

Abstract
Sleep is an essential physiological process, which has been divided into rapid eye movement sleep (REMS) and non-REMS (NREMS) in higher animals. REMS is a unique phenomenon that unlike other sleep-waking states is not under voluntary control. Directly or indirectly it influences or gets influenced by most of the physiological processes controlled by the brain. It has been proposed that REMS serves housekeeping function of the brain. Extensive research has shown that during REMS at least noradrenaline (NA) -ergic neurons must cease activity and upon REMS loss, there are increased levels of NA in the brain, which then induces many of the REMS loss associated acute and chronic effects. The NA level is controlled by many bio-molecules that are regulated at the molecular and transcriptional levels. Similarly, NA can also directly or indirectly modulate the synthesis and levels of many molecules, which in turn may affect physiological processes. The burgeoning field of behavioral neuroepigenetics has gained importance in recent years and explains the regulatory mechanisms underlying several behavioral phenomena. As REMS and its loss associated changes in NA modulate several pathophysiological processes, in this review we have attempted to explain on one hand how the epigenetic mechanisms regulating the gene expression of factors like tyrosine hydroxylase (TH), monoamine oxidase (MAO), noradrenaline transporter (NAT) control NA levels and on the other hand, how NA per se can affect other molecules in neural circuitry at the epigenetic level resulting in behavioral changes in health and diseases. An understanding of these events will expose the molecular basis of REMS and its loss-associated pathophysiological changes; which are presented as a testable hypothesis for confirmation.
Keywords: Chromatin uncoiling, chromatin remodeling, DNA methylation epigenetic modifications, histone, REMS loss, transcription factors.
1. INTRODUCTION
Basic rest and activity cycle (BRAC) is ubiquitously present in every life form and is associated with normal well being of an individual. Sleep-wakefulness in higher order animals is an evolved form of this BRAC. The evolutionary significance of sleep lies in the fact that it saves energy and is crucial for adjustment of the animal to ecological and environmental factors [1]. Sleep and wakefulness are behavioral phenomena, which have been objectively identified, defined, and classified on the basis of electrophysiological signals recorded from the brain the electroencephalogram (EEG), the muscles, the electromyogram (EMG) and the eye movement, the electrooculogram (EOG). Based on these characteristic electrophysiological parameters sleep has been broadly classified into rapid eye movement sleep (REMS) and non-REMS (NREMS). REMS is a reversible, unique physiological process characterized by simultaneous EEG desynchronization, rapid eye movements in the EOG and complete loss of muscle tone (atonia) in EMG. Since EEG is the most important characteristic feature for identification of REMS, it is natural that this state has been identified in species higher in evolution having a reasonably well developed and evolved brain. However, its presence or absence in lower species could not be confirmed as yet primarily due to lack of identification of a more fundamental characteristic marker.
Ontogenetic studies have shown that REMS is expressed maximum in newborn babies and its quantity reduces with ageing, however, it is never absent in life [2, 3]. Its role in brain development has been postulated by the fact that the period spent in REMS is higher in newborn and in babies than in the adults and it is more in babies who are born immature [4]. It is also important to note that this stage does not have voluntary regulation. Its fundamental regulation is done by the neurons in the brain stem, the site for the neural control of other life- sustaining autonomic physiological processes viz. cardiovascular and respiratory regulation. Phylogenetic studies across species suggest that the brain stem activation is the initial element in the REMS evolution [5]. REMS serve several crucial functions and its loss affects various pathophysiological states and processes [6, 7]. It is affected in most pathological conditions, including neurodegenerative diseases, e.g. Parkinson’s, Alzheimer’s, narcolepsy, epilepsy and psychiatric disorders [8-11]. Experimental deprivation of REMS in humans and in animals reported elevated aggressiveness, irritability, confusion, hypersexuality, loss of concentration, impairment of memory processing and memory consolidation [7, 12, 13].
The development and establishment/maturation of any behavioral phenomenon are influenced by several environmental factors like nutrition, social experiences, hormones, etc. [14] and sleep is no exception to it. One of the important factors (if not the most important factor) within the biological system that exerts sustained biological and neurobehavioral manifestations is through the chemical modifications of DNA and histone protein molecules within the cells together known as epigenetic modifications [15]. The field of biology studying the interplay between gene and environmental signals that trigger molecular changes in cells is known as behavioral epigenetics [14]. Epigenetic mechanisms decide the pattern in which environment regulates/influences the genomic organization of living beings. Increasing evidence (mostly indirect though) suggests that epigenetic changes are crucial for chronic or accumulated sleep-loss associated disorders including behavioral changes and also possibly in the regulation of sleep-wake states [16]. Genomic imprinting, which is established by epigenetic processes, also extends its effects to sleep–wake regulation [17]. REMS and NREMS are regulated by separate sets of imprinted genes and these genes are differentially expressed in brain regions [18]. In support, it has been shown that the maternally expressed imprinted gene, Gnas for example, modulates the expression of sleep-wake states [19]. Epigenetic changes have therefore attracted great attention in recent years as researchers are exploring the molecular circuitry underlying several behavioral phenomena including those associated with sleep and its loss. In this review, we have attempted to gain insights into the role of epigenetic changes in the regulation of REMS in particular and its loss-associated disorders/dysfunctions with particular emphasis on noradrenaline (NA).
2. REMS REGULATION AND NORADRENALINE
It has been recently proposed that REMS serves housekeeping function of the brain [20]. The locus coeruleus (LC) possesses mostly the NA-ergic neurons, which project throughout the brain. The NA-ergic neurons in LC cease activity during REMS and are known as REM-OFF neurons while presumably cholinergic REM-ON neurons increase activity during REMS and are located in the dorsolateral pontine region. The REM-OFF neurons, which normally cease activity during REMS, continue firing upon REMS deprivation (REMSD) [21] causing increased levels of NA in the brain [22]. The elevated levels of NA associated with REMS loss have been correlated with many patho-physiological conditions leading to expression of altered behavior and symptoms associated with various disorders [20, 23]. The increased NA also leads to decreased intracellular [Ca2+] which in turn increases Na-K ATPase activity, which would alter brain excitability [20]. Therefore, understanding of the factors regulating the activity of NA-ergic neurons, which in turn would modulate the levels of NA in the brain, is of great significance. The NA levels in the brain may be modulated by a) rate and period of LC neuronal activity which are modulated by various inputs and neurotransmitters, and b) synthesis, release and effective removal of NA from released sites (i.e. projections mostly from the LC neurons).
2.1. NA Neuronal Activity Regulation
REMS is generated and maintained by the neurons located in the core of the brain stem; the neurons from different other brain regions modulate REMS through the former [24]. The LC neurons (the major source of NA in the brain) show a progressive decrease in their activities as the subject moves from wakefulness to sleep and they remain virtually silent during REMS [25]. In rats, when LC neurons were activated using continuous low frequency, mild electrical stimulation [26] or by blocking gamma amino butyric acid (GABA) -ergic receptors [27-29] or by neutralizing Na-K ATPase inhibitors by application of antisera for endoubain [30], REMS was reduced and there was a rebound increase in REMS upon recovery. These, along with other supporting evidence suggested that cessation of LC neurons is a pre-requisite for REMS occurrence and their activation results in REMS loss [31]. The recent finding that optogenetic stimulation of LC neurons enhanced wakefulness and reduced REMS as well as NREMS [32] also supports our contention.
2.2. Maintenance of Effective NA Levels in the Brain
The process of biosynthesis, release, reuptake and degradation is responsible for maintenance of neuro-transmitter levels at the synaptic cleft and neuronal surroundings in the brain. Inputs from various regions of the brain on the LC-NA-ergic neurons exert excitatory and inhibitory influences [33] and accordingly have facilitatory and inhibitory effects, respectively, on NA release from the NA-ergic neurons at the target sites. Tyrosine hydroxylase (TH), dopamine β-hydroxylase, monoamine oxidase-A (MAO-A), noradrenaline transporter (NAT) are some of the important molecules directly modulating effective NA levels in the brain. REMSD has been reported to modulate many of these molecules and their activities, which in turn are likely to affect NA levels in the brain. For example, TH level was elevated [22] while MAO-A was decreased [34] after REMSD and the excess of catecholamines is also known to inhibit the activity of TH by negative feedback regulation.
After NA release and activation of its receptors at the synaptic cleft, part of the unused NA is re-uptaken from the extracellular space into the presynaptic terminal by NAT. The transporter also helps preventing excessive build-up of NA concentration at the synaptic cleft. NAT is a monoamine transporter responsible for sodium–chloride dependent re-uptake of NA. This re-uptake is essential for maintenance of NA level at the synaptic cleft and its biological effects, including adrenergic neurotransmission in the brain, heart and peripheral organs [35]. Various adrenoreceptor isoforms e.g. α1, α2, β are expressed on neurons in different brain areas and are responsible for NA mediated effects including during REMSD. The following may be cited in support as example; adrenergic receptor agonist and antagonist injected intraperitoneally or locally in the brain have been reported to modulate REMS [36, 37]; monoamine re-uptake blockers suppress REMS possibly due to elevated NA levels at the synaptic sites [38]; and elevated NAT mRNA has been reported during REMSD [39].
3. MOLECULAR FACTORS REGULATING NA AT THE GENE LEVEL AND ITS RELEVANCE TO REMS
REMS is an essential phenomenon necessary for the maintenance of normal brain function. However, the detailed mechanisms regulating REMS are yet to be completely understood. Nevertheless, it is known that NA-ergic neurons must cease activity for the generation of REMS [33] and NA levels increase upon REMSD [22]. Independent studies have shown that the bio-molecules (factors) controlling NA levels and its action in the brain, for instance, TH [40-43], α1 adrenergic receptor [44, 45] and MAO [46] are transcriptionally regulated. These factors are encoded by specific genes at the molecular level; however, the details of their modulation and transcriptional regulation in association with REMS and its loss, if any, are still lacking. In addition, the binding of specific transcription factors (TFs) to the specific gene promoter is essential for the homeostatic maintenance of NA level. Therefore, for a comprehensive understanding of the regulation of levels of NA in the brain it is essential to understand the mechanisms controlling transcription of the genes regulating the synthesis and degradation of NA including in relation to REMS, its loss and associated pathophysiological states.
