

Abstract
Undernourished children exhibit impaired gut microbiota development. Transplanting microbiota from 6- and 18-month old healthy or undernourished Malawian donors into young germ-free mice fed a Malawian diet revealed that immature microbiota from undernourished infants/children transmit impaired growth phenotypes. The representation of several age-discriminatory taxa in recipient animals correlated with lean body mass gain, liver, muscle, and brain metabolism, plus bone morphology. Co-housing mice shortly after receiving microbiota from healthy (H) or severely stunted/underweight (Un) infants demonstrated that invasion of age-/growth-discriminatory taxa from H to Un cagemates’ microbiota ameliorates growth faltering. Adding two invasive species, Ruminococcus gnavus and Clostridium symbiosum, to the Un microbiota also ameliorated growth and metabolic abnormalities. These results provide evidence that microbiota immaturity is causally related to undernutrition, and reveal potential therapeutic targets and agents.
Undernutrition is a leading cause of infant and childhood mortality worldwide (1–5). The mechanisms that underlie disease, ranging from persistent abnormalities in growth, immune function, to cognitive deficits, remain obscure. Childhood undernutrition is not due to food insecurity alone, but also results from a combination of factors including diets with low nutrient density/bioavailability, pathogen burden and gut mucosal barrier dysfunction (6–8).
The World Health Organization (WHO) has used a cohort of 8440 healthy children living in six countries to develop anthropometric standards that define nutritional status [weight-for-height Z-Score (WHZ), weight-for-age Z-Score (WAZ), and height-for-age Z-Score (HAZ)] (9, 10). A recent study (11) of infants and children with healthy growth phenotypes living in Mirpur, an urban slum in Dhaka, Bangladesh, involved monthly fecal collection from birth through the end of the second year of life. In this study, 16S rRNA analysis of bacterial membership in their gut microbiota and application of a machine learning method [Random Forests (12)] revealed 24 ‘age-discriminatory’ taxa whose changes in relative abundance over time defined a program of normal ‘maturation’ of the microbiota across biologically unrelated individuals (11). This model served as the basis for computing two related metrics, ‘relative microbiota maturity’ and microbiota-for-age Z-score (MAZ), that significantly correlated with the chronological age of children with healthy growth phenotypes. Applying these metrics revealed that children living in Mirpur with moderate acute malnutrition (MAM) or severe acute malnutrition (SAM) have gut microbiota that are ‘immature’; in other words, the representation of the age-discriminatory taxa in their gut communities was more similar to younger rather than age-matched healthy individuals from the same locale. Moreover, the degree of this ‘immaturity’ was greater in SAM than MAM (11). Treatment of children with SAM with either one of two ready-to-use therapeutic foods (RUTFs) produced incomplete and only transient improvement in this immaturity, and no improvement in their HAZ scores (11).
These findings raise several key questions. To what extent are these age-discriminatory taxa also indicative of the normal development of the gut microbiota of infants and children elsewhere? Are the age-discriminatory taxa biomarkers of gut microbiota development or are they mediators of healthy growth? If the latter, can their introduction into immature microbiota prevent disease?
A Malawian model of gut microbiota development
To address these questions, we first developed a Random Forests-based model of gut microbial community development from a serially sampled cohort of Malawian twins concordant for healthy growth. We investigated correlations between nutritional status and microbiota maturation to ascertain whether an MAZ score could predict future growth performance.
In a previous study, we followed a cohort of 317 twin pairs and three sets of triplets, living in five rural villages in southern Malawi from birth to 36 months of age, with periodic sampling of their fecal microbiota (13). Thus, to define healthy microbiota development in this population, we took the PCR amplicons generated from variable region 4 (V4) of bacterial 16S rRNA genes and sequenced them. We used 220 fecal samples collected from 27 twin pairs and two sets of triplets whose serial anthropometric measurements were indicative of consistently healthy growth [WHZ 0.09±0.93; fecal collection over an age range of 0.6 to 33.5 months; 3.96±1.6 samples (mean±SD)/individual; see Tables S1A and S2A for microbiota donor characteristics and a summary of sequencing datasets]. V4-16S rRNA reads with ≥97% nucleotide sequence identity were grouped into operational taxonomic units (97%ID OTUs) and taxonomic assignments were made (14, 15).
We modeled microbiota maturation in these healthy Malawian infants/children by regressing OTUs against chronological age in a subset (‘training set’) of the healthy twin cohort using Random Forests (Fig. 1A). Application of the training set-based model to the subset excluded from model building (‘test set’) yielded a positive and significant correlation between host chronological age and predicted microbiota age (r2=0.80, p<0.0001) (Fig. 1C, Fig. S1). The model was further validated using an additional randomized nutritional interventional trial that took place in the Mangochi district of southern Malawi and where subjects were studied at 6, 12, and 18 months of age (iLiNS-DYAD-M) (16). We again saw a positive and significant correlation between chronological age and predicted microbiota age (r2=0.71, p<0.0001). Remarkably, multiple OTUs from the sparse Malawian model had ≥97% sequence identity to OTUs in the sparse Bangladeshi model (Fig. 1A, Table S3B) (11).
Fig. 1. Sparse Random Forests-derived model of gut microbiota maturation obtained from concordant healthy Malawian twins/triplets.
