new AD | CRYBB2 NM_000496.2 | c.343C>A | p.Pro115Thr | 3rd Greek key | D, D, B, high, nil | Yes: de novo in proband | Yes | LP |
| CRYBB2 NM_000496.2 | c.355G>A | p.Gly119Arg | 3rd Greek key | D, D, P, mod, nil | Yes: de novo in proband | Yes | LP | ||
| Family 37 (II:1) | #Sporadic/likely new AD | CRYBB2 NM_000496.2 | c.583T>G | p.Trp195Gly | C‐terminal | D, D, P, mod, nil | Likely de novo; mutation not found in mother, father unavailable | Yes | LP |
| Family 39 (II:1) | AD/AD | CRYBB3 NM_004076.4 | c.634T>C | p.*212Argext*40 | C‐terminal | Nil | Yes: present in affected father | Yes | LP |
| Family 40 (III:3) | #AD/X‐linked | NHS NM_198270.3 | c.3624C>A | p.Cys1208* | Nil | Nil | Yes: hemizygous in affected male, heterozygous in mother and sister | Liao et al. (2011) | P |
| Family 41 (II:1) | #Sporadic/new AD | CRYGD NM_006891.3 | c.448dup | p.Asp150Glyfs*3 | 4th Greek key | Nil | Yes: de novo in proband | Yes | P |
| Family 44 (II:1) | #Sporadic/new X‐linked | BCOR NM_001123385.1 | c.1136_1139del | p.Val379Alafs*62 | Nil | Nil | Yes: de novo in proband | Yes | P |
| Family 45 (II:1) | #Sporadic/new AD | GJA8 NM_005267.4 | c.151G>A | p.Asp51Asn | EC1 | D, D, P, high, nil | Yes: de novo in proband | Yes | LP |
| Family 46 (II:2) | AR/AR | BFSP1 NM_001195.4 | c.1492del | p.Ser498Leufs*24 | Tail | Nil | Mother heterozygous | Yes | P |
| BFSP1 NM_001195.4 | c.812T>C | p.Ile271Thr | 2nd coil | D, D, P, high, rs147718368 0.0003625 (44/121392) | Not found in mother, father unavailable | Yes | LP |
Variants are heterozygous unless otherwise specified. # In the “inheritance, before/after testing” column indicate families where new information was obtained regarding the inheritance pattern in the family from the next‐generation sequencing work. Protein domains: SP, signal peptide; TM, transmembrane; EC, extracellular; IC, intracellular domains. In Silico: D, damaging; P, pathogenic; B, benign; T, tolerated. The computational calculations of SIFT, MutationTaster, PolyPhen, and PhyloP were performed in Alamut‐Visual (Version 2.6, Jan 2015; Interactive Biosoftware, http://www.interactive‐biosoftware.com/alamut‐visual/). ExAC rs number and minor allele frequency provided where available. Segregation: N/K, not known. ACMG classification as per Richards et al. (2015); P, pathogenic; LP, likely pathogenic.
Variants with coverage <15X were removed. Variants were annotated for minor allele frequencies in the dbSNP [Smigielski et al., 2000] (Build 144, May 2015, http://www.ncbi.nlm.nih.gov/projects/SNP), 1000genomes [Abecasis et al., 2012] (Phase 3, 2013, http://www.1000genomes.org/), Exome Variant Server (NHLBI GO Exome Sequencing Project [ESP], Seattle, WA; Version 2, July 2013, http://evs.gs.washington.edu/EVS/) and Exome Aggregation Consortium (ExAC) databases (Version 0.3, January 2015, Cambridge, MA, http://exac.broadinstitute.org/), and heterozygous variants with minor allele frequencies >0.01 were filtered out. Variants were analyzed for possible pathogenic clinical significance according to the 2015 American College of Medical Genetics and Genomics (ACMG) guidelines [Richards et al., 2015], based on a combination of previous reports in the literature and computational, functional, and population data. Nonsense, frameshift, and canonical splice‐site variants were considered strongly indicative of pathogenicity. For missense mutations, Alamut Visual (Version 2.6, Jan 2015; Interactive Biosoftware, http://www.interactive‐biosoftware.com/alamut‐visual/) was used for the individual variant analyses, providing computational algorithms for SIFT [Ng and Henikoff, 2001] (Version 1, http://sift.jcvi.org/), PolyPhen‐2 [Adzhubei et al., 2010] (Version 2.2.2, 2012, http://genetics.bwh.harvard.edu/pph2/) and MutationTaster [Schwarz et al., 2010] (Version 2, 2012, http://www.mutationtaster.org/), as well as conservation with PhyloP scores [Pollard et al., 2010]. Variants that were classified as pathogenic or likely pathogenic according to the ACMG guidelines were validated using Sanger sequencing in the proband and segregation was performed in family members when available. Paternity testing in Family 6 used the PowerPlex 16 HS System (Promega, Madison, WI) run on an ABI3730 capillary electrophoresis system (Australian Genome Research Facility (AGRF), Westmead, Australia). Primers for Sanger sequencing were designed on Primer3 (http://biotools.umassmed.edu/bioapps/primer3_www.cgi), and are available upon request. Nucleotide numbering uses +1 as the A of the ATG translation initiation codon in the reference sequence, with the initiation codon as codon 1. All variants reported in this manuscript have been submitted to ClinVar (http://www.ncbi.nlm.nih.gov/clinvar/).
Results
Targeted Region Analysis
The samples using the TruSeq custom amplicon approach (probands 1–38) had a mean depth of coverage of the targeted regions of 940X across all samples, with on average 91% of amplicon regions being covered by ≥ 15X (Supp. Table S3A). The 26 patients sequenced using the TruSight clinical exome approaches had an average coverage of 160X across the targeted regions, and 95% of captured regions covered ≥ 15X (Supp. Table S3B).
While the TruSeq amplicon‐based method achieved a very high average coverage of 940X, there were several gaps with poor coverage (Supp. Table S3C and D). These gaps were mainly in genes that were very GC rich, or contained repetitive regions, which were not adequately covered by the amplicon PCR‐based TruSeq method. Compared with this, the capture‐based TruSight library approach had a lower average coverage of 160X, but this was more even, with fewer and smaller gaps (Supp. Table S3D). Gaps in GJA3 and MAF underwent Sanger sequencing, due to the importance of gap junction genes in cataracts, and our discovery of three new mutations in MAF in this cohort. This led to detection of only one additional mutation, a GJA3 mutation in Family 29.
Likely Causative Variants in 73% of Familial and 68% of Sporadic Congenital Cataract Cases
Overall, likely disease‐causative variants were found in 33 cases in the 46 families we studied, indicating a mutation detection rate of over 70% using NGS (Table 1). This included one family (Family 6), where a mutation in a different cataract gene was found in the affected father, with confirmed paternity, compared with the causative disease gene identified in his two sons. There was a mutation detection rate in the familial cases of ∼73% (16/22), and a detection rate of 68% in the sporadic cases (17/25, including the affected father with a separate mutation in Family 6). Out of the 34 mutations found, 23 (∼68%) were novel. The mutations were spread over 17 out of the 32 genes analyzed (Fig. 1A). Variants in seven crystallin (CRYAA, CRYAB, CRYBB1, CRYBB2, CRYBB3, CRYGC, and CRYGD) and both gap junction genes (GJA3 and GJA8) were the most frequently identified in our cohort, accounting for 20/33 (∼61%) cases (Table 1; Fig. 1A). In addition, there were likely causative variants found in: MAF in three cases; PAX6, MIP, NHS, and BCOR in two cases each; and VIM and BFSP1 in one case each. The majority of familial cases were due to autosomal‐dominant mutations in crystallin or gap junction genes, with two autosomal‐recessive cases due to mutations in these genes, and two families were found to have X‐linked NHS mutations (Fig. 1B). Sporadic cases were mostly due to de novo autosomal‐dominant mutations in a wide variety of genes including crystallins, gap junctions, MAF, MIP, and VIM, and there were two de novo X‐linked BCOR cases (Fig. 1B). In almost two‐thirds (20/33) of the cases where likely causative variants were identified, this resulted in new information about the inheritance pattern for the family (Table 1). Eight variants of unknown significance were found, five of which were the only variants in that family (Supp. Table S4). According to the ACMG mutation guidelines, five of these were reclassified as of uncertain significance, one was likely benign, and two were benign. Nine families had no variants of interest.