Regulation of gene expression at the transcriptional level involves several processes; the epigenetic modification is among the early ones and also for sustained effects. Epigenetic regulation involves DNA methylation and chromatin remodeling through histone modifications. These regulate chromatin uncoiling and thus allow access to TFs and activation of the transcriptional machinery. In a recent study, it was found that DNA methylation modulates the transcriptional and synaptic responses of neuron to sleep loss [47]. The upcoming field of epigenetics has broadened our basic understanding about the regulation of several essential physiological phenomenon and is likely to serve as the much needed fundamental background mechanism regulating innumerable cellular processes. In this review, an attempt has been made to discuss important findings related to epigenetic changes in the neuronal system associated with REMS loss and its related symptoms. Also, the possibilities of epigenetic changes after REMS loss, especially in the regulation of NA-ergic machinery in the brain have been discussed. Below we explain epigenetic modifications and how they may be useful to our understanding of sleep research and sleep-loss associated symptoms.
4. EPIGENETIC MODIFICATIONS
The term epigenetics was first coined by Conrad Waddington (1942), who defined the term as “the fundamental interactions between genes and their products, which brings the phenotype into being”. It includes the processes involving chemical modifications of genomic DNA and histone proteins without affecting the DNA sequence [48]. The non-coding RNA (ncRNA) based epigenetic modifications have been described recently, which as yet is the least understood [49]. Below first we discuss, in brief, the possible mechanisms of epigenetic modifications.
4.1. DNA Methylation
DNA methylation occurs predominantly across regions of genome having a high frequency of –cytosine–phosphate–guanine– (CpG) sequence, known as CpG island, often found in the promoter region of a gene [50-51]. It constitutes the covalent modification of cytosine residues (5’-position) in the CpG dinucleotide sequence resulting in the formation of 5-methylcytosine as shown in Fig. 1A. The enzymes that catalyze the transfer of the methyl group from S-adenosyl methionine to cytosine residues are known as DNA methyl transferases (DNMT) [52]. Another regulatory protein molecule, the methyl CpG-binding-domain (MBD or MeCP2), recognizes and binds to the methylated DNA. The MeCP2 further recruits additional epigenetic modulatory factors, including transcriptional co-repressors and histone deacetylases (HDAC) [53, 54]. As a result, the chromatin becomes densely packed which hinders the accessibility of TFs and other regulatory molecules, which results in the switching off the gene expression [55]. The DNA methylation is a dynamic process and continues to coordinate the transcriptional programming throughout the lifespan of a cell [56].
Fig. (1).
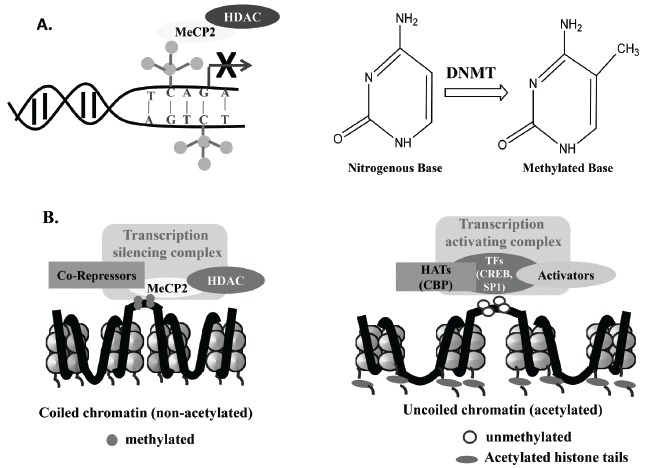
Mechanisms of epigenetic modifications have been represented in this figure. DNA methylation (A) and histone deacetylation (B, left panel) will promote chromatin coiling and inhibit the binding of transcriptional machinery; while histone acetylation (B, right panel) will facilitate uncoiling of chromatin and enhance binding of the transcriptional machinery. Abbreviations are as in the text.
4.2. Histone Modifications
The fundamental structural unit of eukaryotic chromatin is nucleosome, which comprises of an octamer of the four core histones- H2A, H2B, H3 and H4 wrapped with 147 bp of genomic DNA. The nucleosome assembles in chromatin in varying degree of condensation that plays a key role in controlling accessibility of the DNA to the replication and transcriptional machinery and thus regulates gene expression. In loosely packed chromatin, the DNA sequences are easily accessible to a wide range of molecules, including the TFs and other regulatory molecules [57]. The uncoiling of chromatin and binding of TFs to DNA are regulated by two interlinked processes a) chromatin remodeling, which involves ATP-dependent mobilization of nucleosome and exposure of genomic DNA wrapped around the histone proteins in the nucleosome core; and b) specific histone tail modifications [58]. The histone modifying enzymes catalyze the post-translational modifications, including acetylation, methylation, phosphorylation, SUMOylation, ubiquitylation, ADP-ribosylation, etc. at N-terminal domains of histone protein. Each of these modifications affects the interaction of DNA with histone proteins possibly in a unique manner resulting into distinct transcriptional and downstream events culminating in functional implications [59].
Acetylation occurs at specific lysine residues in the N-terminal tails of the histone H3 and H4 and neutralizes their positive charges. This disrupts the interaction of histones with the negatively charged DNA molecules, which results in a more relaxed and accessible chromatin structure [59, 60] (Fig. 1B). Deacetylation of these residues results in condensation of chromatin and heterochromatin formation. The acetylation and deacetylation reactions are catalyzed by histone acetyltransferases (HAT) and HDAC. Different histone modifications have differential effects on gene transcription. In comparison to acetylation, histone methylation can have activating or inhibiting effect(s) on the gene expression and it depends exclusively on the localization of the lysine residue that is being covalently modified. For example, methylation of lysine 4 and 36 in histone H3 (H3K4, H3K36) is associated with decondensed chromatin and active gene expression, while methylation of the lysine 9 and 27 in histone H3 results in compaction of chromatin. Histone lysine residues can carry up to three methyl groups [61]. The cumulative effect of all these histone modifications work in concert and bring about a particular effect on transcription. The cooperative interactions amongst different histone modifications establish functional domains in the nucleus referred to as “histone code” [62]. The histone modifications and DNA methylation work hand in hand to ensure efficient gene transcription or its silencing. An intricate balance between these finally regulates gene expression leading to cellular to systemic and behavioral expressions in health and diseases.
5. EPIGENETIC MODIFICATIONS MODULATING BRAIN FUNCTIONING
Neuroepigenetics or behavioral epigenetics has recently gained recognition and involves “unique mechanisms that allow dynamic experience-dependent regulation of the neuronal epigenome” [63]. Epigenetic regulation allows living cells, including neurons, individually as well as in system to adjust transcriptional changes in response to specific inputs arising from internal as well as external environment from birth until death, through the development, growth and maintenance phases [64]. The synthesis of neurotransmitters and other molecules for the growth, development and sustenance of neural circuitry in response to internal and external signals dynamically are controlled at multiple levels. The crucial neurobiological processes viz. development, neural stem cell maintenance, differentiation generating neural cell identity, neural network connectivity and plasticity have been suggested to be mediated through epigenetic modifications [65]. Thus, the epigenetic regulation constitutes an essential fundamental molecular step, which is necessary for brain functions. Therefore, it is reasonable and timely to understand epigenetic modifications associated with fundamental physiological processes in higher animals, including those in relation to the REMS and its loss.
6. EMERGING ROLE OF EPIGENETIC MODIFICATIONS IN NEUROLOGICAL DISORDERS AND COGNITIVE PROCESSES, ESPECIALLY ASSOCIATED WITH REMS LOSS
Sleep disorders have been classified broadly into conditions of excessive wakefulness (insomnia) and/or excessive sleepiness (narcolepsy, shift work disorder, jet lag) [66]. It has been observed that patients with psychosomatic disorders suffering from depression, Parkinson’s and Alzheimer’s diseases experience co-morbid sleep disturbances [67-69]. In the following section, we provide an account of the role of epigenetic modifications in the regulation of gene expressions in some representative sleep-loss associated pathophysiological and neurobehavioral dysfunctions.
6.1. Alzheimer’s Disease
Alzheimer’s disease (AD) is possibly the most common form of dementia and is characterized by severe memory loss, confusion, depression, apathy, agitation, anxiety and abnormal motor behavior [70]. The brain histology of patients with AD shows two major characteristics viz. neuritic plaques and neurofibrillary tangles due to amyloid β-peptide, amyloid precursor protein and hyper-phosphorylated tau protein, respectively [71, 72]. Epigenetic changes have been correlated with the pathophysiology of AD [73]. In the human cerebral cortex, the age- related alterations in DNA methylation of amyloid precursor protein and microtubule-associated tau protein gene promoter region influence the transcriptional activity of these genes [74, 75]. The amyloid precursor protein is known to form a complex with HAT enzyme, the lysine acetyltransferase 5 (KAT5/Tip60) [76]. Presenilin 1, the gene involved in β-amyloid processing, affects the function of another HAT enzyme, the cAMP response element -binding protein (CREB) -binding protein (CBP/p300) [77].
Further, significantly higher amyloid β- peptide plaque deposition in multiple sub-regions of the cortex was observed after daily sleep restriction for 20 hrs for 21 days [78]. This suggested that sleep disturbances also might be associated with the pathogenesis of AD. Independent studies showed increased apoptosis of neurons by NA after REMSD [79], degeneration of NA-ergic neurons in the LC in AD [23, 80, 81], reduced NAT level in the LC neurons in AD [82] and reduced tissue level of NA in a transgenic mouse model of AD [83] indicate the possibility of involvement of NA in the pathogenesis of AD. However, how at the molecular level the epigenetic modifications affect these changes, which directly or indirectly affect the NA levels in the brain, especially in relation to REMS and its loss, are still unknown.
6.2. Depression
Epigenetic modifications of many genes in neurons have been associated with depression [84]. Transcription of brain-derived neurotrophic factor (BDNF), a product of memory-related gene, is decreased in patients with major depression disorder [85] and DNA methylation has been shown to regulate its gene expression [86]. BDNF is also known to regulate neuronal differentiation and growth [87, 88] and its decreased expression in depressed patients may lead to reduced hippocampal volume [85]. Furthermore, histone acetylation (H3K14Ac) is transiently decreased and then consistently increased in the nucleus accumbens after chronic social defeat stress in depressed patients [89]. Decreased acetylation (H4K12Ac) and phosphoacetylation (H3K9S10) were seen in CA3 and dentate gyrus region in the rat depression model [90].
