


Abstract
Activation of NF-κB-dependent transcription represents an important hallmark of inflammation. While the acute inflammatory response is per se beneficial, it can become deleterious if its spatial and temporal profile is not tightly controlled. Classically, NF-κB activity is limited by cytoplasmic retention of the NF-κB dimer through binding to inhibitory IκB proteins. However, increasing evidence suggests that NF-κB activity can also be efficiently contained by direct ubiquitination of NF-κB subunits. Here, we identify the HECT-domain ubiquitin ligase HERC3 as novel negative regulator of NF-κB activity. We find that HERC3 restricts NF-κB nuclear import and DNA binding without affecting IκBα degradation. Instead HERC3 indirectly binds to the NF-κB RelA subunit after liberation from IκBα inhibitor leading to its ubiquitination and protein destabilization. Remarkably, the regulation of RelA activity by HERC3 is independent of its inherent ubiquitin ligase activity. Rather, we show that HERC3 and RelA are part of a multi-protein complex containing the proteasome as well as the ubiquitin-like protein ubiquilin-1 (UBQLN1). We present evidence that HERC3 and UBQLN1 provide a link between NF-κB RelA and the 26S proteasome, thereby facilitating RelA protein degradation. Our findings establish HERC3 as novel candidate regulating the inflammatory response initiated by NF-κB.
INTRODUCTION
Activation of the NF-κB transcription factor is critically involved in executing inflammatory and immune reactions. On a molecular level, under resting conditions, NF-κB proteins are rendered inactive by association with IκBα and IκBα-related inhibitor proteins, which retains the transcription factor in the cytoplasm (1). Upon stimulation, proteolysis of IκBα is induced (2), which allows NF-κB to enter the nucleus, where it initiates the transcription of different classes of genes, such as growth factors, pro- and anti-inflammatory cytokines and adhesion molecules (3). As important as its activation is the down-regulation of NF-κB signaling once the inflammation or immune challenge is overcome. If the shutdown mechanism fails, maintenance of tissue homeostasis is jeopardized and remaining NF-κB activity can drive cancer and inflammatory pathology (4). Termination of the NF-κB response is mainly achieved by re-association with its inhibitor IκBα, whose synthesis itself is dependent on NF-κB (5). Newly synthesized IκBα enters the nucleus, where it binds to NF-κB, which leads to removal from its cognate DNA binding sites (6). Although IκBα undoubtedly functions as main repressor of NF-κB, more recent findings indicate that NF-κB-dependent transcription can also be efficiently restricted through alternative mechanisms involving degradative and non-degradative deactivation of NF-κB subunits by ubiquitin targeting (7–12). Several ubiquitin ligases that target the NF-κB dimer have been identified. SOCS-1, part of a multi-subunit RING ubiquitin ligase, acts in concert with COMMD1 and GCN5 to promote poly-ubiquitination and degradation of RelA, RelB and p52 (13,14). The nuclear ubiquitin ligase PDLIM2, which contains a LIM domain structurally similar to RING domains, was found to terminate NF-κB activity in myeloid cells through poly-ubiquitination and degradation of RelA (12). More recently, the peroxisome proliferator activated receptor gamma (PPARγ) (8) and the tumor suppressor protein ING4 (15) were added to the growing list of NF-κB-targeting ubiquitin ligases.
HERC3 represents a HECT domain ubiquitin ligase belonging to the class of human homologous to E6AP carboxyl-terminus (HECT) and regulator of chromosome condensation (RCC)-1 containing subfamily (16). Apart from reports that HERC3 can form thioester bonds with ubiquitin, a prerequisite for functional HECT domain ubiquitin ligases (17,18) and that it itself undergoes ubiquitination and proteasomal degradation (17), it remains largely unstudied. Recently we found that HERC3 interacts with the ubiquitin-like proteins hPLIC1/UBQLN1 and hPLIC2/UBQLN2 (19), however a functional connection remains to be established.
Here, we show that HERC3 negatively regulates NF-κB signaling by enhancing RelA subunit degradation. While its ubiquitin ligase activity is dispensable for this function, we find that HERC3 together with UBQLN1 facilitates RelA degradation by serving as bridge to the 26S proteasome.
MATERIALS AND METHODS
Antibodies
The following primary antibodies were used in this study: β-actin (AC-15; Sigma-Aldrich) c-myc (9E10), flag (M2; Agilent Technologies), hemagglutinin (HA; 12CA5; Roche Applied Science), HERC3 (HPA039170; Sigma-Aldrich), IκBα (6A920, Imgenex), myc (71D10; Cell Signaling), PSMA4 (MCP34; Enzo Life Sciences), PSMC2 (MSS1–104; Enzo Life Sciences), PSMD4 (D17E4; Cell Signaling), RelA (C-20 and F-6; Santa Cruz Biotechnology), UBQLN1 (D3T7F; Cell Signaling), mono- and polyubiquitin (ubi-1; Life Technologies), lysine-48 ubiquitin (Apu2; EMD Millipore) and lysine-63 ubiquitin (D7A11; Cell Signaling).
Cell culture and reagents
Human embryonic kidney (HEK) 293T cells, obtained from the American Type Culture Collection (ATCC), and RelA−/− 3T3 fibroblasts, kindly provided by Dr Amer A. Beg, were maintained in DMEM (MediaTech) containing 10% (v/v) fetal bovine serum (FBS; Atlanta Biologicals). Bovine aortic endothelial cells (BAEC) and human umbilical vein endothelial cells (HUVEC) were purchased from VecTechnologies and cultured in DMEM supplemented with 10% (v/v) FBS or MCDB131 medium (VecTechnologies), respectively. All cells were cultured in a humidified atmosphere containing 5% CO2. Plasmid transfections into HEK293T and BAEC were unless otherwise stated achieved with Lipofectamine 2000 (Life Technologies) at about 85 and 65%, respectively, as observed by green fluorescent protein co-expression. Where indicated, cells were treated with MG132 (50 μM; EMD Millipore), leptomycin B (LMB; 20 ng/ml; Enzo Life Sciences) or human recombinant tumor necrosis factor (TNF; 10 ng/ml; Life Technologies).
Plasmid constructs
pCMV-myc-HERC3, pcDNA3-flag-HERC3, pcDNA3-HA-IκBα, pcDNA3 expressing myc-tagged RelA, as well as pcDNA3-his-ubiquitin have been described (7,19,20). pCMV-myc-HERC3 CA was obtained by mutation of the active site cysteine residue at position 1018 to alanine. To construct pCMV-myc-HERC3 ΔHECT (aa 1–845) and RLD (aa 1–367), pCMV-myc-HERC3 full length was cut with SmaI-NotI and BglII-NotI, respectively, and overhang ends of the residual vector backbone were filled with T4 DNA polymerase before re-ligation. The un-blunted BglII-NotI fragment was in parallel cloned into pCMV-myc (Clontech) to retrieve pCMV-myc-HERC3 ΔRLD (aa 368–1050). pCMV-flag-HERC3 and pcDNA3-flag-RelA were obtained by exchanging the myc cassette with a flag cassette through pre-annealed primers harboring the classic flag amino acid (aa) sequence DYKDDDDK. pcDNA3-myc RelA lysine to arginine (K195R, K315R and K195/315R) mutants were generated by site-directed mutagenesis (Agilent Technologies) according to manufacturer's instructions. The NF-κB luciferase reporter gene constructs 3x κB-, IL8-, ICAM1-, IκBα- and MHC-I-luc were described elsewhere (20–22). pCMV2-flag-IKK2 K44M (23) (Addgene plasmid 11104), flag-UBQLN1 (Addgene plasmid 8663) (24), pHM830 (GFP-LacZ) and pHM840 (GFP-SV40 NLS-LacZ) (25) (Addgene plasmids 20702 and 20701) were provided by Drs Anjana Rao, Peter M. Howley and Thomas Stamminger, respectively. All plasmid constructs were verified by automated DNA sequencing.
Co-immunoprecipitation assay in cells
Cells were collected and lysed in RIPA buffer (50 mM Tris-HCl pH8, 150 mM NaCl, 1 mM EDTA pH8, 1% (v/v) Igepal CA-630, 0.5% (v/v) sodium deoxycholate, 0.1% (v/v) SDS, 20 mM N-ethylmaleimide, protease inhibitors). Whole cell extracts were cleared by centrifugation, 1/20 was kept for expression control, and rest of supernatants was incubated with respective antibodies coupled to protein A-sepharose or agarose matrices. Immunoprecipitates were washed thrice with lysis buffer, and eluted from beads by addition of SDS-sample buffer and boiling for 10 min. Proteins were resolved on SDS-PAGE and detected by Western Blotting with respective antibodies.
In vitro translation and co-immunoprecipitation assay
Plasmids containing myc-RelA, flag-HERC3 and HA-IκBα were linearized to enable efficient transcription/translation via T7 RNA polymerase. Coupled in vitro transcription and translation was carried out with the TNT T7 Coupled Wheat Germ Extract System (Promega) according to manufacturer's instructions. Flag-UBQLN1 was transcribed/translated from a circular plasmid with SP6 RNA polymerase (New England Biolabs). For immunoprecipitation, reactions were mixed after translation was finished and 1/10 was removed for input control. RIPA buffer was added and IκBα and HERC3 were pulled down with HA and flag agarose beads, respectively. Precipitates were washed 3 times with RIPA buffer, eluted from beads by boiling in SDS-sample buffer, resolved on SDS-PAGE and detected by Western Blotting with appropriate antibodies.
Ubiquitination assay under native conditions
HEK293T cells transfected with his-ubiquitin and myc-RelA with or without myc-HERC3 were harvested in RIPA buffer. Presence of ubiquitinated RelA was determined after precipitation of total RelA with RelA-coupled protein A-sepharose, and Western Blotting with respective anti-ubiquitin antibodies.
Denaturating ubiquitination assay
HEK293T or BAEC were transfected with histidine-tagged ubiquitin as well as expression constructs for RelA and HERC3. Histidine–ubiquitin was precipitated under denaturating conditions (6 M guanidine hydrochloride) via a nickel-NTA-coupled agarose resin (Qiagen) from either whole cell extracts or subcellular fractions as previously described (19). Ubiquitinated RelA was detected after pull down by Western Blotting with RelA-specific antibody.
RelA stability in HUVEC
HUVEC were transfected with siRNAs targeting human Herc3 and Ubqln1, or a non-targeting control. Three days post-transfection, cells were either left untreated or stimulated with TNF for 0.5 and 6 h, after which they were harvested in RIPA buffer. Protein levels of RelA, HERC3 and UBQLN1 were assessed by Western Blotting with respective antibodies. β-actin served as a loading control.