Figure 1.
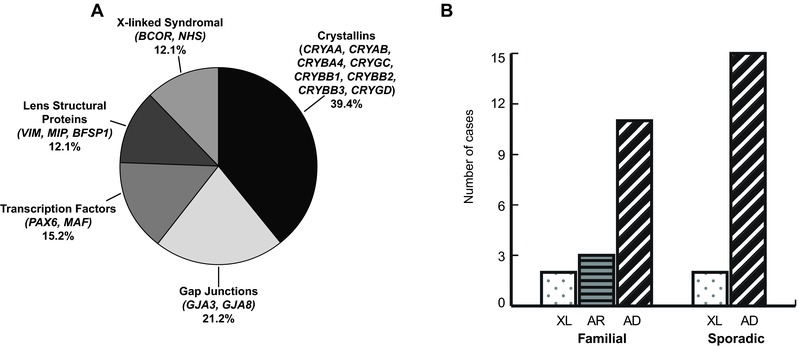
Genes and inheritance patterns identified with NGS in apparently nonsyndromic sporadic and familial congenital cataract cases. A: Mutations were found in 17 different genes in apparently nonsyndromic sporadic and familial congenital cataract cases. These were in genes that encoded crystallins, gap junction, transcription factor, other structural proteins, and X‐linked syndromal proteins. The relative proportions are illustrated in this diagram. B: Mutation‐positive cases in familial and sporadic cases and the inheritance patterns. The 16 mutation‐positive familial cases comprised two X‐linked (XL, dotted), three autosomal‐recessive (AR, horizontal stripes), and 11 autosomal‐dominant (AD, diagonal stripes) diagnoses. In the sporadic cases with mutations, the inheritance pattern was revised to two cases of de novo X‐linked mutations in BCOR, and 15 de novo AD cases, including the father from Family 6 who had a separate genetic answer (mutation in MAF) to his two affected sons with NHS.
Novel Missense, “Run‐On,” and Frameshift Mutations in Key Domains of the Crystallin Genes
Thirteen of the patients with likely causative mutations were found to have variants in the crystallin genes (13/33, 39%), eight in familial and five in sporadic cases, including 10 novel variants (Fig. 1A and B; Table 1). Seven of the eight familial cases were AD, with one AR. The crystallin gene mutation contribution in our AD families (50%, 7/14), is consistent with previous reports [Hansen et al, 2007; Reis et al 2013]. All sporadic cases were shown to be likely new AD cases. Most of these mutations disrupted the Greek Key domains of the crystallin proteins, vital in the protein's folding and maintenance of lens transparency [Vendra et al., 2013].
Two novel missense mutations on the same allele were found in CRYBB2 in Patient II.1 from Family 35 (Fig. 2). Both missense mutations (c.343C>A p.(Pro115Thr) and c.355G>A p.(Gly119Arg)), occurred on the same allele, as revealed by the NGS short read data and were de novo in this sporadic case (Fig. 2D and E). These mutations occurred in exon 5 of CRYBB2 encoding the third Greek Key domain. A number of single missense mutations have been identified in this region of CRYBB2, as well as heterozygous nonsense mutations, but our patient is more severely affected than other reported cases [Litt et al., 1997; Gill et al., 2000; Vanita et al., 2001; Hilal et al., 2002; Yao et al., 2005; Bateman et al., 2007; Devi et al., 2008; Lou et al., 2009; Santhiya et al., 2010] with marked reduction in eye size (HCD 7–8mm and AXL 17mm at age 2 years) and moderately severe visual impairment with VA 6/80 and bilateral nystagmus. In our patient, it is likely that the mutation of both amino acids p.([(Pro115Thr; Gly119Arg)]), in particular the very small nonpolar glycine to a hydrophilic large arginine, within a short distance in this Greek Key motif may have a more severe impact on protein folding than only one mutation alone.
Figure 2.
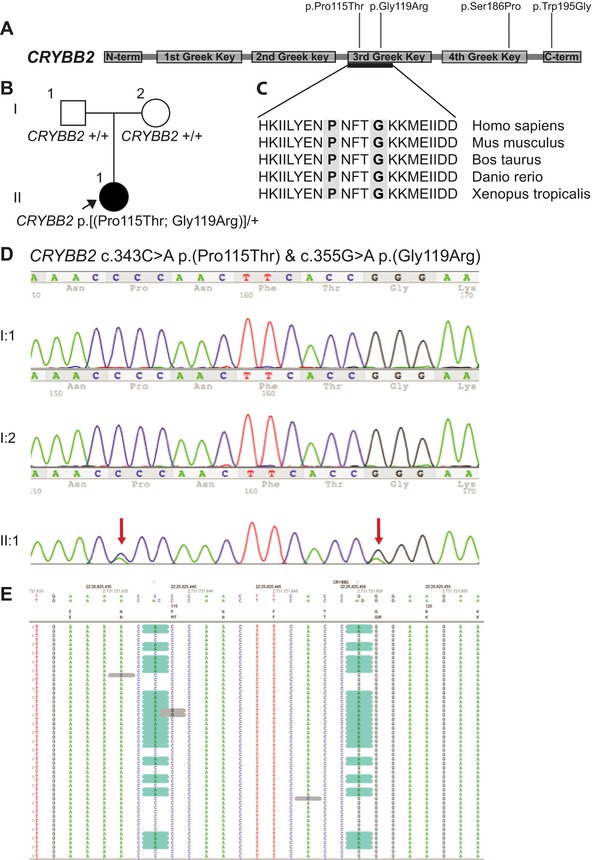
Two novel missense CRYBB2 mutations on the one allele in proband in Family 35, and other CRYBB2 mutations found in this study. A: This schematic shows the encoded domain structure of CRYBB2 (NM_000496.2). This protein contains four “Greek Key” domains between the N and C terminals. Mutations found in this study are illustrated above the schematic, with the two novel missense p.(Pro115Thr) and p.(Gly119Arg) found in Family 35, p.(Ser186Pro) found in Family 13, and p.(Trp195Gly) in Family 37. B: Pedigree from Family 35, with proband highlighted in II:1. C: Protein sequence alignments show that both affected residues, highlighted in gray, are in a highly conserved part of the third Greek Key domain. D: The proband had two de novo missense CRYBB2 mutations, confirmed on Sanger sequencing (red arrows). E: Next‐generation reads reveal that the variants (highlighted in blue) were both consistently in the same reads, showing that they are in cis.
In our series, we report the first identified “run‐on” mutation affecting CRYBB3 [Graw, 2009]. In an affected father and son in Family 39 (Supp. Fig. S1; Table 1), the novel heterozygous missense mutation changed the stop codon of CRYBB3 to arginine and was followed by “run on” into the 3′UTR and an additional 40 residues (c.634T>C p.(*212Argext*40)). This is predicted to cause a lengthened, abnormal CRYBB3 protein likely to disrupt protein folding and function due to the abnormally large product. Two novel heterozygous frameshift mutations in crystallins were identified, one in CRYGC and the other in CRYGD. The novel heterozygous deletion and insertion mutation in CRYGC (c.328_329delinsT p.(Pro110Serfs*37)) is predicted to lead to a frameshift mutation causing familial autosomal‐dominant congenital lamellar cataracts in Family 22 (Supp. Fig. S1; Table 1). The mutation leads to an abnormally short protein, as does a premature stop codon mutation in the same region in another autosomal‐dominant cataract family [Yao et al., 2008], with both mutations predicting loss of the third and fourth Greek Key domains of CRYGC. In the sporadic case in Family 41, a novel de novo heterozygous frameshift mutation was found in CRYGD, (c.448dup p.(Asp150Glyfs*3)) leading to a premature stop codon and deletion of most of the fourth Greek Key and all of the C‐terminal domains of the protein.