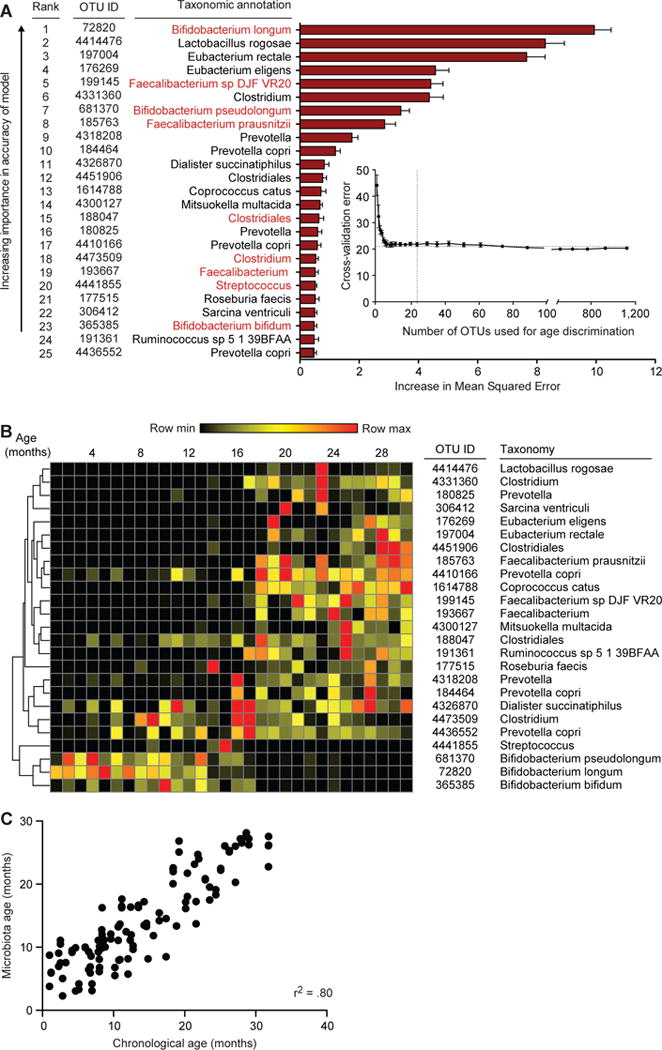
(A) Random-Forests regression of fecal bacterial 97%ID OTUs from a training set of healthy Malawian infants/children (n=31) to chronological age yielded a rank order of age-discriminatory taxa. Random Forests assigns a mean squared error (MSE), or feature importance score, to each OTU that indicates the extent to which each OTU contributes to the accuracy of the model. The 25 most age-discriminatory taxa, ranked by MSE, yielded a sparse model that predicted microbiota age and accounted for ~80% of the observed variance in the healthy cohort (see Table S3A for a complete list of OTUs and MSE values). The top 25 most discriminatory OTUs with their taxonomic assignments are shown ranked by feature importance (mean±SD of the MSE). The insert shows the results of 10-fold cross-validation; as OTUs are added to the model in order of their feature importance rank, the model’s error decreases. Taxa highlighted in red indicate OTUs with >97% nucleotide sequence identity with an OTU present in a sparse Random Forests-based model of microbiota maturation in healthy Bangladeshi infants/children (11). (B) A heatmap of changes over time in relative abundances of the 25 OTUs in fecal microbiota collected from healthy Malawian infants/children comprising the test set (n=29). OTUs are hierarchically clustered according to pairwise distances by Pearson correlation. (C) Predictions of chronological age using the sparse 25 OTU model of microbiota age in healthy children comprising the test set cohort. r2 calculated from Pearson correlation.
We then used the Malawian Random Forests-derived model to analyze the relationship between microbiota maturity and growth/nutritional status in 259 children enrolled in iLiNS-DYAD-M who had fewer than three reported days of antibiotic consumption from 6 to 18 months of age (Table S1B). There was a significant positive correlation between their MAZ scores and weight-for-height Z-scores (ρ=0.1664, p=0.0073, Spearman’s rank correlation) as well as weight-for-age Z-scores (ρ=0.1715, p=0.0056) but not their height-for-age Z-scores (ρ=0.1022, p=0.1) (Table S2B). There was a significant correlation between MAZ at 12 months and anthropometry at 18 months [Spearman’s rank correlation ρ=0.1406 (p=0.02) for WHZ and ρ=0.1373 (p=0.02) for WAZ] [Note that ρ=0.1184 (p=0.05) for HAZ]. These results suggest that MAZ may be useful for predicting future (ponderal) growth. Further studies and analyses are required to discern the effects of several variables, such as duration of prior antibiotic use, enteropathogen load, number of diarrheal days, geography, and various nutritional interventions, on the relationship between MAZ measurements at various postnatal ages, anthropometry and other metrics of healthy growth (e.g., cognitive testing and immunization responses).
Identification of age-discriminatory taxa that are growth-discriminatory
To test whether there is a causal relationship between microbiota maturity and growth, we selected fecal samples from 19 Malawian infants representing either healthy or undernourished growth phenotypes for transplantation into 5-week old, actively growing germ-free C57Bl/6J mice. Of the 19 donor samples, nine were from 6-month old infants [four classified as healthy with WHZ 1.45±0.57 (mean±SD), WAZ 1.75±0.56, and HAZ 1.27±0.45, and five moderately or severely underweight and stunted (WHZ −1.27±0.49, WAZ −3.90±1.82 and HAZ −4.36 ±2.07)]. Ten samples were from 18-month old children, four with healthy weights (WHZ 1.44±0.08, WAZ −0.3±0.57 and HAZ −2.84±0.99) and six who were moderately or severely underweight and stunted (WHZ −1.75±0.17, WAZ −2.88±0.42 and HAZ −3.28±0.82) (see Table S4A for MAZ metrics; note that all 18-month old donors were members of twin pairs where HAZ scores are typically lower than in singletons, and all 6-month old donors were singletons). Fecal samples from the 18-month old children were from the Malawi twin cohort while the 6-month old microbiota donors were members of the iLiNS-DYAD-M study who had not yet received a nutritional supplement.
Each microbiota sample was transplanted into a separate group of 5-week old male germ-free mice (n=5 animals/sample). Three days before transplantation, all mice were switched onto a sterile (irradiated) Malawian diet formulated based on the results of a dietary survey of the complementary feeding practices for 9-month old Malawian children enrolled in the iLiNS-DOSE study (#NCT00945698) that took place in the Mangochi district. We selected eight ingredients to produce a cooked, representative Malawian diet consumed by children (‘M8’; see Materials and Methods); its micro- and macronutrient content does not fulfill the needs of humans or mice (see Table S5A,B for a list of ingredients and the results of a direct nutritional analysis).
Following a single gavage of the donor microbiota, recipient mice were followed for 4–5 weeks (see Fig. 2A for experimental design). Fecal samples were collected weekly for bacterial V4-16S rRNA analysis. Growth was monitored by serial measurements of total body weight and body composition [lean mass and fat mass as defined by quantitative magnetic resonance (qMR)], while following euthanasia femurs were removed for micro-computed tomographic (micro-CT) characterization of bone morphology.
