


Abstract
NUCKS1 (nuclear casein kinase and cyclin-dependent kinase substrate 1) is a 27 kD chromosomal, vertebrate-specific protein, for which limited functional data exist. Here, we demonstrate that NUCKS1 shares extensive sequence homology with RAD51AP1 (RAD51 associated protein 1), suggesting that these two proteins are paralogs. Similar to the phenotypic effects of RAD51AP1 knockdown, we find that depletion of NUCKS1 in human cells impairs DNA repair by homologous recombination (HR) and chromosome stability. Depletion of NUCKS1 also results in greatly increased cellular sensitivity to mitomycin C (MMC), and in increased levels of spontaneous and MMC-induced chromatid breaks. NUCKS1 is critical to maintaining wild type HR capacity, and, as observed for a number of proteins involved in the HR pathway, functional loss of NUCKS1 leads to a slow down in DNA replication fork progression with a concomitant increase in the utilization of new replication origins. Interestingly, recombinant NUCKS1 shares the same DNA binding preference as RAD51AP1, but binds to DNA with reduced affinity when compared to RAD51AP1. Our results show that NUCKS1 is a chromatin-associated protein with a role in the DNA damage response and in HR, a DNA repair pathway critical for tumor suppression.
INTRODUCTION
DNA double-strand breaks (DSBs) are highly toxic and induced exogenously by ionizing radiation (IR) or inter-strand crosslinking agents. DSBs also arise from the attack by endogenous reactive oxygen species or from the encounter of unrepaired single-strand breaks by DNA replication forks, and are created during normal development of the immune system. DSBs elicit a cellular response that involves the activation of cell cycle checkpoints to facilitate double-strand break repair (DSBR), or the activation of genes involved in promoting apoptosis or senescence. Defects in the cellular response to DSBs underpin a number of human diseases, including disorders associated with cancer predisposition, immune dysfunction, radiosensitivity, neurodegeneration and premature aging (1–4). When DSBs occur, cells repair these DNA ends either by homologous recombination (HR) or by non-homologous end joining (NHEJ), and these two biochemically distinct pathways are used differently throughout the cell cycle (for review see (5)).
Previously, we discovered and characterized RAD51-associated protein 1 (RAD51AP1) with regards to its function in HR and DSB repair (6–9). Specifically, we showed that RAD51AP1 interacts with and stimulates the activity of both RAD51 (6) and DMC1 (7,8), the two conserved recombinase enzymes that mediate the homologous DNA pairing reaction during HR. We also showed that RAD51AP1 is essential for maintaining DNA replication fork progression (10), as described for other proteins in the HR pathway (11,12). In addition, and as expected for a major player in HR, we (6,10) and others (13,14) showed that gene-specific knockdown of RAD51AP1 by RNA interference leads to increased cellular sensitivity to DNA damaging agents and to elevated levels of chromatid breaks.
Nuclear casein kinase and cyclin-dependent kinases substrate 1 (NUCKS1) is a nuclear and highly phosphorylated protein (15–17) which also is acetylated, methylated, ubiquitylated and formylated ((18); http://www.phosphosite.org/). Only limited functional data exist for NUCKS1, even though this protein was discovered three decades ago (19). However, emerging clinical evidence establishes NUCKS1 as a biomarker for several human diseases, including cancer and metabolic syndrome (20–29). Interestingly, NUCKS1 has been picked up in several screens aimed at identifying changes to the nuclear phosphoproteome in response to DNA damage induction. In 293T cells, NUCKS1 (i.e. Ser14) was shown to be a substrate of either the ataxia telangiectasia mutated serine/threonine-protein kinase (ATM) or the ataxia telangiectasia and Rad3-related serine/threonine-protein kinase (ATR) following exposure to ionizing radiation (30), and Ser54 and Ser181 were identified as ATM-dependent phosphorylation sites in G361 human melanoma cells following treatment with the radiomimetic drug neocarzinostatin (31).
Here, we show that NUCKS1 and RAD51AP1 share extensive sequence homology throughout and are therefore paralogs. Since paralogs frequently serve a similar biological function, we have tested NUCKS1 for a possible role in DSBR by HR. We report that, in human cells, targeted inactivation of NUCKS1 by RNA interference largely phenocopies knockdown of RAD51AP1. We show that NUCKS1 is epistatic with both RAD51AP1 and XRCC3, thus revealing NUCKS1 as a new player in the HR pathway. Knockdown of NUCKS1 in human cells has no apparent effect on DNA damage-induced RAD51 focus formation, indicative of a function of NUCKS1 downstream of RAD51-single-stranded DNA (ssDNA) nucleoprotein filament formation. Our findings are the first to demonstrate the biological function in DSBR for NUCKS1. Our results are of particular interest in the context of several expression-array studies that report on the altered expression of NUCKS1 mRNA and protein levels in various types of cancer (20–27). We suggest that elevated levels of NUCKS1 may provide a selective advantage in precancerous cells and during cancer development by overcoming the consequences of replication stress, which leads to DSBs and genomic instability (32,33).
MATERIALS AND METHODS
Cell culture, transfection and siRNAs
HeLa cells and U2OS cells from ATCC were maintained as recommended. HCA2-hTERT human foreskin fibroblasts were a kind gift from Dr J. Campisi and were maintained as described previously (34,35); HCA2-hTERT cells were used between 52 and 55 cumulative population doublings in this study. U2OS-DRGFP (i.e. DR-U2OS) cells were a kind gift from Dr M. Jasin and were maintained as described previously (36–38).
Small interfering RNAs (siRNAs) were obtained from Qiagen (Valencia, CA). The target siRNA sequences to knockdown NUCKS1, XRCC3, RAD51AP1 and PALB2 were as follows: 5′- AAGAACCTACTTAAGATAGAA-3′ (here: NUCKS1 #1), 5′-AACCGGAAAGCCCGCCAGAAA-3′ (here: NUCKS1 #2), 5′-AAGAAGGATGATTCTCACTCA-3′ (here: NUCKS1 #3) for NUCKS1, and 5′-CAGAATTATTGCTGCAATTAA-3′ for XRCC3, 5′-AGTCATTTGGATGTCAAGAAA-3′ (here: PALB2#1) and 5′-CTTAGAAGAGGACCTTATTGT-3′ (here: PALB2#2) for PALB2, as described (37), and 5′-AACCTCATATCTCTAATTGCA-3′ for RAD51AP1, as previously described (9,14). For non-depleting negative control siRNA, the following target sequence was used: 5′-GATTCGAACGTGTCACGTCAA-3′, as previously described (39). Where not further specified, a pool of NUCKS1 siRNAs #1, #2 and #3 was used. BLAST sequence analyses of the human genome database were carried out to ensure that all used siRNAs would not target other gene transcripts. SiRNA forward transfections were performed using lipofectamine RNAiMAX (Invitrogen, Carlsbad, CA, USA) on two consecutive days and according to the instructions of the manufacturer, combined siRNA and plasmid transfections were carried out using Lipofectamine2000 (Invitrogen, Carlsbad, CA). SiRNA/lipofectamine RNAiMAX complexes were formed at a 1:1 ratio, and the concentration of siRNAs in transfections was 40 nM. Cells were treated with drugs or ionizing radiation at 72 h after the first transfection, and the extent of protein knockdown or expression was assessed by western blot analysis.
Recombination assay and flow cytometry
U2OS-DRGFP (i.e. DR-U2OS) cells have been described elsewhere (36–38). I-SceI was expressed transiently in U2OS-DRGFP cells from the pCβASCE expression vector (40) at 0.8 μg/150 000 cells and co-transfected with siRNA using Lipofectamine2000 (Invitrogen, Carlsbad, CA). Transfected cells were kept in regular growth medium for 72 h, after which time they were analyzed by flow cytometry to measure the percentage of viable cells expressing GFP, as previously described (6,41).
Exposure to genotoxic agents and western blot analysis
Exposure of cells to MMC for cell survival assays (Sigma, St. Louis, MO, USA) occurred in regular growth medium at 37°C for 1 h at the concentrations indicated. Cells were trypsinized, washed twice and re-suspended in fresh medium, plated and incubated 11 days for colony formation. Exposure to camptothecin (Sigma, St. Louis, MO) for camptothecin (CPT)-washout experiments was carried out at 20 nM in regular growth medium for 24 h. Control cells were exposed to the equivalent concentrations of DMSO only. Exposure to hydroxyurea (HU; Sigma, St. Louis, MO) was carried out at 2 mM in regular growth medium for 5 h to determine the levels of FANCD2 ubiquitylation. For cell survival assays using CPT or HU, 500–1000 cells were plated in 10-cm dishes, allowed to attach for 24 h before treatment with either CTP or HU for 24 h and at the concentrations indicated. The medium was removed after 24 h and the monolayers were washed with PBS once, before incubation in regular growth medium for colony formation. Exposure to X-rays and cell synchronization with mimosine occurred as previously described (6). Western blot analysis was carried out as previously described (42). The primary antibodies that were used are: α-RAD51AP1 (NB100-1129; Novus; 1:5000; and our own α-RAD51AP1 antibody, as previously described in (43)), α-XRCC3 (NB100-165; Novus; 1:10 000); α-QM (C-17; Santa Cruz Biotechnology; 1:3000) and α-PALB2 (A301-246; Bethyl Lab.; 1:2000), α-KU80 (kind gift from Dr S.M. Yannone; 1:5000), α-Tubulin (CP06; EMD Millipore; 1:3000), α-HA (MMS101P; Covance; 1:3000), α-NUCKS1 ((44); 1:10 000), α-pRPA32-S4/S8 (A300-245A; Bethyl Labs; 1:5000), α-RPA32 (Ab-3; EMD Millipore; 1:3000), α-RAD51 (Ab-1; EMD Millipore; 1:3000), α-RAD51 (H-92; Santa Cruz Biotechnology; 1:1000), α-ATM (NB100–104; Novus; 1:1000), α-MSH2 (ab52266; Abcam; 1:8000), α-FANCD2 (sc-20022; Santa Cruz Biotechnology; 1:1000), α-H3 (ab1791; Abcam; 1:5000), α-pATM-S1981 (clone 10H11.E12; Millipore; 1:1000), α-GAPDH (3E8AD9; Abcam; 1:1000), α-HMGB2 (D1P9V; Cell Signaling; 1:2000). The rabbit pNUCKS1-S54 antibody was generated to a peptide with the following sequence: CKNKRRSGKN(pSer)QEDS (GeneScript), and used at 1:6000. HRP-conjugated goat anti-rabbit or goat anti-mouse IgG (Jackson ImmunoResearch Laboratories, West Grove, PA; 1:10000) were used as secondary antibodies.
To assess phosphorylated NUCKS1 (pNUCKS1-S54) by Western blot analysis, protein extracts were prepared using perchloric acid (PCA; Sigma, St. Louis, MO) extraction, essentially as described (19). Briefly, PCA-soluble proteins were obtained from ∼5 × 106 cells in 1.5 ml 5% ice-cold PCA using dounce homogenization followed by precipitation of proteins on ice over night in 25% trichloric acid solution (Sigma, St. Louis, MO). After centrifugation, pellets were washed in 2 ml acetone/ 5 μl HCl first, followed by a second wash in 2 ml acetone only. Pellets were dried and resuspended in 40 μl H2O prior to western blot analysis.
Cell cycle analysis
Briefly, 1 × 106 cells were pelleted, washed in ice-cold PBS, pelleted and resuspended in 50 μl PBS. Cells were fixed by adding 10 ml of cold 70% ethanol/PBS and kept at 4°C for at least 2 days. Fixed cells were pelleted and resuspended in 5 ml 30% ethanol/PBS for 2 min at 4°C, pelleted and resuspended in 1 ml 0.05% BSA/PBS for 2 min at 4°C. Cells were then resuspended in staining solution (40 μg/ml RNase A and 30 μg/ml propidium iodide in PBS; Sigma, St. Louis, MO) before analysis using a FACS Calibur flow cytometer (BD Biosciences, San Jose, CA) with 488 nm excitation and 630 nm long pass filter. FlowJo version X (Tree Star, Ashland, OR) was used to fit the data for cell cycle analysis.
Analysis of chromosomal aberrations and cell fractionation
HCA2-hTERT human fibroblasts were treated with 50 nM MMC for 1 h and with colcemid (100 ng/ml; Sigma, St. Louis, MO) for 4 h prior to fixation. Fixation was carried out at 24 h post MMC treatment. Cells were detached from the culture vessel, washed in PBS and allowed to swell in 0.075 M KCl at 37°C for 10 min. Cells were fixed twice in methanol:acetic acid (3:1), and dropped on wet slides, air dried, and stained in 3% Giemsa solution in Sørensen phosphate buffer for 10 min. Slides were covered with mounting media (Vectashield 60; Vector Laboratories, Burlingame, CA) and analyzed with a Zeiss Axioskop using a 100× lens with oil and 2000× magnification. For each sample 50–100 metaphases were analyzed and all visible aberrations were counted. Gaps were counted as chromatid break (ctb), when the gap was larger than the chromatid was wide. Cell fractionation was carried out using the Subcellular Protein Fractionation Kit (Thermo Scientific, Waltham, MA) and as described by the manufacturer.
