

Abstract
Primary tumors of the brain account for 2 % of all cancers with malignant gliomas taking the lion’s share at 70 %. Malignant gliomas (high grade gliomas WHO° III and °IV) belong to one of the most threatening tumor entities known for their disappointingly short median survival time of just 14 months despite maximum therapy according to current gold standards. Malignant gliomas manifest various factors, through which they adapt and manipulate the tumor microenvironment to their advantage. Epigenetic mechanisms operate on the tumor microenvironment by de- and methylation processes and imbalances between the histone deacetylases (HDAC) and histone acetylases (HAT). Many compounds have been discovered modulating epigenetically controlled signals. Recent studies indicate that xCT (system xc-, SLC7a11) and CD44 (H-CAM, ECM-III, HUTCH-1) functions as a bridge between these epigenetic regulatory mechanisms and malignant glioma progression. The question that ensues is the extent to which therapeutic intervention on these signaling pathways would exert influence on the treatment of malignant gliomas as well as the extent to which manipulation of HDAC activity can sensitize tumor cells for chemotherapeutics through ‘epigenetic priming’. In light of considering the current stagnation in the development of therapeutic options, the need for new strategies in the treatment of gliomas has never been so pressing. In this context the possibility of pharmacological intervention on tumor-associated genes by epigenetic priming opens a novel path in the treatment of primary brain tumors.
Keywords: Glutamate, HDAC inhibitors, malignant gliomas, supra-complete resection, tumor zone model, xCT.
EPIGENETIC REGULATORY MECHANISMS IN ONCOLOGY
The acetylation and deacetylation of histones represent an important epigenetic regulatory mechanism of gene expression [1]. Histone acetylation leads to relaxation of the chromatin structure through cancellation of electrostatic affinity between histones and DNA. In consequence, this leads to stimulation of gene transcription, whereas histone deacetylation is associated with chromatin condensation and mediates suppression of gene activity [2]. This is primarily achieved through addition or removal of negatively charged lysine residues from histones facilitated by histone acetyltransferases (HAT) on the one hand, and through histone deacetylases (HDAC) as counterparts of HATs on the other. The HATs are classified into the three groups termed GNAT, MYST and p300/CBP. The family of HDACs is so far formed of at least four subclasses: zinc-dependent class I, II and IV, and the NAD-dependent class III [3-5] (Fig.1).
Fig. (1).

Overview of the HDAC classes and corresponding HDAC-inhibitors. HDACSs are divided into four different classes with localization in various cell compartments. Abbreviations used: Api = Apicidine, But = Butyrate, Dep = Depsipeptide, GW = GW5074, LAQ = LAQ824, LBH = LBH589, MS = MS-275, PXD = PXD101, Sal = Salermide, SB = SB-379872-A, Sir = Sirtinol, SK1 = SK-7041, SK2 = SK-7068, Thio = Thieno[3,2-d]pyrimidine-6-carboxamide, Tub = Tubacin, VPA = valproic acid.
Balanced activity between the corresponding HATs and HDACs is critical to physiological cell differentiation and commensurate cell metabolism. Disturbance in this fine equilibrium is associated with oncogenic transformation [6]. This primarily concerns the HATs, as evidenced by their structural destruction through deletion or inactivation of mutations in numerous malignant tumor entities [7, 8]. Inhibition of HATs results in a dominance of HDAC-activity, leading in turn to the inactivation of several tumor suppressor and cell-cycle regulatory genes [2, 9]. A further mechanism behind oncogenic transformation lies in HDAC-associated activation of the transcription factor c-myc, leading to stimulation of cell proliferation and inhibition of apoptosis through repression of the tumor suppressive miRNA miR-29 [10-12]. In this manner, oncogenic transformation is facilitated by inactivation of tumor suppressing genes as well as activation of oncogenic signaling pathways as a consequence of dominance in HDAC-activity [13]. The option of blocking the activity of HDACs through specific inhibitors opens up a completely new treatment front with the possibility of influencing gene transcription through drug-induced epigenetic regulation, thereby permitting counteraction of the previously described oncogenic imbalance. Various promising HDAC inhibitors with cytostatic properties have been developed, with those characterized by primary action on HDACs of the zinc-dependent class I, IIa, IIb and IV currently in the clinical test phase [14] (Fig.1).
Recent studies have further shown that class III NAD-dependent HDACs also decisively control oncogenic processes [15, 16]. It is anticipated to be able to induce additional signaling pathways for differentiation and/or cell death mechanisms through the implementation of HDAC inhibitors beyond direct cytotoxicity towards tumor cells [17, 18]. This process is designated as ‘epigenetic priming’, describing sensitizing tumor cells in order to render them more susceptible to cytostatic drugs [19, 20]. In this manner it has been shown that individual HDAC-inhibitors are in a position to induce the expression of death receptors such as death receptor DR5, leading to subsequent activation of caspases in epigenetically primed cells [21-24]. These special properties render HDAC-inhibitors highly relevant to neuro-oncology, especially in the case of patients with high grade gliomas (WHO°III and WHO°IV) where they would constitute a new treatment concept to the backdrop of the currently severely limited therapy options.