Fig. 2. Transplantation of microbiota from 6- and 18-month old donors to young germ-free mice provides evidence of a causal relationship between gut microbiota maturity and growth phenotypes.
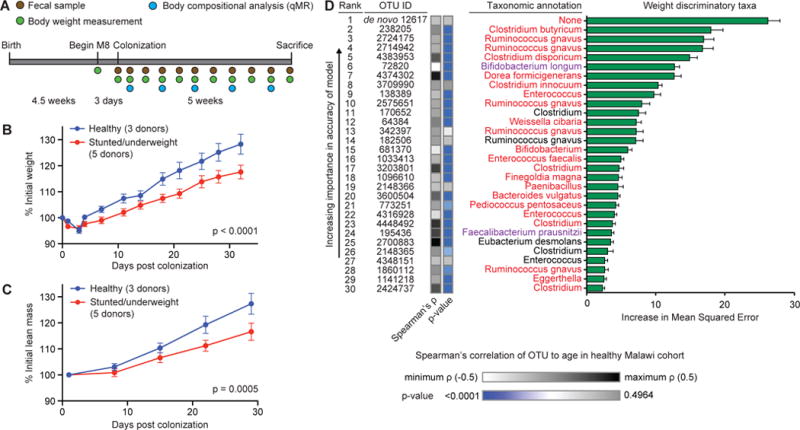
(A) Experimental design of microbiota screen. Mice (4.5-weeks old) were switched to the M8 diet three days prior to gavage with the selected microbiota donor’s fecal sample (n=5 mice per donor). Fecal samples, body weight and body composition were defined at the indicated time. (B,C) Gnotobiotic mice colonized with fecal samples from healthy children gain more total body weight (panel B) and lean mass (panel C) than mice colonized with microbiota from undernourished donors (mean±SEM shown; p-values shown for donor status effect based on 2-way ANOVA). All recipient mice harbor microbiota that represent >50% of OTU diversity present in the intact uncultured donor’s sample. (D) The 30 most weight-gain discriminatory OTUs and their taxonomic assignments, ranked by feature importance (mean MSE±SD values are plotted). The weight gain model explained ~66% of the observed phenotypic variation (p<0.0001, permutation test; 999 permutations). Taxa in red indicate OTUs that appear within the 30 most discriminatory OTUs for both the weight and lean mass gain Random Forests-based models. Taxa in purple indicate species that appear in the 25-member sparse Random Forests-derived model of Malawian gut microbiota maturation. Bars to the right of the OTU ID numbers represent Spearman’s rank correlation of the same OTU ID to chronological age within the healthy Malawian infant/child cohort (see Table S9).
Mice harboring donor microbiota from healthy infants (n=8) gained significantly more weight and lean body mass than mice colonized with microbiota from undernourished donors (n=11) (p<0.0001 and p=0.0001, respectively; 2-way ANOVA; Tables S6A,B), yet there was no significant difference in food consumption between the groups (p>0.05, Student’s t-test, Table S7). 16S rRNA sequencing of fecal samples obtained from recipient mice revealed variable transplantation efficiency. Of the 19 donor samples, eight produced transplantation efficiencies of ≥50%. When we restricted our analysis to the eight donor samples producing >50% transplantation efficiency, the discordant growth phenotypes between recipients of healthy versus undernourished microbiota were pronounced (weight gain, p<0.0001; lean mass gain, p=0.0005; 2-way ANOVA). Importantly, there was no significant difference in fat mass (p=0.78, 2-way ANOVA, Table S6B) or food consumption (p>0.05, Student’s t-test; Fig. 2B,C; Tables S6A,B, and S7) between the groups.
For both the 6-month and 18-month age bins, mice colonized with healthy donor microbiota gained significantly more total body weight and lean mass than those colonized with undernourished donor microbiota (weight for recipients of the 6- and 18-month donor microbiota, p=0.0003 and p=0.0043, respectively; lean body mass, p=0.03 and p=0.0013, respectively; 2-way ANOVA; Tables S6A,B). Recipients of the healthy and undernourished 6-month old donors’ fecal samples both grew more than recipients of the corresponding 18-month old healthy and undernourished donor microbiota (p<0.0001 for healthy; p<0.0001 for undernourished, based on lean mass gain; 2-way ANOVA).
Growth over the course of the 5-week experiment in the 19 different groups of recipient mice ranged from 105% to 152% of starting weight (averaged per group; Table S6A). There was no significant relationship between growth phenotypes and bacterial diversity in the fecal microbiota of recipient animals (Tables S6A,B). We applied Random Forests to regress the growth phenotypes of recipient gnotobiotic mice against 97%ID OTUs identified in their fecal microbiota. Two models were generated, one based on weight gain, the other based on lean body mass gain (see Fig. 2D and Fig. S2, respectively, plus Table S8A,B). Two of the growth-discriminatory species represented in the weight and lean mass gain Random Forests-based models, Bifidobacterium longum and Faecalibacterium prausnitzii, were highly age-discriminatory, ranking 1st and 8th, respectively, in the Malawian model shown in Fig. 1A, and 5th and 1st in the Bangladeshi model (11). In the healthy Malawian and Bangladeshi populations used to create these models, B. longum is an early colonizer with highest mean relative abundance at 5 months of age, while F. prausnitzii becomes more prominent later (highest mean relative abundance achieved at 19 months) (see Fig. 1B (11)).
The relative abundances of 13 OTUs with significant correlations to weight gain- and seven OTUs with significant correlations to lean mass gain also had significant correlations to chronological age in concordant healthy Malawian twins/triplets, including three OTUs for F. prausnitzii (p<0.0001; see Fig. 2D, Fig. S2, plus Table S9 for a complete list of OTUs with their Spearman’s rank correlation ρ–values). Moreover, 15 OTUs positively and significantly correlated to weight gain and 11 OTUs positively and significantly correlated to lean mass gain were also significantly correlated with chronological age for the 259 infants/children from iLiNS-DYAD-M, including the same three OTUs for F. prausnitzii that were significantly correlated with age in the cohort of healthy Malawian twins and triplets (p<0.0001 for all correlations; Spearman’s rank correlation; Table S9).