Treatment with PI3K-related kinase inhibitors
To inhibit ATM and/or DNA-PK, cells were incubated with 5 μM KU60019 (Tocris Bioscience, Bristol, UK) and/or 5 μM NU7026 (Sigma, St. Louis, MO), respectively, in regular growth medium 5 h prior to radiation exposure or MMC treatment. Following treatment, medium was exchanged and fresh inhibitor was added for post-treatment incubation times, as indicated.
Indirect immunofluorescence
Cells, grown on four-well slides, were treated with 8 Gy X-rays 3 days after the first transfection with siRNA, and fixed with 4% paraformaldehyde at room temperature for 10 min at the times post radiation exposure, as indicated. Fixed cells were permeabilized in 0.5% Triton X-100 in PBS for 5 min, washed in PBS, blocked in 2% BSA for 2 h, and incubated in blocking solution with primary antibody to RAD51 (H-92; 1:500; Santa Cruz Biotechnology, Santa Cruz, CA) at 4°C overnight. After further washes with PBS, the cells were incubated for 1 h with AlexaFluor488 goat anti-rabbit IgG (Invitrogen, Carlsbad, CA) at 4 μg/ml in blocking solution. The slides were mounted in Vectashield mounting medium containing 1.5 μg/ml DAPI.
Microscopy and image analysis
For image capture, Z-stack section images (slices with 0.7 μm intervals) were taken using a confocal Zeiss LSM-710 laser-scanning microscope wit 100× magnification. For computational analysis, Z-stacks were collapsed down to the maximum intensity projections. All images were annotated with the respective ionizing radiation dose, post-exposure time and siRNA treatment condition, and registered with the BioSig (Biological Signature) Imaging Bioinformatics Platform, as described (45). Image analysis included nuclear segmentation using convexity (46). Each detected nucleus provided the context for foci analysis, following maximum projection of foci on a nucleus-by-nucleus basis.
DNA fiber assay
Exponentially growing cells were pulse-labeled with 25 μM CldU (Sigma, St. Louis, MO) and 250 μM IdU (Sigma, St. Louis, MO) for the times specified. Where indicated, the cells were exposed to 2 mM hydroxyurea (HU; Sigma, St. Louis, MO) or 0.5 μM topotecan (TPT; Sigma, St. Louis, MO) for the times specified. Labeled cells were harvested and DNA fiber spreads were prepared from 0.5 × 106 cells/ml, as previously described (47). Slides were incubated in 2.5 M HCl for 90 min and then washed several times in PBS, followed by incubation in blocking buffer (2% BSA, 0.1% Tween in PBS) for 1 h. Acid-treated fiber spreads were stained with monoclonal rat anti-BrdU antibody (1:1000; Oxford Biotechnology Waltham, MA) to detect CldU, followed by monoclonal mouse anti-BrdU antibody (1:15 000; Becton Dickinson, Franklin Lakes, NJ) to detect IdU. Secondary antibodies were goat anti-rat AlexaFluor555 and goat anti-mouse AlexaFluor488 (1:500; Invitrogen, Carlsbad, CA). Primary antibodies were diluted in blocking buffer. Incubations with rat anti-BrdU antibody were carried out for 1 h, and incubations with mouse anti- BrdU antibody were carried out overnight. Secondary antibodies were applied for 1.5 h, and slides were mounted in Immuno-Fluor mounting medium (MP Biomedicals, Santa Ana, CA). Fiber tracts were examined using an AxioVert 200M fluorescence microscope (Zeiss, Thornwood, NY) using a 63× objective. Pictures were taken from randomly selected fields with untangled fibers and analyzed using ImageJ software package. For structure analyses, the frequencies of the different classes of fiber tracks were classified as follows: red–green (elongating fork), red (stalled or terminated forks), green–red–red–green (first pulse origin) and green (second pulse origin). For fork speed analyses, the lengths of CldU and IdU tracks were measured and micrometer values were converted into kilobases. A conversion factor for the length of a labeled track of 1 μm = 2.59 kb was used (48). A minimum of 100 individual fibers was analyzed for each experiment and the means of at least three independent experiments are presented.
Purification of recombinant proteins
The cDNA for human NUCKS1 was introduced into the pENTR™/TEV/D-TOPO® entry vector (Invitrogen, Carlsbad, CA) and an amino-terminal GST-tag was engineered into pDEST20-NUCKS1. pDEST20-GST-NUCKS1 was introduced into DH10Bac Escherichia coli cells for bacmid generation. The bacmids were verified by PCR and used to transfect SF9 insect cells to generate recombinant baculoviruses. After amplification in SF9 cells, the viruses were used to infect Hi5 insect cells for expression of GST-NUCKS1 (8 ml virus per 300 ml cells). After a 44-h incubation at 27°C, cells were harvested by centrifugation, frozen in liquid nitrogen, and stored at −80°C until cell lysis. All purification steps were carried out at 0°C to 4°C. To prepare extract, the frozen cell paste (5 g, from 300 ml culture) was thawed and suspended in 40 ml of cell breakage buffer A (25 mM Tris–HCl, pH 7.5, 300 mM KCl, 0.5 mM EDTA, 1 mM 2-mercaptoethanol, 0.01% Igepal CA-630, 5 mM MgCl2, 2 mM ATP and the following protease inhibitors: aprotinin, chymostatin, leupeptin and pepstatin A at 3 μg/ml each, and 1 mM PMSF) for sonication. After centrifugation (100 000 × g for 90 min), the clarified lysate was diluted three times with buffer B (25 mM Tris–HCl, pH 7.5, 0.5 mM EDTA, 1 mM 2-mercaptoethanol and 0.01% Igepal CA-630) and loaded on a Q Sepharose column (10 ml total). The column was washed with 30 ml buffer B containing 75 mM KCl before being eluted with 50 ml buffer B containing 600 mM KCl. The eluate was mixed with 1 ml Glutathione Sepharose 4 Fast Flow resin for 1 h. The resin was washed sequentially with 10 ml of buffer B containing 600 mM KCl, and then with 10 ml buffer C (25 mM Tris–HCl, pH 7.5, 0.5 mM EDTA, 300 mM KCl, 1 mM 2-mercaptoethanol and 0.01% Igepal CA-630), before being eluted five times with 1 ml of 20 mM glutathione in buffer C. The combined eluates were concentrated in a Centricon-30K concentrator (Amicon), and then fractionated in a Superdex 200 10/300 GL gel filtration column. The NUCKS1 protein peak fractions were pooled and concentrated before being frozen in liquid nitrogen and stored at −80°C. RAD51AP1 was purified as previously described (6). RAD51 was purified as reported earlier (49).
DNA binding assay
32P-labeled DNA substrates (2.5 nM) were mixed with the indicated amounts of GST-NUCKS1 or GST-RAD51AP1 at 37°C in 10 μl of Buffer D (25mM Tris–HCl, pH7.5, 90 mM KCl, 1 mM dithiothreitol, and 100 μg/ml bovine serum albumin) for 10 min. After the addition of gel loading buffer (50% glycerol, 20 mM Tris–HCl, pH7.4, 0.5 mM EDTA, 0.05% orange G), the protein–DNA complexes were resolved by 8% native polyacrylamide gel electrophoresis in 1× TAE buffer (40 mM Tris, pH8.0, 20 mM acetic acid and 1 mM EDTA) at 4°C. The gels were dried, and the products were visualized by autoradiography and quantified using the Molecular Imager FX and Quantity One 4.6 software (Bio-Rad).
Immobilized template assay to determine chromatin binding
A biotin-labeled 588-bp PCR product was gel purified and quantified before coupling to streptavidin-coated magnetic Dynabeads (Invitrogen, Carlsbad, CA), according to the manufacturer's instructions. One half of the reaction was used to assemble chromatin using human histone octamers (a kind gift from the Core Facility of Protein Expression and Purification, Department of Biochemistry and Molecular Biology, CSU) by salt deposition, as described previously (50). Both immobilized DNA and chromatin were used to assess the binding affinity of purified NUCKS1 protein in comparative assays. Briefly, either DNA or chromatin (1 pmol/reaction) and 0.5 or 1 pmol of purified GST-NUCKS1, (His)6-RAD51AP1 or RAD51 were incubated in binding buffer (20 mM K2HPO4/KH2PO4, 0.05% NP-40, 150 mM KCl, 20% glycerol, 0.5 mM EDTA and 2 mM DTT) with constant shaking at 30°C for 30 min. Following binding, the supernatant was removed, the resin was washed three times with binding buffer, and the bound protein was assessed by 10% SDS-PAGE and western blot analysis. We also included as control streptavidin-coated Dynabeads without DNA or chromatin.
Affinity pull-down assays
GST-tagged NUCKS1 (3 μg) or GST-tagged RAD51AP1 (5 μg) was incubated with human RAD51 (5 μg) in 30 μl buffer B (25 mM Tris–HCl at pH 7.5, 10% glycerol, 0.5 mM EDTA, 0.01% Igepal, 1 mM DTT, 100 mM KCl) on ice for 30 min, and then 15 μl glutathione resin (GE Healthcare) was added. The GST-tagged protein and associated proteins were captured on the resin by gentle mixing at 4°C for 1 h. The resin was then washed three times with buffer B and treated with 30 μl of 2% SDS to elute the proteins. The supernatant, final wash and SDS-eluted fractions (10 μl each) were analyzed by 10% SDS-PAGE and Coomassie Blue staining.
Statistical analysis
The statistical analysis was performed using Prism 6 (GraphPad Software, Inc., La Jolla, CA) on the data from at least three independent experiments, as specified. Unless stated otherwise, statistical significance was assessed by two-tailed unpaired Student's t-test. P ≤ 0.05 was considered significant.
RESULTS
NUCKS1 and RAD51AP1 are paralogs
In our attempt to identify more proteins in the RAD51AP1 family, we discovered NUCKS1, a protein with poorly understood biological function (15–17,23,28,44,51), as a paralog. The ClustalW sequence alignment of human RAD51AP1 and NUCKS1 is depicted in Figure 1A. NUCKS1 shares 28% amino acid sequence identity and 11% sequence similarity with RAD51AP1. We conclude that NUCKS1 and RAD51AP1 can be considered paralogs of each other. Interestingly, human NUCKS1 does not share the RAD51AP1 C-terminal domain (Figure 1A) that mediates the interaction with the recombinase enzyme RAD51 (6,52). According to our analysis, no NUCKS1 homolog from any other vertebrate species possesses this RAD51-interacting domain (Supplementary Figure S1). The large gap in the N-terminal region of human NUCKS1, as compared to human RAD51AP1, is variable in length between different species, as can be seen in a comparison of these two paralogs in Xenopus tropicalis (Figure 1B). Of note, no function for the N-terminal segment of human RAD51AP1 that is absent in NUCKS1 has yet been determined.
Figure 1.
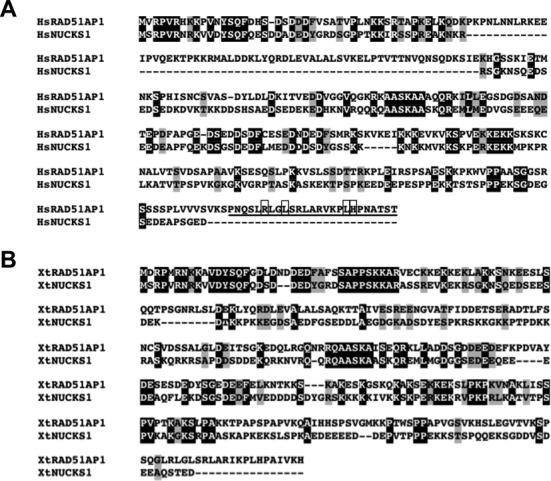
NUCKS1 and RAD51AP1 are paralogs. (A) ClustalW multiple sequence alignment of human NUCKS1 and human RAD51AP1. There is a 28% amino acid sequence identity (black) and 11% similarity (gray) between both proteins. The C-terminal domain (encompassing the last 25 amino acids), identified in human RAD51AP1 to facilitate the interaction with RAD51 (52), is underlined. Critical residues within the domain, as identified by mutational analysis (52), are shown in boxes. (B) ClustalW multiple sequence alignment of NUCKS1 and RAD51AP1 from Xenopus tropicalis. These two proteins share 29% identity (black) and 11% similarity (grey).