Metabolic pulse chase labeling
HUVEC, transfected HEK293T or BAEC cells were washed twice and then incubated for 30 min in labeling medium (methionine/cysteine-free DMEM supplemented with 2 mM L-glutamine and 0.5% (v/v) dialyzed FBS). Cells were subsequently pulsed with 400 μCi/ml [35S]-methionine (Perkin Elmer) for 1 h (in case of HUVEC in presence of TNF), washed twice with labeling medium and further incubated with complete DMEM + 10% (v/v) FBS and 2 mM L-methionine/2 mM L-cysteine for indicated times. The proteasome inhibitor MG132 (50 μM) was added for chase time span where indicated. Upon harvesting, cells were washed in ice-cold PBS and RelA was pulled down by immunoprecipitation from total cell lysates (50 mM Tris-HCl pH8, 150 mM NaCl, 1 mM EDTA pH8, 1% (v/v) Igepal CA-630, 0.5% (v/v) sodium deoxycholate, 0.1% (v/v) SDS, protease inhibitors). Precipitates were resolved by SDS-PAGE, gels were dried and exposed to a phosphor screen (GE Healthcare Life Sciences) over night prior to analysis on a Phosphoimager (Typhoon Trio, GE Healthcare Life Sciences).
Identification of ubiquitinated residues by nanoLC-ESI-MS/MS analysis
HEK293T cells were transfected with histidine-tagged ubiquitin, myc-RelA and myc-HERC3. Cell lysates were subjected to Ni-NTA pull-down, SDS-PAGE and SYPRO Ruby (Molecular Probes) stain. Gel bands in the range of 80–150 kDa were excised, treated with trypsin and resulting peptides were extracted as reported previously (26). Ubiquitin-conjugated RelA residues were determined by nanoLC-ESI-MS/MS analysis as described in detail elsewhere (7).
Identification of HERC3–RelA interaction partners by nanoLC-MS/MS
Flag-HERC3 in presence of myc-RelA was precipitated from transfected HEK293T cells in RIPA buffer (50 mM Tris-HCl pH8, 150 mM NaCl, 1 mM EDTA pH8, 1% (v/v) Igepal CA-630, 0.5% (v/v) sodium deoxycholate, 0.1% (v/v) SDS, protease inhibitors) with flag-specific antibody conjugated to agarose matrix. Precipitation with IgG isotype control was used to identify non-specific hits. The precipitate was eluted from the matrix by competition with 3xflag peptide (Sigma) and samples were filtered through 0.45 μm filter units (Spin-X; Corning) to remove residual beads. Flag peptides were removed by Zeba spin desalting columns with 7K MWCO (Thermo Fisher Scientific) following vendor's recommended protocol. Flag peptide-free samples were collected for subsequent in-solution digestion. Samples were reduced with 10 mM TCEP at 56°C for 45 min, alkylated with 10 mM iodoacetamide for 1 h at room temperature in the dark, and precipitated with a 6-fold volume of cold acetone overnight at −20°C. After washing the pellet twice with cold acetone, samples were reconstituted in 45 μl 100 mM TEAB pH8.5 and digested by incubating with 300 ng trypsin (Promega) at 37°C for 16 h. Digests were then desalted using solid phase extraction (SPE) on Sep-Pak Cartridges (Waters) and the eluted tryptic peptides were evaporated to dryness before analysis. Protein identifications were conducted by nanoLC-MS/MS analysis as described previously (7). All MS and MS/MS raw spectra files were converted to MGF files by Proteome Discoverer 1.4 (Thermo Fisher Scientific) for subsequent database search using in-house license Mascot Daemon (version 2.3, Matrix Science) against Human RefSeq database downloaded from NCBInr. The database search was performed with two-missed cleavage sites by trypsin allowed. The peptide tolerance was set to 10 ppm and MS/MS tolerance was set to 0.8 Da. A fixed carbamidomethyl modification of cysteine, variable modifications on methionine oxidation and deamidation of asparagine/glutamine were set. Only significant scores for the peptides defined by Mascot probability at 99% CI greater than ‘identity’ and peptide expectation value less than 0.05 were considered for the peptide identification. The final protein list contains only proteins, of which at least two distinct peptides were identified meeting the above criteria.
Luciferase reporter gene assays
For RelA-induced reporter activity, RelA−/− 3T3 or BAEC were grown in 12-well plates, and unless otherwise stated in the figure legend, exposed to 400 ng of DNA (200 ng of reporter plasmid, 20 ng of p65 wt or mutant expression plasmid, 140 ng of control or HERC3 vectors, and 40 ng of cytomegalovirus/β-galactosidase (CMV-β-gal) plasmid) and 1.6 μl of Lipofectamine (Life Technologies) in DMEM without serum for 6 h. Medium was exchanged with complete DMEM + 10% (v/v) FBS, and cells were allowed to recover for 40 h. For TNF-induced activity, BAEC were transfected in 12-well plates with 600 ng DNA (300 ng reporter plasmid, 40 ng CMV-β-gal plasmid and 260 ng of control or HERC3 vectors) and 1.8 μl Lipofectamine in DMEM no serum for 5 h. Medium was replaced with complete DMEM + 10% (v/v) FBS. After 24 h, TNF was added for 16 h. Cells were lysed in 1x passive lysis buffer (Promega) and supernatants were assayed for luciferase and β-galactosidase activity as described (22). For detection of AP-1 activity, cells were transfected with 200 ng AP-1-Luc reporter plasmid together with 20 ng pFC-MEKK (both from Agilent Technologies), 140 ng HERC3 vector and 40 ng of respiratory syncytial virus/β-galactosidase (RSV-β-gal) plasmid.
Small interference RNA (siRNA) knock down
HUVEC were seeded on 0.2% gelatin-coated plates and grown overnight in antibiotic-free medium. Introduction of siRNA into HUVEC was performed with Oligofectamine (Life Technologies) according to manufacturer's instructions. Cells were assayed 72 h after transfection. The siRNA sequence for knock down of human HERC3 was 5′- GGA GUG AUU GAA CAG AAG AUU-3′. Non-targeting control and UBQLN siRNA was purchased from Dharmacon (siGENOME control siRNA #1 and SMARTpool ON-TARGET plus human UBQLN1 siRNA, respectively).
Immunofluorescence
For determination of nuclear RelA presence, BAEC cells were seeded on glass-cover-slips and transfected with control vector or myc-HERC3. For assessment of GFP-LacZ localization, either GFP-LacZ or GFP-NLS-LacZ was co-transfected with control vector or myc-HERC3. Cells were either left untreated or stimulated with TNF/LMB for 30 min and 1 h, followed by fixation with 4% paraformaldehyde and permeabilization with 0.5% (v/v) Triton X100 in 1xPBS. RelA and HERC3 were visualized by staining with mouse monoclonal RelA and rabbit polyclonal myc primary, followed by Alexa568 anti-mouse IgG and Alexa488 anti-rabbit IgG secondary antibodies (all Molecular Probes), respectively. GFP was detected by direct green fluorescence. 4′,6-diamidino-2-phenylindole (DAPI) was used as nuclear reference. Subcellular distribution of RelA and HERC3 was analyzed using a Nikon Eclipse TE2000 microscope equipped with a 40x objective. Pictures were acquired with a charge-coupled device using identical acquisition parameters.
Gene expression analysis
RNA was isolated from control-, RelA- and RelA/HERC3-transfected BAEC or from control, HERC3 and UBQLN1 siRNA-transfected, untreated or TNF-treated HUVEC with the RNeasy Plus Kit (Qiagen). Real-time quantitative PCR (qPCR) was carried out as described previously (20). For BAEC, expression levels were calculated relative to control-transfected cells, which were set to 1. For HUVEC, expression levels in un-stimulated, control and HERC3/UBQLN1 siRNA-treated cells were set to 1, and fold induction for TNF-treated groups was calculated. Sequences of primer pairs used for amplification of human genes were: Icam1 5′-CCA AGA GGA AGG AGC AAG ACT-3′ (forward), 5′-CCA ATA GGC AGC AAG TTT CAG-3′ (reverse); Vcam1 5′-CAT GTA GTG TCA TGG GCT GTG-3′ (forward), 5′-CTC AAA ACT CAC AGG GCT CAG-3′ (reverse); Sele 5′-TTC CAA GCA AAG GTG AAG AGA-3′ (forward), 5′-CAG CTG TCG AAA CAC TGT GAA-3′ (reverse); Hprt 5′-TTC TGT GGC CAT CTG CTT AGT-3′ (forward), 5′-GCC CAA AGG GAA CTG ATA GTC-3′ (reverse).
Electrophoretic mobility shift assay (EMSA)
Whole cell extracts were obtained from transfected BAEC by lysis in buffer containing 50 mM HEPES.NaOH pH7, 250 mM NaCl, 5 mM EDTA, 0.1% (v/v) Igepal CA-630, protease inhibitor tablets (Roche Applied Science). Five micrograms of cleared lysates were incubated for 30 min at room temperature with 60 000 cpm of double-stranded purified γ-32P ATP (Perkin Elmer)-labeled Ig κ light chain enhancer oligonucleotide (5′-AGT TGA GGG ACT TTC CCA GGC-3′). For competition, either non-labeled sense oligonucleotide (co Ig κB) or non-labeled scrambled oligonucleotide (5′-ACA GTA TCA AAG GCT CAC ATG-3′) (co sc Ig κB) was added. For the super-shift, complexes were incubated for 1 h on ice with 1 μg of mouse monoclonal p65 antibody (clone F-6; Santa Cruz Biotechnology). Protein–DNA complexes were separated on 5% Tris/glycine/EDTA poly-acrylamide gels, bands were visualized by autoradiography and band intensities were quantified.
Blue native polyacrylamide gel electrophoresis (BN-PAGE)
HEK293T cells were transfected with flag-RelA and/or myc- or flag-HERC3 and extracts were obtained through lysis with modified RIPA buffer (50 mM Tris-HCl pH8, 150 mM NaCl, 1 mM EDTA pH8, 1% (v/v) Igepal CA-630, 0.5% (v/v) sodium-deoxycholate, 0.1% (v/v) SDS, 20 mM N-ethylmaleimide, protease inhibitors). RelA and HERC3 were precipitated with myc- or flag-coupled agarose beads, and bound proteins were eluted from matrices in blue native buffer (20 mM Bis/Tris, 20 mM NaCl, 1 mM EDTA pH8, 10% (v/v) glycerol, 0.1% (v/v) Igepal CA-630, 20 mM N-ethylmaleimide, protease inhibitors) with respective peptides (3xflag peptide or myc peptide, both Sigma). Lysates were purified from residual beads by centrifugation through 0.45 μm cellulose acetate filter columns and loaded on NativePAGE Novex 3–12% Bis/Tris protein gels (Life Technologies). BN-PAGE was executed according to a previously published protocol (27). Molecular weight of proteins was indicated by NativeMark unstained protein standard (Life Technologies).