Depression with or without stress disorder, which may affect the hypothalamic- pituitary-adrenal axis, is also associated with the dysregulation of the NA-ergic LC system [23, 91]. Interestingly, sleep deprivation (SD), including REMSD, reduces the symptoms of depression and improves mood disorder [92]. It appears that increased LC neuronal activity resulting in elevated NA is involved in the antidepressant effects of SD. Our contention may be supported by the fact that dysregulation of NAT has been described in depression and antidepressant drugs are known to inhibit the NAT and increase the NA levels [93, 94]. However, the role of epigenetic modifications in regulating the antidepressant effects of SD needs detailed study.
6.3. Schizophrenia
It is a complex neuropsychiatric disorder characterized by hallucinations, delusions and working memory deficits [95]. A deficit in GABA-ergic local interneurons is thought to be one of the important correlates in this disease [96, 97]. Decreased expression of glutamic acid decarboxylase 1 (GAD1), which encodes the 67-kDa glutamate decarboxylase, the GABA synthesizing enzyme is commonly seen in schizophrenic postmortem brains [98, 99].
It has been reported that in the normal brain there is a progressive increase in H3K4 methylation at GABA-ergic gene promoters, while in the schizophrenic patients there is a decrease in histone methylation [100]. Changes in acetylation levels across histone proteins have been correlated with gene expression of several schizophrenia-related genes [101], including GAD1. Treatment of the rat with methionine for 15 days decreased GAD1 mRNA and its protein expression. This suggests that GAD1 promoter methylation is involved in the transcriptional repression of this gene [102]. It is pertinent to highlight that REMS latency is reduced in schizophrenia [103], GABA levels change through sleep-waking-REMS [104, 105] as well as upon REMSD (our unpublished data) and GAD levels also change upon REMSD [106]. The above correlation studies, although indicate REMSD associated epigenetic modifications causing changes in GAD, it needs to be studied for confirmation. Further, as NA is increased during REMSD, it may influence GAD or vice versa through GABA (a product of the GAD), particularly in relation to REMS and its loss i.e. REMSD.
6.4. Cognitive Processes
An understanding of the cellular and molecular mechanisms of learning and memory processes has been an important area of molecular neuroscience research. It is only recently that epigenetic changes have gained attention as regulators of memory formation [107]; any alteration in these processes is likely to affect (directly or indirectly) various cognitive disorders [108, 109]. DNA methylation was proposed to be the fundamental mechanism required to propagate memory over generations [110]. An increased de novo DNMT expression and methylation across memory–suppressor gene, protein phosphatase I (PPI) was observed in the hippocampus in response to contextual fear conditioning [111]. The PPI is reported to be regulated by NA [112], interacts with HDACs and histone demethylases to increase their activities and thus favors transcriptional silencing. Nuclear PPI promotes memory suppression through dephosphorylation of serine 10 on histone H3 [113]. Learning-induced changes in DNA methylation of BDNF, arc and calcineurin genes are important for formation and maintenance of memory [114, 115]. Hippocampus-dependent tasks were seen to be associated with the global increase of euchromatin-related post-translational modifications of histones. During memory consolidation, H3S10 phosphorylation, H3K14 and H4K5 acetylation, H3K36 trimethylation are rapidly and transiently activated in the hippocampus, whereas they occur with a delay and persist longer in the prefrontal cortex [116].
Sleep including REMS, has been implicated in neuronal plasticity in the brain that underlies the basic mechanism of learning and memory consolidation [117]. Indications are plenty that sleep participates in the consolidation of fresh memory traces arising out of a wide range of experimental conditions [118].NA is an essential modulator of memory formation because of its ability to regulate synaptic plasticity [112]. It is released during arousal and has a central role to play in the emotional regulation of memory [119]. The levels of NA decrease during REMS and is increased upon REMSD [13, 22] and this elevated NA induces many of the REMSD associated molecular changes [20].
In addition to the above, many other neurological disorders, including ageing, attention deficit/hyperactivity disorder, anxiety and post-traumatic stress disorder, are also associated with REMS loss or its dysregulation. A closer look revealed that NA and LC-NA-ergic system are common factors to be dysregulated during these neurological disorders. However, compelling direct evidences relating the role of epigenetic modifications of the NA-ergic system in these neurological disorders, especially in relation to REMS-loss or -dysfunction, are lacking. Nevertheless, it may be summarized that the findings from isolated, independent studies are suggestive of epigenetic modifications in the regulation of expressions of specific genes or bio-molecules associated with neurobehavioral disorders, which are directly or indirectly related to total sleep or REMS disruption and associated imbalance in neurotransmitter levels, particularly that of NA (Fig. 2). REMS loss is reported to modulate several neurotransmitters, NA being a prominent and most studied one. As NA has been shown to affect many REMSD-associated cellular, molecular and physiological processes [20], we propose a testable hypothesis and a model explaining the role of epigenetic modifications responsible for changes in NA levels and its role in modulating the expressions of genes or bio-molecules (factors) which regulate REMS or are affected during REMS-loss and are possibly responsible for REMSD-associated changes.
Fig. (2).
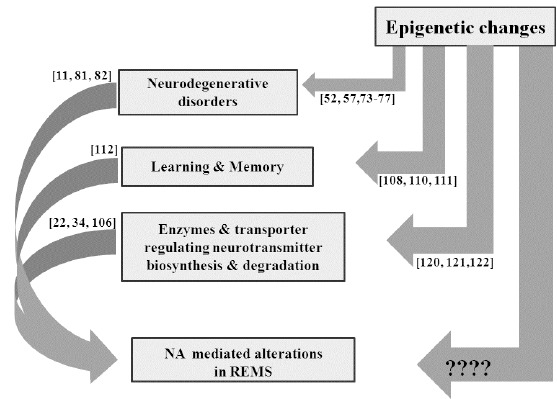
This figure summarizes that epigenetic modifications modulate various behaviours and bio-molecules, including NA;all these changes are known to affect REMS. However, the role of epigenetic modifications in NA mediated REMS modulation is unknown.
7. A PROPOSED MODEL FOR REMS AND ITS LOSS ASSOCIATED EPIGENETIC REGULATION
So far we have discussed that neural regulation of REMS is a complex process involving several neurotransmitters and other bio-molecules. We now know that the NA-ergic neurons in the LC must cease activity for the appearance of REMS, their continuous activation causes REMS loss [33] and elevated NA levels due to REMS loss is a key factor for inducing many of the REMS loss associated symptoms [20]. Therefore, we propose that REMS is affected by several bio-molecules (factors) which are regulated by the epigenetic changes and in turn modulate i) the levels of NA at the synaptic site, and ii) NA-induced pathophysiological changes. We now discuss the possible epigenetic regulation of the bio-molecules.
7.1. Epigenetic Control of Modulation of NA Levels at the Synaptic Site
The maintenance of levels of neurotransmitters, including NA at the synaptic site is a dynamic state. The NA levels at the synaptic site would depend on its release from the presynaptic terminal, which in turn is dependent on the various inputs on the LC-NA-ergic neurons. The NA levels and its physiological actions are dependent on its synthesis, degradation, re-uptake and the density of NA-receptors at the pre- and post-synaptic sites. Each of these processes is dependent on various enzymes and transporters like TH, MAO, NAT and NA-ergic receptor; a common factor being the transcriptional regulation of synthesis of these molecules. For example, as explained earlier, epigenetic modulation of TH [120], NAT [121, 122] and NA-ergic receptors [44, 123, 124] have been reported; however their relationship with REMS and its loss needs to be studied. We propose that factors directly or indirectly modulating transcription and effectiveness of these molecules may affect REMS, or conversely REMS and its loss may modulate some factors, which in turn may affect these molecules and induce REMS-loss associated changes.
Neuronal activity and release of neurotransmitters are dependent on depolarization of the neurons. In response to depolarization of neurons, voltage gated Ca2+ channels in the neuronal membranes open [125] and increase Ca2+ influx into the intracellular space. This cytosolic Ca2+ plays a significant role in regulating many intracellular signaling processes [126, 127], including neuronal activity-dependent gene expressions [128, 129]. The Ca2+ stimulates secondary messengers causing activation and translocation of kinase pathway, including CREB kinase into the nucleus, which results in phosphorylation and activation of CREB [127]. The Ca2+ influx into the neurons is known to recruit calmodulin kinase (CaMK) IV and it is important to note that like N-methyl D- aspartate receptor, Ca2+ is equally potent in causing nuclear transport and activation of HAT protein p300/CBP mediated gene expression [126]. The activation and binding of HAT protein can also mediate the export of HDAC2 and HDAC5 from the nucleus [130]. It is known that pCREB binds to the promoter regions of NA regulated genes [120, 132] and mediates the downstream signaling pathway of NA via PKA/PKC pathway [131]; it also binds to the NAT promoter region [133]. It has been reported that upon REMSD there are alterations in the activity of the NA-ergic neurons [21], the intracellular Ca2+-level [134], TH level [106], ATPase [135] and MAO [34] activities. Thus, upon REMS loss it is likely that the elevated intracellular [Ca2+] concentration would recruit CaMK IV, which would phosphorylate and activate CBP and possibly other factors as well (Fig. 3). The CBP acts as both CREB coactivator as well as HAT protein, which gets activated after phosphorylation by CaMK IV [136]. The p300 is also a HAT protein involved in the chromatin uncoiling. The activated CBP and p300 cause relaxation of the chromatin structure through their intrinsic HAT activity, that increase accessibility of the TF, like CREB and cofactors to the TH genomic DNA and thus upregulates the gene transcription.
Fig. (3).