The relationship between age- and growth-discriminatory taxa and femoral bone phenotypes
We used micro-computed tomography (micro-CT) to assay the morphology of cancellous and cortical regions of femurs obtained from mice harboring microbiota with >50% transplantation efficiency. There were strong trends to higher femoral cortical ratios of bone volume to tissue volume (BV/TV) and volumetric bone mineral density (vBMD) in recipients of undernourished donor gut microbial communities (p=0.05, p=0.07, respectively, Mann-Whitney test). Differences between mice harboring microbiota from 6- and 18-month old donors were evident in cancellous rather than cortical bone, regardless of donor nutritional status; mice colonized with 6-month old donor communities had significantly higher BMD, BV/TV, trabecular connectivity and number, and significantly lower trabecular spacing irrespective of their nutritional status (Fig. S3, Mann-Whitney test, p<0.001 for all bone metrics). We extended our Random Forests and ranked Spearman correlation analyses to identify OTUs that discriminate these femoral bone phenotypes (Fig. S4, Table S10). Six of the OTUs represented in both of the growth discriminatory Random Forests models, including one assigned to B. longum, were also represented among the top 20 most discriminatory features in at least three of the five Random Forests-based models of bone metrics (Fig. S4). Although our sample size is small, these results provide evidence for microbiota-dependent regulation of bone morphology (17), with the effects being influenced by the age and nutritional status of the donor.
Repairing impaired growth phenotypes
We next determined whether age- and growth-discriminatory bacterial species were capable of repairing the growth abnormalities associated with stunted/underweight microbiota. We selected gnotobiotic recipients of two 6-month old donor microbiota that transmitted the most discordant growth phenotypes in the initial screen of 19 microbiota (Fig. S5A). One was a healthy infant from Mangochi (HAZ 1.49, WAZ 1.43, WHZ 0.9) with age appropriate microbiota maturity (microbiota age of 6.7 months); the other was a severely stunted and underweight infant (HAZ −3.35, WAZ −3.08, WHZ −0.79) from Malindi (located 20km from Mangochi) with microbiota immaturity (microbiota age of 4.6 months). The configurations of these transplanted microbiota were quite distinct: mice colonized with the healthy donor community were dominated by F. prausnitzii (33±19% relative abundance) while mice colonized with the undernourished donor microbiota were dominated by Clostridium neonatale (37±11% relative abundance) (n=5 mice/donor microbiota). Of the 27 species (66 97%ID OTUs) present in recipients of the healthy donor community and the 22 species (33 OTUs) present in recipients of the undernourished immature donor microbiota, 13 OTUs, representing 13 different species were present in both (see Fig. S5B and Table S11).
Taking advantage of the coprophagic behavior of mice, we gavaged 5-week old male germ-free C57BL/6J animals with either the healthy or stunted/underweight donor’s microbiota. Four days later, before phenotypic differences were apparent, we combined (dually housed) mice containing the healthy (H) donor microbiota (H-H controls). We also combined mice that received the undernourished (Un) donor community (Un-Un controls). The experimental group consisted of H and Un mice that were co-housed with one another (HCH-UnCH) (see Fig. 3A). All animals were fed the M8 diet beginning three days before colonization and throughout the course of the 3-week experiment. Animals were weighed twice a week while body composition and fecal samples were assayed once a week.
Fig. 3. Co-housing results in transfer of species from the microbiota of cagemates colonized with the healthy donor’s community into the microbiota of cagemates containing the severely stunted/underweight donor’s community and prevention of growth faltering.
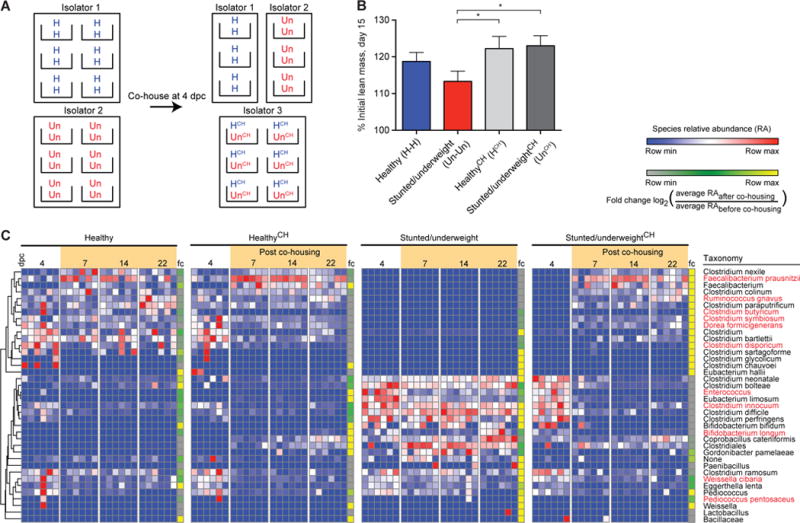
(A) Experimental design for co-housing experiments. Dually housed 4.5-week old mice were switched to the M8 diet and colonized 3 days later with either the intact uncultured healthy or stunted/underweight donor microbiota. Four days after gavage, subsets of the mice were co-housed (HCH and UnCH, respectively), while healthy and stunted/underweight control mice remained in their original isolators and were paired with a new cagemate from that isolator (H-H and Un-Un controls). Fecal samples were collected throughout the experiment; growth was assayed by changes in total body weight and body composition (the latter by qMR). Mice were sacrificed three weeks after colonization. (B) HCH and UnCH mice have increased lean mass gain relative to the Un-Un controls 15 days post colonization (Un-Un vs. HCH p = 0.0447, Un-Un vs. UnCH p=0.0121, Mann-Whitney test; n=6 cages of co-housed mice, 3 cages of each dually housed control group/experiment; two independent experiments). To quantify invasion further, we used the mean and standard deviation of the null distribution of invasion scores (defined as the scores from recipients of the H or Un microbiota that had never been co-housed with each other) to calculate a z-value and a Benjamini-Hochberg adjusted p-value for the invasion score of each species in HCH and UnCH mice (see Materials and Methods). We defined a taxon as a successful invader if it (i) had a Benjamini-Hochberg adjusted p≤0.05, and (ii) had a relative abundance of ≤0.05% before cohousing and ≥0.5% in the fecal microbiota at the time of sacrifice. Fig. 3C and Table S12 provide information about the direction and success of invasion. (C) Heatmap showing results of the invasion assay. Each row represents a species-level taxon, while each column represents a mouse at a given day post colonization (dpc); rows of the heatmap were hierarchically clustered according to pair-wise distances using Pearson correlation. Bars at the right side of each experimental arm represent the fold-change (fc) in that species’ relative abundance before and after cohousing (fold-change defined by the log2[(average relative abundance of species post cohousing (days 7 through 22))/(average relative abundance of species before cohousing (day 4))]. Species in red represent those identified as one of the top 30 growth-discriminatory taxa by the weight or lean mass gain Random Forests-based models shown in Fig. 2.