THE 3 TUMOR ZONES (TZ) MODEL IN NEURO-ONCOLOGY AND HDAC INHIBITION
Belonging to the group of primary malignant brain tumors which account for a staggering 70 % of the most commonly occurring primary brain processes, glioblastomas (GBM, WHO grade IV gliomas) can be counted amongst the most malignant of all tumor entities with a median survival time of just 14 months [25]. The poor prognosis of this tumor entity is significantly facilitated by heightened proliferation, diffuse invasive growth, increased angiogenesis, and the ability to manipulate the tumor microenvironment to their own advantage [26-28]. These characteristics imply that clinicians are continually faced with imminent tumor recurrence bordering on certainty in glioma patients. Due to the lack of a current cure, this represents a fulcrum for treatment with HDAC inhibitors, which are in a position to exert specific and crucial influence on tumor cells (Fig. 2) [11, 29, 30].
Fig. (2).
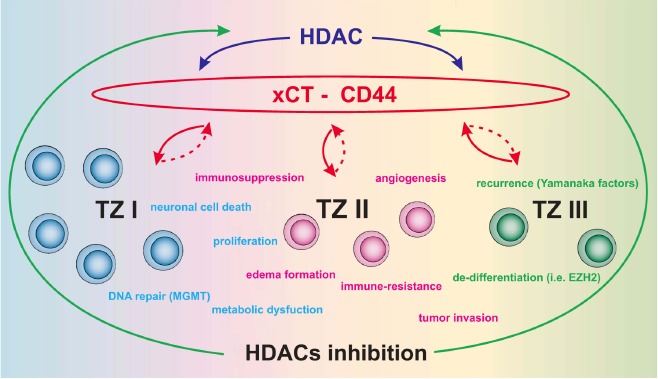
Conceptual scheme of the tumor zone model (TZI-III) and epigenetic regulation. The diagram depicts the known aspects of interaction between HDACs and the xCT system. Hallmarks of malignant glioma cells are color-coded in blue, while green and pink correspond to the various tumor zones (TZI-III). HDACs regulate various properties of malignant gliomas via the xCT (SLC7a11/ system Xc-). Since the glycoprotein CD44 (H-CAM/Hyaluronate receptor) has been previously shown to regulate and stabilize xCT, we suggest this complex as a therapeutical target for HDAC inhibitors. Inhibition of HDAC activity has been shown to render glioma cells vulnerable by diminishing the potency of their destructive properties. The xCT-CD44 complex could represent a crucial starting point for targeting xCT function. Furthermore, transcription factors critical for inducing pluripotent stem cells (Yamanaka factors) could be another vital target of HDAC inhibitors.
A peculiarity of malignant gliomas lies in their ability to reorganize their microenvironment according to their needs [31]. These pathophysiological features are governed according to the so-called tumor zone model comprising 3 zones which encompass all clinical properties and pathophysiological traits of malignant gliomas [32]. Tumor zone I corresponds to the contrast-agent enhancing area in MRI scans, comprising the primary tumor bulk, and consists of the so-called “core cells”. Tumor zone II, also known as the peritumoral zone, corresponds to the zone of perifocal edema in MRI scans and surrounds the tumor zone I. Tumor cells in this area are known as “transitory cells” since they exhibit many but not necessarily all histological and molecular hallmarks of the “core cells” (Fig. 2). Clinical and experimental investigations conducted point to an increased accumulation of microglia, reactively altered astrocytomas and endothelial cells. It is assumed that this area represents the biologically active area of the tumor. Although tumor zone III appears to be pathophysiologically inactive due to its macroscopically unremarkable appearance and consists of healthy brain parenchyma per se, it still harbors solitary tumor cells corresponding to precursor or stem cells collectively designated as “partisan cells” according to the model presented here. Despite belonging to the group of malignant glioma cells, they appear not to be same in character and differ in their molecular profile from the remainder of the tumor cell population. The heterogeneity of intra-glioma cell architecture is reflected by the polyclonal properties of this tumor entity [33]. Thus, HDAC inhibition in such a hetero-geneous tumor microenvironment may affect tumor biology at various levels, i.e. from simple cytostatic/cytotoxic growth arrest to inhibition of their pluripotent nature (Fig. 2).
VISUALIZATION OF THE 3 TUMOR ZONES (TZ) IN NEURO-ONCOLOGY
Tumor visualization is limited due to a very diffuse and infiltrative growth pattern. For example, it is possible to identify the tumor zone I well both intraoperatively as well as in MRI scans [34]. 5-ALA is an amino acid from the group of ketocarbonic acids, which is a precursor of Hem in porphyrin synthesis and normally metabolized through protoporphyrin IX into Hem. This transformation is not completed in tumor cells however, leading to intracellular accumulation of intermediary protoporphyrin IX. This leads to fluorescent excitation following exposure to a blue light source and enables direct intraoperative visualization of tumor cells. 5-ALA guided surgery of brain tumors therefore represents a direct biochemical approach enabling differentiation between tumor and healthy tissue. Differentiation here between auto-fluorescence and a vague fluorescence signal from solitary tumor cells in transition zones represents a technical challenge for the neurosurgeon [35]. An alternative method here is indirect visualization involving the property of increased angiogenesis in malignant gliomas. The identification of zones with a tumor cell density below a threshold permitting visualization of the fluorescing metabolites tumor-occupied zones is possible through visualization of hyper-vascularized areas, the so-called “angiogenic hotspots” [36]. This would correspond most closely to the tumor zone II according to the above described tumor zone model. In contrast, visualization of tumor cells in the tumor zone III is still difficult even following histopathological evaluation due to low numbers and growth activity [35]. This zone is in all probability therefore the most difficult area to tackle from the neuro-oncological perspective, since current therapy options target tumor cells in the individual tumor zones either poorly or so effectively that healthy brain parenchyma would be collaterally damaged.