Knockdown of NUCKS1 sensitizes human cells to mitomycin C (MMC)
Being intrigued that NUCKS1 is a paralog of RAD51AP1, we asked if these two proteins serve similar biological functions. RAD51AP1 is needed for HR-mediated repair of DNA containing interstrand crosslinks (6,13,14). We therefore exposed cells to MMC to induce DNA crosslinks and tested whether gene-specific knockdown of NUCKS1 would enhance the cytotoxicity of HeLa cells to such treatment. Indeed, we found that HeLa cells depleted for NUCKS1 (individually by two different NUCKS1 targeting siRNAs: N #1 or N #2) show decreased survival after MMC treatment compared to HeLa cells treated with a non-depleting negative control siRNA (Figure 2A, left panel, and Supplementary Figure S2A). These results suggest that, similar to RAD51AP1, NUCKS1 is required for the repair of DNA damage introduced by MMC, which is known to challenge the HR pathway (53). The effects of NUCKS1 depletion on MMC-induced cytotoxicity were also observed when the protein was depleted using a third siRNA (N #3), and the increase in MMC sensitivity could not be alleviated by pretreatment with chloroquine (Supplementary Figure S2B and S2E), which promotes chromatin relaxation (54–56). These results suggest that the role of NUCKS1 in DNA crosslink repair is unrelated to its proposed role in relaxing chromatin during transcriptional processes (28,29).
Figure 2.
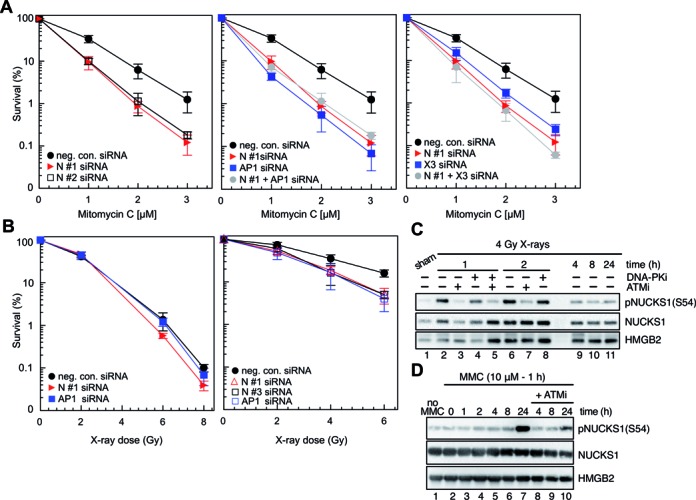
NUCKS1 knockdown sensitizes HeLa cells to mitomycin C (MMC) and X-rays, and NUCKS1 is phosphorylated after DNA damage. (A) Survival curves obtained in control cells (transfected by neg. con. siRNA) or after NUCKS1 depletion following transfection with either NUCKS1 siRNA N #1 or NUCKS1 siRNA N #2 after treatment with MMC (left panel). NUCKS1- and/or RAD51AP1-depleted HeLa cells show the same increased cellular sensitivity to MMC treatment compared to control transfected cells (middle panel). NUCKS1- and/or XRCC3-depleted HeLa cells show similarly increased cellular sensitivity to MMC treatment compared to control transfected cells (right panel). Data points are the means from 3 independent experiments ± 1SD. (B) Cell survival curves obtained after exposure of asynchronous HeLa cells with NUCKS1 or RAD51AP1 depletion to graded doses of X-rays (left panel). Cell survival curves obtained after exposure of HeLa cells in S-phase and with NUCKS1 or RAD51AP1 depletion to graded doses of X-rays (right panel). Data are the mean from 3 independent experiments ± SD. (C) Western blots of PCA extracts obtained from HeLa cells to show that NUCKS1 is phosphorylated in an ATM-dependent manner on serine 54 (S54) after exposure to X-rays. HMGB2 serves as loading control. (D) Western blots of PCA extracts obtained from HeLa cells to show that NUCKS1 is phosphorylated by ATM on serine 54 (S54) after exposure to MMC. HMGB2 serves as loading control.
To test for the epistatic relationship between NUCKS1 and RAD51AP1, we performed MMC cell survival assays with HeLa cells simultaneously depleted for both NUCKS1 and RAD51AP1, and compared their sensitivity to MMC to that of cells singly depleted. In accord with our previous report (6), we find that knockdown of RAD51AP1 sensitizes HeLa cell ∼2-fold (based on D10 values) to the cytotoxic effects of MMC (Figure 2A, middle panel). Interestingly, single knockdown of NUCKS1 or in combination with RAD51AP1, similarly decreases HeLa cell survival ∼2-fold after MMC treatment (Figure 2A, middle panel, and Supplementary Figure S2A). These results show that NUCKS1 and RAD51AP1 are part of the same epistasis group. In addition, knockdown of NUCKS1 in combination with knockdown of XRCC3, another important protein in the HR pathway, also confers no additive or synergistic effect on cell survival after MMC treatment compared to single knockdown of NUCKS1 (Figure 2A, right panel, and Supplementary Figure S2A). Taken together, these results support the notion that NUCKS1 is epistatic with both RAD51AP1 and XRCC3, and that this protein likely is a new factor important for the proper functioning of the HR pathway.
Knockdown of NUCKS1 sensitizes human cells in S-phase to X-rays
Next we tested if functional loss of NUCKS1 would affect the sensitivity of HeLa cells to ionizing radiation. In asynchronous cells, depletion of NUCKS1 only had a very mild effect on further sensitizing HeLa cells to X-rays (Figure 2B, left panel). However, NUCKS1 depleted cells that, following synchronization with mimosine, were in S-phase during the radiation exposure showed increased cellular sensitivity when compared to control cells (Figure 2B, right panel). These results support the notion that NUCKS1 function in DNA damage repair following ionizing radiation exposure largely is confined to S-phase cells, as was also reported for other proteins in the HR pathway (6,14,57). NUCKS1 knockdown also increases cellular sensitivity to the topoisomerase I inhibitor camptothecin (CPT; Supplementary Figure S2C), and to higher concentrations of hydroxyurea (Supplementary Figure S2D) which leads to depleted dNTP pools. These results further support the notion that the role of the NUCKS1 protein in DNA repair is linked to DNA replication.
NUCKS1 is phosphorylated in response to X-rays and MMC treatment
Phospho-Ser54 (pSer54) in NUCKS1 was identified as an ATM-dependent, DNA damage-induced phosphorylation site in response to neocarzinostatin treatment (31). To follow up on these findings, we had an antibody made to the NUCKS1 peptide sequence encompassing pSer54 (i.e. CKNKRRSGKN(pSer)QEDS). As determined by western blot analysis, this antibody was specific for detecting pNUCKS1(S54) in DNA-PK-deficient M059J cells after exposure to 10 Gy X-rays (Supplementary Figure S2F). In HeLa cells, phosphorylation of NUCKS1 at Ser54 was dependent on ATM, independent of DNA-PK and detected as early as 1 h post exposure to 4 Gy X-rays (Figure 2C, lanes 2–4). The phosphorylation persisted for ∼2 h post-radiation treatment (Figure 2C, lanes 6–8), and was clearly diminished by 4 h post exposure (Figure 2C, lane 9). In accordance with the delayed activation of ATM following interstrand crosslink damage by MMC (58), a clear signal for pNUCKS1(S54) was detected at 24 h after treatment with MMC (Figure 2D, lane 7). Treatment with the ATM inhibitor clearly diminished pNUCKS1(S54), but did not fully abrogate this phosphorylation (Figure 2D, lane 10).
NUCKS1 depletion promotes chromatid type aberrations
To investigate whether chromosome damage after NUCKS1 depletion carries the signature of an HR defect, we tested NUCKS1-depleted cells for their levels of chromatid breaks (ctbs), both spontaneously and after the induction of DNA damage by MMC. Chromosome spreads of NUCKS1 depleted hTERT-immortalized human HCA2 fibroblasts (HCA2-hTERT; Figure 3C) were compared to control cells (i.e. transfected with negative control siRNA) 24 h after a mock or an acute 1-h exposure to 50 nM MMC. After MMC treatment, a ∼1.5- to 2-fold increase in the level of induced ctbs was observed for NUCKS1-depleted cells 24 h after the 1 h MMC treatment period (Figure 3A and B). In addition, spontaneous ctbs were slightly elevated in NUCKS1-depleted HCA2-hTERT cells compared to HCA2-hTERT cells transfected with negative control siRNA (Figure 3A). After MMC treatment, we also observed a >2-fold increase in ctbs in RAD51AP1-depleted HCA2-hTERT cells compared to control cells (Figure 3A). This is very consistent with our previous results for RAD51AP1-depleted HeLa cells (6). Taken together, these findings reveal that NUCKS1 is required for the repair of DNA damage in S-phase cells, suggestive of a role for NUCKS1 in HR-mediated DNA repair. Next, we asked whether NUCKS1 knockdown would affect FANCD2 ubiquitylation in response to HU treatment. Similar to the effects of RAD51AP1 depletion, we found that the conversion of FANCD2 to FANCD2-Ub in HeLa cells is not impaired by NUCKS1 depletion (Figure 3D), suggesting that NUCKS1 does not affect the early steps of the Fanconi Anemia pathway.
Figure 3.
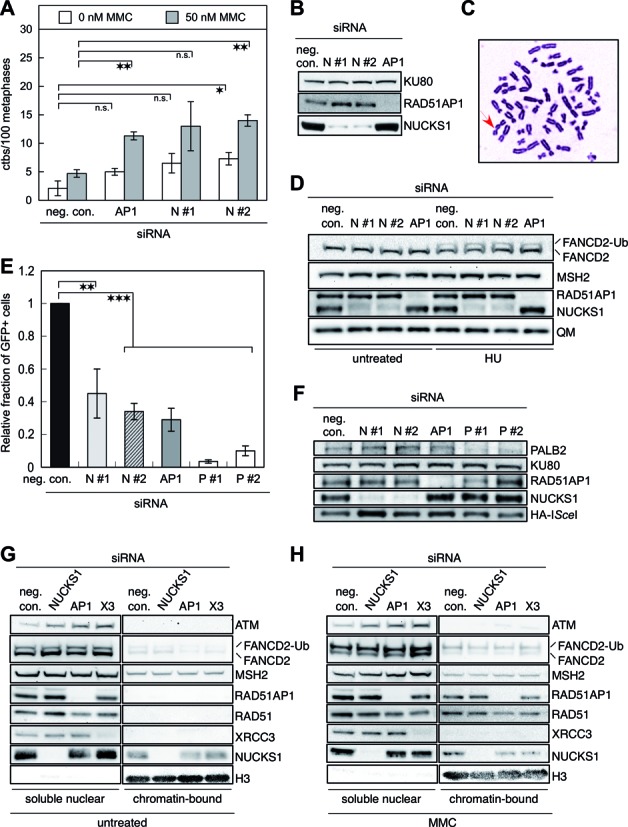
NUCKS1 is required for genome stability and HR repair, is localized to the chromatin fraction and dispensable for FAND2 ubiquitylation. (A) Chromatid-type aberrations are elevated after NUCKS1 knockdown in HCA2-hTERT fibroblasts. The effects of RAD51AP1 depletion (AP1) are shown for comparison purposes. **P ≤ 0.01; *P ≤ 0.05. Data are the mean from 3 (neg. con., AP1, and N #1 siRNA) and 2 (N #2 siRNA) independent experiments ± 1 SEM. (B) Representative Western blots obtained after knockdown of NUCKS1 or RAD51AP1 in HCA2-hTERT cells for the experiments shown in A. The signal for KU80 serves as a loading control. (C) Representative metaphase spread of NUCKS1-depleted HCA2-hTERT cells obtained after MMC treatment. Arrow: indicates chromatid break. (D) Western blots to show that the ubiquitylation of FANCD2 is not impaired in HeLa cells exposed to 2 mM HU and depleted for NUCKS1 by siRNA N #1 or N#2, or depleted for RAD51AP1 (AP1). Extracts from HU-treated cells were generated 5 h after treatment. The signals for MSH2 and QM serve as loading controls. (E) Homology-directed repair at DR-GFP is reduced in DR-U2OS cells depleted for NUCKS1 by one of two different siRNAs (N #1 and N #2). **P ≤ 0.01; ***P ≤ 0.001. The effects of RAD51AP1 depletion (AP1) or of PALB2 depletion by one of two different siRNAs (P #1 and P #2) are shown for comparison purposes. Data are from eight independent experiments ± 1 SD. (F) Representative Western blots obtained for the experiments shown in (E), ensuring similar expression of HA-ISceI in all cell populations. The signal for KU80 serves as a loading control. (G) Western blots obtained after cell fractionation to show that some NUCKS1 protein is associated with the chromatin fraction in HeLa cells under spontaneous conditions. The signals for MSH2 and histone H3 serve as loading controls. To obtain NUCKS1 knockdown, a pool of NUCKS1 siRNAs (N #1, #2 and #3) was used. (H) Western blots obtained after cell fractionation to show that NUCKS1 protein is associated with the chromatin fraction in HeLa cells that were treated with 1 μM MMC for 24 h. The signals for MSH2 and histone H3 serve as loading controls.