Size-exclusion chromatography (SEC)
Protein precipitates from transfected HEK293T cells were obtained as described under BN-PAGE, except that elution was performed in modified RIPA buffer. A 300 µl sample was injected onto an equilibrated Sephacryl S-300 HiLoad 16/60 column (GE Healthcare) and run was executed at a rate of 500 μl/min in 50 mM Tris-HCl pH7.5, 150 mM NaCl, 1 mM phenylmethylsulfonyl fluoride, 1 mM DTT, 0.1% (v/v) Triton X100. Forty-two x 1 ml fractions were collected after one column volume had passed. A mix of thyroglobulin, aldolase and ovalbumin with calculated molecular masses (Mr) of 6.69 × 105, 1.58 × 105 and 4.4 × 104, respectively, was used as molecular weight marker. Proteins were precipitated with 12% (v/v) TCA and 5% (v/v) sodium-deoxycholate, and all fractions were separated on 7% SDS-PAGE.
Statistical analysis
Comparisons between two groups were statistically evaluated by the Student t test. Comparisons between multiple groups were performed with 1-way ANOVA followed by Bonferroni post hoc test. Differences were considered significant at P < 0.05.
RESULTS
HERC3 negatively regulates NF-κB transcriptional activity
In addition to its canonical regulation by inhibitor IκBα, NF-κB activity is also altered by post-translational modifications at the level of the transcription factor dimer (28). Using a 3x κB luciferase reporter system we found that co-expression of the ubiquitin ligase HERC3 leads to significant repression of RelA-induced NF-κB reporter activity in a dose-dependent manner in BAEC as well as in RelA knock out fibroblasts (RelA−/− 3T3) (Figure 1A). HERC3 did not negatively influence the activity of the transcription factor AP-1 implying a specific effect on NF-κB (Supplementary Figure S2). HERC3 comparably reduced NF-κB-dependent luciferase production from different NF-κB consensus sites (Figure 1B), indicating an upstream rather than promoter site-specific effect of HERC3. HERC3 not only influenced RelA-triggered NF-κB activation, but also significantly attenuated TNF-induced reporter activity in endothelial cells (Figure 1C). Finally, we tested the impact of HERC3 on endogenous NF-κB-regulated gene expression in BAEC. Production of Icam1-, Vcam1-, Sele- and IκBα-mRNA was induced by transiently transfecting cells with RelA. Co-transfection of HERC3 decreased the RelA-dependent induction of gene expression on all genes tested (Icam1 and Vcam1 *P < 0.05; Sele p = 0.11; IκBα p = 0.06) (Figure 1D). In summary, we show here that the ubiquitin ligase HERC3 represses RelA- and TNF-stimulated NF-κB transcriptional activity.
Figure 1.
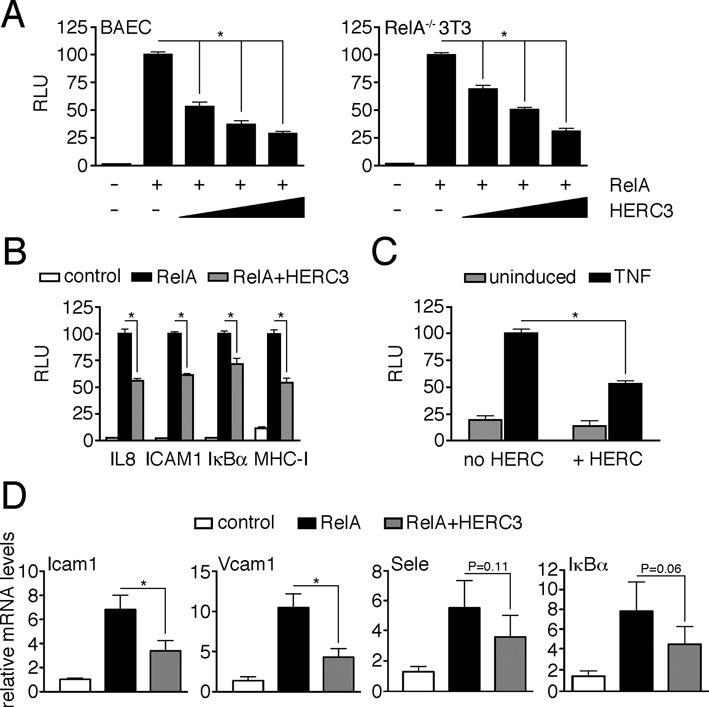
HERC3 suppresses NF-κB transcriptional activity. (A) BAEC and RelA−/− 3T3 were transfected with κB-luciferase reporter (200 ng) and CMV-β-galactosidase control (40 ng) plasmids together with RelA (20 ng) and increasing amounts of HERC3 (BAEC: 80, 160 and 320 ng; RelA−/- 3T3: 35, 70 and 140 ng). Luciferase activities were measured 3 days after transfection and normalized to β-galactosidase activities. Bars represent mean + SEM (n = 9, derived from 3 independent experiments). RelA-induced reporter activity was set to 100 and activity of all other data sets was calculated relative to this value. (B) Assay was carried out in RelA−/− 3T3, transfected as in (A) with different NF-κB decameric consensus binding sites and constant amounts of HERC3. Sites were derived from the human IL8, ICAM1, IκBα and MHC class I enhancer sequences. (C) BAEC were transfected with reporter plasmids, control vector or HERC3, and either left untreated or 2 days later stimulated with TNF for 16 h. Bars represent mean + SEM (n = 12, derived from four independent experiments). TNF-induced reporter activity without HERC3 co-expression was set to 100. (D) BAEC transiently transfected with control, RelA or RelA plus HERC3 were harvested, RNA was isolated and expression levels of endogenous Icam1, Vcam1, Sele and IκBα were evaluated by qRT-PCR. Expression levels of control-transfected cells were set to 1, and relative induction for all other experimental groups was calculated. Bars represent mean + SEM (n = 5, derived from 5 independent experiments). In all experiments appropriate protein expression was monitored by Western Blotting (see Supplementary Figure S1A–D). Data were considered significant at *P < 0.05. RLU, relative luciferase units.
HERC3 limits NF-κB nuclear presence and DNA-binding
Nuclear translocation is critical for activation of NF-κB-dependent transcription (29), hence we next observed RelA subcellular localization in absence and presence of HERC3 co-expression. Indeed, HERC3 significantly reduced RelA nuclear presence in TNF-stimulated BAEC (Figure 2A). This effect was still apparent after addition of the nuclear export inhibitor leptomycin B (LMB), suggesting that reduced nuclear RelA levels are not a result of accelerated nuclear export. Next we assessed whether HERC3 has a general impact on the nuclear import machinery or rather affects NF-κB specifically. RelA nuclear translocation is governed by a classical monopartite nuclear localization sequence (NLS) (30). Hence we tested HERC3 effects on the nuclear presence of GFP-LacZ fused to a similar prototypic monopartite NLS derived from the SV40 large T-antigen, which would be taken up into the nucleus by the same mechanism as RelA (25). However, co-transfection of HERC3 did not alter nuclear translocation of GFP-NLS-LacZ (Figure 2B), suggesting that HERC3 does not generally impair nuclear import. To verify that nuclear NF-κB was reduced, we measured RelA DNA-binding levels in BAEC transfected with RelA with or without HERC3 by electrophoretic mobility shift assay (EMSA). While RelA expression markedly induced RelA DNA-binding as compared to the control, co-expression of HERC3 attenuated this effect (Figure 2C), indicating that the reduced nuclear translocation of RelA in presence of HERC3 is indeed reflective of less DNA-bound NF-κB. The observed signal was eliminated with a non-labeled Ig κB oligonucleotide (co) and super-shifted with RelA antibody, ensuring its NF-κB specificity (Figure 2D).
Figure 2.
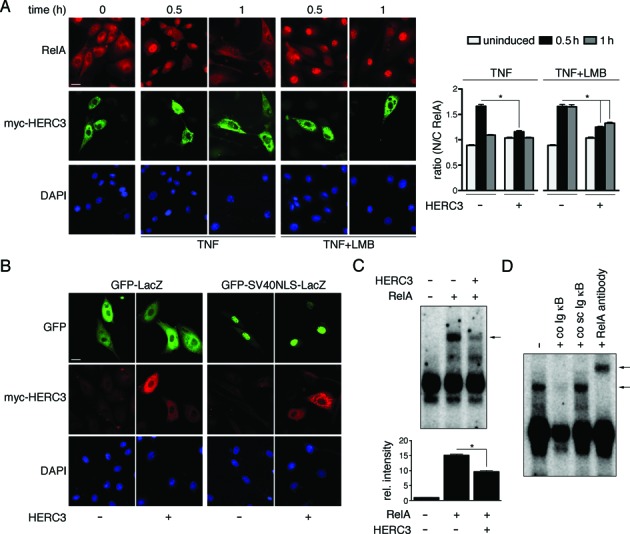
HERC3 reduces NF-κB DNA-binding by limiting its nuclear import. (A) BAEC were transfected with control vector or myc-HERC3, stimulated for 0, 0.5 or 1 h with TNF or TNF/LMB and processed for immunofluorescence. Nuclei were stained with DAPI. Bar represents 10 μm. The graph shows ratios of cytosolic and nuclear fluorescence obtained by automatic quantification of RelA compared to DAPI staining (n = 61–147 cells, derived from 2–3 experiments). Values N/C > 1 indicate predominantly nuclear RelA. (B) Subcellular localization of GFP-LacZ harboring a SV40-derived nuclear localization sequence (NLS) was tested in absence and presence of myc-HERC3 in BAEC. GFP-LacZ, not containing an active NLS, was used as control. DAPI staining served as nuclear reference. Size bar = 10 μm. Data from 18–33 cells for each condition, derived from two independent experiments, were quantified. Percent of nuclear GFP staining was: for GFP-LacZ –HERC3 0 ± 0; +HERC3 0.12 ± 0.02, and for GFP-SV40NLS-LacZ –HERC3 100 ± 0; +HERC3 100 ± 0. (C) RelA DNA-binding in absence and presence of HERC3 was observed by electrophoretic mobility shift assay (EMSA). Total cell extracts from transfected BAEC were incubated with double-stranded purified γ-32P ATP-labeled Igκ light chain enhancer oligonucleotide. Protein–DNA complexes were separated on 5% Tris/glycine/EDTA-PAGE, bands were visualized by autoradiography and results from three experiments were quantified. Equal RelA protein expression with and without HERC3 was ensured by parallel observation of protein levels by Western Blotting (see Supplementary Figure S1E). (D) RelA binding was verified by competition with either non-labeled sense oligonucleotide (co Ig κB) or non-labeled scrambled oligonucleotide (co sc Ig κB), and by super-shift with RelA antibody. Experiments were performed in triplicates. The arrows in (C) and (D) indicate the specific NF-κB band. Values were considered significant at *P < 0.05. h, hours.