GJA3 and GJA8 Mutations Including a Novel Missense Variant with a Severe Phenotype, and a Novel Recessive Frameshift Mutation
Seven of the patients with likely causative mutations in our study were found to have variants in the gap junction genes, GJA3 and GJA8 (7/33, 21%), three in familial and four in sporadic cases, including four novel variants (Table 1; Figs. 1 and 3C). The novel de novo heterozygous GJA8 missense mutation (c.151G>A p.(Asp51Asn)) in the proband in Family 45 (Supp. Fig. S1; Table 1; Fig. 3A–D), was associated with the most severe phenotype yet described in a patient with a gap junction mutation. This patient was markedly affected with bilateral microphthalmia, left more than right, congenital cataracts and sclerocornea of the left eye. The GJA8 missense mutation (c.151G>A p.(Asp51Asn)) occurred in a highly conserved amino acid of the first extracellular domain (EC1) of GJA8 (Fig. 3D). All the other reported mutations in GJA8, including those in the same domain, are associated with congenital cataracts ± microcornea, with a variety of phenotypes, intrafamilial variability, and none with associated sclerocornea or this degree of microphthalmia [Minogue et al., 2009; Beyer et al., 2013; Ge et al., 2014]. We postulate that the p.(Asp51Asn) mutation in this patient may highlight the importance of the Asp 51 amino acid in the EC1 domain in the function of GJA8, and note that the Gja8 p.Ser50Pro mutation in this region in mice is associated with altered interaction with the normal allele, interrupting the process of primary lens fiber cell formation [Xia et al., 2006]. Also, it broadens the phenotypic correlation of GJA8 mutations, highlighting the importance of screening this gene, not only in cataract patients, but also where the main phenotype may be listed as anterior segment abnormality or microphthalmia.
Figure 3.
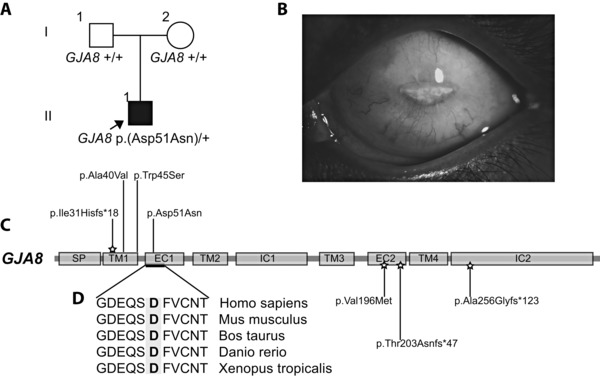
GJA8 mutation in Family 45 caused a severe cataract, sclerocornea, microphthalmia phenotype, and other GJA8 mutations found in this study. A: Pedigree of family 45, with the proband (II:1) highlighted. He was found to have a heterozygous missense mutation in the first extracellular domain (EC1) of GJA8, p.(Asp51Asn), and has the most severe GJA8 ocular phenotype reported to date. B: Left eye image of the proband in Family 45, showing his severe microphthalmia and sclerocornea. C: The schematic shows the encoded domain structure of GJA8 (NM_005267.4). This protein spans the cellular membrane and has a signal peptide (SP), four transmembrane (TM), two extracellular (EC), and two intracellular (IC) domains. Mutations found in GJA8 in this study are shown above the schematic, including the p.(Asp51Asn) mutation in Family 45 in the EC1 domain. The p.(Ile31Hisfs*18) recessive mutation found in Family 14 is the earliest frameshift mutation yet reported in this gene. Other recessive mutations reported in the literature are shown below the gene, and the star highlights recessive mutations. The p.(Ala40Val) and p.(Trp45Ser) heterozygous mutations were found in Families 2 and 20, respectively. D: Protein sequence alignments show that the affected residue in the proband of Family 45, highlighted in gray, lies in a highly conserved region of the EC1 domain.
A novel GJA8 frameshift mutation was identified in Family 14 (Supp. Fig. S1; Table 1). The affected mother and daughter were referred with suspected autosomal‐dominant cataracts, although it was noted that the daughter's parents were consanguineous. The affected daughter was found to have a novel homozygous (c.89dupT p.(Ile31Hisfs*18)) mutation in the first transmembrane domain of GJA8 (Table 1; Fig. 3C). She had congenital cataracts and microcornea, whereas her heterozygous mother had a milder phenotype with nuclear dot opacities. The father was unavailable for analysis. While both dominant and recessive mutations in GJA8 can cause cataracts, recessive mutations are less frequently reported. Interestingly, two out of the three reported recessive mutations are frameshift mutations [Ponnam et al., 2007; Schmidt et al., 2008], and one is a missense mutation [Ponnam et al., 2013] (Fig. 3C). The frameshift mutation we have identified is the earliest recessive mutation in the protein (Fig. 3C). As in our case, affected homozygous individuals in the literature all had severe dense congenital cataracts. The extent to which heterozygous carriers in the literature were affected is variable, with some reported as unaffected [Ponnam et al., 2007], or variably affected with mild nuclear dot opacities [Schmidt et al., 2008; Ponnam et al., 2013] as in the heterozygous mother in our family.
Mutations in X‐Linked Syndromal Cataract Genes NHS and BCOR in Four Families
The diagnosis of mutations in the X‐linked syndromal cataract genes NHS and BCOR highlights the genetic heterogeneity of congenital cataracts. Probands from Families 6 and 40 were diagnosed with Nance Horan syndrome (NHS) due to NHS mutations found in this study (Supp. Fig. S1; Table 1; Figs. 4A–E and 5A) after initial referrals with a presumed diagnosis of autosomal‐dominant cataracts. The proband from Family 6 was the elder of two brothers, aged 9 and 4 years, with congenital cataracts and microcornea (Fig. 4A). The father had cerulean cataracts (Fig. 4S and T), the mother had clear lenses and the family was offered clinical genetics review but declined. The proband in Family 6 had a novel frameshift hemizygous mutation in NHS (c.2707delG, p.(Glu903Asnfs*4)) (Fig. 5A). The mutation was also present in his affected brother and was heterozygous in their mother. When the brothers were reviewed, the typical facial and dental features were apparent, and they were beginning to exhibit intellectual delay and attention‐deficit consistent with a diagnosis of NHS. The male proband in Family 40 (Supp. Fig. S1; Fig. 4B) had congenital cataracts, left foot insertional polydactyly, and mild learning difficulties. His older sister and mother had been found to have juvenile‐onset cataracts (Fig. 4C–E). On clinical genetics review, he was found to have facial and dental features suggestive of NHS. A previously reported nonsense mutation (c.3624C>A, p.(Cys1208*)) in NHS [Huang et al., 2007] (Fig. 5A) was found in the proband and segregated appropriately in an X‐linked pattern in his sister and mother, again providing the correct diagnosis and inheritance pattern information for this family. While the digital anomaly of brachymetacarpalia has been seen in NHS [Ding et al., 2009], this is the first report of insertional or any form of polydactyly in this condition.
Figure 4.
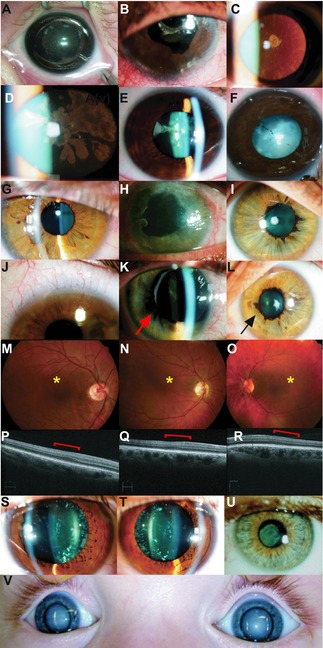
Phenotypic variability of the cataracts and associated ocular phenotypes in this study. This figure shows images of congenital cataracts and other ocular features found in this study and highlights the diverse heterogeneity of congenital cataracts, as well as the importance of accurate phenotyping, fundoscopy, and electrophysiology to clarify the diagnosis. Images are from families diagnosed with mutations in NHS, Families 6(A) and 40(B‐E); PAX6, Families 10(F) and 18(G‐R); MAF, father from Family 6(S&T); MIP, Family 32(U); and VIM, family 25(V). Image A displays the right sided cortical cataract from the proband (II:1) from Family 6, diagnosed with NHS. The other family with NHS (Family 40) is represented in images B–E. B: Right eye from proband (III:3, Family 40), with postsurgical changes following cataract extraction. The variable features demonstrated in obligate carrier females from this family are demonstrated in C, with subcapsular cataract from the mother (II:3) and D (right) and E (left) coralliform cataracts in the sister (III:1). Image F: Cortical congenital cataracts of proband (II:1) from Family 10, diagnosed with a missense PAX6 mutation in the paired domain. Images from Family 18, also diagnosed with a PAX6 mutation, are represented in images G–R, revealing the panocular features of PAX6‐related disease. These are from the proband (III:3) showing: G: anterior polar cataract; J: mild limbal stem cell failure with peripheral corneal pannus; M: altered macular reflex (*); P: OCT of foveal hypoplasia (red bracket) with loss of the foveal pit. The mother in Family 18 (II:2) also demonstrates multiple PAX6‐associated abnormalities with: H: limbal stem cell failure with increased corneal pannus; K: mild iris hypoplasia and mild ectropion uvea (arrow); N: reduced macular reflex (*); Q: OCT showing foveal hypoplasia with loss of the foveal pit (red bracket). The sister in Family 18 (III:2) also has: I: anterior polar cataract; L: ectropion uvea (arrow); O: reduced macular reflex (*); and R: OCT showing loss of foveal pit (red bracket). S (right) and T (left): Cerulean blue dot cataracts from the father (I:1) from Family 6, found to have a novel missense MAF mutation. U: Lamellar cataract of proband (II:1) in Family 32, found to have a novel frameshift mutation in MIP. V: The image demonstrates bilateral total cataracts as they appeared on presentation at 11 months of age in the proband of Family 25 (II:1), found to have a novel frameshift mutation in VIM.