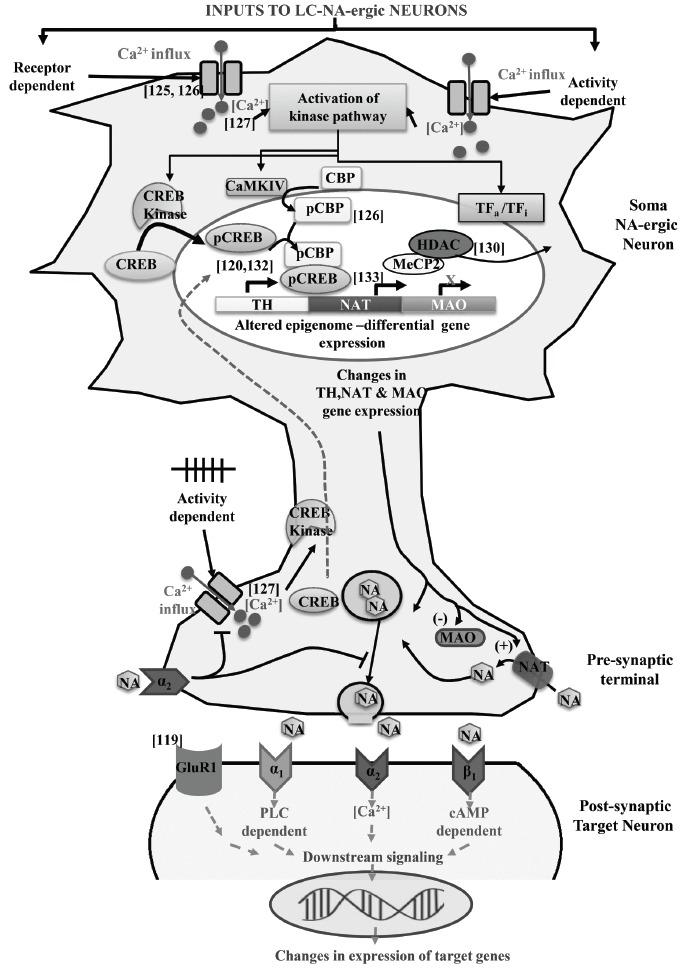
A proposed model explaining how epigenetic modifications in LC-neurons would regulate NA levels in the brain up on REMS-loss. Also, the NA levels in turn might modulate factors for transcriptional regulation of other bio-molecules in different neurons in the brain. Abbreviations are as in the text.
The proposition given above may be supported by the fact that epigenetic regulation of TH gene promoter through methylation has been observed in the hypothalamic region of the brain [137]. Synaptic activity dependent Ca2+ signaling can also deactivate the MeCP2 protein via its phosphorylation by CaMKs [138]. The MeCP2 protein also recruits HDAC to inhibit gene expression [139]. Thus, MeCP2 suppression through phosphorylation by CaMKs will prevent its interaction with CpG residues across the TH gene promoter which further would augment TH gene expression. DNA methylation was seen to alter the function of many elements linked to sleep need and synaptic partners of neuroligin, which are regulators of sleep intensity following SD [47].
Thus, epigenetic modulation within the NA-ergic neurons provide an environment for co-repressors and co-activators to bind to the modified histone tails and regulate the transcription of one or more genes (e.g. TH, MAO, NAT) involved in the synthesis, release, degradation and re-uptake of NA in the LC-NA-ergic REM-OFF neurons. Our model may further be supported by the observation that sleep loss has been shown to up- or down-regulate several genes [140] and it has also been proposed to affect microRNA levels in the brain [141, 142]. Therefore, epigenetic modulation in NA-ergic neurons in response to synaptic activity, specifically for TH regulation, due to altered HATs activity and DNA methylation across the TH gene promoter would open chromatin template. This will make the DNA accessible for the binding of TFs and other co-activator proteins required for altered TH gene expression leading to modulation of NA synthesis. Similarly, expressions of NAT, MAO, adrenoceptors also might get modulated. However, the specific changes taking place due to REMS loss need to be studied.
7.2. REMS Loss Associated NA-induced Epigenetic Modification
We have argued above that how modulation of various factors may alter the synthesis and release of NA. The altered levels of NA may then modulate gene expression in the projected neurons or in itself due to the presence of collateral inputs onto itself [143, 144]. Increased NA-ergic signaling would activate adrenoceptor and mediate signaling by activation or inhibition of various TFs. For example, the NA induces phosphorylation of glutamate receptor1 (GluR1) by inhibiting PPI [119] or activation of pCREB following the PKA pathway [131]. Similarly, increased NA can modulate the intracellular Ca2+ [20, 145, 146] and induce epigenetic modifications (as explained above) to modulate GluR1- and PPI-gene expressions. Thus, the altered NA levels, including upon REMS loss, may modulate the chromatin uncoiling, phosphorylation and expression of many genes including that of GluR1.
Such changes may directly or indirectly modulate expressions of many molecules including neurotransmitters, which are known to affect REMS and its loss-associated memory impairment [117], psycho-behavioral changes e.g. AD, mood disorders, depression and apoptosis [79]. Modulation of the gene expression e.g. by DNMTs and HDAC inhibitors, which may precipitate or predispose to psychiatric disorders, are potential targets of epigenetic drug therapy. For example, HDAC inhibitors like valproic acid and sodium butyrate have been used in the treatment of sleep loss associated disorders like depression [147], learning and memory [148], epilepsy [121], schizophrenia [149]. Also, ‘MS-275’, which is known to cross blood brain barrier and has selectivity for HDAC1, has been developed as one such potential drug molecule [149].
CONCLUSION
REMS loss is reported to elevate the levels of NA in the brain. The modulation of the levels of NA at the synaptic cleft is a dynamic process and it is regulated by many biomolecules, which are transcriptionally regulated. The NA per se may modulate transcription of many molecules, which then may be responsible for REMS loss associated patho-physio-psycho-behavioral changes. Obviously, dynamicity and equilibrium of these processes are very complex and diverse and therefore, studying REMS loss associated changes have been difficult. Although the investigation of epigenetic mechanisms in REMS regulation is still at its infancy, using modern technology, these molecular mechanisms need to be explored for better understanding of REMS regulation in normal and diseased conditions.
ACKNOWLEDGEMENTS
Research funding to BNM from Indian funding agencies under BUILDER program; Council of Scientific and Industrial Research (CSIR); Department of Biotechnology (DBT) and J C Bose fellowship (DST) is acknowledged.
LIST OF ABBREVIATIONS
- AD
= Alzheimer’s disease
- BDNF
= brain derived neurotrophic factor
- CaMKs
= calmodulin kinase
- CpG
= cytosine-phosphate-guanine
- CREB
= cAMP response element-binding protein
- CBP
= CREB binding protein
- DNMT
= DNA methyl transferases
- EEG
= electroencephalogram
- EMG
= electromyogram
- EOG
= electrooculogram
- GABA
= gamma-amino butyric acid
- GAD1
= glutamate decarboxylase1
- GluR1
= glutamate receptor1
- HAT
= histone acetyltransferase
- HDAC
= histone deacetylase
- LC
= locus coeruleus
- MAOA
= monoamine oxidase A
- MBD/MeCP2
= methyl-CpG-binding domain/protein
- NA
= noradrenaline
- NAT
= noradrenaline transporter
- NREMS
= non REMS
- PPI
= protein phosphatase I
- REMS
= rapid eye movement sleep
- REMSD
= REMS deprivation
- SD
= sleep deprivation
- TFs
= transcription factors
- TH
= tyrosine hydroxylase
CONFLICT OF INTEREST
The authors confirm that this article content has no conflict of interest.
References
- 1.Siegel J.M. Sleep viewed as a state of adaptive inactivity. Nat. Rev. Neurosci. 2009;10(10):747–753. doi: 10.1038/nrn2697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Roffwarg H.P., Muzio J.N., Dement W.C. Ontogenetic development of the human sleep-dream cycle. Science. 1966;152(3722):604–619. doi: 10.1126/science.152.3722.604. [DOI] [PubMed] [Google Scholar]
- 3.Frank M.G., Heller H.C. The ontogeny of mammalian sleep: a reappraisal of alternative hypotheses. J. Sleep Res. 2003;12(1):25–34. doi: 10.1046/j.1365-2869.2003.00339.x. [DOI] [PubMed] [Google Scholar]
- 4.Mirmiran M., Yolanda M.G. The function of Fetal/ Neonatal REM sleep. In: Dekker M., editor. Rapid Eye Movement Sleep, Mallick, B.N., Inoue, S. New York; 1999. [Google Scholar]
- 5.Siegel J.M. Sleep in monotremes: implication for the evolution of REM sleep. In: Hayaishi O., Inoué S., editors. Sleep and Sleep Disorders: from Molecule to Behavior. Tokyo: Acadamic Press; 1997. pp. 113–128. [Google Scholar]
- 6.Stickgold R., Walker M.P. Memory consolidation and reconsolidation: what is the role of sleep? Trends Neurosci. 2005;28(8):408–415. doi: 10.1016/j.tins.2005.06.004. [DOI] [PubMed] [Google Scholar]
- 7.Stickgold R. Sleep-dependent memory consolidation. Nature. 2005;437(7063):1272–1278. doi: 10.1038/nature04286. [DOI] [PubMed] [Google Scholar]
- 8.Matos G., Tufik S., Scorza F.A., Cavalheiro E.A., Andersen M.L. Sleep and epilepsy: exploring an intriguing relationship with a translational approach. Epilepsy Behav. 2013;26(3):405–409. doi: 10.1016/j.yebeh.2012.12.003. [DOI] [PubMed] [Google Scholar]
- 9.Lima M.M. Sleep disturbances in Parkinson's disease: the contribution of dopamine in REM sleep regulation. Sleep Med. Rev. 2013;17(5):367–375. doi: 10.1016/j.smrv.2012.10.006. [DOI] [PubMed] [Google Scholar]
- 10.García-Alberca J.M., Lara J.P., Cruz B., Garrido V., Gris E., Barbancho M.A. Sleep disturbances in Alzheimer’s disease are associated with neuropsychiatric symptoms and antidementia treatment. J. Nerv. Ment. Dis. 2013;201(3):251–257. doi: 10.1097/NMD.0b013e3182848d04. [DOI] [PubMed] [Google Scholar]
- 11.Gagnon J.F., Petit D., Latreille V., Montplaisir J. Neurobiology of sleep disturbances in neurodegenerative disorders. Curr. Pharm. Des. 2008;14(32):3430–3445. doi: 10.2174/138161208786549353. [DOI] [PubMed] [Google Scholar]
- 12.Kushida C.A. New York: marcel-dekker; 2005. Sleep Deprivation; basic science, physiology and behavior. [Google Scholar]
- 13.Mallick B.N., Majumdar S., Faisal M., Yadav V., Madan V., Pal D. Role of norepinephrine in the regulation of rapid eye movement sleep. J. Biosci. 2002;27(5):539–551. doi: 10.1007/BF02705052. [DOI] [PubMed] [Google Scholar]