HERC3 does not effect IκBα degradation
Next we set out to determine the mechanism by which HERC3 limits NF-κB nuclear appearance. Given the exclusive presence of HERC3 in the cytoplasm (Figure 2A) and (17,19), it is highly likely that HERC3 exerts its effect on RelA before it enters the nucleus. The main regulator of NF-κB subcellular localization is IκBα, which is removed in the cytoplasm upon stimulation by ubiquitin-dependent proteolysis (31,32). To test whether HERC3 is suppressing IκBα degradation to inhibit NF-κB nuclear translocation, we observed IκBα protein levels with and without HERC3 expression after TNF stimulation. As shown in Figure 3A, IκBα levels were decreased regardless of HERC3 over-expression, indicating that HERC3 does not interfere with IκBα degradation. In contrast, over-expression of dominant-negative IKK2 (23) and pre-treatment of cells with MG132 proteasome inhibitor totally blocked IκBα removal. Additionally, unlike MG132 treatment HERC3 expression did not promote accumulation of ubiquitinated IκBα (Figure 3B), which likely would be observed if IκBα degradation was inhibited. Taken together, the reduced RelA nuclear translocation in presence of HERC3 cannot be attributed to stabilization of IκBα.
Figure 3.
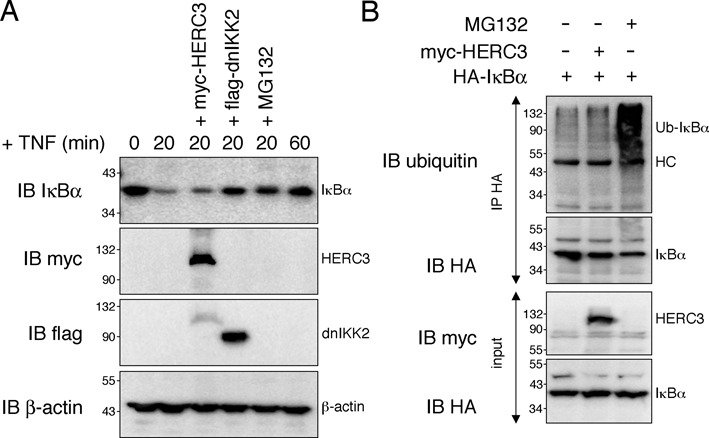
HERC3 has no effect on IκBα degradation. (A) IκBα turnover was monitored in HEK293T cells transfected with empty vector, myc-HERC3 or flag-dnIKK2 after stimulation with 10 ng/ml TNF for 0, 20 or 60 min. Where indicated cells were pre-treated with 50 μM MG132. Presence of transfected proteins was verified by immunoblotting with myc- or flag-specific antibodies. (B) HEK293T cells were transfected with HA-IκBα together with either empty vector or myc-HERC3. Cells were treated with 10 ng/ml TNF for 20 min and where indicated pre-treated for 3 h with 50 μM MG132. IκBα was pulled down from cell lysates with HA-coupled agarose and its ubiquitination was assessed by detection with an anti-ubiquitin antibody. All experiments were carried out in triplicates. HC, heavy chain; IB, immunoblot; IP, immunoprecipitation; min, minutes.
HERC3 binds to NF-κB RelA and promotes its ubiquitination
HERC3 constitutes a putative HECT-domain ubiquitin ligase (17) and therefore may directly induce RelA ubiquitination, which in turn might affect NF-κB subcellular localization and/or protein stability. We found by co-immunoprecipitation assays that HERC3 binds to RelA (Figure 4A, B), a prerequisite for promoting RelA ubiquitination. To assess whether HERC3 is able to regulate ubiquitination of RelA we co-expressed the two proteins together with histidine-tagged ubiquitin and performed an ubiquitination assay under denaturing conditions. While no ubiquitinated RelA was detected when RelA was expressed alone, HERC3 co-expression readily induced RelA ubiquitination (Figure 4C). Further, we detected a notable increase in endogenous RelA ubiquitination in presence of HERC3 in TNF-stimulated BAEC (Figure 4D). Interestingly, we find that presence of IκBα significantly reduces HERC3 binding to RelA (Supplementary Figure S3A) and as a consequence abates HERC3-mediated RelA ubiquitination (Supplementary Figure S3B), supporting the interpretation that HERC3 exerts its effect on NF-κB after its liberation from IκBα inhibitor.
Figure 4.
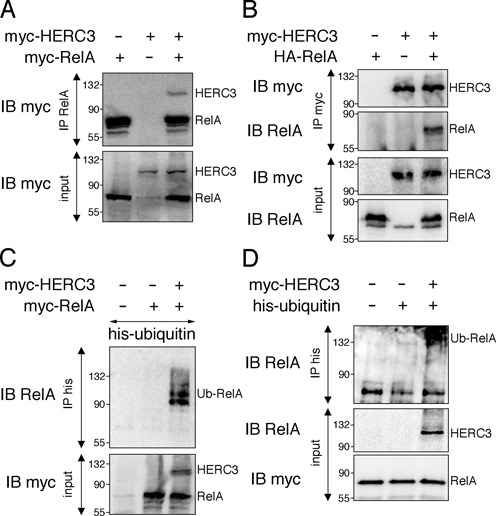
HERC3 interacts with RelA and mediates its ubiquitination. (A) RelA was precipitated with RelA antibody from total HEK293T cell lysates transfected with myc-RelA and myc-HERC3. RelA and HERC3 co-precipitation was assessed by Western Blotting with anti-myc antibody. (B) HEK293T cells were transfected with HA-RelA and myc-HERC3. HERC3 was pulled down from cell lysates with myc-specific antibody and RelA was detected in the precipitate with anti-RelA antibody. (C) HEK293T were transfected with myc-RelA, his-ubiquitin and myc-HERC3. Ubiquitinated RelA was detected by pull down of his-ubiquitin under denaturing conditions, followed by Western Blotting with RelA-specific antibody. (D) BAEC were transfected with his-ubiquitin and myc-HERC3. After 24 h cells were treated with TNF for 30 min and assay was performed as described in (C). All experiments were carried out in triplicates. IB, immunoblot; IP, immunoprecipitation.
Confirmative of RelA ubiquitination in presence of HERC3 we identified two ubiquitinated RelA lysine (K) residues, K195 and K315, with HERC3 co-expression by tandem mass spectrometry (nanoLC-ESI-MS/MS) (Supplementary Figure S4A). Interestingly the same residues among others were shown to be modified with ubiquitin before (7,33). Confirming the MS data, substitution of lysine to arginine at positions 195 and 315 markedly decreased RelA ubiquitination levels in presence of HERC3 (Supplementary Figure S4B). However, mutation of the same lysines only minimally reverted HERC3 effects on RelA transcriptional activity (Supplementary Figure S4C). Since residual ubiquitination is detected with the RelA double KR mutant (Supplementary Figure S4B), it is likely that additional RelA lysines might be targeted by HERC3. In summary, our data support HERC3 as novel protein mediating NF-κB RelA ubiquitination after liberation from IκBα at a minimum of two lysine acceptor sites.
HERC3 induces K48-linked RelA ubiquitination and protein destabilization
We have shown that HERC3 mediates the ubiquitination of RelA, which could potentially impact its protein stability. Ubiquitin has seven lysine residues (K6, K11, K27, K29, K33, K48, and K63). Any one of them can be conjugated to another ubiquitin resulting in the formation of polyubiquitin chains of different linkages. While K48 chains mainly target proteins for degradation by the 26S proteasome, K63-linked ubiquitin chains influence target proteins in a non-degradative manner (34). By using antibodies detecting exclusively K48- or K63-linked ubiquitin molecules we evaluated the type of ubiquitin chain conjugated to immunoprecipitated RelA in presence of HERC3. Staining was only observed with K48-specific antibody (Figure 5A), indicating that HERC3 might target RelA for proteasomal degradation. Therefore, we next tested whether HERC3 influences RelA protein stability. As shown in Figure 5B and C, RelA protein was significantly less stable in presence of HERC3 in HEK293T and endothelial cells, resulting in a more than 50% reduction in RelA protein half-life. This effect was partially reverted by administration of a proteasome inhibitor (Figure 5C, black bars). Taken together, our data so far suggest that HERC3 mediates RelA ubiquitination to induce its degradation before it can enter the nucleus.
Figure 5.
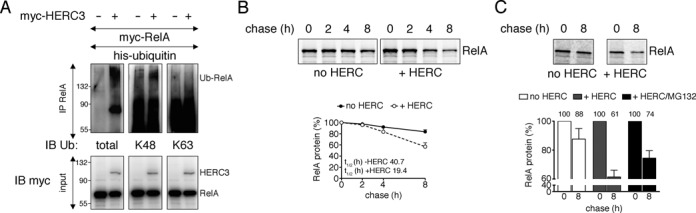
HERC3 mediates RelA K48 ubiquitination and protein destabilization. (A) His-ubiquitin, myc-RelA, with or without myc-HERC3 were transiently introduced into HEK293T cells. RelA was precipitated from whole cell lysates with RelA-specific antibody and the nature of RelA ubiquitination was examined by Western Blotting with anti-ubiquitin antibodies recognizing total, K48 or K63 ubiquitin, respectively. (B) HEK293T cells were transfected with myc-RelA with or without myc-HERC3. Twenty four hours after transfection, cells were pulse-labeled with [35S]-methionine for 1 h and chased for 0, 2, 4 and 8 h. RelA was precipitated from total cell extracts with myc-affinity agarose, and protein levels were detected by autoradiography. (C) RelA was precipitated from transfected BAEC total cell extracts after pulse-labeling with [35S]-methionine for 1 h and chasing for 0 and 8 h. Protein levels in precipitates were assessed by separation on SDS-PAGE and autoradiography. Average percent protein remaining before and after chase is indicated. All experiments were carried out at least in triplicates. h, hours; IB, immunoblot; IP, immunoprecipitation.