Figure 5.
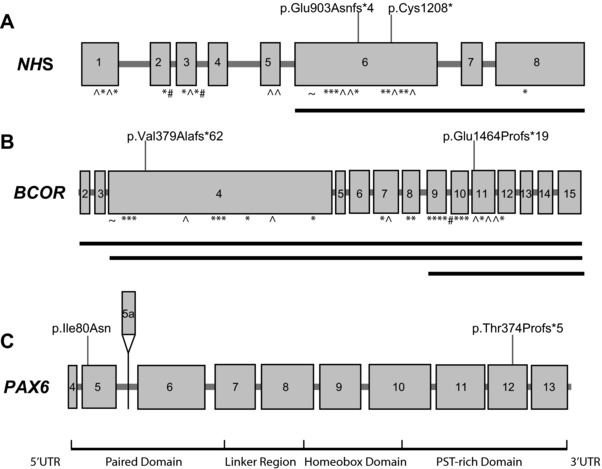
Mutations in NHS and BCOR revealed syndromic conditions, and PAX6 mutations indicated more complex ocular phenotypes. A: Exonic structure of NHS (NM_198270.3) with previously reported mutations marked by symbols below the gene. KEY: ^, nonsense; *, frameshift; #, splice site; ∼, missense mutations. The solid black lines delineate reported exonic deletions. Mutations found in this study are demonstrated above the gene, from Families 6 and 40. B: Exonic structure of BCOR (NM_001123385.1) with previously reported mutations marked by symbols below the gene. KEY: ^, nonsense; *, frameshift; #, splice site; ∼. missense mutations. The solid black lines delineate reported exonic deletions. Mutations found in this study are demonstrated above the gene, from Families 44 and 7. C: Exonic and protein domain structure of PAX6 (NM_0001604.4). The exons are numbered, with the alternatively spliced exon 5a highlighted above the gene. Below the gene, the protein domains are highlighted (UTR, untranslated region; PST, Proline, Serine, Threonine rich domain). The two mutations found in this study, from Families 18 and 10, are marked above the gene. Due to the large number of mutations found in this gene, they are not illustrated in the figure. A curated database of PAX6 mutations can be found at LOVD (http://www.lovd.nl/PAX6).
In two families, de novo novel frameshift mutations were found in BCOR, the gene responsible for oculo‐facio‐cardio‐dental (OFCD) syndrome (Fig. 5B). In Family 44 (Supp. Fig. S1), the female proband had sporadic cataracts and cleft palate. She was found to have a novel de novo heterozygous frameshift deletion in BCOR (c.1136_1139del p.(Val379Alafs*62)). She was subsequently diagnosed with atrial septal defect and was later found to have dental and facial features consistent with OFCD. In Family 7, the female proband with microphthalmia, cataracts and glaucoma, was found to have a de novo novel frameshift BCOR mutation (c.4390_4393del p.(Glu1464Profs*19)). This mutation was not found in her son who had aniridia and had been found in a separate study to have a paternally inherited heterozygous mutation in PAX6 [Willcock et al., 2006] (Supp. Fig. S2).
Mutations in PAX6 in Two Families with Complex Cataract Phenotypes
Two heterozygous variants were found in PAX6, a familial frameshift mutation in Family 10 and a de novo novel missense mutation in Family 18 (Supp. Fig. S1; Table 1; Figs. 4F–R and 5C). They add to the increasing evidence that patients with a variety of eye phenotypes such as cataract, glaucoma, anterior segment dysgenesis (ASD), and nystagmus should have PAX6 testing performed, and this gene should be included in NGS panels for congenital cataracts. This is particularly important if clinical findings are atypical or visual impairment more severe than expected. The proband from Family 10 was referred with an atypical presentation of cataracts, with additional nystagmus and congenital ptosis and astigmatism. A frameshift mutation in PAX6 (c.1119del, p.(Thr374Profs*5)) was identified and his affected sister and mother had the same PAX6 mutation and displayed similar clinical features, although they lacked nystagmus (Fig. 4G–R). This mutation is recently reported in a family with a similarly complex and variable cataract phenotype including cataracts, ptosis, iris hypoplasia, corneal opacification, and diabetes [Peter et al., 2013]. A novel de novo PAX6 missense mutation (c.239T>A p.(Ile80Asn)) was identified in Patient II.1 in Family 18 (Supp. Fig. S1; Table 1; Figs. 4F and 5C). He also had nystagmus and reduced visual acuity and required strabismus surgery. The PAX6 mutation explains his complex phenotype and may provide an explanation for the poorer than expected outcome following his cataract surgery.
Three New Mutations Found in the Basic Zipper Region of MAF
Three novel heterozygous mutations in MAF were identified in Families 6, 27, and 30 (Fig. 1; Table 1), one in the EHR (extended homology region, EHR), and two in the bZIP (b‐zipper) domain (Fig. 6A and B). The father in Family 6 had cerulean cataracts (Fig. 4S and T), and we undertook NGS in this man when the X‐linked cause of his boys’ cataracts was found. This led to identification of a novel heterozygous missense mutation in MAF (c.819G>C p.(Glu273Asp)), the first identified in the EHR. The proband from Family 30 had sporadic congenital cataracts and a heterozygous missense c.880C>T p.(Arg294Trp) mutation in the basic region of MAF, very close to where previous mutations were reported (Fig. 6B). Another novel heterozygous mutation (c.915C>T p.(Cys305Trp)) was found in Patient II.1, from Family 27 (Fig. 6B). This patient had sporadic congenital cataracts and severe glaucoma requiring surgical intervention.
Figure 6.
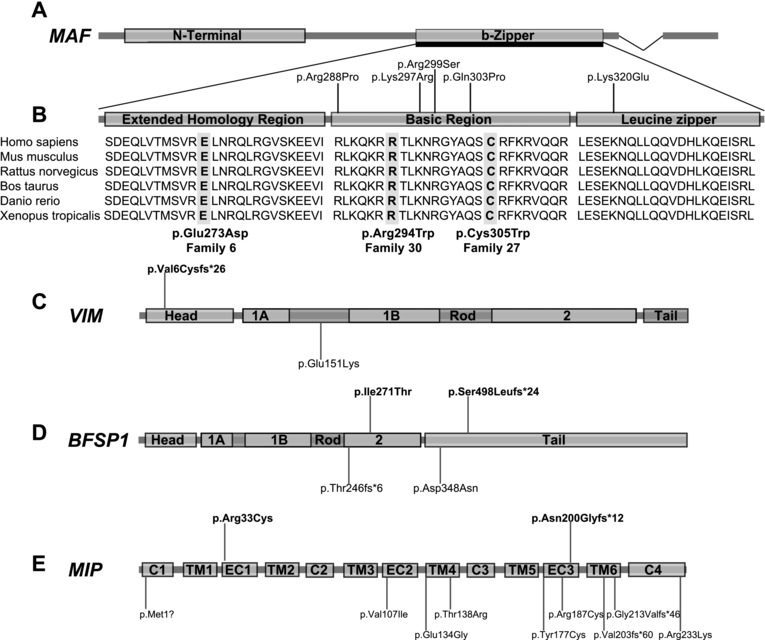
Novel mutations identified in MAF, VIM, BFSP1, and MIP. A: Schematic showing the encoded structure of MAF (NM_001031804.2). MAF has an N‐terminal and b‐zipper domain, and the detailed section of b‐ZIP reveals three evolutionarily conserved subdomains with the EHR, basic and leucine zipper regions. B: The b‐zipper domain is expanded to reveal these three subregions. Previously reported mutations in the basic and leucine zipper regions are demonstrated above the figure. The extensive amino acid conservation of the b‐zipper region is revealed, and the highly conserved three new mutations found in this study in MAF, highlighted in gray, from Families 6, 30, and 27. C: Schematic of encoded structure of VIM (NM_003380.3), which comprises a head, rod, and tail domain. The rod domain has three coil subdomains; 1A, 1B, and 2. A novel frameshift mutation was found in the proband of Family 25 (II:1), in the head domain (labeled above head domain). The only other mutation, a heterozygous missense mutation in the rod domain, is also marked, below the rod domain. D: Schematic of encoded structure of BFSP1 (NM_001195.4), which comprises a head, rod, and tail domain. Similar to VIM, the rod domain of BFSP1 also has coil subdomains 1A, 1B, and 2. Compound heterozygous mutations were found in the proband from Family 46, highlighted above the second coil and tail domains. The other mutations reported in the literature are labeled below the gene; a recessive frameshift mutation in the rod domain, and a heterozygous missense mutation in the tail domain in an autosomal‐dominant family. E: Schematic of encoded structure of MIP (NM_012064.3), which is a protein with four cytoplasmic (C) domains, six transmembrane (TM) domains, and three extracellular (EC) domains. The two mutations found in this study from Families 28 and 32 are illustrated above the gene, along with other mutations reported in the literature to date below the gene.