- 14.Powledge T.M. Behavioral Epigenetics: How Nurture Shapes Nature. Bioscience. 2011;61(8):588–592. doi: 10.1525/bio.2011.61.8.4. [DOI] [Google Scholar]
- 15.Keverne E.B., Curley J.P. Epigenetics, brain evolution and behaviour. Front. Neuroendocrinol. 2008;29(3):398–412. doi: 10.1016/j.yfrne.2008.03.001. [DOI] [PubMed] [Google Scholar]
- 16.Qureshi I.A., Mehler M.F. Epigenetics of sleep and chronobiology. Curr. Neurol. Neurosci. Rep. 2014;14(3):432. doi: 10.1007/s11910-013-0432-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McNamara P. Genomic Imprinting and Neurodevelopmental Disorders of Sleep. Sleep and Hypnosis. 2004;6:2:82–90. [Google Scholar]
- 18.Tucci V., Nolan P.M. Evolution of Sleep: Phylogenetic and Functional Perspectives. Cambridge University Press; 2009. Toward an understanding of the function of sleep: New insights from mouse genetics. pp. 218–237. [DOI] [Google Scholar]
- 19.Lassi G., Ball S.T., Maggi S., Colonna G., Nieus T., Cero C., Bartolomucci A., Peters J., Tucci V. Loss of Gnas imprinting differentially affects REM/NREM sleep and cognition in mice. PLoS Genet. 2012;8(5):e1002706. doi: 10.1371/journal.pgen.1002706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mallick B.N., Singh A. REM sleep loss increases brain excitability: role of noradrenaline and its mechanism of action. Sleep Med. Rev. 2011;15(3):165–178. doi: 10.1016/j.smrv.2010.11.001. [DOI] [PubMed] [Google Scholar]
- 21.Mallick B.N., Siegel J.M., Fahringer H. Changes in pontine unit activity with REM sleep deprivation. Brain Res. 1990;515(1-2):94–98. doi: 10.1016/0006-8993(90)90581-U. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Porkka-Heiskanen T., Smith S.E., Taira T., Urban J.H., Levine J.E., Turek F.W., Stenberg D. Noradrenergic activity in rat brain during rapid eye movement sleep deprivation and rebound sleep. Am. J. Physiol. 1995;268(6 Pt 2):R1456–R1463. doi: 10.1152/ajpregu.1995.268.6.R1456. [DOI] [PubMed] [Google Scholar]
- 23.Gary S. Brain Norepinephrine Neurobiology and Therapeutics. Cambridge University Press; 2007. Aston-Jones; Gonzalez, M.; Doran, S. Role of the locus coeruleus-norepinephrine system in arousal and circadian regulation of the sleep–wake cycle. pp. 157–195. [Google Scholar]
- 24.Mallick B.N., Pandi-Permual S.R., McCarley R.W., Morrison A.R. Rapid Eye Movement Sleep – Regulation and Function. Cambridge University Press; 2011. [DOI] [Google Scholar]
- 25.McCarley R.W., Hobson J.A. Discharge patterns of cat pontine brain stem neurons during desynchronized sleep. J. Neurophysiol. 1975;38(4):751–766. doi: 10.1152/jn.1975.38.4.751. [DOI] [PubMed] [Google Scholar]
- 26.Singh S., Mallick B.N. Mild electrical stimulation of pontine tegmentum around locus coeruleus reduces rapid eye movement sleep in rats. Neurosci. Res. 1996;24(3):227–235. doi: 10.1016/0168-0102(95)00998-1. [DOI] [PubMed] [Google Scholar]
- 27.Kaur S., Saxena R.N., Mallick B.N. GABA in locus coeruleus regulates spontaneous rapid eye movement sleep by acting on GABAA receptors in freely moving rats. Neurosci. Lett. 1997;223(2):105–108. doi: 10.1016/S0304-3940(97)13410-3. [DOI] [PubMed] [Google Scholar]
- 28.Kaur S., Panchal M., Faisal M., Madan V., Nangia P., Mallick B.N. Long term blocking of GABA-A receptor in locus coeruleus by bilateral microinfusion of picrotoxin reduced rapid eye movement sleep and increased brain Na-K ATPase activity in freely moving normally behaving rats. Behav. Brain Res. 2004;151(1-2):185–190. doi: 10.1016/j.bbr.2003.08.011. [DOI] [PubMed] [Google Scholar]
- 29.Kaur S., Saxena R.N., Mallick B.N. GABAergic neurons in prepositus hypoglossi regulate REM sleep by its action on locus coeruleus in freely moving rats. Synapse. 2001;42(3):141–150. doi: 10.1002/syn.1109. [DOI] [PubMed] [Google Scholar]
- 30.Jaiswal M.K., Dvela M., Lichtstein D., Mallick B.N. Endogenous ouabain-like compounds in locus coeruleus modulate rapid eye movement sleep in rats. J. Sleep Res. 2010;19(1 Pt 2):183–191. doi: 10.1111/j.1365-2869.2009.00781.x. [DOI] [PubMed] [Google Scholar]
- 31.Pal D., Mallick B.N. Neural mechanism of rapid eye movement sleep generation with reference to REM-OFF neurons in locus coeruleus. Indian J. Med. Res. 2007;125(6):721–739. [PubMed] [Google Scholar]
- 32.Carter M.E., Yizhar O., Chikahisa S., Nguyen H., Adamantidis A., Nishino S., Deisseroth K., de Lecea L. Tuning arousal with optogenetic modulation of locus coeruleus neurons. Nat. Neurosci. 2010;13(12):1526–1533. doi: 10.1038/nn.2682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mallick B.N., Singh A., Khanday M.A. Activation of inactivation process initiates rapid eye movement sleep. Prog. Neurobiol. 2012;97(3):259–276. doi: 10.1016/j.pneurobio.2012.04.001. [DOI] [PubMed] [Google Scholar]
- 34.Thakkar M., Mallick B.N. Effect of rapid eye movement sleep deprivation on rat brain monoamine oxidases. Neuroscience. 1993;55(3):677–683. doi: 10.1016/0306-4522(93)90433-G. [DOI] [PubMed] [Google Scholar]
- 35.Tellioglu T., Robertson D. Genetic or acquired deficits in the norepinephrine transporter: current understanding of clinical implications. Expert Rev. Mol. Med. 2001;2001:1–10. doi: 10.1017/S1462399401003878. [DOI] [PubMed] [Google Scholar]
- 36.Hilakivi I. The role of beta- and alpha-adrenoceptors in the regulation of the stages of the sleep-waking cycle in the cat. Brain Res. 1983;277(1):109–118. doi: 10.1016/0006-8993(83)90912-5. [DOI] [PubMed] [Google Scholar]
- 37.Mallick B.N., Singh S., Pal D. Role of alpha and beta adrenoceptors in locus coeruleus stimulation-induced reduction in rapid eye movement sleep in freely moving rats. Behav. Brain Res. 2005;158(1):9–21. doi: 10.1016/j.bbr.2004.08.004. [DOI] [PubMed] [Google Scholar]
- 38.Hilakivi L.A., Taira T., Hilakivi I. Early postnatal deprivation of active sleep with desipramine or zimeldine impairs later behavioural reactivity to auditory stimuli in rats. Acta Physiol. Scand. 1988;132(2):191–198. doi: 10.1111/j.1748-1716.1988.tb08317.x. [DOI] [PubMed] [Google Scholar]
- 39.Basheer R., Magner M., McCarley R.W., Shiromani P.J. REM sleep deprivation increases the levels of tyrosine hydroxylase and norepinephrine transporter mRNA in the locus coeruleus. Brain Res. Mol. Brain Res. 1998;57(2):235–240. doi: 10.1016/S0169-328X(98)00088-6. [DOI] [PubMed] [Google Scholar]
- 40.Kumer S.C., Vrana K.E. Intricate regulation of tyrosine hydroxylase activity and gene expression. J. Neurochem. 1996;67(2):443–462. doi: 10.1046/j.1471-4159.1996.67020443.x. [DOI] [PubMed] [Google Scholar]
- 41.Lewis E.J., Tank A.W., Weiner N., Chikaraishi D.M. Regulation of tyrosine hydroxylase mRNA by glucocorticoid and cyclic AMP in a rat pheochromocytoma cell line. Isolation of a cDNA clone for tyrosine hydroxylase mRNA. J. Biol. Chem. 1983;258(23):14632–14637. [PubMed] [Google Scholar]
- 42.Lewis E.J., Harrington C.A., Chikaraishi D.M. Transcriptional regulation of the tyrosine hydroxylase gene by glucocorticoid and cyclic AMP. Proc. Natl. Acad. Sci. USA. 1987;84(11):3550–3554. doi: 10.1073/pnas.84.11.3550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lewis-Tuffin L.J., Quinn P.G., Chikaraishi D.M. Tyrosine hydroxylase transcription depends primarily on cAMP response element activity, regardless of the type of inducing stimulus. Mol. Cell. Neurosci. 2004;25(3):536–547. doi: 10.1016/j.mcn.2003.10.010. [DOI] [PubMed] [Google Scholar]
- 44.Michelotti G.A., Brinkley D.M., Morris D.P., Smith M.P., Louie R.J., Schwinn D.A. Epigenetic regulation of human alpha1d-adrenergic receptor gene expression: a role for DNA methylation in Sp1-dependent regulation. FASEB J. 2007;21(9):1979–1993. doi: 10.1096/fj.06-7118com. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Razik M.A., Lee K., Price R.R., Williams M.R., Ongjoco R.R., Dole M.K., Rudner X.L., Kwatra M.M., Schwinn D.A. Transcriptional regulation of the human alpha1a-adrenergic receptor gene. Characterization Of the 5′-regulatory and promoter region. J. Biol. Chem. 1997;272(45):28237–28246. doi: 10.1074/jbc.272.45.28237. [DOI] [PubMed] [Google Scholar]
- 46.Zhu Q.S., Chen K., Shih J.C. Bidirectional promoter of human monoamine oxidase A (MAO A) controlled by transcription factor Sp1. J. Neurosci. 1994;14(12):7393–7403. doi: 10.1523/JNEUROSCI.14-12-07393.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Massart R., Freyburger M., Suderman M., Paquet J., El Helou J., Belanger-Nelson E., Rachalski A., Koumar O.C., Carrier J., Szyf M., Mongrain V. The genome-wide landscape of DNA methylation and hydroxymethylation in response to sleep deprivation impacts on synaptic plasticity genes. Transl. Psychiatry. 2014;4:e347. doi: 10.1038/tp.2013.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Berger S.L. The complex language of chromatin regulation during transcription. Nature. 2007;447(7143):407–412. doi: 10.1038/nature05915. [DOI] [PubMed] [Google Scholar]