NUCKS1 and RAD51AP1 are equally important for DNA homology-directed DSB repair
We used the recombinational reporter cell line U2OS-DRGFP (i.e. DR-U2OS) (36–38) to directly examine the repair of DSBs induced by I-SceI endonuclease in the context of suppressed expression of NUCKS1. In this system, gene conversion HR triggered by DSB formation generates a functional GFP gene, expression of which can be conveniently monitored by flow cytometry. Importantly, depletion of NUCKS1 by one of two different siRNAs (N #1 or N #2) led to a significantly reduced level of gene conversion (Figure 3E and Supplementary Figure S2G). The level of impaired homology-mediated DSBR in NUCKS1-depleted cells is comparable to that engendered by RAD51AP1 knockdown (Figure 3E; (6)). However, we also found that knockdown of PALB2 by one of two different siRNAs elicits a more dramatic effect on gene conversion frequency than depletion of either NUCKS1 or RAD51AP1 (Figure 3E). This is consistent with the multiple roles of PALB2 in HR (see ‘Discussion’ section). Notably, a comparable level of transiently expressed I-SceI nuclease was observed in all cell populations assessed (Figure 3F). Taken together, these results show that the NUCKS1 protein is involved in homology-directed DSB repair.
A subpopulation of nuclear NUCKS1 protein is associated with the chromatin
To assess if NUCKS1 is associated with the chromatin fraction and if its depletion would affect the recruitment of other HR proteins to chromatin after DNA damage induction, we fractionated HeLa cells depleted for NUCKS1, RAD51AP1 or XRCC3 and monitored the levels of soluble nuclear and chromatin-associated ATM, FANCD2, MSH2, RAD51AP1, RAD51, XRCC3 and NUCKS1 without and with MMC treatment by Western blot analysis. Under spontaneous conditions, the level of soluble nuclear RAD51 was slightly diminished in HeLa cells depleted for RAD51AP1 but not diminished in HeLa cells depleted for NUCKS1 (Figure 3G). Compared to cells transfected with the control siRNA, the soluble nuclear level of ATM was increased in NUCKS1-, RAD51AP1- and in XRCC3-depleted cell populations. NUCKS1 was associated with the chromatin in control-, RAD51AP1- and in XRCC3-depleted cells (Figure 3G). Under spontaneous conditions, this assay was not sensitive enough to detect chromatin-bound ATM, FANCD2, RAD51AP1, RAD51 or XRCC3 (Figure 3G). However, after DNA damage induction (1 μM MMC for 24 h), chromatin-bound RAD51AP1 and RAD51 were readily detectable in control-, in NUCKS1- and in XRCC3-depleted cell populations (Figure 3H). Compared to control-depleted cells, NUCKS1-depletion did not affect the levels of recruited and chromatin-bound RAD51 or RAD51AP1, whereas RAD51AP1 depletion reproducibly led to reduced amounts of chromatin-associated RAD51 (Figure 3H). Notably, NUCKS1 itself was again associated with the chromatin and this was independent of RAD51AP1 or XRCC3 knockdown (Figure 3H). These results show that a small fraction of the NUCKS1 protein available in the nucleus is associated with the chromatin both spontaneously and after DNA damage induction.
Depletion of NUCKS1 results in elevated pRPA(S4/S8) phosphorylation after DNA damage
To further investigate the role of NUCKS1 in DSBR, we tested if the depletion of NUCKS1 in U2OS cells would affect the level of pRPA(S4/S8) phosphorylation after long-term exposure to a low concentration of the topoisomerase I inhibitor CPT (59). To do so, we treated NUCKS1- and RAD51AP1-depleted U2OS cells and U2OS cells transfected with the control siRNA with 20 nM CPT for 24 h. We collected cell pellets from unexposed cell populations, and from cell populations at 0, 24, 48 and 72 h post CPT treatment and assessed their level of pRPA(S4/S8) by western blot analysis. Compared to U2OS cells transfected with the control siRNA, a higher level of phosphorylated RPA (i.e. pRPA(S4/S8)) was detected in both NUCKS1- and RAD51AP1-depleted cells immediately (i.e. at 0 h) and at 24 h following CPT wash out (Figure 4A). These findings could be recapitulated by assessing the fraction of HeLa cell nuclei with pRPA32(S4/S8) foci using immunocytohistochemistry (Supplementary Figure S2H). By western blot analysis, elevated pRPAS4/S8 was also detected in XRCC3-depleted U2OS cells compared to control cells at 0 and 24 h post CPT wash out (Supplementary Figure S2I). Analysis of the cell cycle distributions at 24 h post CPT washout in transfected U2OS cell populations revealed a larger fraction of cells in G2/M-phase after NUCKS1- and RAD51AP1-depletion when compared to control cells (Figure 4B).
Figure 4.
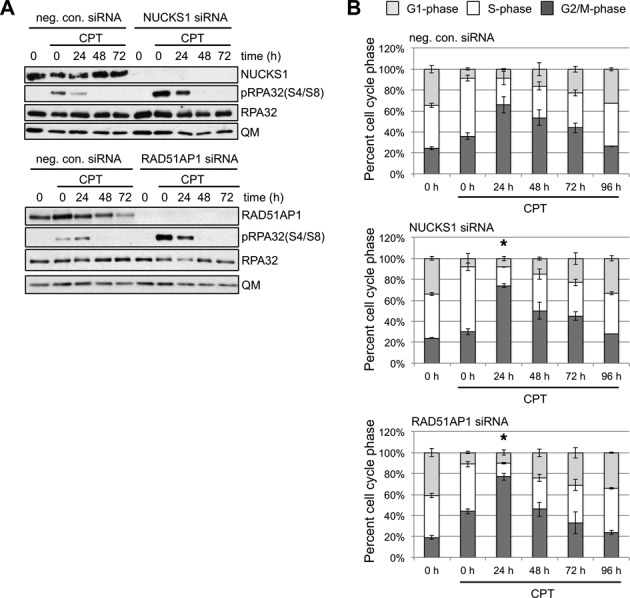
Wild type levels of both NUCKS1 and RAD51AP1 are required to prevent increased pRPA(S4/S8) levels, indicative of increased replication stress, after a 24 h low dose camptothecin (CPT) treatment. (A) Western blots to show increased levels of pRPA(S4/S8) in NUCKS1-depleted (upper panel) and RAD51AP1-depleted (lower panel) U2OS cells, when compared to U2OS cells transfected with negative control siRNA, immediately (0 h) and at 24 h after a 24-h incubation in 20 nM CPT. (B) Analysis of the cell cycle distributions shows a larger fraction of G2/M-phase cells for NUCKS1- and RAD51AP1-depleted cells than in control cells transfected with negative control siRNA at 24 h post CPT wash out. Bars represent the means from 3 independent experiments ± 1 SEM. *P ≤ 0.05.
NUCKS1 is required for efficient recovery from spontaneously occurring DNA replication stress
Our results show that NUCKS1 is required for gene conversion (as measured by homology-directed repair at DRGFP) and affects the replication stress response (as measured by pRPA(S4/S8) phosphorylation following CPT treatment). As HR is the preferred repair pathway for DSBs and other lesions associated with the replication fork in vertebrate cells (12,60,61), and since it has been shown that a spontaneous reduction in replication fork elongation rate and/or an increased level of origin firing can occur in HR-defective cells (10,11,62), we tested if NUCKS1 is needed to maintain elongation rates during DNA replication. To do so, we used the DNA fiber technique to assess DNA replication speed and origin firing by labeling exponentially growing U2OS cells in two consecutive rounds of 45 min each with CldU first and then with IdU (Figure 5A). Quantitative analysis of individual replication fibers revealed an almost two-fold and statistically significant decrease in DNA replication speed in NUCKS1-depleted U2OS cells (i.e. by NUCKS1 siRNA N #2 or N #3) compared to control cells (i.e. untransfected or transfected with negative control siRNA) (Figure 5B and Supplementary Figure S3A and B). These results demonstrate that, similar to RAD51AP1, BRCA2 and PALB2, NUCKS1 is important for maintaining the proper elongation rate of DNA replication. In addition, we observed a significant effect on the discontinuity in fork progression toward smaller IdU/CldU length ratios in NUCKS1-depleted cells (Figure 5C), suggesting that NUCKS1 helps overcome spontaneous DNA damage that is inhibitory to replication fork progression. Upon further analysis of these DNA fibers, we noticed a trend toward a higher number of DNA replication origins in NUCKS1-depleted cells compared to untransfected cells or to U2OS cells transfected with the control siRNA. These observations led us to test if, under unpertubed conditions, increased origin firing occurs in NUCKS1-depleted cells to compensate for reduced fork progression and discontinuity in fork progression. Indeed, when compared to control cells, the combined fractions of first and second pulse origins were higher in NUCKS1-depleted cells compared to control cells (Figure 5D). However, this phenotype was not as pronounced as when RAD51AP1 was depleted (Figure 5D). Importantly, cell cycle progression was unaffected in unperturbed NUCKS1-depleted cells, as shown in Figure 4B.
Figure 5.
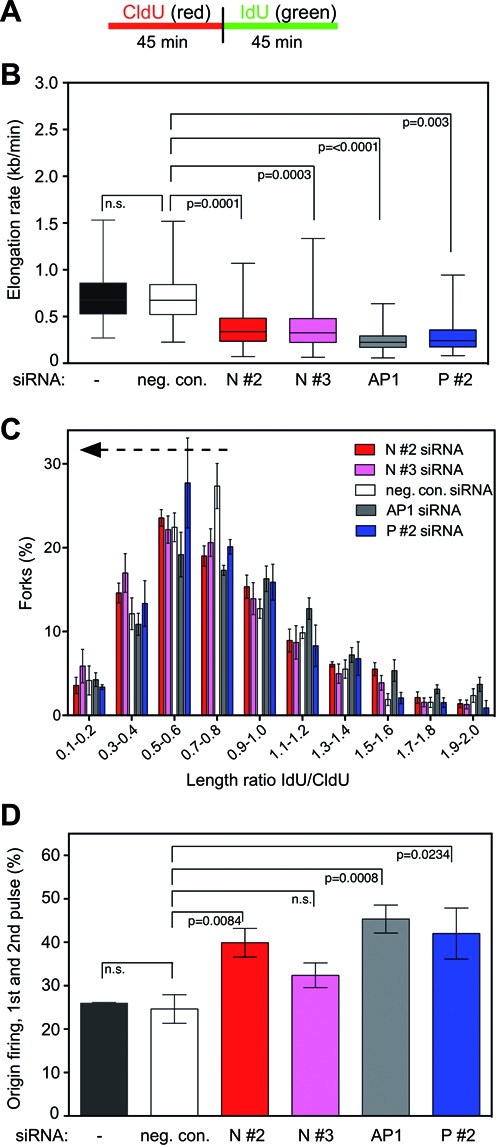
NUCKS1 is required for unperturbed and continuous DNA replication fork progression. (A) The labeling protocol used in this experiment. (B) Knockdown of NUCKS1 impairs DNA elongation rates under unperturbed conditions. U2OS cells were transfected with one of two different siRNAs targeting NUCKS1 (N #2, N #3), or with negative control (neg. con.), RAD51AP1 (AP1), or PALB2 siRNA (P #2) for comparison purposes. Untransfected cells (-) were also assessed. In box plots, boxes encompass the 25th–75th percentile, with error bars defining the minima and maxima. The black horizontal bars within the boxes indicate the means. The data are from seven independent experiments, except for P #2 siRNA the data for which are from 3 independent experiments. (C) Knockdown of NUCKS1 causes discontinuity in replication fork progression. U2OS cells were transfected as in (B), and elongation rates of red- (CldU) and green-labeled (IdU) fiber tracts were determined. The data show the distribution of IdU/CldU length ratios from 400 individual fibers of 7 (neg. con., N #2, N #3, AP1 siRNA) and 3 (P #2 siRNA) different experiments. Compared to control cells (neg. con. siRNA), IdU/CdU length ratios ≤ 0.84 are significantly shorter for N #2 (P = 0.0023), N #3 (P = <0.0001) and AP1 depletion (P = 0.0207), but not for P #2 depletion (P = 0.1553; one-tailed Mann Whitney U test). (D) Knockdown of NUCKS1 leads to elevated levels of first and second pulse replication origins under unperturbed conditions. U2OS cells were transfected as in (B) and first and second pulse replication origins were determined by quantification of green–red–red–green and green-only structures, respectively. The data are the means ± 1 SEM of 7 (neg. con., N #2, N #3, AP1 siRNA) and 3 (P #2 siRNA) independent experiments.