HDAC INHIBITORS AS AN OPTION IN THE TREATMENT OF MALIGNANT GLIOMAS
HDAC inhibitors are characterized by the singular property of being able to interact specifically with tumor cells [37]. As opposed to conventional chemotherapeutic agents, they selectively act upon transformed cells instead of globally on brain parenchyma [1, 38]. Although a mechanism fundamental to this selectiveness is known to lie in influencing the balance between thioredoxin and reactive oxygen species [37], this phenomenon is yet to be fully explained [39]. The specific and tremendous advantage this tumor selectiveness awards to neuro-oncology lies in the fact that HDAC inhibitors can modulate gene expression of exclusively glioma cells in all three tumor zones without noticeable effect on healthy brain parenchyma. A possible growth inhibiting mechanism in glioblastomas lies in the specific up regulation of the cell cycle protein p21/WAF [40]. Through this, tumor cells are pushed into a G0-G1 cell-cycle arrest and consequently into apoptotic cell death [41]. Malignant gliomas are hence characterized by the following properties besides high proliferative activity:
gliomas are characterized by high angiogenic potential [27],
gliomas are accompanied by perifocal edema [31],
gliomas are characterized by high neurotoxic potential and induce neuronal cell death [42, 43], and
gliomas act in an immune-suppressive manner on immune-competent cells [44, 45].
These properties are primarily traced back to the synthesis and secretion of factors in the three tumor zones which manipulate the microenvironment according to the needs and advantage of the tumor [32]. One of these factors is the amino-acid glutamate. It is secreted by tumor cells via the channel protein xCT in exchange for cysteine [46]. Glutamate can potentially increase tumor cell proliferation through the activation of AMPA receptors via an autocrine loop [47]. In malignant gliomas tumor-derived glutamate additionally takes center stage in the induction of perifocal edema, thereby forming the tumor zone II [31]. Glutamate further stimulates microglial activity and leads to their accumulation [48, 49], which can additively trigger neuronal cell death through dendritic retraction [50]. The cytokine macrophage migration inhibitory factor (MIF) is then secreted by glioma cells in tumor zones I and II, leading to paralysis of microglial activity and absence of an immune response to the neoplastic process [44, 51]. In addition, the secreted glutamate concentration is toxic and leads to neuronal cell death, through which room for uninhibited tumor expansion is created [42]. This provides the base for modulatory influence on this process through HDAC inhibitors (Fig. 2). The expression of the glutamate transporter xCT can be specifically modulated through inhibition of HDAC activity in malignant gliomas, which leads to a reduction in tumor associated glutamate release in all three tumor zones [52]. In this manner, it is conceivable to normalize the tumor microenvironment:
microglial activity is normalized and an immune reaction takes place,
astrocyte swelling is reduced,
angiogenesis is impeded and
glutamate associated peritumoral neuronal cell death is considerably reduced
The cell-surface glycoprotein molecule CD44 (formerly termed HCAM, Pgp-11/phagocytic glycoprotein-1, Hermes antigen, lymphocyte homing receptor, ECM-III, HUTCH-1) is important in this respect as it stabilizes and controls the xCT-complex [53]. It is to be noted, that a selective removal of the tumor-specific variants of the CD44 molecule from the complex, i.e. Cd44v6-12, leads to a loss of the xCT transporter from the tumor cell surface with subsequent suppression of tumor growth. Additionally, an increase in the expression of the cell-cycle inhibitor p21/WAF occurs [53] – similar to the phenomenon of treatment with HDAC inhibitors: xCT expression is reduced and the generation of p21/WF increased. The extent to which normalization of the tumor microenvironment also encompasses the partisan cells in tumor zone III as well is yet to be investigated. Since however glioblastomas recur with a penetrance of almost 100 % despite resection with surgical margin (i.e. the 100 + approach) followed by adjuvant radio-chemotherapy, it can be assumed that partisan cells from the tumor zone III may account for this, the sensitivity of which to HDAC inhibitors needs to be investigated in the future. Additionally, CD44 is involved in the formation of resistance to radio-chemo-therapy through regulating anti-oxidation and detoxification pathways [54].
It is known with respect to adult stem cells that individual substances such as SAHA or MS-275 induce a differentiation with a shift in the direction of neurons [55]. The so-called “Yamanaka-factors“ are of clinical interest in this context as just the activity of these four factors or a combination of at least two of these, i.e. Oct3/4, Sox2, c-Myc and Klf4 alone are sufficient to initiate mechanisms which generate embryonic-like stem cells from any cell type including adult differentiated cells [56]. It is meanwhile possible to initiate these mechanisms by utilizing ”small-molecule compounds” [57]. The clinical phenomenon of glioma recurrences could be explained through this “gain-of-function” activity of individual tumor stem cells. Although the role of enhancer of zeste homologue 2 (EZH2) which belongs to the group of polycomb proteins has not yet been defined, EZH2 is over-expressed in glioblastomas and could potentially take part in the self-renewal of tumor cells [58]. Tumor cells are maintained in an undifferentiated state through the increased expression of EZH2. Inhibition of EZH2 expression through the specific inhibitor 3-deazaneplanocin A (DZNep) releases these cells into a differentiated state similar to following treatment with individual HDAC inhibitors such as SAHA. Although these results suggest that partisan cells probably react well to HDAC inhibitors too, this is yet to be demonstrated.