- 49.Vinci M.C., Polvani G., Pesce M. Epigenetic programming and risk: the birthplace of cardiovascular disease? Stem Cell Rev. 2013;9(3):241–253. doi: 10.1007/s12015-012-9398-z. [DOI] [PubMed] [Google Scholar]
- 50.Law J.A., Jacobsen S.E. Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat. Rev. Genet. 2010;11(3):204–220. doi: 10.1038/nrg2719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Prokhortchouk E., Defossez P.A. The cell biology of DNA methylation in mammals. Biochimica Et Biophysica Acta-Mol. Cell Res. 2008;1783(11):2167–2173. doi: 10.1016/j.bbamcr.2008.07.015. [DOI] [PubMed] [Google Scholar]
- 52.Gos M. Epigenetic mechanisms of gene expression regulation in neurological diseases. Acta Neurobiol. Exp. (Warsz.) 2013;73(1):19–37. doi: 10.55782/ane-2013-1919. [DOI] [PubMed] [Google Scholar]
- 53.Jaenisch R., Bird A. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat. Genet. 2003;33(Suppl.):245–254. doi: 10.1038/ng1089. [DOI] [PubMed] [Google Scholar]
- 54.Robertson K.D. DNA methylation and chromatin - unraveling the tangled web. Oncogene. 2002;21(35):5361–5379. doi: 10.1038/sj.onc.1205609. [DOI] [PubMed] [Google Scholar]
- 55.Portela A., Esteller M. Epigenetic modifications and human disease. Nat. Biotechnol. 2010;28(10):1057–1068. doi: 10.1038/nbt.1685. [DOI] [PubMed] [Google Scholar]
- 56.Li X., Wei W., Ratnu V.S., Bredy T.W. On the potential role of active DNA demethylation in establishing epigenetic states associated with neural plasticity and memory. Neurobiol. Learn Mem. 2013;105:125–132. doi: 10.1016/j.nlm.2013.06.009. [DOI] [PubMed] [Google Scholar]
- 57.Qureshi I.A., Mehler M.F. Epigenetic mechanisms governing the process of neurodegeneration. Mol. Aspects Med. 2013;34(4):875–882. doi: 10.1016/j.mam.2012.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Peterson C.L. Chromatin remodeling: nucleosomes bulging at the seams. Curr. Biol. 2002;12(7):R245–R247. doi: 10.1016/S0960-9822(02)00782-0. [DOI] [PubMed] [Google Scholar]
- 59.Kouzarides T. Chromatin modifications and their function. Cell. 2007;128(4):693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 60.Choi J.K., Howe L.J. Histone acetylation: truth of consequences? Biochem. Cell Biol. 2009;87(1):139–150. doi: 10.1139/O08-112. [DOI] [PubMed] [Google Scholar]
- 61.Nimura K., Ura K., Kaneda Y. Histone methyltransferases: regulation of transcription and contribution to human disease. J. Mol. Med. (Berl). 2010;88(12):1213–1220. doi: 10.1007/s00109-010-0668-4. [DOI] [PubMed] [Google Scholar]
- 62.Berger S.L. Histone modifications in transcriptional regulation. Curr. Opin. Genet. Dev. 2002;12(2):142–148. doi: 10.1016/S0959-437X(02)00279-4. [DOI] [PubMed] [Google Scholar]
- 63.Day J.J., Sweatt J.D. DNA methylation and memory formation. Nat. Neurosci. 2010;13(11):1319–1323. doi: 10.1038/nn.2666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pirooznia S.K., Elefant F. Targeting specific HATs for neurodegenerative disease treatment: translating basic biology to therapeutic possibilities. Front. Cell. Neurosci. 2013;7:30. doi: 10.3389/fncel.2013.00030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mehler M.F. Epigenetic principles and mechanisms underlying nervous system functions in health and disease. Prog. Neurobiol. 2008;86(4):305–341. doi: 10.1016/j.pneurobio.2008.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mitchell H.A., Weinshenker D. Good night and good luck: norepinephrine in sleep pharmacology. Biochem. Pharmacol. 2010;79(6):801–809. doi: 10.1016/j.bcp.2009.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Adolfsson R., Gottfries C.G., Roos B.E., Winblad B. Changes in the brain catecholamines in patients with dementia of Alzheimer type. Br. J. Psychiatry. 1979;135:216–223. doi: 10.1192/bjp.135.3.216. [DOI] [PubMed] [Google Scholar]
- 68.Comella C.L. Sleep disturbances and excessive daytime sleepiness in Parkinson disease: an overview. J. Neural Transm. Suppl. 2006;(70):349–355. doi: 10.1007/978-3-211-45295-0_53. [DOI] [PubMed] [Google Scholar]
- 69.Sanchez C., Brennum L.T., Storustovu S., Kreilgard M., Mork A. Depression and poor sleep: the effect of monoaminergic antidepressants in a pre-clinical model in rats. Pharmacol. Biochem. Behav. 2007;86(3):468–476. doi: 10.1016/j.pbb.2007.01.006. [DOI] [PubMed] [Google Scholar]
- 70.Leszek J., Sochocka M., Gasiorowski K. Vascular factors and epigenetic modifications in the pathogenesis of Alzheimer's disease. J. Neurol. Sci. 2012;323(1-2):25–32. doi: 10.1016/j.jns.2012.09.010. [DOI] [PubMed] [Google Scholar]
- 71.Goedert M., Spillantini M.G. A century of Alzheimer’s disease. Science. 2006;314(5800):777–781. doi: 10.1126/science.1132814. [DOI] [PubMed] [Google Scholar]
- 72.Roberson E.D., Mucke L. 100 years and counting: prospects for defeating Alzheimer’s disease. Science. 2006;314(5800):781–784. doi: 10.1126/science.1132813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lee J., Ryu H. Epigenetic modification is linked to Alzheimer’s disease: is it a maker or a marker? BMB Rep. 2010;43(10):649–655. doi: 10.5483/BMBRep.2010.43.10.649. [DOI] [PubMed] [Google Scholar]
- 74.Tohgi H., Utsugisawa K., Nagane Y., Yoshimura M., Ukitsu M., Genda Y. The methylation status of cytosines in a tau gene promoter region alters with age to downregulate transcriptional activity in human cerebral cortex. Neurosci. Lett. 1999;275(2):89–92. doi: 10.1016/S0304-3940(99)00731-4. [DOI] [PubMed] [Google Scholar]
- 75.Tohgi H., Utsugisawa K., Nagane Y., Yoshimura M., Genda Y., Ukitsu M. Reduction with age in methylcytosine in the promoter region -224 approximately -101 of the amyloid precursor protein gene in autopsy human cortex. Brain Res. Mol. Brain Res. 1999;70(2):288–292. doi: 10.1016/S0169-328X(99)00163-1. [DOI] [PubMed] [Google Scholar]
- 76.Cao X., Südhof T.C. A transcriptionally [correction of transcriptively] active complex of APP with Fe65 and histone acetyltransferase Tip60. Science. 2001;293(5527):115–120. doi: 10.1126/science.1058783. [DOI] [PubMed] [Google Scholar]
- 77.Saura C.A., Choi S.Y., Beglopoulos V., Malkani S., Zhang D., Shankaranarayana Rao B.S., Chattarji S., Kelleher R.J., III, Kandel E.R., Duff K., Kirkwood A., Shen J. Loss of presenilin function causes impairments of memory and synaptic plasticity followed by age-dependent neurodegeneration. Neuron. 2004;42(1):23–36. doi: 10.1016/S0896-6273(04)00182-5. [DOI] [PubMed] [Google Scholar]
- 78.Kang J.E., Lim M.M., Bateman R.J., Lee J.J., Smyth L.P., Cirrito J.R., Fujiki N., Nishino S., Holtzman D.M. Amyloid-beta dynamics are regulated by orexin and the sleep-wake cycle. Science. 2009;326(5955):1005–1007. doi: 10.1126/science.1180962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Biswas S., Mishra P., Mallick B.N. Increased apoptosis in rat brain after rapid eye movement sleep loss. Neuroscience. 2006;142(2):315–331. doi: 10.1016/j.neuroscience.2006.06.026. [DOI] [PubMed] [Google Scholar]
- 80.Heneka M.T., Ramanathan M., Jacobs A.H., Dumitrescu-Ozimek L., Bilkei-Gorzo A., Debeir T., Sastre M., Galldiks N., Zimmer A., Hoehn M., Heiss W.D., Klockgether T., Staufenbiel M. Locus ceruleus degeneration promotes Alzheimer pathogenesis in amyloid precursor protein 23 transgenic mice. J. Neurosci. 2006;26(5):1343–1354. doi: 10.1523/JNEUROSCI.4236-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Liu L., Luo S., Zeng L., Wang W., Yuan L., Jian X. Degenerative alterations in noradrenergic neurons of the locus coeruleus in Alzheimer’s disease. Neural Regen. Res. 2013;8(24):2249–2255. doi: 10.3969/j.issn.1673-5374.2013.24.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Tejani-Butt S.M., Yang J., Zaffar H. Norepinephrine transporter sites are decreased in the locus coeruleus in Alzheimer’s disease. Brain Res. 1993;631(1):147–150. doi: 10.1016/0006-8993(93)91201-3. [DOI] [PubMed] [Google Scholar]
- 83.Francis B.M., Yang J., Hajderi E., Brown M.E., Michalski B., McLaurin J., Fahnestock M., Mount H.T. Reduced tissue levels of noradrenaline are associated with behavioral phenotypes of the TgCRND8 mouse model of Alzheimer’s disease. Neuropsychopharmacology. 2012;37(8):1934–1944. doi: 10.1038/npp.2012.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Menke A., Klengel T., Binder E.B. Epigenetics, depression and antidepressant treatment. Curr. Pharm. Des. 2012;18(36):5879–5889. doi: 10.2174/138161212803523590. [DOI] [PubMed] [Google Scholar]