NUCKS1 is required for efficient recovery from induced DNA replication stress
Next, we assessed the role of NUCKS1 under conditions of perturbed DNA replication. To do so, we labeled exponentially growing cells with CldU for 15 min first, then incubated the cells in fresh medium containing 2 mM HU for 30 min, after which time we switched to IdU-containing fresh medium and incubated for 45 min before cell lysis (Figure 6A). We determined the fraction of terminated/stalled forks (i.e. only red-labeled fibers) after replication fork stalling from short-term HU treatment to assess the sensitivity of NUCKS1-depleted cells to HU, and normalized this to the fraction of terminated/stalled forks under unperturbed conditions for the same cell population. After HU treatment, the relative fraction of terminated/stalled forks was significantly increased in NUCKS1-depleted cells compared to control cells (Figure 6B and Supplementary Figure S3C). In this assay and in line with the HU cell survival data (Supplementary Figure S2D), NUCKS1 depletion shows greater effects than RAD51AP1 depletion (Figure 6B), suggesting that NUCKS1 is of greater importance than RAD51AP1 in the recovery from DNA replication fork stalling.
Figure 6.
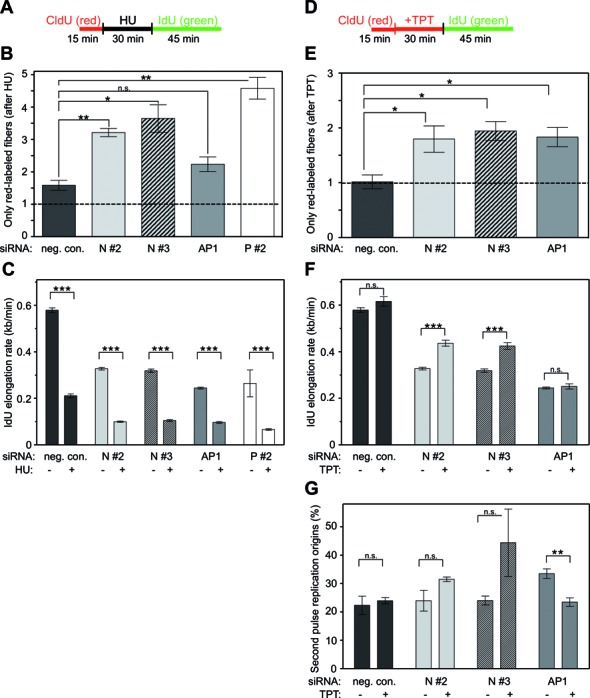
NUCKS1 is required for the restart of replication forks after fork stalling and fork collapse. (A) Schematic of the DNA labeling protocol used in (B) and (C). (B) Knockdown of NUCKS1 causes an increase in only red-labeled fibers after HU treatment. U2OS cells were transfected with one of two different siRNAs for NUCKS1 (N #2, N #3), or with negative control (neg. con.), with RAD51AP1 (AP1) or with PALB2 (P #2) siRNA. The data are the means ± 1 SEM of three independent experiments. The bar graphs show the fraction of red-labeled fibers after HU treatment, normalized to the untreated control for each gene-specific knockdown. *P < 0.05; and **P < 0.01; n.s, not significant. (C) Knockdown of NUCKS1 causes a reduction of DNA elongation rates after HU treatment. U2OS cells were transfected with one of two different siRNAs for NUCKS (N #2, N #3), or with negative control (neg. con.), RAD51AP1 (AP1) or PALB2 siRNA (P #2). IdU elongation rates were determined by measuring the green (IdU) of both red- and green-labeled DNA fibers using Image J, and fiber lengths were converted into kb/min, as described (48). IdU elongation rates are presented as the means ± 1 SEM of three independent experiments. ***P < 0.001 (D) Schematic of the DNA labeling protocol used in (E)–(G). (E) Knockdown of NUCKS1 causes an increase in stalled replication forks after topotecan (TPT) treatment. Red-labeled fibers were quantified using Image J. The normalized data (to untreated cells of each gene-specific knockdown condition) are represented as the means ± 1 SEM of at least three independent experiments. *P < 0.05. (F) After TPT treatment, NUCKS1-depleted cells show faster elongating IdU tracts compared to unperturbed NUCKS1-depleted cells. IdU elongation rates were determined by measuring green tracts (IdU) of both red- and green-labeled DNA tracts using ImageJ and tract lengths were converted into kb/min, as described (48). The data are represented as means ± 1 SEM of three independent experiments. The bar graphs show the IdU elongation rates for untreated cell populations and after TPT treatment. ***P < 0.001; n.s., not significant. (G) Knockdown of NUCKS1 causes an increase in second pulse origin firing after TPT treatment. Second pulse origins were determined by quantification of green tracts in both red- and green-labeled DNA tracts using ImageJ. The data shown are the means ± 1 SEM of three independent experiments. **P < 0.01; n.s., not significant.
We also determined the elongation rate of restarted replication forks in NUCKS1-depleted cells by assessing the progression rate of IdU elongating fibers of CldU-labeled tracts with and without HU treatment. We found that under unperturbed conditions and compared to control cells, the IdU elongation rate is greatly reduced in NUCKS1-, RAD51AP1- and PALB2-depleted cells (Figure 6C), as expected from the aforementioned spontaneously occurring replication stress (see Figure 5). After HU treatment, the IdU elongation rate becomes further diminished in NUCKS1-depleted cells, very similar to the effects of RAD51AP1 or PALB2 knockdown (Figure 6C).
Short-term treatment with topotecan (TPT) leads to the collapse of replication forks and the formation of replication-associated DSBs, which normally are repaired via HR (63). Here, we found that, compared to cells transfected with the control siRNA, NUCKS1- and RAD51AP1-depleted cells showed similarly increased levels of terminated/collapsed forks after a 30 min exposure to TPT (Figure 6D and E, and Supplementary Figure S3D), suggesting that they are equally important for overcoming replication fork collapse in response to TPT treatment. Next, we determined the elongation rate of restarting replication forks for both NUCKS1- and RAD51AP1-depleted cells with TPT treatment. In control-transfected cells, replication forks after TPT treatment progressed with a similar elongation rate as in untreated cells (Figure 6F). The same pattern was observed for RAD51AP1-depleted cells (Figure 6F). Interestingly, in NUCKS1-depleted cells, after TPT treatment, replication forks restarted at a significantly faster rate (Figure 6F). When assessed for the fraction of second pulse replication origins with and without TPT treatment, NUCKS1-depleted cells showed a trend toward increased levels of second pulse replication origins, while RAD51AP1-depleted cells significantly suppressed firing of second pulse replication origins after TPT treatment (Figure 6G).
DNA and chromatin binding by NUCKS1
We added a GST-tag to the amino terminus of the human NUCKS1 protein and expressed the tagged protein in Hi5 insect cells. The tagged NUCKS1 protein is soluble, and a procedure was devised for its purification to near homogeneity (Supplementary Figure S4A). We used a DNA mobility shift assay to examine the DNA binding properties of GST-NUCKS1 to D-loop, dsDNA and ssDNA (Figure 7A–C, Supplementary Figure S4B and C). GST-NUCKS1 had no affinity to ssDNA (Figure 7C and Supplementary Figure S4B), but it bound dsDNA and exhibited a greater preference for the D-loop substrate (Figure 7B and C, Supplementary Figure S4C). However, when compared to the DNA-binding properties of RAD51AP1, we found that RAD51AP1 bound to D-loop and dsDNA with ∼25-fold and ∼50-fold greater affinity than did NUCKS1, respectively (Figure 7B, compare lanes 2 and 12; Figure 7C, compare lanes 4 and 10). Next, we used an immobilized DNA pull-down assay, which allowed us to assemble chromatin in vitro using the purified human histone octamer and a 588 bp biotin-labeled dsDNA fragment bound to streptavidin-coated magnetic beads. In this assay, we compared binding of NUCKS1 to naked dsDNA vs. chromatin (Figure 7D and Supplementary Figure S4D). Interestingly, we found that NUCKS1 greatly preferred the chromatinized substrate over the naked DNA (Figure 7E, compare lanes 3 and 4 to lanes 1 and 2), whereas both RAD51AP1 and RAD51 showed no such preference (Supplementary Figure S4E and F). Although NUCKS1 bound to the D-loop substrate (Figure 7B), it did not stimulate D-loop formation using either naked or chromatinized DNA as a donor (Supplementary Figure S5).
Figure 7.
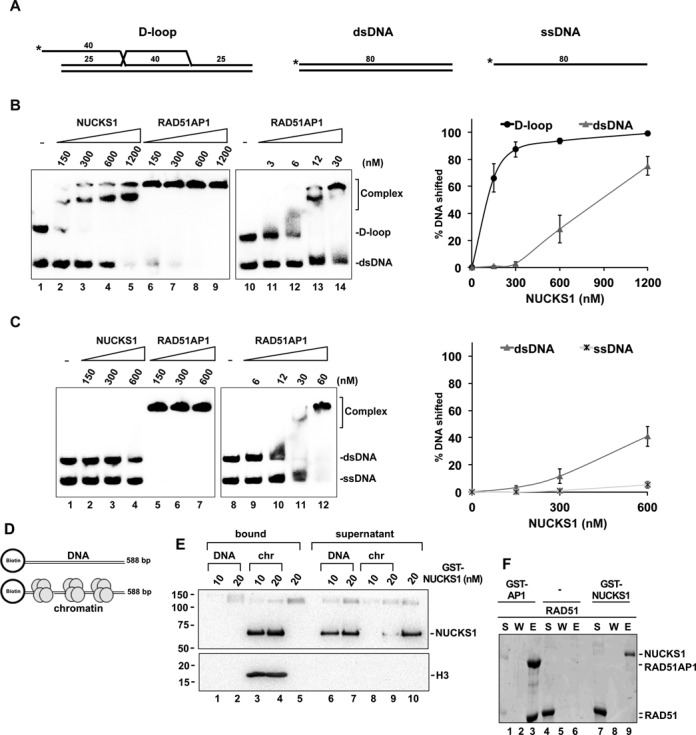
NUCKS1 prefers binding to chromatin over naked DNA, and does not interact with RAD51. (A) Schematics of the DNA substrates used in (B) and (C). Numbers indicate lengths of DNA fragments in bp. Asterisks denote the position of the 5-prime label. (B) GST-NUCKS1 (0.15–1.2 μM) and GST-RAD51AP1 (0.15–1.2 μM, and 0.003–0.03 μM for comparison purposes) were incubated with dsDNA and with the D-loop substrate (2.5 nM each) and analyzed for mobility shifts using 8% polyacrylamide gels (left panels). The results from the quantified mobility shifts were plotted (right panel). Data points are the means from three independent experiments ± 1SD. See also Supplementary Figure S4C. (C) GST-NUCKS1 (0.15–0.6 μM) and GST-RAD51AP1 (0.15–0.6 μM, and 0.006–0.06 μM for comparison purposes) were incubated with ssDNA or with dsDNA (2.5 nM each) and analyzed for mobility shifts using 8% polyacrylamide gels (left panels). The results from the quantified mobility shifts were plotted (right panel). Data points are the means from 3 independent experiments ± 1SD. See also Supplementary Figure S4B and C. (D) Schematics of the immobilized DNA substrates used (E). (E) Western blots obtained after the immobilized DNA pull-down assays using either naked DNA (here: DNA (50 nM/reaction)) or chromatinized DNA (here: chr (50 nM/reaction)) and 10 or 20 nM GST-NUCKS1, as indicated. DNA-bound GST-NUCKS1 (lanes 1 and 2), chromatin-bound GST-NUCKS1 (lanes 3 and 4), bead-only control (lane 5), and unbound GST-NUCKS1 in the supernatants from the reactions in lanes 1–5 (lanes 6–10, respectively). The signal for histone H3 (here: H3) serves as control for successful chromatinization (lanes 3–4) and shows that histones are not disrupted (no H3 signal in lanes 8 and 9). (F) SDS-PAGE to show interaction between GST-RAD51AP1 and RAD51 in GST pull-down assay (lane 3). RAD51 does not precipitate non-specifically (lane 6), and GST-NUCKS1 does not interact with RAD51 (lane 9). S, supernatant containing unbound proteins; W, wash; E, SDS eluate of the glutathione resin.
NUCKS1 does not interact with RAD51
A pull-down assay was used to examine the ability of GST-NUCKS1 to interact directly with human RAD51. As expected from the sequence alignment presented in Figure 1A and in contrast to RAD51AP1 which avidly interacts with RAD51 (Figure 7F, lane 3), purified NUCKS1 does not bind RAD51 (Figure 7F, lane 9). Similarly, in extracts from human cells, we could not detect any interaction of RAD51 with NUCKS1 in immunoprecipitation reactions (data not shown). These results indicate that NUCKS1, unlike RAD51AP1 (6,14,64), does not associate with the RAD51 recombinase.