Novel Mutations in Lens Structural Genes VIM, BFSP1, and MIP
A novel heterozygous frameshift mutation was identified in the Intermediate Filament gene, VIM (c.15del p.(Val6Cysfs*26)) in Patient II.1, Family 25 (Supp. Fig. S1; Table 1; Figs. 4V and 6C). This is only the second reported mutation in VIM. Compound heterozygous mutations in BFSP1 were found in Family 46 (Supp. Fig. S2; Table 1), representing only the second recessive mutations reported in this gene (Fig. 6D). This family had two affected sons with cataract and microcornea from a consanguineous family. The proband was found to be compound heterozygous for mutations in BFSP1 (c.1492del p.(Ser498Leufs*24) and c.812T>C p.(Ile271Thr)). Segregation studies on the mother revealed she was heterozygous for the frameshift mutation, confirming that these are in trans. The other son and father were unavailable for study. A patient with sporadic cataracts (Patient II.2, Family 28) and a mother and daughter (Family 32) with autosomal‐dominant lamellar cataracts (Supp. Fig. S1; Table 1; Fig. 4U) were found to have heterozygous mutations in MIP (Fig. 6E). Our patient from Family 28 had a previously reported de novo missense mutation in MIP (c.97C>T p.(Arg33Cys)) [Gu et al., 2007], affecting the first extracellular loop of the protein (Fig. 6E). The affected individuals in Family 32 had a novel heterozygous variant in MIP, a 14‐bp insertion leading to a premature stop codon, c.597_598insGGGAACATTCCACT p.(Asn200Glyfs*12).
Discussion
This study highlights the benefits of an NGS approach with analysis of a targeted group of genes in the genetically heterogeneous condition of congenital cataracts. We achieved a mutation detection rate of over 70% in 46 sporadic and familial cases with apparently nonsyndromic, bilateral congenital cataracts. Similar detection rates were found in the sporadic and familial cases. Several novel mutations were found in a variety of genes including the crystallin and gap junction genes, more rarely reported disease genes including MAF, VIM, and MIP, and the syndrome‐associated genes, NHS and BCOR. This approach provides a significant advance for patients in provision of genetic diagnosis and new recurrence risk information, as well as identification of clinically unrecognized syndromic associations.
NGS Is a Powerful Tool in Sporadic and Familial Cataract Cases and in Identification of Subtle Syndromal Cases
Sporadic congenital cataract patients present diagnostic challenges when attempting to identify a genetic aetiology, and make up the majority of congenital cataract cases, since only around 18% of congenital cataract cases have a family history [Wirth et al., 2002]. While possible empiric‐based explanations for sporadic cases include de novo mutations in autosomal‐dominant genes, autosomal‐recessive inheritance, or X‐linked mutations, each of these possible modes of inheritance have vastly different recurrence risk likelihoods for the parents and affected individual, and are of limited use without a specific genetic diagnosis. Our study demonstrates that the majority of the mutations identified in sporadic bilateral congenital cataract cases were due to de novo heterozygous mutations in autosomal‐dominant genes (15/17, 88%) (Fig. 1B). In the remaining two sporadic cases, we found de novo X‐linked mutations in BCOR in females. This raises the possibility of asymptomatic or undiagnosed female carriers, which may not be easily diagnosed without the aid of an NGS approach. A high de novo mutation rate in this condition is important information for couples seeking reproductive information after an affected child. It indicates a low recurrence rate, with a small chance of gonadal mosaicism, and may decrease parental anxiety when planning for another child. Prenatal testing can be offered to exclude gonadal mosaicism, further enhancing reproductive confidence. In addition, probands with de novo heterozygous autosomal‐dominant mutations do themselves have a 50% chance of passing on their mutation to future offspring, whereas females with de novo X‐linked mutations are at risk of more severely affected male offspring or recurrent miscarriages in the case of BCOR mutations. In both cases, affected individuals may wish to utilize reproductive options such as prenatal testing or preimplantation genetic diagnosis. These results demonstrate that for sporadic cataracts, a targeted NGS strategy is useful for couples who have had an affected child, as well as the affected individuals themselves, to provide accurate recurrence and transmission risk counselling.
Similar difficulties exist in counseling families with familial cataracts, as pedigree information alone may not be accurate or extensive enough to provide accurate information for inheritance risk. In this study, three familial cases had revised genetic information due to the genetic results from this study (Table 1). This included a case of autosomal‐recessive GJA8 mutations (Family 14) and an X‐linked NHS mutation (Family 40) in families that appeared autosomal‐dominant on pedigree information. In Family 6, an unexpected answer with mutations in two different cataract genes, NHS and MAF, was found. The possibility of two entirely different genes responsible for the same condition running in a family is often overlooked, and we have now demonstrated this in Family 6, as well as in Family 7, where a BCOR mutation was found in the mother of this previously reported family, with a PAX6 mutation in other family members [Willcock et al., 2006]. These cases highlight the ability of an NGS‐based approach to provide accurate diagnosis that cannot be achieved through phenotyping alone.
This study has also shown the benefit of using an NGS approach in young patients and situations where syndromal diagnosis may not be clear‐cut or the clinical signs are still evolving. Male patients with NHS have bilateral severe congenital cataracts, and most have microcornea, as well as nystagmus, and glaucoma, which can worsen postoperatively [Ding et al., 2009]. Our findings in Families 6 and 40 highlight the need for increased clinical attention to this condition in young male patients where the full hand of clinical features may not be immediately obvious. Also, findings in the female carriers in these families demonstrated the variability and difficulty of clinical diagnosis in this condition where females may be entirely disease‐free or have mild lens opacities not obvious on initial review (Fig. 4C–E). Similarly, Families 7 and 44 highlight the importance of X‐linked syndromal diagnosis in females with BCOR mutations, associated with OFCD syndrome. Clinical features can be subtle especially in young patients, and cardiac status should be monitored.
NGS Identifies Novel Variants and Phenotypes Associated with Mutations in Crystallin and Gap Junction Genes, and in Rarely Reported Cataract Genes
Mutations in the crystallin and gap junction genes have a major role in maintaining lens transparency and were the most frequent mutations found in this study (Fig. 1). Two‐thirds (14/21) of these mutations were novel (Table 1), including two missense heterozygous mutations in CRYBB2 on the same allele in the proband from Family 35 with a severe phenotype (Fig. 2). Also, a striking novel phenotype of microphthalmia, sclerocornea, and cataracts was seen in the proband from Family 45, due to a novel missense mutation, p.(Asp51Asn) in GJA8 (Fig. 3).