Individual HDAC-inhibitors influence another epigenetic signaling pathway too, i.e. the MGMT promotor methylation status, although it was assumed that DNA methylation suppresses gene transcription independent of HDAC [59]. This observation is important from the clinical point of view. The established standard adjuvant treatment of glioblastomas according to current studies includes temozolomide (Temodal®), a DNA alkylating agent [60, 61]. Patients with hypermethylated MGMT status profit most from temozolomide treatment [60]. Generally, tumor cells can repair temozolomide-induced DNA damage through the DNA repair enzyme O6-Methylguanin-DNA-Methyltransferase (MGMT). Thus, patients with a non-methylated MGMT promotor status profit little from treatment with DNA alkylating agents such as temozolomide [60]. This group is designated as temozolomide non-responder and comprises about 65 % of all glioblastoma patients [62, 63]. This underlines the importance of being able to offer these patients an alternative therapy option. In this context it is to be examined in future whether the MGMT promotor status can be changed through treatment with HDAC inhibitors, thereby achieving “epigenetic priming” for subsequent treatment with temozolomide. These findings are furthermore especially important in order to arrive at a better understanding of the xCT/CD44/glutamate system. xCT expression is nowadays being considered as a predictive factor similar to MGMT promotor methylation status [64]. Furthermore, CD44 is controlled through methylation processes [65]. It has also been shown that the glutamate-receptor mGlu3 controls the expression of the MGMT-enzyme and can therefore confer resistance against chemo-therapy agents [66]. Inhibition of the mGlu3 receptor here leads to sensitization of tumor cells towards temozolomide. xCT control of the MGMT system through secreted glutamate therefore represents a realistic overlapping of two systems considered to be independent of each other according to current knowledge. This means that normalization of the xCT system could lead to a change in MGMT promotor methylation status.
Recent data suggest that a modulation or intervention in the balanced HAT and HDAC signaling in malignant gliomas could be an effective treatment route. A decisive factor here is to define the particular functions of the 11 HDACs and 7 SIRTs. This is especially important since current HDAC inhibitors in use show certain cross-specificity with several HDACs at the same time.
Hence, HDAC inhibitors are accompanied by numerous side effects as reported in initial clinical studies ranging from weight loss to severe changes in the blood count. The identification of ‘glioma-specific’ HDACs and tumor zone-specific HDACS (TZ-specific HDACs) would be relevant to the development of new inhibitors to target particular glioma functions.
ACKNOWLEDGEMENTS
We express our gratitude to of all members of the cell and neurooncology lab. In particular, we thank Tina Sehm, Ali Ghoochani, Zheng Fan, Daishi Chen, Eduard Yakubov and Nirjhar Hore for their contribution to this work.
CONFLICT OF INTEREST
The authors confirm that this article content has no conflict of interest.
References
- 1.Peart M.J., Smyth G.K., van Laar R.K., Bowtell D.D., Richon V.M., Marks P.A., Holloway A.J., Johnstone R.W. Identification and functional significance of genes regulated by structurally different histone deacetylase inhibitors. Proc. Natl. Acad. Sci U. S. A. 2005;102(10):3697–702. doi: 10.1073/pnas.0500369102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.de Ruijter A.J., van Gennip A.H., Caron H.N., Kemp S., van Kuilenburg A.B. Histone deacetylases (HDACs): characterization of the classical HDAC family. Biochem. J. 2003;370(Pt 3):737–749. doi: 10.1042/bj20021321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Berndsen C.E., Denu J.M. Catalysis and substrate selection by histone/protein lysine acetyltransferases. Curr. Opin. Struct. Biol. 2008;18(6):682–689. doi: 10.1016/j.sbi.2008.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Berndsen C.E., Tsubota T., Lindner S.E., Lee S., Holton J.M., Kaufman P.D., Keck J.L., Denu J.M. Molecular functions of the histone acetyltransferase chaperone complex Rtt109-Vps75. Nat. Struct. Mol. Biol. 2008;15(9):948–956. doi: 10.1038/nsmb.1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Haberland M., Montgomery R.L., Olson E.N. The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nat. Rev. Genet. 2009;10(1):32–42. doi: 10.1038/nrg2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Minucci S., Pelicci P.G. Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer. Nat. Rev. Cancer. 2006;6(1):38–51. doi: 10.1038/nrc1779. [DOI] [PubMed] [Google Scholar]