DISCUSSION
The NUCKS1 gene consists of seven exons and six introns, is ubiquitously expressed in all mammalian tissues, and appears to be a housekeeping gene (44). The NUCKS1 gene belongs to a group of co-expressed genes located on chromosomal region 1q32.1 that often is amplified in breast cancers (23,51,65,66), and other cancers (22,24–26,67,68). Although there is little functional information on NUCKS1, several studies have suggested a linkage between its high expression and breast cancer (18,23,29,66,69). NUCKS1 also was identified as a colorectal cancer prognostic marker and as a biomarker for recurrence-free survival in cervical squamous cell carcinoma (24,68). However, how elevated levels of NUCKS1 may promote progression towards malignancy is currently unknown.
Based on sequence homology to RAD51AP1, a RAD51-binding protein intimately involved in homologous recombination DNA repair (HR) as shown by us and the Kanaar group (6–9,14,43), we have tested and uncovered a role for NUCKS1 in HR and in maintaining DNA replication integrity and genome stability. Using a recombinational reporter, we have shown that NUCKS1 deletion results in a significant decrease in gene conversion levels, which is very similar to the results obtained for either RAD51AP1 or XRCC3 depletion (this study, (6)). From these results, we can conclude that NUCKS1 plays a role in HR DNA repair, as does its paralog RAD51AP1. However, the effects of NUCKS1 or RAD51AP1 knockdown on gene conversion are less severe than what we (this study) and others (37) have observed for PALB2 knockdown. This may relate to the multiple roles in HR of PALB2, which functions as an HR mediator, as a stimulator of the strand invasion step, and also as a key regulator of the DNA synthesis step after strand invasion at replication-dependent DSBs (37,70,71).
While recombinant NUCKS1 shows no affinity for ssDNA, recombinant RAD51AP1 does (6,14). Albeit with very different affinities, both NUCKS1 and RAD51AP1 display the same DNA-binding preference for D-loop and dsDNA. However, while RAD51AP1 stimulates RAD51 activity and D-loop formation in vitro (6,14), NUCKS1 fails to do so. Given our findings that show that NUCKS1 and RAD51 do not interact, the inability of NUCKS1 to stimulate D-loop formation is not surprising, as the functional synergy of RAD51AP1 with RAD51 is reliant on complex formation between them (6,14). While, in light of the many posttranslational modifications of NUCKS1, it is possible that the NUCKS1 protein that we have isolated from insect cells lacks the modifications required for its proper function, it is also very possible that the functions of NUCKS1 and RAD51AP1 in HR have diverged. However, as for RAD51AP1 (6,10,14), we find that NUCKS1 is not required for the formation of RAD51 foci following DNA damage induction by ionizing radiation (Supplementary Figure S6A and B), suggesting that NUCKS1, like RAD51AP1, functions downstream of RAD51-ssDNA nucleoprotein filament formation in HR. Notably, as assessed by cell fractionation assays, NUCKS1 depletion also does not affect the levels of RAD51 recruited to the chromatin after MMC treatment, while RAD51AP1 ablation exerts a small but consistent effect.
Our results further suggest that NUCKS1 behaves like other proteins in the HR pathway, including RAD51AP1, in affecting the tolerance of cells to replication stress. Since many DNA-damaging agents, including those that are used in cancer therapy, act by inducing replication stress, it is important to better understand the complexity of the replication stress response (72). This is particularly useful in anticipation of the development of new cancer treatment regimens and with respect to the fact that a close but poorly understood link exists between elevated NUCKS1 expression and cancer development processes (23,24,26,51,65,73).
Our results show that NUCKS1 is important for the progression of DNA replication forks. Under conditions of unperturbed DNA replication, we have found that the elongation speed of individual replication forks is greatly reduced upon NUCKS1 depletion. Concomitantly, we have observed a trend towards increased replication origin firing in NUCKS1-depleted cells without any notable effect on cell cycle progression, as reported previously for other factors that globally reduce replication fork speeds, including RAD51AP1 (10,74–76). Short-term treatment with HU leads to the depletion of the pool of dNTPs and to replication fork stalling. HU-induced replication fork stalling can be more deleterious to cells when coupled with a HR deficiency (10,77,78), and this also is what we have observed here for cells with NUCKS1 knockdown. In the same manner, the speed at which restarted forks progress becomes shortened significantly in NUCKS1-depleted cells after HU treatment. Taken together, our results show that HR-defective, NUCKS1-depleted U2OS cells are sensitive to replication stress under unperturbed conditions and also after short-term HU treatment stemming from an impaired ability to overcome replication fork stalling. Of note, we have found that cells depleted for PALB2 are greatly impaired in their ability to restart stalled replication forks as well (Figure 6B), a finding that is in agreement with a previous report (11). It is also interesting to note that, despite the fact that fork stalling in response to HU treatment is higher after NUCKS1 knockdown than after RAD51AP1 knockdown, the rates of restarted forks are very similar for NUCKS1- and RAD51AP1-depleted cells. These results suggest that NUCKS1 and RAD51AP1 play an equally important role in the resumption of replication after short-term HU treatment.
Treatment of cells with the topoisomerase I (Topo I) covalent poison TPT results in the formation of a covalent DNA–Topo I conjugate and a single-strand break (SSB). Collision between a DNA polymerase with this lesion leads to replication fork collapse and DSB formation. HR repair is thus critically important for mitigating TPT-induced replication stress. We have shown that NUCKS1- and RAD51AP1-depleted cells are equally sensitive to the effects of TPT treatment. However, NUCKS1-depleted cells are able to overcome TPT treatment more promptly, and while second pulse origin firing (after TPT treatment) is still reduced in RAD51AP1-depleted cells, enhanced firing is actually observed in NUCKS1-depleted cells. We speculate that RAD51-mediated fork reversal (79), which has been described to occur in response to topoisomerase poisoning (80), may occur to some extent in NUCKS1-depleted cells, but may be more severely compromised under RAD51AP1 deficiency.
As measured by the increased accumulation of pRPA(S4/S8) after long-term, low dose CPT treatment upon protein depletion, we further substantiated our findings that NUCKS1 is required to alleviate replication stress. In the same manner, both RAD51AP1 and XRCC3, established HR factors, are required to limit pRPA(S4/S8) accumulation after CPT treatment. Of note, S4/S8 in RPA2 are phosphorylated under conditions that cause DSBs (81). While elevated levels of pRPAS4/S8 are indicative of ongoing replication stress (82), our analyses of the corresponding cell cycle profiles also revealed an extended G2/M checkpoint arrest after CPT treatment in both NUCKS1- and RAD51AP1-depleted cells. It is important to note that increased pRPA(S4/S8) has been shown to contribute to G2/M checkpoint arrest and to HR suppression (83), such that mutagenesis of S4 and S8 to alanines causes increased mitotic entry with enhanced RAD51 foci formation and HR repair (83). Taken together, our pRPA(S4/S8) data further support a defect in HR repair of DSBs stemming from replication stress in cells with reduced expression of NUCKS1.
Off-target RAD51 depletion was noted as a major source of identified false positive genes in HR (84). However, for all NUCKS1 siRNAs tested here, we clearly can exclude such an effect on RAD51. Nonetheless, we have been unable to rescue the increase in cytotoxicity and the decrease in homology-mediated repair of NUCKS1-depleted cells by overexpression of the siRNA-resistant NUCKS1 cDNA from ectopic promoters. We attribute this observation to the fact that NUCKS1, in addition to the phosphorylation at S54 induced by ionizing radiation or MMC exposure as shown here, is heavily post-translationally modified and suspect, that ectopically expressed NUCKS1 may lack some of the modifications that are critical for its homeostasis and function in cells. Of note, the inability to rescue has been observed before for DSBR defects associated with MSH2 or MLH1 deficiency (85).
In summary, we have provided substantial evidence for a role of the NUCKS1 protein in the HR pathway. We surmise that, as has been proposed for other genes in the DNA damage response (32,33), elevated levels of NUCKS1 may provide a selective growth advantage for premalignant cells that experience induced replication stress and may contribute towards the development of malignancy. The mechanism by which NUCKS1 influences HR and attenuates the replication stress response remains to be determined. Although clearly a RAD51AP1 paralog, NUCKS1 may have gained different functions. For example, a recent report implicates a role for NUCKS1 in transcriptional regulation and that it regulates the DNA structure to allow RNA polymerase II access to the promoter of genes involved in the insulin-signaling and other pathways (28). Interestingly, for both BRCA1 and PALB2, two important HR factors, a role in transcriptional regulation, independent of their function in HR, has also been described (86,87). Although we have entertained a comparable role for NUCKS1 in affecting the expression of genes involved in DSB repair, our investigation of a large number of candidate genes have shown no role of NUCKS1 in the regulation of their expression. In addition, based on our experiments using chloroquine to force chromatin relaxation, we can detect no role for NUCKS1 in DNA repair related to major effects on the overall chromatin structure, unlike what was observed for the early HR factor RNF20 (56). It is possible, however, that NUCKS1, which appears to function downstream of RAD51 filament formation, is involved in HR by very locally regulating the accessibility of DNA repair factors to chromatin, and our results from the immobilized DNA pull-down assay using chromatin as a substrate support such a premise. In addition, it is also possible that NUCKS1 is a multi-functional protein, with its potentially diverse biological tasks being governed by its abundant and highly dynamic post-translational modifications.
Supplementary Material
Acknowledgments
We thank Ms Sara Seidler and Ms KaLee Chau for assistance. The human histone octamer was a generous gift from the Core Facility of Protein Expression and Purification, Department of Biochemistry and Molecular Biology, Colorado State University.
Footnotes
Present addresses:
Ann C. Parplys, Campus Science 27, University Medical Centre Hamburg-Eppendorf, 20246 Hamburg, Germany.
Torsten Groesser, Technical University of Denmark at Risø, DTU Nutech, 4000 Roskilde, Denmark.
Eloïse Dray, Australian Institute for Bioengineering and Nanotechnology, The University of Queensland, Brisbane, Queensland 4072, Australia.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institutes of Health [ES021454 to C.W.; CA120315 and CA092584 to D.S.; ES015252 and CA168635 to P.S.]; DOE LBNL LDRD grant [to C.W.]. Funding for open access charge: National Institutes of Health.
Conflict of interest statement. None declared.