This study also found six novel mutations in MAF, VIM, BFSP1, and MIP, highlighting the need to include these rarely reported genes in any multigene congenital cataract panel for examination of patients with this genetically heterogeneous condition. Three of these novel mutations were found in MAF (Fig. 6B). Up until this report, five mutations have been found in the basic region of MAF, in patients with isolated ocular abnormalities, all clustered around the b‐zipper domain [Jamieson et al., 2002, 2003; Vanita et al., 2006; Hansen et al., 2009; Narumi et al., 2014] (Fig. 6B). A variety of cataracts and ocular malformations have been reported in these patients including microcornea, iris coloboma, and ASD. The relative scarcity of reported mutations in this gene may reflect the difficulties in conventional Sanger sequencing, as it is very GC rich. In this report, we identified the first mutation affecting the EHR of MAF, a region that assists with specificity of DNA binding by MAF [Kerppola and Curran, 1994], as well as two novel mutations in the basic region of the DNA binding domain (Fig. 6B). We did not identify any mutations in the transactivating N‐terminal domain of MAF, where mutations have been recently reported in patients with syndromic cataracts, with additional features of deafness, intellectual disability, seizures, and dysmorphism [Niceta et al., 2015]. Our findings highlight an emerging genotype–phenotype correlation associated with MAF mutations, where heterozygous missense mutations in the b‐zipper domain are associated with isolated ocular findings, whereas heterozygous missense mutations in the N‐terminal transactivation domain are associated with a syndromic cataract phenotype.
We report the second cataract‐associated mutation in VIM in Family 25 (Figs. 4V and 6C). Vimentin is a highly conserved type III intermediate filament protein, expressed in mesenchymal cells and tissues including the lens [Herrmann and Aebi, 2004]. The previous Vimentin mutation is in a family with autosomal‐dominant pulverulent cataracts, c.596G>A p.(Glu151Lys) [Muller et al., 2009], and affects the alpha‐helical coil 1B rod segment of VIM. Cell‐based assays suggest this mutation leads to defects in Vimentin assembly [Muller et al., 2009]. In Family 25, a heterozygous frameshift mutation was found in the head domain. This mutation is predicted to lead to premature truncation of the protein proximal to the rod domain, therefore abolishing any functional protein.
In another lens structural protein with a similar domain structure, the beaded filament gene BFSP1, we report the second family with recessive mutations in this gene, in Family 46 (Fig. 6D). The beaded filament proteins BFSP1 and BFSP2 are exclusively expressed in the eye lens. They assemble together to form heteropolymeric filaments, to create the unique cytoskeletal structure of the beaded filament in all vertebrate lenses [Perng et al., 2007]. The first reported mutation was a recessive homozygous deletion of exon 6 [Ramachandran et al., 2007], predicted to lead to a frameshift and loss of part of the rod and full tail region of the protein p.(Thr246fs*6). The second was a heterozygous mutation in a five‐generation dominant Chinese family due to a missense mutation in exon 7 (c.1042G>A p.(Asp348Asn)) in the tail domain. Both mutations point toward the importance of the tail region in the protein's function. In our patient (Family 46), the missense mutation affects the rod domain and the frameshift is predicted to lead to loss of part of the tail domain, highlighting the critical nature of these regions in the functions of BFSP1 required for lens clarity (Fig. 6D).
While approximately 15 heterozygous missense, frameshift, and splice‐site mutations in the water channel gene MIP are reported in cataract patients [Shiels, 2012] (Fig. 6E), it is generally a less frequently investigated gene. In our study, we found two mutations in MIP, a previously reported mutation in a sporadic case (Family 28) and a novel frameshift mutation, p.(Asn200Glyfs*12), in a dominant family (Family 32, Fig. 4U). The frameshift mutation is the most proximal mutation yet identified in MIP, and would be expected to lead to complete loss of the sixth transmembrane domain and C‐terminal of this water transport protein (Fig. 6E).
NGS Affects Management in PAX6‐Associated Cataract Cases
In two congenital cataract cases in this study, mutations were identified in PAX6, leading to significant new information for the families and changes to management. Patient III.1 in Family 10 (Table 1; Figs. 4G–R and 5C; Supp. Fig. S1) was referred with his partner for prepregnancy genetic information, with possible diagnoses of autosomal‐dominant cataracts and congenital motor nystagmus. The diagnosis of a PAX6 frameshift mutation (c.1119del p.(Thr374Profs*5)) led to a unifying diagnosis explaining the patient's nystagmus and cataracts, and further clinical ophthalmic review revealed associated ophthalmic features of limbal stem cell failure and ectropion uvea, features previously identified in a family with “run‐on” PAX6 mutation affecting the stop codon [Willcock et al., 2006]. He also had subtle hypoplasia of the irides and loss of the foveal pit confirmed with optical coherence tomography (OCT) (Fig. 4G, J, M, and P). His affected mother was found to have the same mutation as well as the additional complication of glaucoma. Both the mother (Fig. 4H, K, N, and Q) and sister (Fig. 4I, L, O, and R) were also found subsequently to have evidence of iris and foveal hypoplasia on fundoscopy and OCT. Identification of a heterozygous PAX6 mutation in this family has made a significant difference in their management, with regular monitoring for PAX6‐associated complications such as glaucoma, limbal stem cell‐associated corneal abnormalities, and foveal hypoplasia, which all have a significant impact on long‐term vision. This result also clarified the recurrence risk advice for affected family members for this autosomal‐dominant condition. The proband in Family 18 had a de novo missense mutation in the paired domain of PAX6 (Figs. 4F and 5C), adding to the increasing evidence in the literature that missense mutations in the paired domain of PAX6 can lead to a milder nonaniridia phenotype [Park et al., 2012], possibly by affecting DNA binding to transcriptional targets. Similar to the PAX6 frameshift family, this result has important implications for this patient. He had surgery for a large exotropia before his molecular diagnosis, and following surgery he has persistent diplopia. This failure of fusion may be related to his underlying pan‐ocular disorder due to his PAX6 mutation.
Benefits of Higher Depth and Evenness of Coverage in Cataract Mutation Detection
Our targeted NGS approach highlights the benefits of improved evenness and depth of coverage to maximize the mutation detection rate. The capture‐based TruSight library approach had a lower average coverage, but this was more even with fewer and smaller gaps, compared with the TruSeq amplicon‐based approach (Supp. Table S3). Using the expanded TruSight One library, five additional mutations were found in the 17 original “TruSeq Negative” patients, including in key genes with gaps on the amplicon approach including BCOR, GJA3, and MAF. The higher average coverage and read depth, able to be achieved economically with a targeted approach, led to a greater detection rate than a previous study using WES [Reis et al., 2013] in familial congenital cataract patients (mutations found in 39% of families). A study using targeted NGS in a cohort with more consanguineous and overtly syndromic congenital cataract patients [Gillespie et al., 2014] compared with our study, found a similar mutation detection rate to our study. Our work shows that apparently nonsyndromic, sporadic, and familial cases of congenital cataracts, also benefit from a targeted NGS approach for improved diagnosis and management.
Conclusion
This study highlights the clinical utility of targeted NGS in the setting of genetically heterogeneous congenital cataract patients. The 70% mutation detection rate shows that NGS is a useful test for both familial and sporadic apparently nonsyndromic cases. This has led to revision of diagnoses, accurate recurrence risk counseling, with an impact on management for each family. Overall, it demonstrates that in patients with congenital cataracts, whether sporadic or familial, a NGS‐based test provides significant additional diagnostic information and is warranted to improve management and provide accurate genetic counseling.
Supporting information
Disclaimer: Supplementary materials have been peer‐reviewed but not copyedited.
Supporting Information
Acknowledgment
We would like to thank the families involved in this study, and the supporting clinicians and molecular genetics staff for their time and efforts.
Contract grant sponsor: Ophthalmic Research Institute of Australia.