HCH and UnCH cagemates both gained significantly more lean mass compared to Un-Un controls (Fig. 3B; UnCH p=0.0121; HCH p=0.0447, Mann-Whitney test), but had no significant difference to lean mass gain in H-H controls. We characterized invasion using a previously described approach (18) that uses Microbial SourceTracker (19) (also see Materials and Methods). The fecal microbiota of H-H or Un-Un controls sampled 4 days after gavage (just prior to co-housing) were used as source communities. The fecal communities belonging to each HCH and UnCH mouse were then traced to these sources (see Materials and Methods); results indicated significant invasion of members of the healthy microbiota into the microbiota of undernourished cagemates, but not vice versa (Fig. 3C). Nine OTUs from the HCH cagemate microbiota were consistent invaders (total of 12 co-housed mice). Based on the rank order of their invasion scores, the two most successful invaders were F. prausnitzii, an age- and growth-discriminatory taxon in the Random Forests models and the most abundant OTU in the fecal microbiota of UnCH mice at the conclusion of the co-housing experiment, and Ruminococcus gnavus, a growth-discriminatory taxon. In contrast, only two OTUs from the stunted/underweight microbial community successfully invaded the gut community of HCH mice; one was assigned to Enterococcus and the other to Eubacterium limosum, which represented 2.7±1.7% of the community after co-housing [note that the Enterococcus OTU 4316566 had a significant negative correlation to lean mass gain in the initial screen of 19 donor microbiota (ρ=−0.14, p=0.0065; Spearman’s rank correlation)].
Culturing discriminatory taxa and characterizing their effects on growth
Acquisition of HCH-derived bacterial taxa in the microbiota of UnCH cagemates was accompanied by reductions in the relative abundances of 19 OTUs, six of which were below the limits of detection (<0.01%) by the end of the co-housing experiment. These observations raised two key questions. Which invasive OTUs mediated the observed effects on growth phenotypes? Did the reduction in other OTUs improve growth in UnCH cagemates? Therefore, we attempted to culture F. prausnitzii and R. gnavus from the fecal microbiota of healthy Malawian infants from the iLiNS-DYAD-M cohort. These efforts yielded three additional strains: Clostridium nexile (positively correlated to lean mass gain), Clostridium symbiosum (lean mass gain discriminatory), and Dorea formicigenerans (weight and lean mass gain discriminatory).
Analogous to the co-housing experiment, male germ-free C57Bl/6J mice were placed on the M8 diet at 4.5 weeks of age: three days later 5 mice were each given a single gavage with the intact microbiota from the Un donor, with or without an equivalent mixture of the 5-member consortium (Fig. 4A). The 5-member consortium produced a significant increase in body weight and lean mass gain compared to the untreated group (p=0.03, p=0.01, respectively, at the time of sacrifice 21 days after gavage, Mann-Whitney test) (Fig. 4B). Only two members of the consortium, R. gnavus strain TS8243C and C. symbiosum strain TS8243C, successfully colonized recipient mice [27±3% (mean±SD) and 2.6±0.3% relative abundance, respectively, in feces obtained at sacrifice] (Fig. 4C; see Fig. S6 and Table S13 for the results of reconstructions of selected metabolic subsystems in R. gnavus TS8243C and C. symbiosum TS8243C). None of the other members were detected in recipients sampled on days 4, 7 and 14 after gavage. Moreover, the efficiency of incorporation of members of the Un donor’s microbiota into recipient mice was indistinguishable between the two treatment arms; i.e., (i) R. gnavus and C. symbiosum did not result in extirpation of any major constituents of the community (the only species that fell below 0.1% of the community’s composition was Bifidobacterium bifidum, whose relative abundance was only 0.6±0.1% (mean±SD) at time of sacrifice in the control group); and (ii) the proportional representation of OTUs in the Un community was similar with respect to one another in the two treatment arms (Fig. 4C, Table S14). These results provide direct evidence that the cultured strains of R. gnavus and C. symbiosum ameliorate the impaired growth phenotype transmitted by an immature undernourished donor’s microbiota.
Fig. 4. A consortium of cultured growth-discriminatory OTUs augments the growth of mice colonized with the 6-month severely stunted/underweight donor’s microbiota.