HERC3 effects on RelA-dependent transcription and ubiquitination are independent of its catalytic domains
Small HERC proteins contain two potential catalytic domains, the amino-terminal RCC1-like domain (RLD) and the carboxyl-terminal HECT domain. While ubiquitin transfer is generally attributed to the HECT domain, for the transfer of the ubiquitin-like molecule ISG15 by HERC5, e.g. both HECT and RLD domains are required (35). To determine the domains needed for HERC3-mediated NF-κB transcriptional repression and ubiquitination we manufactured HERC3 truncation mutants lacking parts of the RLD or HECT domains (Figure 6A). As shown in Figure 6B, RelA-dependent transcription was still efficiently repressed by HERC3 mutants carrying an inactive or truncated HECT domain (C1018A or ΔHECT) or missing RLD domain (ΔRLD). Solely a mutant lacking everything but the RLD domain (RLD) was unable to suppress NF-κB transcription. Correspondingly, ubiquitination of RelA was apparent with all tested HERC3 mutants except the RLD domain alone (Figure 6C). These data indicate that HERC3 mediates RelA ubiquitination independently of its catalytic domains. While HERC3 is needed for RelA ubiquitination, it may not actively transfer ubiquitin to NF-κB. Since HECT domain proteins need to directly bind their targets in order to transfer ubiquitin (36), we tested interaction of in vitro translated HERC3 and RelA. Supporting the idea that HERC3 is not directly ubiquitinating RelA, we found that while HERC3 and RelA interact in cells (Figure 4A, B), they do not bind to each other in vitro (Supplementary Figure S5A).
Figure 6.
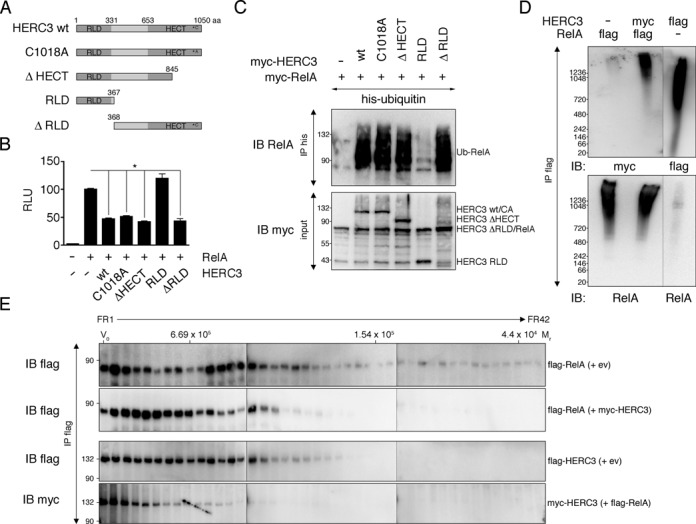
HERC3 affects RelA ubiquitination independently of its catalytic domains as part of a larger molecular weight complex. (A) Schematic representation of HERC3 constructs used in (B and C). (B) Transcriptional activity of RelA was examined in presence of different HERC3 constructs as described in Figure 1A. Bars represent mean + SEM (n = 9–18, derived from 3–6 independent experiments). Values were significant at *P < 0.05. (C) HEK293T cells were transfected with his-ubiquitin, myc-RelA and indicated myc-HERC3 wild type, mutant or truncation constructs. Ubiquitinated RelA was pulled down with Ni-agarose beads bound to his-tagged ubiquitin and detected in immunoblotting with a RelA-specific antibody. (D) HEK293T cells were transfected with flag-RelA alone, flag-RelA and myc-HERC3 or flag-HERC3 alone. Lysates were subjected to precipitation with flag-agarose beads, followed by flag-peptide elution. Eluates were loaded on 3–12% Native Bis-Tris Gels and separated by Blue Native Polyacrylamide Gel Electrophoresis (BN-PAGE). (E) HEK293T cell transfection, lysate precipitation and elution were performed as outlined in (D). Eluates were loaded on a Sephacryl S-300 HiPrep 16/60 column for size exclusion chromatography (SEC). Thyroglobulin, aldolase and ovalbumin with calculated relative molecular masses (Mr) of 669, 158 and 44, respectively, served as molecular weight indication. All experiments were carried out in triplicates. aa, amino acids; CA, cysteine to alanine; ev, empty vector; FR, fraction; IB, immunoblot; IP, immunoprecipitation; Mr, relative molecular mass; RLU, relative luciferase units; V0, column void volume; wt, wild type.
RelA and HERC3 co-sediment at high-molecular weight (MW) under native conditions
Since HERC3 is not directly involved in ubiquitination of RelA, we hypothesized that HERC3 functions as a scaffolding protein that facilitates the interaction of RelA and other proteins involved in ubiquitination or degradation processes. Beginning to assess this possibility, we immunoprecipitated HERC3 and RelA either when transfected alone or together under native conditions and examined their size by blue native polyacrylamide gel electrophoresis (BN-PAGE; Figure 6D) and size-exclusion chromatography (SEC; Figure 6E). NF-κB RelA was located in slow migrating protein bands (Figure 6D, lanes 1 and 2) and higher MW fractions (Figure 6E, first two rows) with and without HERC3, indicating that it is part of a larger protein complex regardless of HERC3 presence. Similarly, HERC3 migrated/fractionated at high MW when not bound to RelA (Figure 6D, lane 3 and Figure 6E, third row). Interestingly, co-expression led to a shift of both proteins to even higher MW fractions, suggesting a change in RelA-containing complexes with HERC3, and vice versa (Figure 6D, lane 2 and Figure 6E, second and fourth row). In summary, we find native HERC3 and RelA proteins associated with high MW fractions, indicating multimerization and/or presence of other proteins.
HERC3/RelA associate with the 26S proteasome and the ubiquitin-like protein UBQLN1
To reveal proteins bound to HERC3 and RelA we performed LC-MS/MS analysis of control and HERC3 precipitates from RelA/HERC3-transfected cells. While not present in the control pull down, we found a considerable number of proteasomal subunits precipitated with HERC3, suggesting a specific association with HERC3 and possibly with RelA (listed in Figure 7A). To confirm the interaction of HERC3/RelA with the proteasome we performed co-immunoprecipitation assays with various proteasomal subunits found by mass spectrometry. The 19S ATPase subunit PSMC2 and the 20S core subunit PSMA4 were pulled down with HERC3 (Figure 7B). We also found HERC3 co-precipitation with the 19S non-ATPase subunit PSMD4 (Figure 7C), further confirming the data obtained by LC-MS/MS. Notably, HERC3 binding to proteasomal subunits was detected regardless of RelA presence, while RelA when expressed alone was unable to efficiently bind the proteasome (Figure 7B, C).
Figure 7.
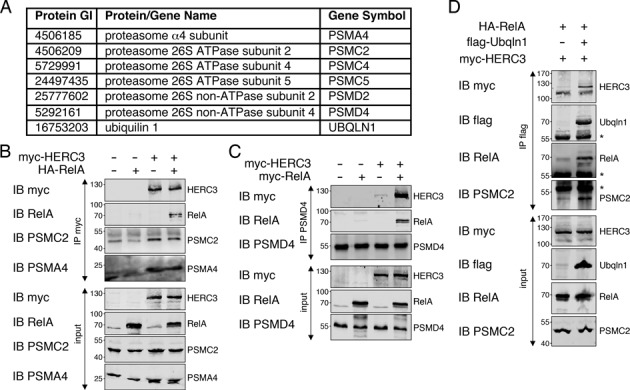
HERC3/RelA associate with the ubiquitin-like protein UBQLN1 and the 26S proteasome. (A) List of ubiquitination/degradation-associated proteins found by MS/MS analysis to bind to HERC3/RelA. (B) HEK293T cells transfected with indicated constructs were lysed and immunoprecipitation was performed with myc-specific antibody to pull down HERC3. RelA, PSMC2 and PSMA4 co-association was detected with specific antibodies recognizing these proteins. (C) HEK293T cells were transfected with control, myc-RelA, myc-HERC3 or myc-RelA/myc-HERC3 together. The endogenous proteasomal subunit PSMD4 was precipitated with PSMD4 antibody and association of RelA and HERC3 was tested in Western Blotting with respective antibodies. (D) HA-RelA, flag-tagged UBQLN1 and myc-HERC3 were introduced into HEK293T cells. Association of HERC3 and RelA, as well as the proteasomal subunit PSMC2 with UBQLN1 was tested by immunoprecipitation of UBQLN1 with flag-beads, followed by Western Blot detection. All experiments were performed 3 times. The asterisks mark the heavy chain detected by Western Blotting after immunoprecipitation. IB, immunoblot; IP, immunoprecipitation.
In addition to proteasomal proteins, LC-MS/MS detected UBQLN1, a protein that was previously found to interact with HERC3 (19) and the proteasome (24) (listed in Figure 7A). A pull down assay with UBQLN1 not only confirmed the interaction of UBQLN1 with HERC3, but also found associated RelA and proteasomal subunit PSMC2 (Figure 7D). In summary, we identify UBQLN1 and the 26S proteasome as binding partners of HERC3/RelA.