- 7.Lin R.J., Nagy L., Inoue S., Shao W., Miller W.H., Jr, Evans R.M. Role of the histone deacetylase complex in acute promyelocytic leukaemia. Nature. 1998;391(6669):811–814. doi: 10.1038/35895. [DOI] [PubMed] [Google Scholar]
- 8.Marks P., Rifkind R.A., Richon V.M., Breslow R., Miller T., Kelly W.K. Histone deacetylases and cancer: causes and therapies. Nat. Rev. Cancer. 2001;1(3):194–202. doi: 10.1038/35106079. [DOI] [PubMed] [Google Scholar]
- 9.Marks P.A., Richon V.M., Miller T., Kelly W.K. Histone deacetylase inhibitors. Adv. Cancer Res. 2004;91:137–168. doi: 10.1016/S0065-230X(04)91004-4. [DOI] [PubMed] [Google Scholar]
- 10.Fabbri M., Garzon R., Cimmino A., Liu Z., Zanesi N., Callegari E., Liu S., Alder H., Costinean S., Fernandez-Cymering C., Volinia S., Guler G., Morrison C.D., Chan K.K., Marcucci G., Calin G.A., Huebner K., Croce C.M. MicroRNA-29 family reverts aberrant methylation in lung cancer by targeting DNA methyltransferases 3A and 3B. Proc. Natl. Acad. Sci. USA. 2007;104(40):15805–15810. doi: 10.1073/pnas.0707628104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang X., Zhao X., Fiskus W., Lin J., Lwin T., Rao R., Zhang Y., Chan J.C., Fu K., Marquez V.E., Chen-Kiang S., Moscinski L.C., Seto E., Dalton W.S., Wright K.L., Sotomayor E., Bhalla K., Tao J. Coordinated silencing of MYC-mediated miR-29 by HDAC3 and EZH2 as a therapeutic target of histone modification in aggressive B-Cell lymphomas. Cancer Cell. 2012;22(4):506–523. doi: 10.1016/j.ccr.2012.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 12.Zhao J.J., Lin J., Lwin T., Yang H., Guo J., Kong W., Dessureault S., Moscinski L.C., Rezania D., Dalton W.S., Sotomayor E., Tao J., Cheng J.Q. microRNA expression profile and identification of miR-29 as a prognostic marker and pathogenetic factor by targeting CDK6 in mantle cell lymphoma. Blood. 2010;115(13):2630–2639. doi: 10.1182/blood-2009-09-243147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bolden J.E., Peart M.J., Johnstone R.W. Anticancer activities of histone deacetylase inhibitors. Nat. Rev. Drug Discov. 2006;5(9):769–784. doi: 10.1038/nrd2133. [DOI] [PubMed] [Google Scholar]
- 14.Azad N., Zahnow C.A., Rudin C.M., Baylin S.B. The future of epigenetic therapy in solid tumours--lessons from the past. Nat. Rev. Clin. Oncol. 2013;10(5):256–266. doi: 10.1038/nrclinonc.2013.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bruzzone S., Parenti M.D., Grozio A., Ballestrero A., Bauer I., Del Rio A., Nencioni A. Rejuvenating sirtuins: the rise of a new family of cancer drug targets. Curr. Pharm. Des. 2013;19(4):614–623. doi: 10.2174/138161213804581954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jung-Hynes B., Reiter R.J., Ahmad N. Sirtuins, melatonin and circadian rhythms: building a bridge between aging and cancer. J. Pineal Res. 2010;48(1):9–19. doi: 10.1111/j.1600-079X.2009.00729.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Butler L.M., Zhou X., Xu W.S., Scher H.I., Rifkind R.A., Marks P.A., Richon V.M. The histone deacetylase inhibitor SAHA arrests cancer cell growth, up-regulates thioredoxin-binding protein-2, and down-regulates thioredoxin. Proc. Natl. Acad. Sci. U. S. A. 2002;99(18):5–11700. doi: 10.1073/pnas.182372299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shao Y., Gao Z., Marks P.A., Jiang X. Apoptotic and autophagic cell death induced by histone deacetylase inhibitors. Proc. Natl. Acad. Sci. USA. 2004;101(52):18030–18035. doi: 10.1073/pnas.0408345102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bangert A., Häcker S., Cristofanon S., Debatin K.M., Fulda S. Chemosensitization of glioblastoma cells by the histone deacetylase inhibitor MS275. Anticancer Drugs. 2011;22(6):494–499. doi: 10.1097/CAD.0b013e32834631e0. [DOI] [PubMed] [Google Scholar]
- 20.Scandura J.M., Roboz G.J., Moh M., Morawa E., Brenet F., Bose J.R., Villegas L., Gergis U.S., Mayer S.A., Ippoliti C.M., Curcio T.J., Ritchie E.K., Feldman E.J. Phase 1 study of epigenetic priming with decitabine prior to standard induction chemotherapy for patients with AML. Blood. 2011;118(6):1472–1480. doi: 10.1182/blood-2010-11-320093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huang S.K., Scruggs A.M., Donaghy J., Horowitz J.C., Zaslona Z., Przybranowski S., White E.S., Peters-Golden M. Histone modifications are responsible for decreased Fas expression and apoptosis resistance in fibrotic lung fibroblasts. Cell Death Dis. 2013;4:e621. doi: 10.1038/cddis.2013.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nebbioso A., Clarke N., Voltz E., Germain E., Ambrosino C., Bontempo P., Alvarez R., Schiavone E.M., Ferrara F., Bresciani F., Weisz A., de Lera A.R., Gronemeyer H., Altucci L. Tumor-selective action of HDAC inhibitors involves TRAIL induction in acute myeloid leukemia cells. Nat. Med. 2005;11(1):77–84. doi: 10.1038/nm1161. [DOI] [PubMed] [Google Scholar]