Communicated by Daniel Schorderet
References
- Abecasis GR, Auton A, Brooks LD, DePristo MA, Durbin RM, Handsaker RE, Kang HM, Marth GT, McVean GA. 2012. An integrated map of genetic variation from 1,092 human genomes. Nature 491:56–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. 2010. A method and server for predicting damaging missense mutations. Nat Methods 7:248–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bateman JB, von‐Bischhoffshaunsen FR, Richter L, Flodman P, Burch D, Spence MA. 2007. Gene conversion mutation in crystallin, beta‐B2 (CRYBB2) in a Chilean family with autosomal dominant cataract. Ophthalmology 114:425–432. [DOI] [PubMed] [Google Scholar]
- Bennett TM, Mackay DS, Knopf HL, Shiels A. 2004. A novel missense mutation in the gene for gap‐junction protein alpha3 (GJA3) associated with autosomal dominant "nuclear punctate" cataracts linked to chromosome 13q. Mol Vis 10:376–382. [PubMed] [Google Scholar]
- Beyer EC, Ebihara L, Berthoud VM. 2013. Connexin mutants and cataracts. Front Pharmacol 4:43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burdon KP, Wirth MG, Mackey DA, Russell‐Eggitt IM, Craig JE, Elder JE, Dickinson JL, Sale MM. 2004. Investigation of crystallin genes in familial cataract, and report of two disease associated mutations. Br J Ophthalmol 88:79–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devi RR, Yao W, Vijayalakshmi P, Sergeev YV, Sundaresan P, Hejtmancik JF. 2008. Crystallin gene mutations in Indian families with inherited pediatric cataract. Mol Vis 14:1157–1170. [PMC free article] [PubMed] [Google Scholar]
- Ding X, Patel M, Herzlich AA, Sieving PC, Chan CC. 2009. Ophthalmic pathology of Nance‐Horan syndrome: case report and review of the literature. Ophthalmic Genet 30:127–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster A, Gilbert C, Rahi J. 1997. Epidemiology of cataract in childhood: a global perspective. J Cataract Refract Surg 23 Suppl 1:601–604. [DOI] [PubMed] [Google Scholar]
- Ge XL, Zhang Y, Wu Y, Lv J, Zhang W, Jin ZB, Qu J, Gu F. 2014. Identification of a novel GJA8 (Cx50) point mutation causes human dominant congenital cataracts. Sci Rep 4:4121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert C, Foster A. 2001. Childhood blindness in the context of VISION 2020—the right to sight. Bull World Health Organ 79:227–232. [PMC free article] [PubMed] [Google Scholar]
- Gilbert CE, Canovas R, Hagan M, Rao S, Foster A. 1993. Causes of childhood blindness: results from west Africa, south India and Chile. Eye (Lond) 7:184–188. [DOI] [PubMed] [Google Scholar]
- Gill D, Klose R, Munier FL, McFadden M, Priston M, Billingsley G, Ducrey N, Schorderet DF, Heon E. 2000. Genetic heterogeneity of the Coppock‐like cataract: a mutation in CRYBB2 on chromosome 22q11.2. Invest Ophthalmol Vis Sci 41:159–165. [PubMed] [Google Scholar]
- Gillespie RL, O'Sullivan J, Ashworth J, Bhaskar S, Williams S, Biswas S, Kehdi E, Ramsden SC, Clayton‐Smith J, Black GC, Lloyd IC. 2014. Personalized diagnosis and management of congenital cataract by next‐generation sequencing. Ophthalmology 121:2124–2137e2. [DOI] [PubMed] [Google Scholar]
- Graw J. 2009. Genetics of crystallins: cataract and beyond. Exp Eye Res 88:173–189. [DOI] [PubMed] [Google Scholar]
- Gu F, Zhai H, Li D, Zhao L, Li C, Huang S, Ma X. 2007. A novel mutation in major intrinsic protein of the lens gene (MIP) underlies autosomal dominant cataract in a Chinese family. Mol Vis 13:1651–1656. [PubMed] [Google Scholar]
- Guleria K, Vanita V, Singh D, Singh JR. 2007. A novel "pearl box" cataract associated with a mutation in the connexin 46 (GJA3) gene. Mol Vis 13:797–803. [PMC free article] [PubMed] [Google Scholar]
- Hansen L, Mikkelsen A, Nurnberg P, Nurnberg G, Anjum I, Eiberg H, Rosenberg T. 2009. Comprehensive mutational screening in a cohort of Danish families with hereditary congenital cataract. Invest Ophthalmol Vis Sci 50:3291–3303. [DOI] [PubMed] [Google Scholar]
- Hansen L, Yao W, Eiberg H, Kjaer KW, Baggesen K, Hejtmancik JF, Rosenberg T. 2007. Genetic heterogeneity in microcornea‐cataract: five novel mutations in CRYAA, CRYGD, and GJA8. Invest Ophthalmol Vis Sci 48:3937–3944. [DOI] [PubMed] [Google Scholar]
- Hejtmancik JF. 2008. Congenital cataracts and their molecular genetics. Semin Cell Dev Biol 19:134–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrmann H, Aebi U. 2004. Intermediate filaments: molecular structure, assembly mechanism, and integration into functionally distinct intracellular Scaffolds. Annu Rev Biochem 73:749–789. [DOI] [PubMed] [Google Scholar]
- Hilal L, Nandrot E, Belmekki M, Chefchaouni M, El Bacha S, Benazzouz B, Hajaji Y, Gribouval O, Dufier J, Abitbol M, Berraho A. 2002. Evidence of clinical and genetic heterogeneity in autosomal dominant congenital cerulean cataracts. Ophthalmic Genet 23:199–208. [DOI] [PubMed] [Google Scholar]
- Huang B, He W. 2010. Molecular characteristics of inherited congenital cataracts. Eur J Med Genet 53:347–357. [DOI] [PubMed] [Google Scholar]
- Huang KM, Wu J, Brooks SP, Hardcastle AJ, Lewis RA, Stambolian D. 2007. Identification of three novel NHS mutations in families with Nance‐Horan syndrome. Mol Vis 13:470–474. [PMC free article] [PubMed] [Google Scholar]
- Jamieson RV, Munier F, Balmer A, Farrar N, Perveen R, Black GC. 2003. Pulverulent cataract with variably associated microcornea and iris coloboma in a MAF mutation family. Br J Ophthalmol 87:411–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamieson RV, Perveen R, Kerr B, Carette M, Yardley J, Heon E, Wirth MG, van Heyningen V, Donnai D, Munier F, Black GC. 2002. Domain disruption and mutation of the bZIP transcription factor, MAF, associated with cataract, ocular anterior segment dysgenesis and coloboma. Hum Mol Genet 11:33–42. [DOI] [PubMed] [Google Scholar]
- Kerppola TK, Curran T. 1994. A conserved region adjacent to the basic domain is required for recognition of an extended DNA binding site by Maf/Nrl family proteins. Oncogene 9:3149–3158. [PubMed] [Google Scholar]
- Kumar M, Agarwal T, Khokhar S, Kaur P, Roy TS, Dada R. 2011. Mutation screening and genotype phenotype correlation of alpha‐crystallin, gamma‐crystallin and GJA8 gene in congenital cataract. Mol Vis 17:693–707. [PMC free article] [PubMed] [Google Scholar]
- Liao HM, Niu DM, Chen YJ, Fang JS, Chen SJ, Chen CH. 2011. Identification of a microdeletion at Xp22.13 in a Taiwanese family presenting with Nance‐Horan syndrome. J Hum Genet 56:8–11. [DOI] [PubMed] [Google Scholar]
- Litt M, Carrero‐Valenzuela R, LaMorticella DM, Schultz DW, Mitchell TN, Kramer P, Maumenee IH. 1997. Autosomal dominant cerulean cataract is associated with a chain termination mutation in the human beta‐crystallin gene CRYBB2. Hum Mol Genet 6:665–668. [DOI] [PubMed] [Google Scholar]
- Lou D, Tong JP, Zhang LY, Chiang SW, Lam DS, Pang CP. 2009. A novel mutation in CRYBB2 responsible for inherited coronary cataract. Eye (Lond) 23:1213–1220. [DOI] [PubMed] [Google Scholar]
- Ma ZW, Zheng JQ, Li J, Li XR, Tang X, Yuan XY, Zhang XM, Sun HM. 2005. Two novel mutations of connexin genes in Chinese families with autosomal dominant congenital nuclear cataract. Br J Ophthalmol 89:1535–1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minogue PJ, Tong JJ, Arora A, Russell‐Eggitt I, Hunt DM, Moore AT, Ebihara L, Beyer EC, Berthoud VM. 2009. A mutant connexin50 with enhanced hemichannel function leads to cell death. Invest Ophthalmol Vis Sci 50:5837–5845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muhit M, Gilbert C. 2003. A review of the epidemiology and control of childhood blindness. Trop Doct 33:197–201. [DOI] [PubMed] [Google Scholar]
- Muller M, Bhattacharya SS, Moore T, Prescott Q, Wedig T, Herrmann H, Magin TM. 2009. Dominant cataract formation in association with a vimentin assembly disrupting mutation. Hum Mol Genet 18:1052–1057. [DOI] [PubMed] [Google Scholar]