HERC3 and UBQLN1 regulate NF-κB activity by promoting RelA degradation by the 26S proteasome
Our data so far suggest that HERC3 reduces NF-κB transcriptional activity by binding to the proteasome and delivering RelA for degradation (Figures 1, 4 and 6). UBQLN1 was previously shown to regulate ubiquitin-mediated proteasomal proteolysis (24,37). To test the role of UBQLN1 in HERC3-mediated RelA degradation, we knocked down UBQLN1 protein and assessed HERC3 binding to RelA and the proteasome. While depletion of UBQLN1 had no effect on HERC3 binding to the proteasome, it reduced HERC3 binding to RelA (Figure 8A), a finding essentially reproduced when RelA was pulled down (Figure 8B). Interestingly, in addition we detected less proteasome bound to RelA without UBQLN1 (Figure 8B), indicating that UBQLN1 is not only required for efficient RelA binding to HERC3, but also for tethering it to the proteasome. To assess whether the observed decrease in HERC3-RelA-proteasome interaction with UBQLN1 depletion has any effect on RelA protein degradation, we performed pulse chase analyses in presence and absence of UBQLN1. As seen before, addition of HERC3 in presence of UBQLN1 significantly decreased RelA protein stability (Figure 8C). However, RelA levels partly recovered when UBQLN1 was depleted (Figure 8C), suggesting that UBQLN1 is needed for HERC3-mediated RelA degradation. To test whether endogenous RelA was regulated by HERC3 and/or UBQLN1, we studied RelA protein levels in endothelial cells after siRNA mediated knock down of HERC3 and UBQLN1. RelA was analyzed in metabolically labeled HUVEC transfected with siRNA and treated with TNF for 7 h to induce NF-κB activity. As expected RelA protein was markedly reduced in control siRNA cells (Figure 8D), reflecting a high protein turn over after NF-κB stimulation. HERC3 and UBQLN1 knock down were able to slightly stabilize RelA under these conditions. Remarkably, knock down of both HERC3 and UBQLN1 at the same time potentiated the rescue of RelA protein levels (Figure 8D), suggesting an additive effect. We also observed RelA levels in presence and absence of HERC3 and UBQLN1 in HUVEC with and without TNF treatment. In line with pulse chase data we found that knock down of HERC3 and UBQLN1 increased RelA levels in untreated as well as TNF-treated cells (Figure 8E). To investigate whether the stabilization of RelA by HERC3 and UBQLN1 depletion affects NF-κB transcription factor activity we tested the expression of NF-κB-dependent genes after 6 and 16 h TNF stimulation in control and HERC3/UBQLN1 siRNA-treated HUVEC. In concordance with RelA protein stability data we find that knock down of either HERC3 or UBQLN1 leads to a notable increase in NF-κB transcriptional activity at 6 h TNF stimulation (Icam1: si-Herc3 P = 0.65, si-Ubqln1 P = 0.40; Vcam1: si-Herc3 p = 0.12, si-Ubqln1 *P < 0.05; Sele: si-Herc3 P = 0.59, si-Ubqln1 *P < 0.05) (Figure 8F). Again, knock down of both proteins amplified this effect (Icam1: P = 0.24; Vcam1: *P < 0.05; Sele: *P < 0.05) (Figure 8F). Sixteen hours TNF stimulation led to similar results (Icam1: si-Herc3 p = 0.41, si-Ubqln1 P = 0.45, si-Herc3+si-Ubqln1 P = 0.41; Vcam1: si-Herc3 P = 0.20, si-Ubqln1 *P < 0.05, si-Herc3 + si-Ubqln1 *P < 0.05; Sele: si-Herc3 P = 0.48, si-Ubqln1 P = 0.09, si-Herc3 + si-Ubqln1 P = 0.07). In conclusion, we show here that HERC3 and UBQLN1 regulate the turn over of RelA protein, thereby altering NF-κB transcriptional profiles.
Figure 8.
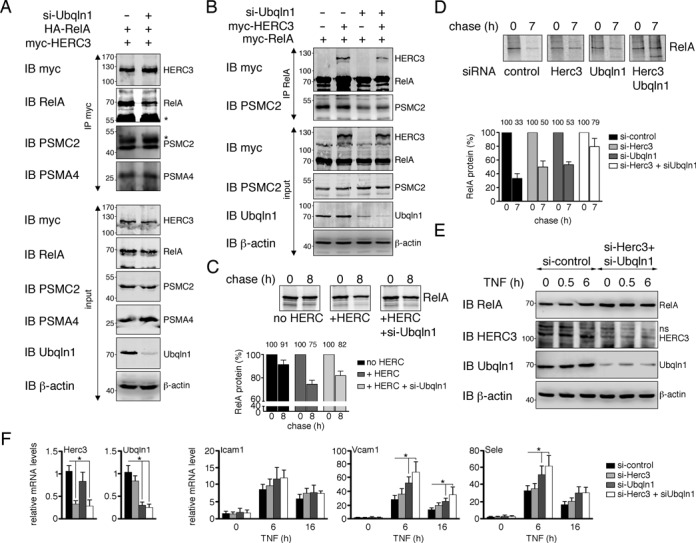
HERC3 and UBQLN1 conjointly affect NF-κB activity by linking RelA to the proteasome and promoting its degradation. (A) UBQLN1 expression was knocked down by siRNA transfection into HEK293T cells. At the same time myc-HERC3 and HA-RelA were introduced and interaction between these two proteins was studied by co-immunoprecipitation approach. Association of endogenous proteasomal subunits PSMC2 and PSMA4 was also tested. The asterisks point out the antibody heavy chains detected by Western Blotting after pull down. (B) HEK293T were transfected with siRNA targeting UBQLN1, myc-HERC3 and myc-RelA. Binding of proteins to RelA was examined by pull down with anti-RelA antibody and detection of HERC3 and PSMC2 by Western Blotting. (C) Stability of RelA protein in presence of HERC3 and absence of UBQLN1 was determined in HEK293T by metabolic labeling as described in Figure 5B. Results from 4 independent experiments were quantified. Average percent of protein before and after chase is noted on top of the respective graph columns. (D) Endogenous RelA protein stability was assessed in HUVEC exposed to siRNA targeting HERC3 and/or UBQLN1. Non-targeting siRNA was used as control. HUVEC were incubated in [35S]-methionine-containing growth medium for 2 h on day 3 after siRNA exposure. Pulse medium was removed and cells were left in culture for another 7 h in normal growth medium substituted with 10 ng/ml TNF. HUVEC were harvested and RelA was precipitated from total cell extracts with RelA-specific antibody. Data from four experiments were plotted on a graph. Average percent protein remaining before and after chase is indicated. (E) HUVEC transfected with non-targeting control or Herc3/Ubqln1-targeting siRNAs were stimulated with 10 ng/ml TNF for 0, 0.5 and 6 h. Protein levels of endogenous RelA, HERC3, UBQLN1 and β-actin were determined. Shown blots are representative of 2 independent experiments. (F) mRNA expression of three NF-κB-regulated genes Icam1, Vcam1 and Sele was examined in siRNA-treated HUVEC. Three days after siRNA exposure NF-κB-dependent transcription was stimulated with TNF for 6 and 16 h. RNA was isolated and gene expression was assessed by qRT-PCR. HERC3 and UBQLN1 knock down efficiency was tested in un-stimulated cells and plotted in the first two panels. Data were normalized to each group and represent mean +SEM from 5–9 independent experiments. Values were considered significant at *P < 0.05. IB, immunoblot; IP, immunoprecipitation; si, small interfering RNA.
DISCUSSION
We identify the ubiquitin ligase HERC3 as novel negative regulator of NF-κB-dependent transcription. HERC3 achieves this regulation by mediating RelA subunit ubiquitination and destabilization, leading to less nuclear NF-κB. So far all identified ubiquitin ligases involved in NF-κB RelA ubiquitination, including the COMMD1/SOCS1/GCN5 complex, ING4, PPARγ and PDLIM2, are predominantly nuclear proteins and hence act on nuclear RelA (8,12–15,38). HERC3 on the contrary is a cytoplasmic protein (Figure 2 and (17,19)) that affects NF-κB before it enters the nucleus. Interestingly, HERC3 binding and subsequent RelA ubiquitination are inhibited by IκBα (Supplementary Figure S3), suggesting that it targets NF-κB after liberation from or in absence of its inhibitor. NF-κB regulatory proteins that influence NF-κB fate in the spatial window between IκB removal and nuclear translocation are rare. One example known to date is the tumor suppressor Sef, which after stimulus-induced IκB elimination continues to inhibit NF-κB nuclear import and activity by binding to p50 (39). This mechanism is believed to counterbalance and modulate the extent of the NF-κB response, a process also conceivable for HERC3. There is another interesting parallel between Sef and HERC3. Like Sef (39–41), HERC3 co-localizes with endomembranes (17,19). When visualized by immuno-fluorescence HERC3 resembles a fine punctuate staining especially in perinuclear regions. Whether HERC3 together with RelA also localizes to endocytic vesicles remains to be established. Until then it cannot be excluded that associated endomembranes play some role in the HERC3-driven retention of NF-κB in the cytoplasm. Interestingly the identified RelA/HERC3 interaction partner UBQLN1 resides not only at proteasomes, but has also been found associated with the endocytic pathway (42,43). Further investigation will be necessary to clarify whether endocytic vesicles are involved in HERC3 and UBQLN1 regulation of RelA, possibly by extending its cytosolic retention, where it becomes a substrate for the proteasome.
Unlike Sef, which suppresses NF-κB nuclear levels solely by cytoplasmic sequestration, HERC3 mediates its ubiquitination and degradation. Although HERC3 is equipped with a functional HECT domain containing a catalytically active cysteine residue embedded in a characteristic 4-amino acid motif (44), our data indicate that HERC3 intrinsic ligase activity is not required for RelA ubiquitination. Even catalytically inactive HERC3 mutants efficiently induce RelA ubiquitination (Figure 6C). HECT domain ubiquitin ligases need to directly bind their targets to execute their function, therefore, we also assessed HERC3 direct binding to RelA with cell free in vitro binding assays. In contrast to in vivo binding studies (Figure 4A, B) these assays resulted in no detectable interaction between the two proteins (Supplementary Figure S5A). These data together with the ubiquitination assay strongly suggest that another unidentified ligase must act on RelA in presence of HERC3. Although HECT domain ubiquitin ligases contrary to most RING domain ubiquitin ligases are traditionally known to act as monomers, the large HERC family member HERC2 was shown to serve as an adaptor protein for the assembly of a multi-ubiquitin ligase complex independently of its ubiquitin ligase activity (45). There, binding of HERC2 to RNF8/RNF168 ubiquitin ligases facilitated Ubc13 recruitment and production of K63-linked ubiquitin chains, which tethered repair factors like p53BP1, RAP80 and BRCA1 to DNA damage sites (45). To identify the ubiquitin ligase that is actually responsible for RelA ubiquitination in presence of HERC3 we performed mass spectrometry analyses of HERC3-associated proteins. However, we did not reveal any ligase associated with HERC3 in presence of RelA, suggesting that it might either be of very low abundance and therefore hard to detect by MS analysis or HERC3 indirectly activates a RelA-targeting ligase without physically binding to it. In the latter case a mass spectrometry approach identifying RelA- instead of HERC3-bound proteins would potentially be useful in future studies to reveal the identity of the acting ligase.
Our analyses of HERC3 and RelA precipitates under non-denaturing conditions indicate that HERC3 and RelA are part of a MW complex larger than 1000 kDa (Figure 6D, E). Assuming that the complex does not consist of HERC3 and RelA multimers, we predicted that other proteins are likely to be present. Although we could not detect an ubiquitin ligase, mass-spectrometry analysis of HERC3-binding proteins in presence of RelA revealed components of the 26S proteasome, which could total up to 2000 kDa (46). Interestingly, even when expressed individually, HERC3 and RelA are barely detected at monomeric sizes of 115 and 65 kDa, respectively. Minor parts of RelA are found around 100 kDa, which may reflect homo- or heterodimers with other NF-κB proteins (p50, cRel, RelB). The vast majority of RelA protein, however, is detected within two molecular weight peaks around 600–1000 kDa (Figure 6D, E). This might represent its association with IKK complexes, which have been shown to reach a size of approximately 900 kDa (23), paired with other NF-κB-associated factors, like IκBα, p105 or p50. HERC3 expressed alone is found starting at a molecular weight of 600 kDa, suggesting that HERC3 like RelA is primarily found in multi-protein complexes. Although it would be undoubtedly interesting to assess the protein composition of these individual complexes, we did not pursue this endeavor for this study.