- 85.Dwivedi Y., Rizavi H.S., Conley R.R., Roberts R.C., Tamminga C.A., Pandey G.N. Altered gene expression of brain-derived neurotrophic factor and receptor tyrosine kinase B in postmortem brain of suicide subjects. Arch. Gen. Psychiatry. 2003;60(8):804–815. doi: 10.1001/archpsyc.60.8.804. [DOI] [PubMed] [Google Scholar]
- 86.Martinowich K., Hattori D., Wu H., Fouse S., He F., Hu Y., Fan G., Sun Y.E. DNA methylation-related chromatin remodeling in activity-dependent BDNF gene regulation. Science. 2003;302(5646):890–893. doi: 10.1126/science.1090842. [DOI] [PubMed] [Google Scholar]
- 87.Benraiss A., Chmielnicki E., Lerner K., Roh D., Goldman S.A. Adenoviral brain-derived neurotrophic factor induces both neostriatal and olfactory neuronal recruitment from endogenous progenitor cells in the adult forebrain. J. Neurosci. 2001;21(17):6718–6731. doi: 10.1523/JNEUROSCI.21-17-06718.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Pencea V., Bingaman K.D., Wiegand S.J., Luskin M.B. Infusion of brain-derived neurotrophic factor into the lateral ventricle of the adult rat leads to new neurons in the parenchyma of the striatum, septum, thalamus, and hypothalamus. J. Neurosci. 2001;21(17):6706–6717. doi: 10.1523/JNEUROSCI.21-17-06706.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Covington H.E., III, Maze I., LaPlant Q.C., Vialou V.F., Ohnishi Y.N., Berton O., Fass D.M., Renthal W., Rush A.J., III, Wu E.Y., Ghose S., Krishnan V., Russo S.J., Tamminga C., Haggarty S.J., Nestler E.J. Antidepressant actions of histone deacetylase inhibitors. J. Neurosci. 2009;29(37):11451–11460. doi: 10.1523/JNEUROSCI.1758-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ferland C.L., Schrader L.A. Regulation of histone acetylation in the hippocampus of chronically stressed rats: a potential role of sirtuins. Neuroscience. 2011;174:104–114. doi: 10.1016/j.neuroscience.2010.10.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Roy A., Pickar D., De Jong J., Karoum F., Linnoila M. Norepinephrine and its metabolites in cerebrospinal fluid, plasma, and urine. Relationship to hypothalamic-pituitary-adrenal axis function in depression. Arch. Gen. Psychiatry. 1988;45(9):849–857. doi: 10.1001/archpsyc.1988.01800330081010. [DOI] [PubMed] [Google Scholar]
- 92.Benca R.M. Kryger M.H., Roth T., Dement W.C., editors. Mood disorders. Principles and practice of sleep medicine. 2000:1140–1157. [Google Scholar]
- 93.Moret C., Briley M. The importance of norepinephrine in depression. Neuropsychiatr. Dis. Treat. 2011;7(Suppl. 1):9–13. doi: 10.2147/NDT.S19619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Ryu S.H., Lee S.H., Lee H.J., Cha J.H., Ham B.J., Han C.S., Choi M.J., Lee M.S. Association between norepinephrine transporter gene polymorphism and major depression. Neuropsychobiology. 2004;49(4):174–177. doi: 10.1159/000077361. [DOI] [PubMed] [Google Scholar]
- 95.Akbarian S., Cheung I., Connor C., Jakovcevski M., Jiang Y. Petronis A., Mill J., editors. Posttranslational Histone Modifications and the Neurobiology of Psychosis. Brain, Behavior and epigenetics, Epigenetics and Human Health. 2011:1–21. doi: 10.1007/978-3-642-17426-1_1. [DOI] [Google Scholar]
- 96.Belforte J.E., Zsiros V., Sklar E.R., Jiang Z., Yu G., Li Y., Quinlan E.M., Nakazawa K. Postnatal NMDA receptor ablation in corticolimbic interneurons confers schizophrenia-like phenotypes. Nat. Neurosci. 2010;13(1):76–83. doi: 10.1038/nn.2447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Lewis D.A., Cho R.Y., Carter C.S., Eklund K., Forster S., Kelly M.A., Montrose D. Subunit-selective modulation of GABA type A receptor neurotransmission and cognition in schizophrenia. Am. J. Psychiatry. 2008;165(12):1585–1593. doi: 10.1176/appi.ajp.2008.08030395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Akbarian S., Kim J.J., Potkin S.G., Hagman J.O., Tafazzoli A., Bunney W.E., Jr, Jones E.G. Gene expression for glutamic acid decarboxylase is reduced without loss of neurons in prefrontal cortex of schizophrenics. Arch. Gen. Psychiatry. 1995;52(4):258–266. doi: 10.1001/archpsyc.1995.03950160008002. [DOI] [PubMed] [Google Scholar]
- 99.Dong E., Agis-Balboa R.C., Simonini M.V., Grayson D.R., Costa E., Guidotti A. Reelin and glutamic acid decarboxylase67 promoter remodeling in an epigenetic methionine-induced mouse model of schizophrenia. Proc. Natl. Acad. Sci. USA. 2005;102(35):12578–12583. doi: 10.1073/pnas.0505394102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Huang H.S., Matevossian A., Whittle C., Kim S.Y., Schumacher A., Baker S.P., Akbarian S. Prefrontal dysfunction in schizophrenia involves mixed-lineage leukemia 1-regulated histone methylation at GABAergic gene promoters. J. Neurosci. 2007;27(42):11254–11262. doi: 10.1523/JNEUROSCI.3272-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Tang B., Dean B., Thomas E.A. Disease- and age-related changes in histone acetylation at gene promoters in psychiatric disorders. Transl. Psychiatry. 2011;1:e64. doi: 10.1038/tp.2011.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Tremolizzo L., Carboni G., Ruzicka W.B., Mitchell C.P., Sugaya I., Tueting P., Sharma R., Grayson D.R., Costa E., Guidotti A. An epigenetic mouse model for molecular and behavioral neuropathologies related to schizophrenia vulnerability. Proc. Natl. Acad. Sci. USA. 2002;99(26):17095–17100. doi: 10.1073/pnas.262658999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Cohrs S. Sleep disturbances in patients with schizophrenia : impact and effect of antipsychotics. CNS Drugs. 2008;22(11):939–962. doi: 10.2165/00023210-200822110-00004. [DOI] [PubMed] [Google Scholar]
- 104.Nitz D., Siegel J.M. GABA release in the locus coeruleus as a function of sleep/wake state. Neuroscience. 1997;78(3):795–801. doi: 10.1016/S0306-4522(96)00549-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Watson C.J., Lydic R., Baghdoyan H.A. Sleep and GABA levels in the oral part of rat pontine reticular formation are decreased by local and systemic administration of morphine. Neuroscience. 2007;144(1):375–386. doi: 10.1016/j.neuroscience.2006.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Majumdar S., Mallick B.N. Increased levels of tyrosine hydroxylase and glutamic acid decarboxylase in locus coeruleus neurons after rapid eye movement sleep deprivation in rats. Neurosci. Lett. 2003;338(3):193–196. doi: 10.1016/S0304-3940(02)01404-0. [DOI] [PubMed] [Google Scholar]
- 107.Zovkic I.B., Sweatt J.D. Epigenetic mechanisms in learned fear: implications for PTSD. Neuropsychopharmacology. 2013;38(1):77–93. doi: 10.1038/npp.2012.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Levenson J.M., Sweatt J.D. Epigenetic mechanisms in memory formation. Nat. Rev. Neurosci. 2005;6(2):108–118. doi: 10.1038/nrn1604. [DOI] [PubMed] [Google Scholar]
- 109.Sananbenesi F., Fischer A. The epigenetic bottleneck of neurodegenerative and psychiatric diseases. Biol. Chem. 2009;390(11):1145–1153. doi: 10.1515/BC.2009.131. [DOI] [PubMed] [Google Scholar]
- 110.Zovkic I.B., Guzman-Karlsson M.C., Sweatt J.D. Epigenetic regulation of memory formation and maintenance. Learn. Mem. 2013;20(2):61–74. doi: 10.1101/lm.026575.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Miller C.A., Sweatt J.D. Covalent modification of DNA regulates memory formation. Neuron. 2007;53(6):857–869. doi: 10.1016/j.neuron.2007.02.022. [DOI] [PubMed] [Google Scholar]
- 112.Tully K., Bolshakov V.Y. Emotional enhancement of memory: how norepinephrine enables synaptic plasticity. Mol. Brain. 2010;3:15. doi: 10.1186/1756-6606-3-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Koshibu K., Gräff J., Mansuy I.M. Nuclear protein phosphatase-1: an epigenetic regulator of fear memory and amygdala long-term potentiation. Neuroscience. 2011;173:30–36. doi: 10.1016/j.neuroscience.2010.11.023. [DOI] [PubMed] [Google Scholar]
- 114.Lubin F.D., Roth T.L., Sweatt J.D. Epigenetic regulation of BDNF gene transcription in the consolidation of fear memory. J. Neurosci. 2008;28(42):10576–10586. doi: 10.1523/JNEUROSCI.1786-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Penner M.R., Roth T.L., Chawla M.K., Hoang L.T., Roth E.D., Lubin F.D., Sweatt J.D., Worley P.F., Barnes C.A. Age-related changes in Arc transcription and DNA methylation within the hippocampus. Neurobiol. Aging. 2011;32(12):2198–2210. doi: 10.1016/j.neurobiolaging.2010.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Gräff J., Woldemichael B.T., Berchtold D., Dewarrat G., Mansuy I.M. Dynamic histone marks in the hippocampus and cortex facilitate memory consolidation. Nat. Commun. 2012;3:991. doi: 10.1038/ncomms1997. [DOI] [PubMed] [Google Scholar]
- 117.Stickgold R., Hobson J.A., Fosse R., Fosse M. Sleep, learning, and dreams: off-line memory reprocessing. Science. 2001;294(5544):1052–1057. doi: 10.1126/science.1063530. [DOI] [PubMed] [Google Scholar]