- 23.Rahmani M., Yu C., Reese E., Ahmed W., Hirsch K., Dent P., Grant S. Inhibition of PI-3 kinase sensitizes human leukemic cells to histone deacetylase inhibitor-mediated apoptosis through p44/42 MAP kinase inactivation and abrogation of p21(CIP1/WAF1) induction rather than AKT inhibition. Oncogene. 2003;22(40):6231–6242. doi: 10.1038/sj.onc.1206646. [DOI] [PubMed] [Google Scholar]
- 24.Shankar S., Singh T.R., Fandy T.E., Luetrakul T., Ross D.D., Srivastava R.K. Interactive effects of histone deacetylase inhibitors and TRAIL on apoptosis in human leukemia cells: involvement of both death receptor and mitochondrial pathways. Int. J. Mol. Med. 2005;16(6):1125–1138. doi: 10.3892/ijmm.16.6.1125. [DOI] [PubMed] [Google Scholar]
- 25.Van Meir E.G., Hadjipanayis C.G., Norden A.D., Shu H.K., Wen P.Y., Olson J.J. Exciting new advances in neuro-oncology: the avenue to a cure for malignant glioma. CA Cancer J. Clin. 2010;60(3):166–193. doi: 10.3322/caac.20069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hanahan D., Weinberg R.A. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 27.Jain R.K., di Tomaso E., Duda D.G., Loeffler J.S., Sorensen A.G., Batchelor T.T. Angiogenesis in brain tumours. Nat. Rev. Neurosci. 2007;8(8):610–622. doi: 10.1038/nrn2175. [DOI] [PubMed] [Google Scholar]
- 28.Rao J.S. Molecular mechanisms of glioma invasiveness: the role of proteases. Nat. Rev. Cancer. 2003;3(7):489–501. doi: 10.1038/nrc1121. [DOI] [PubMed] [Google Scholar]
- 29.Di Micco R., Sulli G., Dobreva M., Liontos M., Botrugno O.A., Gargiulo G., dal Zuffo R., Matti V., d’Ario G., Montani E., Mercurio C., Hahn W.C., Gorgoulis V., Minucci S., d’Adda di Fagagna F. Interplay between oncogene-induced DNA damage response and heterochromatin in senescence and cancer. Nat. Cell Biol. 2011;13(3):292–302. doi: 10.1038/ncb2170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kim M.S., Kwon H.J., Lee Y.M., Baek J.H., Jang J.E., Lee S.W., Moon E.J., Kim H.S., Lee S.K., Chung H.Y., Kim C.W., Kim K.W. Histone deacetylases induce angiogenesis by negative regulation of tumor suppressor genes. Nat. Med. 2001;7(4):437–443. doi: 10.1038/86507. [DOI] [PubMed] [Google Scholar]
- 31.Savaskan N.E., Heckel A., Hahnen E., Engelhorn T., Doerfler A., Ganslandt O., Nimsky C., Buchfelder M., Eyüpoglu I.Y. Small interfering RNA-mediated xCT silencing in gliomas inhibits neurodegeneration and alleviates brain edema. Nat. Med. 2008;14(6):629–632. doi: 10.1038/nm1772. [DOI] [PubMed] [Google Scholar]
- 32.Eyüpoglu I.Y., Buchfelder M., Savaskan N.E. Surgical resection of malignant gliomas-role in optimizing patient outcome. Nat. Rev. Neurol. 2013;9(3):141–151. doi: 10.1038/nrneurol.2012.279. [DOI] [PubMed] [Google Scholar]
- 33.Glas M., Rath B.H., Simon M., Reinartz R., Schramme A., Trageser D., Eisenreich R., Leinhaas A., Keller M., Schildhaus H.U., Garbe S., Steinfarz B., Pietsch T., Steindler D.A., Schramm J., Herrlinger U., Brüstle O., Scheffler B. Residual tumor cells are unique cellular targets in glioblastoma. Ann. Neurol. 2010;68(2):264–269. doi: 10.1002/ana.22036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Eyüpoglu I.Y., Hore N., Savaskan N.E., Grummich P., Roessler K., Buchfelder M., Ganslandt O. Improving the extent of malignant glioma resection by dual intraoperative visualization approach. PLoS One. 2012;7(9):e44885. doi: 10.1371/journal.pone.0044885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Valdés P.A., Kim A., Brantsch M., Niu C., Moses Z.B., Tosteson T.D., Wilson B.C., Paulsen K.D., Roberts D.W., Harris B.T. δ-aminolevulinic acid-induced protoporphyrin IX concentration correlates with histopathologic markers of malignancy in human gliomas: the need for quantitative fluorescence-guided resection to identify regions of increasing malignancy. Neuro-oncol. 2011;13(8):846–856. doi: 10.1093/neuonc/nor086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Eyüpoglu I.Y., Hore N., Fan Z., Buslei R., Merkel A., Buchfelder M., Savaskan N.E. Intraoperative vascular DIVA surgery reveals angiogenic hotspots in tumor zones of malignant gliomas. Sci. Rep. 2015;5:7958. doi: 10.1038/srep07958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ungerstedt J.S., Sowa Y., Xu W.S., Shao Y., Dokmanovic M., Perez G., Ngo L., Holmgren A., Jiang X., Marks P.A. Role of thioredoxin in the response of normal and transformed cells to histone deacetylase inhibitors. Proc. Natl. Acad. Sci. USA. 2005;102(3):673–678. doi: 10.1073/pnas.0408732102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Atadja P., Gao L., Kwon P., Trogani N., Walker H., Hsu M., Yeleswarapu L., Chandramouli N., Perez L., Versace R., Wu A., Sambucetti L., Lassota P., Cohen D., Bair K., Wood A., Remiszewski S. Selective growth inhibition of tumor cells by a novel histone deacetylase inhibitor, NVP-LAQ824. Cancer Res. 2004;64(2):689–695. doi: 10.1158/0008-5472.CAN-03-2043. [DOI] [PubMed] [Google Scholar]