HERC3 is a poorly characterized protein. Apart from its cytoplasmic, vesicle-associated localization and suggested involvement in ubiquitination (by virtue of the presence of a HECT domain) it has been shown to interact with two proteins, UBQLN1 and UBQLN2 (19), however, the functional impact of this interaction remained elusive. Interestingly, we again found UBQLN1 in the current MS analysis as HERC3/RelA-interacting protein. UBQLN proteins contain one ubiquitin-associated (UBA) and one ubiquitin-like (UBL) domain, which have been previously shown to associate with ubiquitinated proteins, the ubiquitin ligases HERC3, βTRCP and E6AP and the proteasome (19,24,47). The ability of UBQLNs to interact with substrates, as well as components of the ubiquitination and degradation machineries puts it into a prime spot for regulation of these processes. However, whether UBQLNs stimulate or inhibit degradation of proteasomal targets has been controversial. While on the one hand it has been reported that UBQLNs cause inhibition of ubiquitin-dependent proteasomal degradation (24), it has also been shown that they enhance degradation of ubiquitinated protein substrates (37). The underlying cause for this discrepancy is not known. One possible explanation could be that UBQLNs specifically interfere with degradation of some substrates, while they promote the degradation of others. This might be regulated at the level of the ubiquitin ligase or at the proteasome itself. Our model supports a degradation-enhancing role of UBQLN1 for NF-κB, as its knock down rescues HERC3-induced RelA destabilization (Figure 8C). Although we show that UBQLN1 binds the proteasome in presence of HERC3 and RelA (Figure 7D), the regulation of RelA stability by UBQLN1 may only be partly dependent on its proteasome binding activity. Rather our data suggest that UBQLN1 strengthens the binding of HERC3 to RelA (Figure 8A, B), which is important to deliver RelA to the proteasome. The weakened binding between RelA and HERC3 in absence of UBQLN1 is consistent and leads to effects on RelA stability and NF-κB transcriptional activity, however the detected changes are modest. To test whether UBQLN1, HERC3 and RelA form a ternary complex via direct interactions we assessed their binding to each other in vitro. As already suspected from the in vivo data, we found no detectable interaction between the three proteins (Supplementary Figure S5B). Therefore, it seems likely that additional proteins are present in the HERC3/UBQLN1/proteasome complex and future studies will be necessary to further address the involvement of currently unidentified players. The more pronounced effect of HERC3-UBQLN1 double versus each single knock down on RelA stability and transcriptional activity indicates that the two proteins are not positioned to affect NF-κB in a linear fashion. Instead, HERC3 and UBQLN1 may act in concert to strengthen RelA association with the proteasome, thereby altering its degradation profile and as a result its activity.
We show that HERC3 mediates ubiquitination of RelA on two distinct lysine residues, K195 and K315 (Supplementary Figure S4). Interestingly, the same sites were found to be ubiquitinated before in our studies and also by others. Previously, we detected the ubiquitination of RelA K62, K123 and K315 in response to proteasomal inhibition (7). Similarly, Li et al. discovered the ubiquitination of RelA after proteasomal blockade on seven lysines, among them again K62, K123, K315 and also K195 (33). Both studies found that RelA ubiquitination sites appear to be extremely promiscuous, as site-directed mutagenesis of either one or combinations of MS-identified RelA lysines did not result in any reduction of RelA ubiquitination. In contrast to above described studies, which used proteasomal inhibition as inducer for RelA ubiquitination, we do see a reduction in HERC3-driven RelA ubiquitination when lysines 195 and 315 are mutated (Supplementary Figure S4B). This indicates that although similar RelA lysines are targeted with different stimuli, the preference for certain lysines may vary. Nevertheless, like observed with proteasomal blockade, HERC3 is also likely to induce RelA ubiquitination on more than the currently identified lysines. First, while RelA single and double K195 and K315 mutant ubiquitination is diminished, it is not completely abolished. And second, the effect of K195 and K315 mutation on RelA transcriptional recovery is minimal.
The identification and characterization of new mediators in the NF-κB pathway is important to improve our knowledge and understanding of mechanisms involved in inflammation and associated diseases, and may potentially provide new targets for therapy. We present here two proteins, HERC3 and UBQLN1, which have not been previously connected with the NF-κB pathway. HERC3 regulation of NF-κB exhibits two unique features: (i) HERC3 is the only ligase identified to date that mediates ubiquitination of NF-κB before it enters the nucleus. (ii) HERC3 links NF-κB ubiquitination with its degradation machinery independent of its own enzymatic activity. UBQLN1 takes part in this regulation by stabilizing the interaction of HERC3 and the proteasome with NF-κB. Future studies will be directed at identifying additional partners in the HERC3-UBQLN1-NF-κB regulatory module, and at ways to harness this pathway for potential therapies.
Supplementary Material
Acknowledgments
We would like to thank Reunet Rodney-Sandy and Gianfranco Racchumi (both Weill Cornell Medical College, New York, USA) for technical assistance and performing qPCR, respectively, Dr Wei Chen, James A. McCardle and Elizabeth T. Anderson (Cornell University, Ithaca, USA) for help with the mass spectrometry analysis and Dr Amer A. Beg (Moffitt Cancer Center, Tampa, USA) for providing RelA−/− 3T3 cells.
Footnotes
Present address: Karin Hochrainer, 407 East 61st Street, RR-406, New York, NY10065, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
American Heart Association Scientist Development [SDG102600298 to K.H.] and National Institute of Health Grants (NIH) [HL077308 to J.A. and NS34179 to C.I.]. Funding for open access charge: NIH [NS34179].
Conflict of interest statement. None declared.
REFERENCES
- 1.Beg A.A., Ruben S.M., Scheinman R.I., Haskill S., Rosen C.A., Baldwin A.S. Jr. I kappa B interacts with the nuclear localization sequences of the subunits of NF-kappa B: a mechanism for cytoplasmic retention. Genes Dev. 1992;6:1899–1913. doi: 10.1101/gad.6.10.1899. [DOI] [PubMed] [Google Scholar]
- 2.Henkel T., Machleidt T., Alkalay I., Kronke M., Ben-Neriah Y., Baeuerle P.A. Rapid proteolysis of I kappa B-alpha is necessary for activation of transcription factor NF-kappa B. Nature. 1993;365:182–185. doi: 10.1038/365182a0. [DOI] [PubMed] [Google Scholar]
- 3.Pahl H.L. Activators and target genes of Rel/NF-kappaB transcription factors. Oncogene. 1999;18:6853–6866. doi: 10.1038/sj.onc.1203239. [DOI] [PubMed] [Google Scholar]
- 4.Ruland J. Return to homeostasis: downregulation of NF-kappaB responses. Nat. Immunol. 2011;12:709–714. doi: 10.1038/ni.2055. [DOI] [PubMed] [Google Scholar]
- 5.Sun S.C., Ganchi P.A., Ballard D.W., Greene W.C. NF-kappa B controls expression of inhibitor I kappa B alpha: evidence for an inducible autoregulatory pathway. Science. 1993;259:1912–1915. doi: 10.1126/science.8096091. [DOI] [PubMed] [Google Scholar]
- 6.Arenzana-Seisdedos F., Turpin P., Rodriguez M., Thomas D., Hay R.T., Virelizier J.L., Dargemont C. Nuclear localization of I kappa B alpha promotes active transport of NF-kappa B from the nucleus to the cytoplasm. J. Cell Sci. 1997;110:369–378. doi: 10.1242/jcs.110.3.369. [DOI] [PubMed] [Google Scholar]
- 7.Hochrainer K., Racchumi G., Zhang S., Iadecola C., Anrather J. Monoubiquitination of nuclear RelA negatively regulates NF-kappaB activity independent of proteasomal degradation. Cell. Mol. Life Sci. 2012;69:2057–2073. doi: 10.1007/s00018-011-0912-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hou Y., Moreau F., Chadee K. PPARgamma is an E3 ligase that induces the degradation of NFkappaB/p65. Nat. Commun. 2012;3:1300. doi: 10.1038/ncomms2270. [DOI] [PubMed] [Google Scholar]
- 9.Leidner J., Palkowitsch L., Marienfeld U., Fischer D., Marienfeld R. Identification of lysine residues critical for the transcriptional activity and polyubiquitination of the NF-kappaB family member RelB. Biochem. J. 2008;416:117–127. doi: 10.1042/BJ20080432. [DOI] [PubMed] [Google Scholar]
- 10.Ryo A., Suizu F., Yoshida Y., Perrem K., Liou Y.C., Wulf G., Rottapel R., Yamaoka S., Lu K.P. Regulation of NF-kappaB signaling by Pin1-dependent prolyl isomerization and ubiquitin-mediated proteolysis of p65/RelA. Mol. Cell. 2003;12:1413–1426. doi: 10.1016/s1097-2765(03)00490-8. [DOI] [PubMed] [Google Scholar]
- 11.Saccani S., Marazzi I., Beg A.A., Natoli G. Degradation of promoter-bound p65/RelA is essential for the prompt termination of the nuclear factor kappaB response. J. Exp. Med. 2004;200:107–113. doi: 10.1084/jem.20040196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tanaka T., Grusby M.J., Kaisho T. PDLIM2-mediated termination of transcription factor NF-kappaB activation by intranuclear sequestration and degradation of the p65 subunit. Nat. Immunol. 2007;8:584–591. doi: 10.1038/ni1464. [DOI] [PubMed] [Google Scholar]