REFERENCES
- 1.Katyal S., McKinnon P.J. DNA repair deficiency and neurodegeneration. Cell Cycle (Georgetown, Tex) 2007;6:2360–2365. doi: 10.4161/cc.6.19.4757. [DOI] [PubMed] [Google Scholar]
- 2.Katyal S., McKinnon P.J. DNA strand breaks, neurodegeneration and aging in the brain. Mech. Ageing Dev. 2008;129:483–491. doi: 10.1016/j.mad.2008.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sakata K., Someya M., Matsumoto Y., Hareyama M. Ability to repair DNA double-strand breaks related to cancer susceptibility and radiosensitivity. Radiat. Med. 2007;25:433–438. doi: 10.1007/s11604-007-0161-3. [DOI] [PubMed] [Google Scholar]
- 4.de Villartay J.P. V(D)J recombination deficiencies. Adv. Exp. Med. Biol. 2009;650:46–58. doi: 10.1007/978-1-4419-0296-2_4. [DOI] [PubMed] [Google Scholar]
- 5.Tamulevicius P., Wang M., Iliakis G. Homology-directed repair is required for the development of radioresistance during S phase: interplay between double-strand break repair and checkpoint response. Radiat. Res. 2007;167:1–11. doi: 10.1667/RR0751.1. [DOI] [PubMed] [Google Scholar]
- 6.Wiese C., Dray E., Groesser T., San Filippo J., Shi I., Collins D.W., Tsai M.S., Williams G.J., Rydberg B., Sung P., et al. Promotion of homologous recombination and genomic stability by RAD51AP1 via RAD51 recombinase enhancement. Mol. Cell. 2007;28:482–490. doi: 10.1016/j.molcel.2007.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dray E., Dunlop M.H., Kauppi L., San Filippo J., Wiese C., Tsai M.S., Begovic S., Schild D., Jasin M., Keeney S., et al. Molecular basis for enhancement of the meiotic DMC1 recombinase by RAD51 associated protein 1 (RAD51AP1) Proc. Natl. Acad. Sci. U.S.A. 2011;108:3560–3565. doi: 10.1073/pnas.1016454108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dunlop M.H., Dray E., Zhao W., Tsai M.S., Wiese C., Schild D., Sung P. RAD51-associated protein 1 (RAD51AP1) interacts with the meiotic recombinase DMC1 through a conserved motif. J Biol. Chem. 2011;286:37328–37334. doi: 10.1074/jbc.M111.290015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dunlop M.H., Dray E., Zhao W., San Filippo J., Tsai M.S., Leung S.G., Schild D., Wiese C., Sung P. Mechanistic insights into RAD51-associated protein 1 (RAD51AP1) action in homologous DNA repair. J. Biol. Chem. 2012;287:12343–12347. doi: 10.1074/jbc.C112.352161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Parplys A.C., Kratz K., Speed M.C., Leung S.G., Schild D., Wiese C. RAD51AP1-deficiency in vertebrate cells impairs DNA replication. DNA Repair. 2014;24:87–97. doi: 10.1016/j.dnarep.2014.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nikkila J., Parplys A.C., Pylkas K., Bose M., Huo Y., Borgmann K., Rapakko K., Nieminen P., Xia B., Pospiech H., et al. Heterozygous mutations in PALB2 cause DNA replication and damage response defects. Nat. Commun. 2013;4:2578. doi: 10.1038/ncomms3578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Truong L.N., Li Y., Sun E., Ang K., Hwang P.Y., Wu X. Homologous recombination is a primary pathway to repair DNA double-strand breaks generated during DNA rereplication. J. Biol. Chem. 2014;289:28910–28923. doi: 10.1074/jbc.M114.576488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Henson S.E., Tsai S.C., Malone C.S., Soghomonian S.V., Ouyang Y., Wall R., Marahrens Y., Teitell M.A. Pir51, a Rad51-interacting protein with high expression in aggressive lymphoma, controls mitomycin C sensitivity and prevents chromosomal breaks. Mutat. Res. 2006;601:113–124. doi: 10.1016/j.mrfmmm.2006.06.016. [DOI] [PubMed] [Google Scholar]
- 14.Modesti M., Budzowska M., Baldeyron C., Demmers J.A., Ghirlando R., Kanaar R. RAD51AP1 is a structure-specific DNA binding protein that stimulates joint molecule formation during RAD51-mediated homologous recombination. Mol. Cell. 2007;28:468–481. doi: 10.1016/j.molcel.2007.08.025. [DOI] [PubMed] [Google Scholar]
- 15.Grundt K., Skjeldal L., Anthonsen H.W., Skauge T., Huitfeldt H.S., Ostvold A.C. A putative DNA-binding domain in the NUCKS protein. Arch. Biochem. Biophys. 2002;407:168–175. doi: 10.1016/s0003-9861(02)00513-1. [DOI] [PubMed] [Google Scholar]
- 16.Grundt K., Haga I.V., Aleporou-Marinou V., Drosos Y., Wanvik B., Ostvold A.C. Characterisation of the NUCKS gene on human chromosome 1q32.1 and the presence of a homologous gene in different species. Biochem. Biophys. Res. Commun. 2004;323:796–801. doi: 10.1016/j.bbrc.2004.08.153. [DOI] [PubMed] [Google Scholar]
- 17.Grundt K., Haga I.V., Huitfeldt H.S., Ostvold A.C. Identification and characterization of two putative nuclear localization signals (NLS) in the DNA-binding protein NUCKS. Biochim. Biophys. Acta. 2007;1773:1398–1406. doi: 10.1016/j.bbamcr.2007.05.013. [DOI] [PubMed] [Google Scholar]
- 18.Wisniewski J.R., Zougman A., Kruger S., Ziolkowski P., Pudelko M., Bebenek M., Mann M. Constitutive and dynamic phosphorylation and acetylation sites on NUCKS, a hypermodified nuclear protein, studied by quantitative proteomics. Proteins. 2008;73:710–718. doi: 10.1002/prot.22104. [DOI] [PubMed] [Google Scholar]
- 19.Ostvold A.C., Holtlund J., Laland S.G. A novel, highly phosphorylated protein, of the high-mobility group type, present in a variety of proliferating and non-proliferating mammalian cells. Eur. J. Biochem. 1985;153:469–475. doi: 10.1111/j.1432-1033.1985.tb09325.x. [DOI] [PubMed] [Google Scholar]
- 20.Thompson H.G., Harris J.W., Wold B.J., Quake S.R., Brody J.P. Identification and confirmation of a module of coexpressed genes. Genome Res. 2002;12:1517–1522. doi: 10.1101/gr.418402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sova P., Feng Q., Geiss G., Wood T., Strauss R., Rudolf V., Lieber A., Kiviat N. Discovery of novel methylation biomarkers in cervical carcinoma by global demethylation and microarray analysis. Cancer Epidemiol. Biomarkers Prev. 2006;15:114–123. doi: 10.1158/1055-9965.EPI-05-0323. [DOI] [PubMed] [Google Scholar]
- 22.Sargent L.M., Ensell M.X., Ostvold A.C., Baldwin K.T., Kashon M.L., Lowry D.T., Senft J.R., Jefferson A.M., Johnson R.C., Li Z., et al. Chromosomal changes in high- and low-invasive mouse lung adenocarcinoma cell strains derived from early passage mouse lung adenocarcinoma cell strains. Toxicol. Appl. Pharmacol. 2008;233:81–91. doi: 10.1016/j.taap.2008.01.031. [DOI] [PubMed] [Google Scholar]
- 23.Drosos Y., Kouloukoussa M., Ostvold A.C., Grundt K., Goutas N., Vlachodimitropoulos D., Havaki S., Kollia P., Kittas C., Marinos E., et al. NUCKS overexpression in breast cancer. Cancer Cell Int. 2009;9:19. doi: 10.1186/1475-2867-9-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kikuchi A., Ishikawa T., Mogushi K., Ishiguro M., Iida S., Mizushima H., Uetake H., Tanaka H., Sugihara K. Identification of NUCKS1 as a colorectal cancer prognostic marker through integrated expression and copy number analysis. Int. J. Cancer. 2013;132:2295–2302. doi: 10.1002/ijc.27911. [DOI] [PubMed] [Google Scholar]
- 25.Akbari Moqadam F., Boer J.M., Lange-Turenhout E.A., Pieters R., den Boer M.L. Altered expression of miR-24, miR-126 and miR-365 does not affect viability of childhood TCF3-rearranged leukemia cells. Leukemia. 2014;28:1008–1014. doi: 10.1038/leu.2013.308. [DOI] [PubMed] [Google Scholar]
- 26.Yang M., Wang X., Zhao Q., Liu T., Yao G., Chen W., Li Z., Huang X., Zhang Y. Combined evaluation of the expression of NUCKS and Ki-67 proteins as independent prognostic factors for patients with gastric adenocarcinoma. Tumour Biol. 2014;35:7505–7512. doi: 10.1007/s13277-014-1880-6. [DOI] [PubMed] [Google Scholar]
- 27.Zduniak K., Agrawal S., Symonowicz K., Jurczyszyn K., Ziolkowski P. The comparison of nuclear ubiquitous casein and cyclin-dependent kinases substrate (NUCKS) with Ki67 proliferation marker expression in common skin tumors. Polish J. Pathol. 2014;65:48–54. doi: 10.5114/pjp.2014.42669. [DOI] [PubMed] [Google Scholar]
- 28.Qiu B., Shi X., Wong E.T., Lim J., Bezzi M., Low D., Zhou Q., Akincilar S.C., Lakshmanan M., Swa H.L., et al. NUCKS is a positive transcriptional regulator of insulin signaling. Cell Rep. 2014;7:1876–1886. doi: 10.1016/j.celrep.2014.05.030. [DOI] [PubMed] [Google Scholar]
- 29.Qiu B., Han W., Tergaonkar V. NUCKS: a potential biomarker in cancer and metabolic disease. Clin. Sci. 2015;128:715–721. doi: 10.1042/CS20140656. [DOI] [PubMed] [Google Scholar]
- 30.Matsuoka S., Ballif B.A., Smogorzewska A., McDonald E.R. 3rd, Hurov K.E., Luo J., Bakalarski C.E., Zhao Z., Solimini N., Lerenthal Y., et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science. 2007;316:1160–1166. doi: 10.1126/science.1140321. [DOI] [PubMed] [Google Scholar]
- 31.Bensimon A., Schmidt A., Ziv Y., Elkon R., Wang S.Y., Chen D.J., Aebersold R., Shiloh Y. ATM-dependent and -independent dynamics of the nuclear phosphoproteome after DNA damage. Sci Signal. 2010;3:rs3. doi: 10.1126/scisignal.2001034. [DOI] [PubMed] [Google Scholar]
- 32.Gorgoulis V.G., Vassiliou L.V., Karakaidos P., Zacharatos P., Kotsinas A., Liloglou T., Venere M., Ditullio R.A. Jr, Kastrinakis N.G., Levy B., et al. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature. 2005;434:907–913. doi: 10.1038/nature03485. [DOI] [PubMed] [Google Scholar]
- 33.Halazonetis T.D., Gorgoulis V.G., Bartek J. An oncogene-induced DNA damage model for cancer development. Science. 2008;319:1352–1355. doi: 10.1126/science.1140735. [DOI] [PubMed] [Google Scholar]
- 34.Kumazaki T., Robetorye R.S., Robetorye S.C., Smith J.R. Fibronectin expression increases during in vitro cellular senescence: correlation with increased cell area. Exp. Cell Res. 1991;195:13–19. doi: 10.1016/0014-4827(91)90494-f. [DOI] [PubMed] [Google Scholar]
- 35.Gorbunova V., Seluanov A., Pereira-Smith O.M. Expression of human telomerase (hTERT) does not prevent stress-induced senescence in normal human fibroblasts but protects the cells from stress-induced apoptosis and necrosis. J. Biol. Chem. 2002;277:38540–38549. doi: 10.1074/jbc.M202671200. [DOI] [PubMed] [Google Scholar]
- 36.Nakanishi K., Yang Y.G., Pierce A.J., Taniguchi T., Digweed M., D'Andrea A.D., Wang Z.Q., Jasin M. Human Fanconi anemia monoubiquitination pathway promotes homologous DNA repair. Proc. Natl. Acad. Sci. U.S.A. 2005;102:1110–1115. doi: 10.1073/pnas.0407796102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xia B., Sheng Q., Nakanishi K., Ohashi A., Wu J., Christ N., Liu X., Jasin M., Couch F.J., Livingston D.M. Control of BRCA2 cellular and clinical functions by a nuclear partner, PALB2. Mol. Cell. 2006;22:719–729. doi: 10.1016/j.molcel.2006.05.022. [DOI] [PubMed] [Google Scholar]
- 38.Nakanishi K., Cavallo F., Perrouault L., Giovannangeli C., Moynahan M.E., Barchi M., Brunet E., Jasin M. Homology-directed Fanconi anemia pathway cross-link repair is dependent on DNA replication. Nat. Struct. Mol. Biol. 2011;18:500–503. doi: 10.1038/nsmb.2029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zafar F., Seidler S.B., Kronenberg A., Schild D., Wiese C. Homologous recombination contributes to the repair of DNA double-strand breaks induced by high-energy iron ions. Radiat. Res. 2010;173:27–39. doi: 10.1667/RR1910.1. [DOI] [PubMed] [Google Scholar]
- 40.Richardson C., Moynahan M.E., Jasin M. Double-strand break repair by interchromosomal recombination: suppression of chromosomal translocations. Genes Dev. 1998;12:3831–3842. doi: 10.1101/gad.12.24.3831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wiese C., Pierce A.J., Gauny S.S., Jasin M., Kronenberg A. Gene conversion is strongly induced in human cells by double-strand breaks and is modulated by the expression of BCL-x(L) Cancer Res. 2002;62:1279–1283. [PubMed] [Google Scholar]
- 42.Wiese C., Hinz J.M., Tebbs R.S., Nham P.B., Urbin S.S., Collins D.W., Thompson L.H., Schild D. Disparate requirements for the Walker A and B ATPase motifs of human RAD51D in homologous recombination. Nucleic Acids Res. 2006;34:2833–2843. doi: 10.1093/nar/gkl366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dray E., Etchin J., Wiese C., Saro D., Williams G.J., Hammel M., Yu X., Galkin V.E., Liu D., Tsai M.S., et al. Enhancement of RAD51 recombinase activity by the tumor suppressor PALB2. Nat. Struct. Mol. Biol. 2010;17:1255–1259. doi: 10.1038/nsmb.1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ostvold A.C., Norum J.H., Mathiesen S., Wanvik B., Sefland I., Grundt K. Molecular cloning of a mammalian nuclear phosphoprotein NUCKS, which serves as a substrate for Cdk1 in vivo. Eur. J. Biochem. 2001;268:2430–2440. doi: 10.1046/j.1432-1327.2001.02120.x. [DOI] [PubMed] [Google Scholar]