- 39.Hahnen E., Hauke J., Tränkle C., Eyüpoglu I.Y., Wirth B., Blümcke I. Histone deacetylase inhibitors: possible implications for neurodegenerative disorders. Expert Opin. Investig. Drugs. 2008;17(2):169–184. doi: 10.1517/13543784.17.2.169. [DOI] [PubMed] [Google Scholar]
- 40.Eyüpoglu I.Y., Hahnen E., Buslei R., Siebzehnrübl F.A., Savaskan N.E., Lüders M., Tränkle C., Wick W., Weller M., Fahlbusch R., Blümcke I. Suberoylanilide hydroxamic acid (SAHA) has potent anti-glioma properties in vitro, ex vivo and in vivo. J. Neurochem. 2005;93(4):992–999. doi: 10.1111/j.1471-4159.2005.03098.x. [DOI] [PubMed] [Google Scholar]
- 41.Eyüpoglu I.Y., Hahnen E., Tränkle C., Savaskan N.E., Siebzehnrübl F.A., Buslei R., Lemke D., Wick W., Fahlbusch R., Blümcke I. Experimental therapy of malignant gliomas using the inhibitor of histone deacetylase MS-275. Mol. Cancer Ther. 2006;5(5):55–1248. doi: 10.1158/1535-7163.MCT-05-0533. [DOI] [PubMed] [Google Scholar]
- 42.Eyüpoglu I.Y., Hahnen E., Heckel A., Siebzehnrübl F.A., Buslei R., Fahlbusch R., Blümcke I. Malignant glioma-induced neuronal cell death in an organotypic glioma invasion model. Technical note. J. Neurosurg. 2005;102(4):738–744. doi: 10.3171/jns.2005.102.4.0738. [DOI] [PubMed] [Google Scholar]
- 43.Lee S.G., Kim K., Kegelman T.P., Dash R., Das S.K., Choi J.K., Emdad L., Howlett E.L., Jeon H.Y., Su Z.Z., Yoo B.K., Sarkar D., Kim S.H., Kang D.C., Fisher P.B. Oncogene AEG-1 promotes glioma-induced neurodegeneration by increasing glutamate excitotoxicity. Cancer Res. 2011;71(20):6514–6523. doi: 10.1158/0008-5472.CAN-11-0782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Engelhorn T., Savaskan N.E., Schwarz M.A., Kreutzer J., Meyer E.P., Hahnen E., Ganslandt O., Dorfler A., Nimsky C., Buchfelder M., Eyupoglu I.Y. Cellular characterization of the peritumoral edema zone in malignant brain tumors. Cancer Sci. 2009;100(10):62–1856. doi: 10.1111/j.1349-7006.2009.01259.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nieto-Sampedro M., Valle-Argos B., Gómez-Nicola D., Fernández-Mayoralas A., Nieto-Díaz M. Inhibitors of Glioma Growth that Reveal the Tumour to the Immune System. Clin. Med. Insights Oncol. 2011;5:265–314. doi: 10.4137/CMO.S7685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Savaskan N.E., Seufert S., Hauke J., Tränkle C., Eyüpoglu I.Y., Hahnen E. Dissection of mitogenic and neurodegenerative actions of cystine and glutamate in malignant gliomas. Oncogene. 2011;30(1):43–53. doi: 10.1038/onc.2010.391. [DOI] [PubMed] [Google Scholar]
- 47.Ishiuchi S., Tsuzuki K., Yoshida Y., Yamada N., Hagimura N., Okado H., Miwa A., Kurihara H., Nakazato Y., Tamura M., Sasaki T., Ozawa S. Blockage of Ca(2+)-permeable AMPA receptors suppresses migration and induces apoptosis in human glioblastoma cells. Nat. Med. 2002;8(9):971–978. doi: 10.1038/nm746. [DOI] [PubMed] [Google Scholar]
- 48.Ullrich O., Diestel A., Eyüpoglu I.Y., Nitsch R. Regulation of microglial expression of integrins by poly(ADP-ribose) polymerase-1. Nat. Cell Biol. 2001;3(12):1035–1042. doi: 10.1038/ncb1201-1035. [DOI] [PubMed] [Google Scholar]
- 49.Taylor D.L., Jones F., Kubota E.S., Pocock J.M. Stimulation of microglial metabotropic glutamate receptor mGlu2 triggers tumor necrosis factor alpha-induced neurotoxicity in concert with microglial-derived Fas ligand. J. Neurosci. 2005;25(11):2952–2964. doi: 10.1523/JNEUROSCI.4456-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Eyüpoglu I.Y., Bechmann I., Nitsch R. Modification of microglia function protects from lesion-induced neuronal alterations and promotes sprouting in the hippocampus. FASEB J. 2003;17(9):1110–1111. doi: 10.1096/fj.02-0825fje. [DOI] [PubMed] [Google Scholar]
- 51.Mittelbronn M., Platten M., Zeiner P., Dombrowski Y., Frank B., Zachskorn C., Harter P.N., Weller M., Wischhusen J. Macrophage migration inhibitory factor (MIF) expression in human malignant gliomas contributes to immune escape and tumour progression. Acta Neuropathol. 2011;122(3):353–365. doi: 10.1007/s00401-011-0858-3. [DOI] [PubMed] [Google Scholar]
- 52.Wolf I.M., Fan Z., Rauh M., Seufert S., Hore N., Buchfelder M., Savaskan N.E., Eyüpoglu I.Y. Histone deacetylases inhibition by SAHA/Vorinostat normalizes the glioma microenvironment via xCT equilibration. Sci. Rep. 2014;4:6226. doi: 10.1038/srep06226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ishimoto T., Nagano O., Yae T., Tamada M., Motohara T., Oshima H., Oshima M., Ikeda T., Asaba R., Yagi H., Masuko T., Shimizu T., Ishikawa T., Kai K., Takahashi E., Imamura Y., Baba Y., Ohmura M., Suematsu M., Baba H., Saya H. CD44 variant regulates redox status in cancer cells by stabilizing the xCT subunit of system xc(-) and thereby promotes tumor growth. Cancer Cell. 2011;19(3):387–387. doi: 10.1016/j.ccr.2011.01.038. [DOI] [PubMed] [Google Scholar]