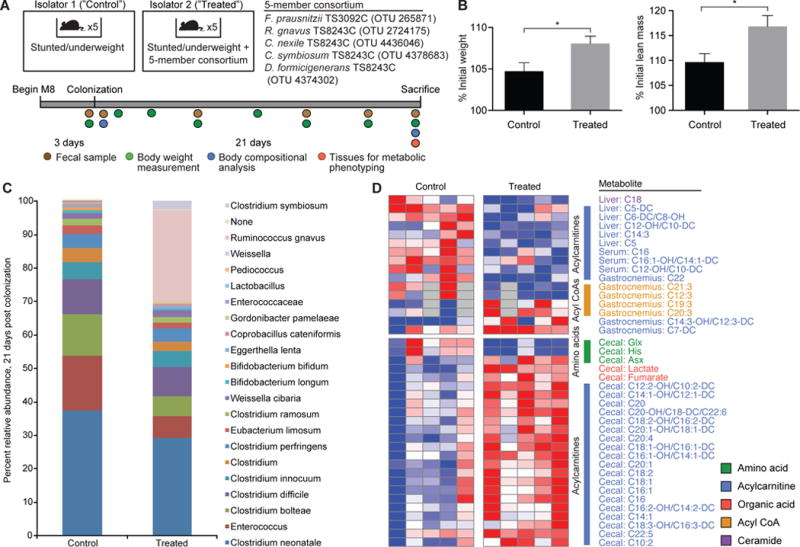
(A) Experimental design including the composition of the 5-member consortium of cultured bacterial strains. (B) Weight gain and lean body mass gain 21 days after gavage of the donor microbiota with or without the cultured consortium. (C) Comparison of the fecal microbiota of mice belonging to untreated control and treated experimental group showing the establishment of OTUs from the consortium at 21 days post colonization. (D) The effects of treatment with the consortium on host metabolism (p<0.05 for all metabolites shown, Student’s t-test). Each row represents a metabolite from a given tissue, while each column represents an individual mouse. Tissues were collected 21 days after colonization.
The presence of these two organisms also affected metabolic phenotypes (Fig. 4D, Table S15). The metabolic features that most discriminated mice with R. gnavus and C. symbiosum from the untreated group were acylcarnitines (C5–C16), which were significantly increased in cecal samples and decreased in the liver and serum in mice harboring the two taxa (p<0.05, Student’s t-test). Some of these acylcarnitines also distinguished mice harboring the H compared to Un donor microbiota (Fig. S7A,B). Among metabolites that were decreased in the livers of treated mice were C5, C5-DC and C6-DC acylcarnitines, all of which can be derived from branched-chain amino acid catabolism. As proposed for the comparison of mice harboring the H versus Un donor microbiota (see Supplementary Results), the decrease in these liver metabolites suggests an impact of the presence of two growth-promoting taxa on host metabolic machinery that drives amino acids away from oxidation in favor of protein synthesis and lean mass formation. The mechanisms by which the gut microbiota communicates metabolically with other tissues remain to be defined.
While generalizability of these effects needs to be directly tested using additional microbiota from other undernourished infants and children, we assessed whether the growth discriminatory taxa identified in our preclinical model correlate with growth in Malawian infants and children. To do so, we used the 220 samples from the healthy Malawian twin cohort to generate two Random Forests-based models: one regressed the 30 most weight gain-discriminatory OTUs against weight-for-height Z-scores (WHZ); the other regressed the 30 most lean mass gain-discriminatory taxa against this metric. We then evaluated how well the models were able to predict WHZ scores in the 259 members of the iLiNS-DYAD-M cohort across all time points surveyed (6, 12, and 18-months for each individual). During this brief 12-month window, there were significant and positive correlations between predicted and observed WHZ using either the weight or lean mass gain discriminatory taxa (weight discriminatory model ρ=0.12, p<0.0008; lean mass gain discriminatory model ρ=0.11, p<0.002; p<0.0001 for both models, permutation test, 999 permutations). In both models, B. longum was the most discriminatory taxon, while F. prausnitzii OTUs followed as the second or third most discriminatory in the weight gain and lean mass gain models, respectively (Fig. S10). The increasing ability to assemble the genomes of bacterial strains from shotgun sequencing of fecal community DNA (20) should allow these types of analyses to be further developed in Malawian as well as other populations of interest surveyed longitudinally at frequent intervals over extended periods of time. This approach would also provide direct information about the representation of the strains that we cultured and subsequently characterized in our preclinical model.
Prospectus
The studies reported here indicate that gut microbiota immaturity is not only associated with undernutrition but also causally related to it. There are several interrelated factors that could disrupt normal gut microbiota succession in infants and children. They include (i) poor maternal nutritional status, (ii) enteropathogen invasion, (iii) the history of consumption of antibiotics, (iv) disturbances in gut mucosal immune system development (21), and/or (v) the history of complementary feeding. Phenotyped birth cohorts provide an opportunity to perform correlation analyses designed to test the significance of the relationship between microbiota maturity (and the representation of specific age-/growth-discriminatory taxa), anthropometry, and the factors listed above, as well as the enigmatic disorder currently described as environmental enteric dysfunction (22).
Our current study showed that microbiota from 6-month old children produced greater effects on growth in recently weaned mice than the microbiota from 18-month old donors, although overall the effects produced by a healthy donor’s age-appropriate community were greater than those produced by an immature undernourished donor microbiota. While it will be important to extend these analyses to microbiota sampled from additional individuals representing this and other geographic sites/cultural traditions, we take these findings to suggest that in healthy children microbiota development is optimized to satisfy the different growth needs of the host at different ages. An immature microbiota appears to cause a form of neoteny that is not conducive to healthy growth when nutrients are limiting. However, how will the host adapt to therapeutic interventions that result in rapid progression to an age-appropriate microbiota? Are there mechanisms (including those involving the mucosal immune system) that feed back to regulate the rate of microbiota development? The answers have implications for designing therapeutic strategies for durable rescue of stunting, neurodevelopmental and/or immunologic abnormalities associated with undernutrition.
So far ready-to-use complementary foods (RUCFs) with and without antibiotics have generally produced only modest effects on growth and the longer-term sequelae of undernutrition (23). Certain locally available complementary foods that are provided after cessation of exclusive breastfeeding may have the ability to promote colonization of growth-discriminatory gut taxa in proportions that are age-appropriate, and these could be tested for their clinical value in systematic trials. In this respect, gnotobiotic mice colonized with microbiota from chronologically age-matched healthy and undernourished donors, and fed diets representative of those consumed by microbiota donors, should permit highly controlled, direct tests of the effects of antibiotics, breast milk components (e.g., bovine mimics of human breast oligosaccharides), and complementary foods on the mechanisms by which growth-promoting bacterial strains influence growth, metabolic, bone, immune and neurologic phenotypes.