- 13.Maine G.N., Mao X., Komarck C.M., Burstein E. COMMD1 promotes the ubiquitination of NF-kappaB subunits through a cullin-containing ubiquitin ligase. EMBO J. 2007;26:436–447. doi: 10.1038/sj.emboj.7601489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mao X., Gluck N., Li D., Maine G.N., Li H., Zaidi I.W., Repaka A., Mayo M.W., Burstein E. GCN5 is a required cofactor for a ubiquitin ligase that targets NF-kappaB/RelA. Genes Dev. 2009;23:849–861. doi: 10.1101/gad.1748409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hou Y., Zhang Z., Xu Q., Wang H., Xu Y., Chen K. Inhibitor of growth 4 induces NFkappaB/p65 ubiquitin-dependent degradation. Oncogene. 2014;33:1997–2003. doi: 10.1038/onc.2013.135. [DOI] [PubMed] [Google Scholar]
- 16.Hochrainer K., Mayer H., Baranyi U., Binder B., Lipp J., Kroismayr R. The human HERC family of ubiquitin ligases: novel members, genomic organization, expression profiling, and evolutionary aspects. Genomics. 2005;85:153–164. doi: 10.1016/j.ygeno.2004.10.006. [DOI] [PubMed] [Google Scholar]
- 17.Cruz C., Ventura F., Bartrons R., Rosa J.L. HERC3 binding to and regulation by ubiquitin. FEBS Lett. 2001;488:74–80. doi: 10.1016/s0014-5793(00)02371-1. [DOI] [PubMed] [Google Scholar]
- 18.Kroismayr R., Baranyi U., Stehlik C., Dorfleutner A., Binder B.R., Lipp J. HERC5, a HECT E3 ubiquitin ligase tightly regulated in LPS activated endothelial cells. J. Cell Sci. 2004;117:4749–4756. doi: 10.1242/jcs.01338. [DOI] [PubMed] [Google Scholar]
- 19.Hochrainer K., Kroismayr R., Baranyi U., Binder B.R., Lipp J. Highly homologous HERC proteins localize to endosomes and exhibit specific interactions with hPLIC and Nm23B. Cell. Mol. Life Sci. 2008;65:2105–2117. doi: 10.1007/s00018-008-8148-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Anrather J., Racchumi G., Iadecola C. cis-acting, element-specific transcriptional activity of differentially phosphorylated nuclear factor-kappa B. J. Biol. Chem. 2005;280:244–252. doi: 10.1074/jbc.M409344200. [DOI] [PubMed] [Google Scholar]
- 21.Anrather J., Csizmadia V., Soares M.P., Winkler H. Regulation of NF-kappaB RelA phosphorylation and transcriptional activity by p21(ras) and protein kinase Czeta in primary endothelial cells. J. Biol. Chem. 1999;274:13594–13603. doi: 10.1074/jbc.274.19.13594. [DOI] [PubMed] [Google Scholar]
- 22.Brostjan C., Anrather J., Csizmadia V., Natarajan G., Winkler H. Glucocorticoids inhibit E-selectin expression by targeting NF-kappaB and not ATF/c-Jun. J. Immunol. 1997;158:3836–3844. [PubMed] [Google Scholar]
- 23.Mercurio F., Zhu H., Murray B.W., Shevchenko A., Bennett B.L., Li J., Young D.B., Barbosa M., Mann M., Manning A., et al. IKK-1 and IKK-2: cytokine-activated IkappaB kinases essential for NF-kappaB activation. Science. 1997;278:860–866. doi: 10.1126/science.278.5339.860. [DOI] [PubMed] [Google Scholar]
- 24.Kleijnen M.F., Shih A.H., Zhou P., Kumar S., Soccio R.E., Kedersha N.L., Gill G., Howley P.M. The hPLIC proteins may provide a link between the ubiquitination machinery and the proteasome. Mol. Cell. 2000;6:409–419. doi: 10.1016/s1097-2765(00)00040-x. [DOI] [PubMed] [Google Scholar]
- 25.Sorg G., Stamminger T. Mapping of nuclear localization signals by simultaneous fusion to green fluorescent protein and to beta-galactosidase. Biotechniques. 1999;26:858–862. doi: 10.2144/99265bm12. [DOI] [PubMed] [Google Scholar]
- 26.Zhang S., Van Pelt C.K., Henion J.D. Automated chip-based nanoelectrospray-mass spectrometry for rapid identification of proteins separated by two-dimensional gel electrophoresis. Electrophoresis. 2003;24:3620–3632. doi: 10.1002/elps.200305585. [DOI] [PubMed] [Google Scholar]
- 27.Swamy M., Siegers G.M., Minguet S., Wollscheid B., Schamel W.W. Blue native polyacrylamide gel electrophoresis (BN-PAGE) for the identification and analysis of multiprotein complexes. Sci. STKE. 2006;345:pl4. doi: 10.1126/stke.3452006pl4. [DOI] [PubMed] [Google Scholar]
- 28.Huang B., Yang X.D., Lamb A., Chen L.F. Posttranslational modifications of NF-kappaB: another layer of regulation for NF-kappaB signaling pathway. Cell. Signal. 2010;22:1282–1290. doi: 10.1016/j.cellsig.2010.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Baeuerle P.A., Baltimore D. I kappa B: a specific inhibitor of the NF-kappa B transcription factor. Science. 1988;242:540–546. doi: 10.1126/science.3140380. [DOI] [PubMed] [Google Scholar]
- 30.Zabel U., Henkel T., Silva M.S., Baeuerle P.A. Nuclear uptake control of NF-kappa B by MAD-3, an I kappa B protein present in the nucleus. EMBO J. 1993;12:201–211. doi: 10.1002/j.1460-2075.1993.tb05646.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sun S.C., Ganchi P.A., Beraud C., Ballard D.W., Greene W.C. Autoregulation of the NF-kappa B transactivator RelA (p65) by multiple cytoplasmic inhibitors containing ankyrin motifs. Proc. Natl. Acad. Sci. U.S.A. 1994;91:1346–1350. doi: 10.1073/pnas.91.4.1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Traenckner E.B., Pahl H.L., Henkel T., Schmidt K.N., Wilk S., Baeuerle P.A. Phosphorylation of human I kappa B-alpha on serines 32 and 36 controls I kappa B-alpha proteolysis and NF-kappa B activation in response to diverse stimuli. EMBO J. 1995;14:2876–2883. doi: 10.1002/j.1460-2075.1995.tb07287.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li H., Wittwer T., Weber A., Schneider H., Moreno R., Maine G.N., Kracht M., Schmitz M.L., Burstein E. Regulation of NF-kappaB activity by competition between RelA acetylation and ubiquitination. Oncogene. 2012;31:611–623. doi: 10.1038/onc.2011.253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pickart C.M., Fushman D. Polyubiquitin chains: polymeric protein signals. Curr. Opin. Chem. Biol. 2004;8:610–616. doi: 10.1016/j.cbpa.2004.09.009. [DOI] [PubMed] [Google Scholar]
- 35.Dastur A., Beaudenon S., Kelley M., Krug R.M., Huibregtse J.M. Herc5, an interferon-induced HECT E3 enzyme, is required for conjugation of ISG15 in human cells. J. Biol. Chem. 2006;281:4334–4338. doi: 10.1074/jbc.M512830200. [DOI] [PubMed] [Google Scholar]
- 36.Scheffner M., Nuber U., Huibregtse J.M. Protein ubiquitination involving an E1-E2-E3 enzyme ubiquitin thioester cascade. Nature. 1995;373:81–83. doi: 10.1038/373081a0. [DOI] [PubMed] [Google Scholar]
- 37.Liu Y., Lu L., Hettinger C.L., Dong G., Zhang D., Rezvani K., Wang X., Wang H. Ubiquilin-1 protects cells from oxidative stress and ischemic stroke caused tissue injury in mice. J Neurosci. 2014;34:2813–2821. doi: 10.1523/JNEUROSCI.3541-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Strebovsky J., Walker P., Lang R., Dalpke A.H. Suppressor of cytokine signaling 1 (SOCS1) limits NFkappaB signaling by decreasing p65 stability within the cell nucleus. FASEB J. 2011;25:863–874. doi: 10.1096/fj.10-170597. [DOI] [PubMed] [Google Scholar]
- 39.Fuchs Y., Brunwasser M., Haif S., Haddad J., Shneyer B., Goldshmidt-Tran O., Korsensky L., Abed M., Zisman-Rozen S., Koren L., et al. Sef is an inhibitor of proinflammatory cytokine signaling, acting by cytoplasmic sequestration of NF-kappaB. Dev. Cell. 2012;23:611–623. doi: 10.1016/j.devcel.2012.07.013. [DOI] [PubMed] [Google Scholar]
- 40.Ren Y., Cheng L., Rong Z., Li Z., Li Y., Zhang X., Xiong S., Hu J., Fu X.Y., Chang Z. hSef potentiates EGF-mediated MAPK signaling through affecting EGFR trafficking and degradation. Cell. Signal. 2008;20:518–533. doi: 10.1016/j.cellsig.2007.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Torii S., Kusakabe M., Yamamoto T., Maekawa M., Nishida E. Sef is a spatial regulator for Ras/MAP kinase signaling. Dev. Cell. 2004;7:33–44. doi: 10.1016/j.devcel.2004.05.019. [DOI] [PubMed] [Google Scholar]
- 42.Bedford F.K., Kittler J.T., Muller E., Thomas P., Uren J.M., Merlo D., Wisden W., Triller A., Smart T.G., Moss S.J. GABA(A) receptor cell surface number and subunit stability are regulated by the ubiquitin-like protein Plic-1. Nat. Neurosci. 2001;4:908–916. doi: 10.1038/nn0901-908. [DOI] [PubMed] [Google Scholar]
- 43.Regan-Klapisz E., Sorokina I., Voortman J., de Keizer P., Roovers R.C., Verheesen P., Urbe S., Fallon L., Fon E.A., Verkleij A., et al. Ubiquilin recruits Eps15 into ubiquitin-rich cytoplasmic aggregates via a UIM-UBL interaction. J. Cell Sci. 2005;118:4437–4450. doi: 10.1242/jcs.02571. [DOI] [PubMed] [Google Scholar]
- 44.Schwarz S.E., Rosa J.L., Scheffner M. Characterization of human hect domain family members and their interaction with UbcH5 and UbcH7. J. Biol. Chem. 1998;273:12148–12154. doi: 10.1074/jbc.273.20.12148. [DOI] [PubMed] [Google Scholar]
- 45.Bekker-Jensen S., Rendtlew Danielsen J., Fugger K., Gromova I., Nerstedt A., Lukas C., Bartek J., Lukas J., Mailand N. HERC2 coordinates ubiquitin-dependent assembly of DNA repair factors on damaged chromosomes. Nat. Cell Biol. 2010;12:80–86. doi: 10.1038/ncb2008. [DOI] [PubMed] [Google Scholar]
- 46.Tanahashi N., Tsurumi C., Tamura T., Tanaka K. Molecular structure of 20S and 26S proteasomes. Enzyme Protein. 1993;47:241–251. doi: 10.1159/000468683. [DOI] [PubMed] [Google Scholar]
- 47.Ko H.S., Uehara T., Tsuruma K., Nomura Y. Ubiquilin interacts with ubiquitylated proteins and proteasome through its ubiquitin-associated and ubiquitin-like domains. FEBS Lett. 2004;566:110–114. doi: 10.1016/j.febslet.2004.04.031. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.