- 54.Nagano O., Okazaki S., Saya H. Redox regulation in stem-like cancer cells by CD44 variant isoforms. Oncogene. 2013;32(44):5191–5198. doi: 10.1038/onc.2012.638. [DOI] [PubMed] [Google Scholar]
- 55.Siebzehnrubl F.A., Buslei R., Eyupoglu I.Y., Seufert S., Hahnen E., Blumcke I. Histone deacetylase inhibitors increase neuronal differentiation in adult forebrain precursor cells. Exp. Brain Res. 2007;176(4):672–678. doi: 10.1007/s00221-006-0831-x. [DOI] [PubMed] [Google Scholar]
- 56.Takahashi K., Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126(4):76–663. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- 57.Hou P., Li Y., Zhang X., Liu C., Guan J., Li H., Zhao T., Ye J., Yang W., Liu K., Ge J., Xu J., Zhang Q., Zhao Y., Deng H. Pluripotent stem cells induced from mouse somatic cells by small-molecule compounds. Science. 2013;341(6146):651–654. doi: 10.1126/science.1239278. [DOI] [PubMed] [Google Scholar]
- 58.Suva M.L., Riggi N., Janiszewska M., Radovanovic I., Provero P., Stehle J.C., Baumer K., Le Bitoux M.A., Marino D., Cironi L., Marquez V.E., Clement V., Stamenkovic I. EZH2 is essential for glioblastoma cancer stem cell maintenance. Cancer Res. 2009;69(24):8–9211. doi: 10.1158/0008-5472.CAN-09-1622. [DOI] [PubMed] [Google Scholar]
- 59.Cameron E.E., Bachman K.E., Myöhänen S., Herman J.G., Baylin S.B. Synergy of demethylation and histone deacetylase inhibition in the re-expression of genes silenced in cancer. Nat. Genet. 1999;21(1):103–107. doi: 10.1038/5047. [DOI] [PubMed] [Google Scholar]
- 60.Hegi M.E., Diserens A.C., Gorlia T., Hamou M.F., de Tribolet N., Weller M., Kros J.M., Hainfellner J.A., Mason W., Mariani L., Bromberg J.E., Hau P., Mirimanoff R.O., Cairncross J.G., Janzer R.C., Stupp R. MGMT gene silencing and benefit from temozolomide in glioblastoma. N. Engl. J. Med. 2005;352(10):997–1003. doi: 10.1056/NEJMoa043331. [DOI] [PubMed] [Google Scholar]
- 61.Stupp R., Mason W.P., van den Bent M.J., Weller M., Fisher B., Taphoorn M.J., Belanger K., Brandes A.A., Marosi C., Bogdahn U., Curschmann J., Janzer R.C., Ludwin S.K., Gorlia T., Allgeier A., Lacombe D., Cairncross J.G., Eisenhauer E., Mirimanoff R.O., European Organisation for Research and Treatment of Cancer Brain Tumor and Radiotherapy Groups. National Cancer Institute of Canada Clinical Trials Group Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005;352(10):987–996. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 62.Quillien V., Lavenu A., Karayan-Tapon L., Carpentier C., Labussière M., Lesimple T., Chinot O., Wager M., Honnorat J., Saikali S., Fina F., Sanson M., Figarella-Branger D. Comparative assessment of 5 methods (methylation-specific polymerase chain reaction, MethyLight, pyrosequencing, methylation-sensitive high-resolution melting, and immunohistochemistry) to analyze O6-methylguanine-DNA-methyltranferase in a series of 100 glioblastoma patients. Cancer. 2012;118(17):4201–4211. doi: 10.1002/cncr.27392. [DOI] [PubMed] [Google Scholar]
- 63.Wick W., Platten M., Meisner C., Felsberg J., Tabatabai G., Simon M., Nikkhah G., Papsdorf K., Steinbach J.P., Sabel M., Combs S.E., Vesper J., Braun C., Meixensberger J., Ketter R., Mayer-Steinacker R., Reifenberger G., Weller M., NOA-08 Study Group of Neuro-oncology Working Group (NOA) of German Cancer Society Temozolomide chemotherapy alone versus radiotherapy alone for malignant astrocytoma in the elderly: the NOA-08 randomised, phase 3 trial. Lancet Oncol. 2012;13(7):707–715. doi: 10.1016/S1470-2045(12)70164-X. [DOI] [PubMed] [Google Scholar]
- 64.Takeuchi S., Wada K., Toyooka T., Shinomiya N., Shimazaki H., Nakanishi K., Nagatani K., Otani N., Osada H., Uozumi Y., Matsuo H., Nawashiro H., Braun C., Meixensberger J., Ketter R., Mayer-Steinacker R., Reifenberger G., Weller M. Increased xCT expression correlates with tumor invasion and outcome in patients with glioblastomas. Neurosurgery. 2013;72(1):33–41. doi: 10.1227/NEU.0b013e318276b2de. [DOI] [PubMed] [Google Scholar]
- 65.Branham M.T., Marzese D.M., Laurito S.R., Gago F.E., Orozco J.I., Tello O.M., Vargas-Roig L.M., Roqué M. Methylation profile of triple-negative breast carcinomas. Oncogenesis. 2012;1:e17. doi: 10.1038/oncsis.2012.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ciceroni C., Bonelli E., Mastrantoni C., Niccolini M. Type-3 metabotropic glutamate receptors regulate chemo resistance in glioma stem cells, and their levels are inversely related to survival in patients with malignant gliomas. Cell Death Differ. 2013;20(3):396–407. doi: 10.1038/cdd.2012.150. [DOI] [PMC free article] [PubMed] [Google Scholar]