- 118.Maquet P. The role of sleep in learning and memory. Science. 2001;294(5544):1048–1052. doi: 10.1126/science.1062856. [DOI] [PubMed] [Google Scholar]
- 119.Hu H., Real E., Takamiya K., Kang M.G., Ledoux J., Huganir R.L., Malinow R. Emotion enhances learning via norepinephrine regulation of AMPA-receptor trafficking. Cell. 2007;131(1):160–173. doi: 10.1016/j.cell.2007.09.017. [DOI] [PubMed] [Google Scholar]
- 120.Lenartowski R., Goc A. Epigenetic, transcriptional and posttranscriptional regulation of the tyrosine hydroxylase gene. Int. J. Develop. Neurosci. 2011;29(8):873–883. doi: 10.1016/j.ijdevneu.2011.07.006. [DOI] [PubMed] [Google Scholar]
- 121.Bayles R., Baker E., Eikelis N., El-Osta A., Lambert G. Histone modifications regulate the norepinephrine transporter gene. Cell Cycle. 2010;9(22):4600–4601. doi: 10.4161/cc.9.22.13888. [DOI] [PubMed] [Google Scholar]
- 122.Bayles R., Harikrishnan K.N., Lambert E., Baker E.K., Agrotis A., Guo L., Jowett J.B.M., Esler M., Lambert G., El-Osta A. Epigenetic Modification of the Norepinephrine Transporter Gene in Postural Tachycardia Syndrome. Arteriosclerosis Thromb. Vas. Biol. 2012;32(8):U1432–1910. doi: 10.1161/ATVBAHA.111.244343. [DOI] [PubMed] [Google Scholar]
- 123.McAlees J.W., Smith L.T., Erbe R.S., Jarjoura D., Ponzio N.M., Sanders V.M. Epigenetic regulation of beta2-adrenergic receptor expression in T(H)1 and T(H)2 cells. Brain Behav. Immun. 2011;25(3):408–415. doi: 10.1016/j.bbi.2010.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Jiang Q., Yuan H., Xing X., Liu J., Huang Z., Du X. Methylation of adrenergic β1 receptor is a potential epigenetic mechanism controlling antihypertensive response to metoprolol. Indian J. Biochem. Biophys. 2011;48(5):301–307. [PubMed] [Google Scholar]
- 125.Dunlap K., Luebke J.I., Turner T.J. Exocytotic Ca2+ channels in mammalian central neurons. Trends Neurosci. 1995;18(2):89–98. doi: 10.1016/0166-2236(95)80030-6. [DOI] [PubMed] [Google Scholar]
- 126.Hardingham G.E., Chawla S., Cruzalegui F.H., Bading H. Control of recruitment and transcription-activating function of CBP determines gene regulation by NMDA receptors and L-type calcium channels. Neuron. 1999;22(4):789–798. doi: 10.1016/S0896-6273(00)80737-0. [DOI] [PubMed] [Google Scholar]
- 127.Rajadhyaksha A., Barczak A., Macías W., Leveque J.C., Lewis S.E., Konradi C. L-Type Ca(2+) channels are essential for glutamate-mediated CREB phosphorylation and c-fos gene expression in striatal neurons. J. Neurosci. 1999;19(15):6348–6359. doi: 10.1523/JNEUROSCI.19-15-06348.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Brosenitsch T.A., Salgado-Commissariat D., Kunze D.L., Katz D.M. A role for L-type calcium channels in developmental regulation of transmitter phenotype in primary sensory neurons. J. Neurosci. 1998;18(3):1047–1055. doi: 10.1523/JNEUROSCI.18-03-01047.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Murphy T.H., Worley P.F., Baraban J.M. L-type voltage-sensitive calcium channels mediate synaptic activation of immediate early genes. Neuron. 1991;7(4):625–635. doi: 10.1016/0896-6273(91)90375-A. [DOI] [PubMed] [Google Scholar]
- 130.McKinsey T.A., Zhang C.L., Olson E.N. Activation of the myocyte enhancer factor-2 transcription factor by calcium/calmodulin-dependent protein kinase-stimulated binding of 14-3-3 to histone deacetylase 5. Proc. Natl. Acad. Sci. USA. 2000;97(26):14400–14405. doi: 10.1073/pnas.260501497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Carriba P., Pardo L., Parra-Damas A., Lichtenstein M.P., Saura C.A., Pujol A., Masgrau R., Galea E. ATP and noradrenaline activate CREB in astrocytes via noncanonical Ca(2+) and cyclic AMP independent pathways. Glia. 2012;60(9):1330–1344. doi: 10.1002/glia.22352. [DOI] [PubMed] [Google Scholar]
- 132.Tinti C., Conti B., Cubells J.F., Kim K.S., Baker H., Joh T.H. Inducible cAMP early repressor can modulate tyrosine hydroxylase gene expression after stimulation of cAMP synthesis. J. Biol. Chem. 1996;271(41):25375–25381. doi: 10.1074/jbc.271.41.25375. [DOI] [PubMed] [Google Scholar]
- 133.Pellegrino M.J., Parrish D.C., Zigmond R.E., Habecker B.A. Cytokines inhibit norepinephrine transporter expression by decreasing Hand2. Mol. Cell. Neurosci. 2011;46(3):671–680. doi: 10.1016/j.mcn.2011.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Mallick B.N., Gulyani S. Alterations in synaptosomal calcium concentrations after rapid eye movement sleep deprivation in rats. Neuroscience. 1996;75(3):729–736. doi: 10.1016/0306-4522(96)00177-7. [DOI] [PubMed] [Google Scholar]
- 135.Gulyani S., Mallick B.N. Possible mechanism of rapid eye movement sleep deprivation induced increase in Na-K ATPase activity. Neuroscience. 1995;64(1):255–260. doi: 10.1016/0306-4522(94)00333-Z. [DOI] [PubMed] [Google Scholar]
- 136.Hagenston A.M., Bading H. Calcium Signaling in Synapse-to-Nucleus Communication. Cold Spring Harbor Perspectives in Biology. 2011;3(11):a004564. doi: 10.1101/cshperspect.a004564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Vakhitova Y.V., Sadovnikov S.V., Yamidanov R.S., Seredenin S.B. Cytosine demethylation in the tyrosine hydroxylase gene promoter in hypothalamus cells of rat brain under the action of 2-aminoadamantane compound Ladasten. Russ. J. Genet. 2006;42(7):795–801. doi: 10.1134/S1022795406070155. [DOI] [PubMed] [Google Scholar]
- 138.Zhou Z.L., Hong E.J., Cohen S., Zhao W.N., Ho H.Y.H., Schmidt L., Chen W.G., Lin Y.X., Savner E., Griffith E.C., Hu L., Steen J.A.J., Weitz C.J., Greenberg M.E. Brain-specific phosphorylation of MeCP2 regulates activity-dependent Bdnf transcription, dendritic growth, and spine maturation. Neuron. 2006;52(2):255–269. doi: 10.1016/j.neuron.2006.09.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Jones P.L., Veenstra G.J., Wade P.A., Vermaak D., Kass S.U., Landsberger N., Strouboulis J., Wolffe A.P. Methylated DNA and MeCP2 recruit histone deacetylase to repress transcription. Nat. Genet. 1998;19(2):187–191. doi: 10.1016/j.neuron.2006.09.037. [DOI] [PubMed] [Google Scholar]
- 140.Tononi G., Cirelli C. Modulation of brain gene expression during sleep and wakefulness: A review of recent findings. Neuropsychopharmacology. 2001;25:S28–S35. doi: 10.1016/S0893-133X(01)00322-0. [DOI] [PubMed] [Google Scholar]
- 141.Davis C.J., Clinton J.M., Taishi P., Bohnet S.G., Honn K.A., Krueger J.M. MicroRNA 132 alters sleep and varies with time in brain. J. Appl. Physiol. 2011;111(3):665–672. doi: 10.1152/japplphysiol.00517.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Davis C.J., Bohnet S.G., Meyerson J.M., Krueger J.M. Sleep loss changes microRNA levels in the brain: a possible mechanism for state-dependent translational regulation. Neurosci. Lett. 2007;422(1):68–73. doi: 10.1016/j.neulet.2007.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Aghajanian G.K., VanderMaelen C.P. alpha 2-adrenoceptor-mediated hyperpolarization of locus coeruleus neurons: intracellular studies in vivo. Science. 1982;215(4538):1394–1396. doi: 10.1126/science.6278591. [DOI] [PubMed] [Google Scholar]
- 144.Aghajanian G.K., Cedarbaum J.M., Wang R.Y. Evidence for norepinephrine-mediated collateral inhibition of locus coeruleus neurons. Brain Res. 1977;136(3):570–577. doi: 10.1016/0006-8993(77)90083-X. [DOI] [PubMed] [Google Scholar]
- 145.Mallick B.N., Adya H.V. Norepinephrine induced alpha-adrenoceptor mediated increase in rat brain Na-K ATPase activity is dependent on calcium ion. Neurochem. Int. 1999;34(6):499–507. doi: 10.1016/S0197-0186(99)00025-X. [DOI] [PubMed] [Google Scholar]
- 146.Mallick B.N., Adya H.V., Faisal M. Norepinephrine-stimulated increase in Na+, K+-ATPase activity in the rat brain is mediated through alpha1A-adrenoceptor possibly by dephosphorylation of the enzyme. J. Neurochem. 2000;74(4):1574–1578. doi: 10.1046/j.1471-4159.2000.0741574.x. [DOI] [PubMed] [Google Scholar]
- 147.Schroeder F.A., Lin C.L., Crusio W.E., Akbarian S. Antidepressant-like effects of the histone deacetylase inhibitor, sodium butyrate, in the mouse. Biol. Psychiatry. 2007;62(1):55–64. doi: 10.1016/j.biopsych.2006.06.036. [DOI] [PubMed] [Google Scholar]
- 148.Wu T.Y., Chen C.P., Jinn T.R. Alzheimer's Disease: Aging, Insomnia and Epigenetics. Taiwanese J. Obstetrics Gynecol. 2010;49(4):468–472. doi: 10.1016/S1028-4559(10)60099-X. [DOI] [PubMed] [Google Scholar]
- 149.Simonini M.V., Camargo L.M., Dong E., Maloku E., Veldic M., Costa E., Guidotti A. The benzamide MS-275 is a potent, long-lasting brain region-selective inhibitor of histone deacetylases. Proc. Natl. Acad. Sci. U.S.A. 2006;103(5):1587–1592. doi: 10.1073/pnas.0510341103. [DOI] [PMC free article] [PubMed] [Google Scholar]