Supplementary Material
Acknowledgments
We are indebted to the parents and children from Malawi for their participation in this study. We thank Sabrina Wagoner, David O’Donnell, Maria Karlsson, and Justin Serugo for their assistance with gnotobiotic mouse husbandry, Daniel Leib and Matt Silva for their assistance with bone morphology assays, Janaki Guruge for help with anaerobic microbiology, Betsy Dankenbring for assistance with maintenance of the biospecimen repository, plus Marty Meier, Su Deng, and Jessica Hoisington-Lopez for their contribution to various facets of the DNA sequencing pipeline. This work was supported by the Bill & Melinda Gates Foundation and the NIH (DK30292) with additional funding from the Office of Health, Infectious Diseases and Nutrition, Bureau for Global Health, USAID under terms of Cooperative Agreement No. AID-OAA-A-12-00005, through the Food and Nutrition Technical Assistance III Project (FANTA) managed by FHI 360. Data management and statistical analysis for the iLiNS-DYAD-M clinical study were also funded by the Academy of Finland (grant 252075), and the Medical Research Fund of Tampere University Hospital (grant 9M004). Micro-CT analysis was performed in the Washington University Musculoskeletal Research Center, which is supported by NIH P30 AR057235. L.V.B. received stipend support from NIH predoctoral training grants NIH T32 AI007172, T32 GM007067, and the Lucille P. Markey Special Emphasis Pathway in Human Pathobiology. D.A.R. and S.A.L. were supported by the Russian Science Foundation (Grant 14-14-00289).
Footnotes
Human studies were conducted using protocols for obtaining informed consent, clinical samples, and clinical metadata that were approved by institutional review boards from the University of Malawi, Pirkanmaa Hospital, and Washington University School of Medicine. Fecal specimens used in this study were covered by a Materials Transfer Agreement (MTA) between The University of Malawi, College of Medicine and Washington University in St. Louis.
Data Deposition:
16S rRNA sequences, generated from fecal samples in raw format prior to post-processing and data analysis, plus shotgun sequencing datasets generated from the R. gnavus TS8243C and C. symbiosum TS8243C genomes have been deposited at the European Nucleotide Archive under accession number PRJEB9853.
Declarations:
J.I.G. is co-founder of Matatu Inc., a company characterizing the role of diet-by-microbiota interactions in animal health. A.L.O. is an Adjunct Vice President for Research for Buffalo BioLabs, LLC.
Author Contributions:
L.V.B., M.R.C. and J.I.G. designed the gnotobiotic mouse studies; L.V.B. and M.R.C. performed the experiments with gnotobiotic animals; I.T. and M.J.M. designed and implemented the clinical monitoring and sampling for the twin study and participated in patient recruitment, sample collection and preservation, and/or clinical evaluations; K.M.M., Y.F., J.M.J., K.G.D. and P.A. designed and oversaw the clinical studies, sample collection and processing, and/or clinical monitoring/evaluations in the iLiNS-DYAD-M study; L.V.B. generated the 16S rRNA data; L.V.B., M.R.C., S.V., and O.I. generated metabolomics data; T.S. and L.V.B. cultured bacterial isolates; B.H., D.A.R., S.A.L., and A.L.O. performed metabolic reconstructions of the R. gnavus and C. symbiosum genomes; L.V.B., M.R.C., M.J.B., S.S., C.B.N., and J.I.G. analyzed the data; L.V.B. and J.I.G. wrote the paper.
References and Notes
- 1.Victora CG, et al. Maternal and child undernutrition: consequences for adult health and human capital. Lancet. 2008;371:340–357. doi: 10.1016/S0140-6736(07)61692-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gaayeb L, et al. Effects of malnutrition on children’s immunity to bacterial antigens in Northern Senegal. Am J Trop Med Hyg. 2014;90:566–573. doi: 10.4269/ajtmh.12-0657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kosek M, et al. Fecal markers of intestinal inflammation and permeability associated with the subsequent acquisition of linear growth deficits in infants. Am J Trop Med Hyg. 2013;88:390–396. doi: 10.4269/ajtmh.2012.12-0549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Waber DP, et al. Impaired IQ and academic skills in adults who experienced moderate to severe infantile malnutrition: a 40-year study. Nutr Neurosci. 2014;17:58–64. doi: 10.1179/1476830513Y.0000000061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Black RE, et al. Maternal and child undernutrition and overweight in low-income and middle-income countries. Lancet. 2013;382:427–451. doi: 10.1016/S0140-6736(13)60937-X. [DOI] [PubMed] [Google Scholar]
- 6.Richard SA, et al. Modeling Environmental Influences on Child Growth in the MAL-ED Cohort Study: Opportunities and Challenges. Clin Infect Dis. 2014;59(Suppl 4):S255–60. doi: 10.1093/cid/ciu436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Keusch GT, et al. Environmental enteric dysfunction: pathogenesis, diagnosis, and clinical consequences. Clin Infect Dis. 2014;59(Suppl 4):S207–12. doi: 10.1093/cid/ciu485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dewey KG. The challenge of meeting nutrient needs of infants and young children during the period of complementary feeding: an evolutionary perspective. J Nutr. 2013;143:2050–2054. doi: 10.3945/jn.113.182527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.de Onis M, et al. The WHO Multicentre Growth Reference Study: planning, study design, and methodology. Food Nutr Bull. 2004;25:S15–26. doi: 10.1177/15648265040251S103. [DOI] [PubMed] [Google Scholar]
- 10.de Onis M, Onyango AW, Van den Broeck J, Chumlea WC, Martorell R. Measurement and standardization protocols for anthropometry used in the construction of a new international growth reference. Food Nutr Bull. 2004;25:S27–36. doi: 10.1177/15648265040251S104. [DOI] [PubMed] [Google Scholar]
- 11.Subramanian S, et al. Persistent gut microbiota immaturity in malnourished Bangladeshi children. Nature. 2014;509:417–421. doi: 10.1038/nature13421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Breiman L. Random Forests. Machine Learning. 2001;45:5–32. [Google Scholar]
- 13.Smith MI, et al. Gut microbiomes of Malawian twin pairs discordant for kwashiorkor. Science. 2013;339:548–554. doi: 10.1126/science.1229000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics. 2010;26:2460–2461. doi: 10.1093/bioinformatics/btq461. [DOI] [PubMed] [Google Scholar]