- 45.Parvin B., Yang Q., Han J., Chang H., Rydberg B., Barcellos-Hoff M.H. Iterative voting for inference of structural saliency and characterization of subcellular events. IEEE Trans. Image Process. 2007;16:615–623. doi: 10.1109/tip.2007.891154. [DOI] [PubMed] [Google Scholar]
- 46.Raman S., Maxwell C.A., Barcellos-Hoff M.H., Parvin B. Geometric approach to segmentation and protein localization in cell culture assays. J. Microsc. 2007;225:22–30. doi: 10.1111/j.1365-2818.2007.01712.x. [DOI] [PubMed] [Google Scholar]
- 47.Henry-Mowatt J., Jackson D., Masson J.Y., Johnson P.A., Clements P.M., Benson F.E., Thompson L.H., Takeda S., West S.C., Caldecott K.W. XRCC3 and Rad51 modulate replication fork progression on damaged vertebrate chromosomes. Mol. Cell. 2003;11:1109–1117. doi: 10.1016/s1097-2765(03)00132-1. [DOI] [PubMed] [Google Scholar]
- 48.Jackson D.A., Pombo A. Replicon clusters are stable units of chromosome structure: evidence that nuclear organization contributes to the efficient activation and propagation of S phase in human cells. J. Cell Biol. 1998;140:1285–1295. doi: 10.1083/jcb.140.6.1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sigurdsson S., Trujillo K., Song B., Stratton S., Sung P. Basis for avid homologous DNA strand exchange by human Rad51 and RPA. J. Biol. Chem. 2001;276:8798–8806. doi: 10.1074/jbc.M010011200. [DOI] [PubMed] [Google Scholar]
- 50.Sharma N., Nyborg J.K. The coactivators CBP/p300 and the histone chaperone NAP1 promote transcription-independent nucleosome eviction at the HTLV-1 promoter. Proc. Natl. Acad. Sci. U.S.A. 2008;105:7959–7963. doi: 10.1073/pnas.0800534105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Soliman N.A., Zineldeen D.H., El-Khadrawy O.H. Effect of NUCKS-1 overexpression on cytokine profiling in obese women with breast cancer. Asian Pac. J. Cancer Prev. 2014;15:837–845. doi: 10.7314/apjcp.2014.15.2.837. [DOI] [PubMed] [Google Scholar]
- 52.Kovalenko O.V., Wiese C., Schild D. RAD51AP2, a novel vertebrate- and meiotic-specific protein, shares a conserved RAD51-interacting C-terminal domain with RAD51AP1/PIR51. Nucleic Acids Res. 2006;34:5081–5092. doi: 10.1093/nar/gkl665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tucker J.D., Jones N.J., Allen N.A., Minkler J.L., Thompson L.H., Carrano A.V. Cytogenetic characterization of the ionizing radiation-sensitive Chinese hamster mutant irs1. Mutat. Res. 1991;254:143–152. doi: 10.1016/0921-8777(91)90005-a. [DOI] [PubMed] [Google Scholar]
- 54.Bakkenist C.J., Kastan M.B. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature. 2003;421:499–506. doi: 10.1038/nature01368. [DOI] [PubMed] [Google Scholar]
- 55.Krajewski W.A. Effect of in vivo histone hyperacetylation on the state of chromatin fibers. J. Biomol. Struct. Dyn. 1999;16:1097–1106. doi: 10.1080/07391102.1999.10508318. [DOI] [PubMed] [Google Scholar]
- 56.Nakamura K., Kato A., Kobayashi J., Yanagihara H., Sakamoto S., Oliveira D.V., Shimada M., Tauchi H., Suzuki H., Tashiro S., et al. Regulation of homologous recombination by RNF20-dependent H2B ubiquitination. Mol. Cell. 2011;41:515–528. doi: 10.1016/j.molcel.2011.02.002. [DOI] [PubMed] [Google Scholar]
- 57.Wilson P.F., Hinz J.M., Urbin S.S., Nham P.B., Thompson L.H. Influence of homologous recombinational repair on cell survival and chromosomal aberration induction during the cell cycle in gamma-irradiated CHO cells. DNA Repair. 2010;9:737–744. doi: 10.1016/j.dnarep.2010.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bae J.B., Mukhopadhyay S.S., Liu L., Zhang N., Tan J., Akhter S., Liu X., Shen X., Li L., Legerski R.J. Snm1B/Apollo mediates replication fork collapse and S Phase checkpoint activation in response to DNA interstrand cross-links. Oncogene. 2008;27:5045–5056. doi: 10.1038/onc.2008.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kim T.M., Son M.Y., Dodds S., Hu L., Hasty P. Deletion of BRCA2 exon 27 causes defects in response to both stalled and collapsed replication forks. Mutat. Res. 2014;766–767:66–72. doi: 10.1016/j.mrfmmm.2014.06.003. [DOI] [PubMed] [Google Scholar]
- 60.Helleday T. Pathways for mitotic homologous recombination in mammalian cells. Mutat. Res. 2003;532:103–115. doi: 10.1016/j.mrfmmm.2003.08.013. [DOI] [PubMed] [Google Scholar]
- 61.Eppink B., Wyman C., Kanaar R. Multiple interlinked mechanisms to circumvent DNA replication roadblocks. Exp. Cell Res. 2006;312:2660–2665. doi: 10.1016/j.yexcr.2006.06.011. [DOI] [PubMed] [Google Scholar]
- 62.Wilhelm T., Magdalou I., Barascu A., Techer H., Debatisse M., Lopez B.S. Spontaneous slow replication fork progression elicits mitosis alterations in homologous recombination-deficient mammalian cells. Proc. Natl. Acad. Sci. U.S.A. 2014;111:763–768. doi: 10.1073/pnas.1311520111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Seiler J.A., Conti C., Syed A., Aladjem M.I., Pommier Y. The intra-S-phase checkpoint affects both DNA replication initiation and elongation: single-cell and -DNA fiber analyses. Mol. Cell. Biol. 2007;27:5806–5818. doi: 10.1128/MCB.02278-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kovalenko O.V., Golub E.I., Bray-Ward P., Ward D.C., Radding C.M. A novel nucleic acid-binding protein that interacts with human rad51 recombinase. Nucleic Acids Res. 1997;25:4946–4953. doi: 10.1093/nar/25.24.4946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Naylor T.L., Greshock J., Wang Y., Colligon T., Yu Q.C., Clemmer V., Zaks T.Z., Weber B.L. High resolution genomic analysis of sporadic breast cancer using array-based comparative genomic hybridization. Breast Cancer Res. 2005;7:R1186–R1198. doi: 10.1186/bcr1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Symonowicz K., Dus-Szachniewicz K., Wozniak M., Murawski M., Kolodziej P., Osiecka B., Jurczyszyn K., Ziolkowski P. Immunohistochemical study of nuclear ubiquitous casein and cyclin-dependent kinase substrate 1 in invasive breast carcinoma of no special type. Exp. Ther. Med. 2014;8:1039–1046. doi: 10.3892/etm.2014.1847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Corson T.W., Huang A., Tsao M.S., Gallie B.L. KIF14 is a candidate oncogene in the 1q minimal region of genomic gain in multiple cancers. Oncogene. 2005;24:4741–4753. doi: 10.1038/sj.onc.1208641. [DOI] [PubMed] [Google Scholar]
- 68.Gu L., Xia B., Zhong L., Ma Y., Liu L., Yang L., Lou G. NUCKS1 overexpression is a novel biomarker for recurrence-free survival in cervical squamous cell carcinoma. Tumour Biol. 2014;35:7831–7836. doi: 10.1007/s13277-014-2035-5. [DOI] [PubMed] [Google Scholar]
- 69.Li Y., Pan J., Li J.L., Lee J.H., Tunkey C., Saraf K., Garbe J.C., Whitley M.Z., Jelinsky S.A., Stampfer M.R., et al. Transcriptional changes associated with breast cancer occur as normal human mammary epithelial cells overcome senescence barriers and become immortalized. Mol. Cancer. 2007;6:7. doi: 10.1186/1476-4598-6-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Buisson R., Dion-Cote A.M., Coulombe Y., Launay H., Cai H., Stasiak A.Z., Stasiak A., Xia B., Masson J.Y. Cooperation of breast cancer proteins PALB2 and piccolo BRCA2 in stimulating homologous recombination. Nat. Struct. Mol. Biol. 2010;17:1247–1254. doi: 10.1038/nsmb.1915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Buisson R., Niraj J., Pauty J., Maity R., Zhao W., Coulombe Y., Sung P., Masson J.Y. Breast cancer proteins PALB2 and BRCA2 stimulate polymerase eta in recombination-associated DNA synthesis at blocked replication forks. Cell Rep. 2014;6:553–564. doi: 10.1016/j.celrep.2014.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Allen C., Ashley A.K., Hromas R., Nickoloff J.A. More forks on the road to replication stress recovery. J. Mol. Cell Biol. 2011;3:4–12. doi: 10.1093/jmcb/mjq049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ziolkowski P., Gamian E., Osiecka B., Zougman A., Wisniewski J.R. Immunohistochemical and proteomic evaluation of nuclear ubiquitous casein and cyclin-dependent kinases substrate in invasive ductal carcinoma of the breast. J. Biomed. Biotechnol. 2009:919645. doi: 10.1155/2009/919645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Petermann E., Helleday T. Pathways of mammalian replication fork restart. Nat. Rev. Mol. Cell Biol. 2010;11:683–687. doi: 10.1038/nrm2974. [DOI] [PubMed] [Google Scholar]
- 75.Ge X.Q., Jackson D.A., Blow J.J. Dormant origins licensed by excess Mcm2–7 are required for human cells to survive replicative stress. Genes Dev. 2007;21:3331–3341. doi: 10.1101/gad.457807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Anglana M., Apiou F., Bensimon A., Debatisse M. Dynamics of DNA replication in mammalian somatic cells: nucleotide pool modulates origin choice and interorigin spacing. Cell. 2003;114:385–394. doi: 10.1016/s0092-8674(03)00569-5. [DOI] [PubMed] [Google Scholar]
- 77.Daboussi F., Courbet S., Benhamou S., Kannouche P., Zdzienicka M.Z., Debatisse M., Lopez B.S. A homologous recombination defect affects replication-fork progression in mammalian cells. J. Cell Sci. 2008;121:162–166. doi: 10.1242/jcs.010330. [DOI] [PubMed] [Google Scholar]
- 78.Parplys A.C., Petermann E., Petersen C., Dikomey E., Borgmann K. DNA damage by X-rays and their impact on replication processes. Radiother. Oncol. 2012;102:466–471. doi: 10.1016/j.radonc.2012.01.005. [DOI] [PubMed] [Google Scholar]
- 79.Zellweger R., Dalcher D., Mutreja K., Berti M., Schmid J.A., Herrador R., Vindigni A., Lopes M. Rad51-mediated replication fork reversal is a global response to genotoxic treatments in human cells. J. Cell Biol. 2015;208:563–579. doi: 10.1083/jcb.201406099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ray Chaudhuri A., Hashimoto Y., Herrador R., Neelsen K.J., Fachinetti D., Bermejo R., Cocito A., Costanzo V., Lopes M. Topoisomerase I poisoning results in PARP-mediated replication fork reversal. Nat. Struct. Mol. Biol. 2012;19:417–423. doi: 10.1038/nsmb.2258. [DOI] [PubMed] [Google Scholar]
- 81.Murphy A.K., Fitzgerald M., Ro T., Kim J.H., Rabinowitsch A.I., Chowdhury D., Schildkraut C.L., Borowiec J.A. Phosphorylated RPA recruits PALB2 to stalled DNA replication forks to facilitate fork recovery. J. Cell Biol. 2014;206:493–507. doi: 10.1083/jcb.201404111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Vassin V.M., Anantha R.W., Sokolova E., Kanner S., Borowiec J.A. Human RPA phosphorylation by ATR stimulates DNA synthesis and prevents ssDNA accumulation during DNA-replication stress. J. Cell Sci. 2009;122:4070–4080. doi: 10.1242/jcs.053702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Liaw H., Lee D., Myung K. DNA-PK-dependent RPA2 hyperphosphorylation facilitates DNA repair and suppresses sister chromatid exchange. PLoS One. 2011;6:e21424. doi: 10.1371/journal.pone.0021424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Adamson B., Smogorzewska A., Sigoillot F.D., King R.W., Elledge S.J. A genome-wide homologous recombination screen identifies the RNA-binding protein RBMX as a component of the DNA-damage response. Nat. Cell Biol. 2012;14:318–328. doi: 10.1038/ncb2426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Villemure J.F., Abaji C., Cousineau I., Belmaaza A. MSH2-deficient human cells exhibit a defect in the accurate termination of homology-directed repair of DNA double-strand breaks. Cancer Res. 2003;63:3334–3339. [PubMed] [Google Scholar]
- 86.Rosen E.M., Fan S., Ma Y. BRCA1 regulation of transcription. Cancer Lett. 2006;236:175–185. doi: 10.1016/j.canlet.2005.04.037. [DOI] [PubMed] [Google Scholar]
- 87.Gardini A., Baillat D., Cesaroni M., Shiekhattar R. Genome-wide analysis reveals a role for BRCA1 and PALB2 in transcriptional co-activation. EMBO J. 2014;33:890–905. doi: 10.1002/embj.201385567. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.