- Narumi Y, Nishina S, Tokimitsu M, Aoki Y, Kosaki R, Wakui K, Azuma N, Murata T, Takada F, Fukushima Y, Kosho T. 2014. Identification of a novel missense mutation of MAF in a Japanese family with congenital cataract by whole exome sequencing: a clinical report and review of literature. Am J Med Genet A 164:1272–1276. [DOI] [PubMed] [Google Scholar]
- Ng PC, Henikoff S. 2001. Predicting deleterious amino acid substitutions. Genome Res 11:863–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niceta M, Stellacci E, Gripp KW, Zampino G, Kousi M, Anselmi M, Traversa A, Ciolfi A, Stabley D, Bruselles A, Caputo V, Cecchetti S, et al. 2015. Mutations impairing GSK3‐mediated MAF phosphorylation cause cataract, deafness, intellectual disability, seizures, and a down syndrome‐like facies. Am J Hum Genet 96:816–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SH, Kim MS, Chae H, Kim Y, Kim M. 2012. Molecular analysis of the PAX6 gene for congenital aniridia in the Korean population: identification of four novel mutations. Mol Vis 18:488–494. [PMC free article] [PubMed] [Google Scholar]
- Perng MD, Zhang Q, Quinlan RA. 2007. Insights into the beaded filament of the eye lens. Exp Cell Res 313:2180–2188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peter NM, Leyland M, Mudhar HS, Lowndes J, Owen KR, Stewart H. 2013. PAX6 mutation in association with ptosis, cataract, iris hypoplasia, corneal opacification and diabetes: a new variant of familial aniridia? Clin Exp Ophthalmol 41:835–841. [DOI] [PubMed] [Google Scholar]
- Pollard KS, Hubisz MJ, Rosenbloom KR, Siepel A. 2010. Detection of nonneutral substitution rates on mammalian phylogenies. Genome Res 20:110–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponnam SP, Ramesha K, Matalia J, Tejwani S, Ramamurthy B, Kannabiran C. 2013. Mutational screening of Indian families with hereditary congenital cataract. Mol Vis 19:1141–1148. [PMC free article] [PubMed] [Google Scholar]
- Ponnam SP, Ramesha K, Tejwani S, Ramamurthy B, Kannabiran C. 2007. Mutation of the gap junction protein alpha 8 (GJA8) gene causes autosomal recessive cataract. J Med Genet 44:e85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prokudin I, Simons C, Grigg JR, Storen R, Kumar V, Phua ZY, Smith J, Flaherty M, Davila S, Jamieson RV. 2014. Exome sequencing in developmental eye disease leads to identification of causal variants in GJA8, CRYGC, PAX6 and CYP1B1. Eur J Hum Genet 22:907–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramachandran RD, Perumalsamy V, Hejtmancik JF. 2007. Autosomal recessive juvenile onset cataract associated with mutation in BFSP1. Hum Genet 121:475–482. [DOI] [PubMed] [Google Scholar]
- Reilich P, Schoser B, Schramm N, Krause S, Schessl J, Kress W, Muller‐Hocker J, Walter MC, Lochmuller H. 2010. The p.G154S mutation of the alpha‐B crystallin gene (CRYAB) causes late‐onset distal myopathy. Neuromuscul Disord 20:255–259. [DOI] [PubMed] [Google Scholar]
- Reis LM, Tyler RC, Muheisen S, Raggio V, Salviati L, Han DP, Costakos D, Yonath H, Hall S, Power P, Semina EV. 2013. Whole exome sequencing in dominant cataract identifies a new causative factor, CRYBA2, and a variety of novel alleles in known genes. Hum Genet 132:761–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riazuddin SA, Yasmeen A, Yao W, Sergeev YV, Zhang Q, Zulfiqar F, Riaz A, Riazuddin S, Hejtmancik JF. 2005. Mutations in betaB3‐crystallin associated with autosomal recessive cataract in two Pakistani families. Invest Ophthalmol Vis Sci 46:2100–2106. [DOI] [PubMed] [Google Scholar]
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier‐Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm H, et al. 2015. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17:405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santhiya ST, Kumar GS, Sudhakar P, Gupta N, Klopp N, Illig T, Soker T, Groth M, Platzer M, Gopinath PM, Graw J. 2010. Molecular analysis of cataract families in India: new mutations in the CRYBB2 and GJA3 genes and rare polymorphisms. Mol Vis 16:1837–1847. [PMC free article] [PubMed] [Google Scholar]
- Schmidt W, Klopp N, Illig T, Graw J. 2008. A novel GJA8 mutation causing a recessive triangular cataract. Mol Vis 14:851–856. [PMC free article] [PubMed] [Google Scholar]
- Schwarz JM, Rodelsperger C, Schuelke M, Seelow D. 2010. MutationTaster evaluates disease‐causing potential of sequence alterations. Nat Methods 7:575–576. [DOI] [PubMed] [Google Scholar]
- Shiels A. 2012. Focus on molecules: major intrinsic protein. Exp Eye Res 101:107–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiels A, Bennett TM, Hejtmancik JF. 2010. Cat‐Map: putting cataract on the map. Mol Vis 16:2007–2015. [PMC free article] [PubMed] [Google Scholar]
- Shiels A, Hejtmancik JF. 2007. Genetic origins of cataract. Arch Ophthalmol 125:165–173. [DOI] [PubMed] [Google Scholar]
- Slavotinek AM. 2011. Eye development genes and known syndromes. Mol Genet Metab 104:448–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smigielski EM, Sirotkin K, Ward M, Sherry ST. 2000. dbSNP: a database of single nucleotide polymorphisms. Nucleic Acids Res 28:352–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun W, Xiao X, Li S, Guo X, Zhang Q. 2011. Mutation analysis of 12 genes in Chinese families with congenital cataracts. Mol Vis 17:2197–2206. [PMC free article] [PubMed] [Google Scholar]
- Swamy BN, Billson F, Martin F, Donaldson C, Hing S, Jamieson R, Grigg J, Smith JE. 2007. Secondary glaucoma after paediatric cataract surgery. Br J Ophthalmol 91:1627–1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanita V, Singh D, Robinson PN, Sperling K, Singh JR. 2006. A novel mutation in the DNA‐binding domain of MAF at 16q23.1 associated with autosomal dominant "cerulean cataract" in an Indian family. Am J Med Genet A 140:558–566. [DOI] [PubMed] [Google Scholar]
- Vanita, Sarhadi V, Reis A, Jung M, Singh D, Sperling K, Singh JR, Burger J. 2001. A unique form of autosomal dominant cataract explained by gene conversion between beta‐crystallin B2 and its pseudogene. J Med Genet 38:392–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vendra VP, Agarwal G, Chandani S, Talla V, Srinivasan N, Balasubramanian D. 2013. Structural integrity of the Greek key motif in betagamma‐crystallins is vital for central eye lens transparency. PLoS One 8:e70336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K, Li M, Hakonarson H. 2010. ANNOVAR: functional annotation of genetic variants from high‐throughput sequencing data. Nucleic Acids Res 38:e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willcock C, Grigg J, Wilson M, Tam P, Billson F, Jamieson R. 2006. Congenital iris ectropion as an indicator of variant aniridia. Br J Ophthalmol 90:658–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wirth MG, Russell‐Eggitt IM, Craig JE, Elder JE, Mackey DA. 2002. Aetiology of congenital and paediatric cataract in an Australian population. Br J Ophthalmol 86:782–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia CH, Liu H, Cheung D, Cheng C, Wang E, Du X, Beutler B, Lo WK, Gong X. 2006. Diverse gap junctions modulate distinct mechanisms for fiber cell formation during lens development and cataractogenesis. Development 133:2033–2040. [DOI] [PubMed] [Google Scholar]
- Yao K, Jin C, Zhu N, Wang W, Wu R, Jiang J, Shentu X. 2008. A nonsense mutation in CRYGC associated with autosomal dominant congenital nuclear cataract in a Chinese family. Mol Vis 14:1272–1276. [PMC free article] [PubMed] [Google Scholar]
- Yao K, Tang X, Shentu X, Wang K, Rao H, Xia K. 2005. Progressive polymorphic congenital cataract caused by a CRYBB2 mutation in a Chinese family. Mol Vis 11:758–763. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Disclaimer: Supplementary materials have been peer‐reviewed but not copyedited.
Supporting Information