- 15.Wang Q, Garrity GM, Tiedje JM, Cole JR. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol. 2007;73:5261–5267. doi: 10.1128/AEM.00062-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ashorn P, et al. The impact of lipid-based nutrient supplement provision to pregnant women on newborn size in rural Malawi: a randomized controlled trial. Am J Clin Nutr. 2015;101:387–397. doi: 10.3945/ajcn.114.088617. [DOI] [PubMed] [Google Scholar]
- 17.Sjögren K, et al. The gut microbiota regulates bone mass in mice. J Bone Miner Res. 2012;27:1357–1367. doi: 10.1002/jbmr.1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ridaura VK, et al. Gut microbiota from twins discordant for obesity modulate metabolism in mice. Science. 2013;341:1241214. doi: 10.1126/science.1241214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Knights D, et al. Bayesian community-wide culture-independent microbial source tracking. Nat Methods. 2011;8:761–763. doi: 10.1038/nmeth.1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cleary B, et al. Detection of low-abundance bacterial strains in metagenomic datasets by eigengenome partitioning. Nat Biotechnol. 2015;33:1053–1060. doi: 10.1038/nbt.3329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kau AL, et al. Functional characterization of IgA-targeted bacterial taxa from undernourished Malawian children that produce diet-dependent enteropathy. Sci Transl Med. 2015;7:276ra24. doi: 10.1126/scitranslmed.aaa4877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Crane RJ, Jones KDJ, Berkley JA. Environmental enteric dysfunction: an overview. Food Nutr Bull. 2015;36:S76–87. doi: 10.1177/15648265150361S113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dewey KG, Adu-Afarwuah S. Systematic review of the efficacy and effectiveness of complementary feeding interventions in developing countries. Matern Child Nutr. 2008;4:24–85. doi: 10.1111/j.1740-8709.2007.00124.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Magoč T, Salzberg SL. FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics. 2011;27:2957–2963. doi: 10.1093/bioinformatics/btr507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Goodman AL, et al. Extensive personal human gut microbiota culture collections characterized and manipulated in gnotobiotic mice. Proc Natl Acad Sci USA. 2011;108:6252–6257. doi: 10.1073/pnas.1102938108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ferrara CT, et al. Genetic networks of liver metabolism revealed by integration of metabolic and transcriptional profiling. PLoS Genet. 2008;4:e1000034. doi: 10.1371/journal.pgen.1000034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.An J, et al. Hepatic expression of malonyl-CoA decarboxylase reverses muscle, liver and whole-animal insulin resistance. Nat Med. 2004;10:268–274. doi: 10.1038/nm995. [DOI] [PubMed] [Google Scholar]
- 28.Jensen MV, et al. Compensatory responses to pyruvate carboxylase suppression in islet beta-cells. Preservation of glucose-stimulated insulin secretion. The Journal of biological chemistry. 2006;281:22342–22351. doi: 10.1074/jbc.M604350200. [DOI] [PubMed] [Google Scholar]
- 29.Deutsch J, Grange E, Rapoport SI, Purdon AD. Isolation and quantitation of long-chain acyl-coenzyme A esters in brain tissue by solid-phase extraction. Anal Biochem. 1994;220:321–323. doi: 10.1006/abio.1994.1344. [DOI] [PubMed] [Google Scholar]
- 30.Magnes C, Sinner FM, Regittnig W, Pieber TR. LC/MS/MS method for quantitative determination of long-chain fatty acyl-CoAs. Anal Chem. 2005;77:2889–2894. doi: 10.1021/ac048314i. [DOI] [PubMed] [Google Scholar]
- 31.Minkler PE, Kerner J, Ingalls ST, Hoppel CL. Novel isolation procedure for short-, medium-, and long-chain acyl-coenzyme A esters from tissue. Anal Biochem. 2008;376:275–276. doi: 10.1016/j.ab.2008.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Merrill AH, Sullards MC, Allegood JC, Kelly S, Wang E. Sphingolipidomics: high-throughput, structure-specific, and quantitative analysis of sphingolipids by liquid chromatography tandem mass spectrometry. Methods. 2005;36:207–224. doi: 10.1016/j.ymeth.2005.01.009. [DOI] [PubMed] [Google Scholar]
- 33.Chevreux B, et al. Using the miraEST assembler for reliable and automated mRNA transcript assembly and SNP detection in sequenced ESTs. Genome Res. 2004;14:1147–1159. doi: 10.1101/gr.1917404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Seemann T. Prokka: rapid prokaryotic genome annotation. Bioinformatics. 2014;30:2068–2069. doi: 10.1093/bioinformatics/btu153. [DOI] [PubMed] [Google Scholar]
- 35.Markowitz VM, et al. IMG: the Integrated Microbial Genomes database and comparative analysis system. Nucleic Acids Res. 2012;40:D115–22. doi: 10.1093/nar/gkr1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Overbeek R, et al. The SEED and the Rapid Annotation of microbial genomes using Subsystems Technology (RAST) Nucleic Acids Res. 2014;42:D206–14. doi: 10.1093/nar/gkt1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rodionov DA. Comparative genomic reconstruction of transcriptional regulatory networks in bacteria. Chem Rev. 2007;107:3467–3497. doi: 10.1021/cr068309+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Novichkov PS, et al. RegPrecise 3.0–a resource for genome-scale exploration of transcriptional regulation in bacteria. BMC Genomics. 2013;14:745. doi: 10.1186/1471-2164-14-745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ravcheev DA, Godzik A, Osterman AL, Rodionov DA. Polysaccharides utilization in human gut bacterium Bacteroides thetaiotaomicron: comparative genomics reconstruction of metabolic and regulatory networks. BMC Genomics. 2013;14:873. doi: 10.1186/1471-2164-14-873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rodionov DA, et al. Transcriptional regulation of the carbohydrate utilization network in Thermotoga maritima. Front Microbiol. 2013;4:244. doi: 10.3389/fmicb.2013.00244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rodionov DA, et al. Genomic encyclopedia of sugar utilization pathways in the Shewanella genus. BMC Genomics. 2010;11:494. doi: 10.1186/1471-2164-11-494. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.