


Abstract
The prediction of novel pre-microRNA (miRNA) from genomic sequence has received considerable attention recently. However, the majority of studies have focused on the human genome. Previous studies have demonstrated that sensitivity (correctly detecting true miRNA) is sustained when human-trained methods are applied to other species, however they have failed to report the dramatic drop in specificity (the ability to correctly reject non-miRNA sequences) in non-human genomes. Considering the ratio of true miRNA sequences to pseudo-miRNA sequences is on the order of 1:1000, such low specificity prevents the application of most existing tools to non-human genomes, as the number of false positives overwhelms the true predictions. We here introduce a framework (SMIRP) for creating species-specific miRNA prediction systems, leveraging sequence conservation and phylogenetic distance information. Substantial improvements in specificity and precision are obtained for four non-human test species when our framework is applied to three different prediction systems representing two types of classifiers (support vector machine and Random Forest), based on three different feature sets, with both human-specific and taxon-wide training data. The SMIRP framework is potentially applicable to all miRNA prediction systems and we expect substantial improvement in precision and specificity, while sustaining sensitivity, independent of the machine learning technique chosen.
INTRODUCTION
MicroRNAs (miRNAs) are short (18–23 nt), non-coding RNAs (ncRNAs) that play central roles in cellular regulation by modulating the post-transcriptional expression of messenger RNA (mRNA) transcripts (1). Most miRNAs are considered to share a similar biogenesis mechanism: they are derived from RNA transcripts (pre-miRNAs) that fold into imperfect hairpin structures (∼70 nt in length) and are subsequently processed by one or more endonucleases (e.g. Drosha and Dicer in animals, DLC1 in plants) to form mature miRNA. After processing and formation, the mature miRNA is incorporated into the RNA-induced silencing complex (miRISC), where the miRNA guides the associated RISC proteins to the targeted mRNA strand, annealing to the target mRNA and promoting either degradation or reversible translational repression (2). It has been previously estimated that 60–90% of all mammalian mRNAs may be targeted by miRNAs (3), and at this time over 2500 mature miRNAs have been identified in the human genome (miRBase v.21.0 released in June 2014 (4)). Through a myriad of comparative expression analyses and gain- and loss-of-function experiments, miRNAs have been shown to be critically involved in regulating the expression of proteins involved in biological development (5), cell differentiation (6), apoptosis (7), cell cycle control (8), stress response (9) and disease pathogenesis (10). Recent studies have also highlighted the role of miRNA in the cellular adaptation to severe environmental stresses (such as freezing, dehydration and anoxia) in tolerant animals (11–13). Due to their biological importance, the ability to accurately predict their sequence in newly sequenced genomes is of great importance.
Computational techniques for the de novo prediction of pre-miRNA sequences within larger genomic sequences—referred to as ‘miRNA prediction’ within this text—can be broadly separated into two categories: homology-based prediction (14–18) and machine-learning-based prediction (19–42). Homology-based methods predict miRNA based on similarity to other known miRNA, with respect to sequence, structure or target site. These techniques are able to confidently identify homologous miRNA across species, but are not able to predict novel miRNAs that differ significantly from known miRNA.
The largest class of de novo miRNA prediction tools are machine-learning-based classifiers which separate true miRNA from miRNA-like structures, based on elements of primary sequence and secondary structure. A wide array of classification techniques have been applied to this problem, including random forests (35,37), hidden Markov models (22,42), naive Bayes classifiers (34) and KNN classifiers (31). The most common technique is support vector machines (SVMs) (32,38–39,41). Recent improvements in classifier selection, feature extraction, class imbalance correction and training data quality have resulted in incremental improvements in both sensitivity (the ability to correctly identify true miRNAs) and specificity (the ability to correctly reject sequences that do not constitute miRNAs). This study focuses on the improvement of machine-learning-based miRNA predictors.
Recently, the decreasing cost of next-generation RNA sequencing experiments has increased the popularity of RNA-seq-based miRNA discovery methods such as miRDeep (43,44). RNA-seq-based methods have shown success in discovering miRNA within RNA expression data. However, these approaches remain time-consuming and expensive relative to computational methods (45). Furthermore, RNA-seq-based methods have also been shown to be biased towards miRNA with higher copy numbers or expression levels (46–48), and RNA-seq data may not contain miRNA of interest which are temporally expressed or stress-, cell- or developmental-specific (46). Lastly, there are species of interest for which only sequence data is available, such as the woolly mammoth or other hypothetical sequences arising from fundamental research. For these reasons, de novo miRNA prediction remains an important approach for the discovery of novel miRNA.
Selection of training data, both positive and negative, is a key component in the development and evaluation of de novo miRNA classification pipelines. Positive training sets for miRNA classification pipelines consist of known pre-miRNA sequences, which are retrieved from miRNA databases. All major studies to date draw positive training sets from the miRBase database (4), however, recent work suggests that the miRTarBase database (49) may provide higher-quality positive training sets (45). Negative training sets typically consist of protein-coding (exonic) sequences that form miRNA-like hairpins, and of ncRNAs which are similar in structure to pre-miRNA. Since these coding and ncRNA sequences are known to play other biological roles, they are assumed not to form miRNAs. The selection of training data can have a large impact on the quality of a classifier; in particular, this study addresses low classification performance which occurs when a classifier is applied to a species that differs from the species for which it was trained.
Figure 1 demonstrates the substantial loss in specificity which occurs when a classifier that is trained using data from species A (here human) is applied to a test species (species B), where A ≠ B. This experiment makes use of the microPred classifier, introduced by Batuwita and Palade (38), which was trained exclusively on Homo sapiens data. The low overall specificity of microPred on hold-out data has been previously observed (24). However, the dramatic drop in specificity which typically occurs when miRNA classifiers are used to predict miRNA in species outside those in their training set has not generally been reported in miRNA prediction studies. Several miRNA prediction studies (e.g. (38)) report sustained sensitivity across multiple species; however, they conspicuously omit reporting the specificity on species which differ from those used to train their classifiers. This is critical, as correctly predicted miRNA arising from a system with low specificity will quickly be overwhelmed by false positive predictions due to the large class imbalance between hairpin-like sequences that form true miRNA versus those that do not (discussed further below).
Figure 1.

Specificity decreases as miRNA classifiers are used to predict pseudo-miRNA on species that are phylogenetically distant from those to which the classifier was trained on. The sensitivity and specificity of predicting miRNA using microPred from classifiers trained against H. sapiens from H. sapiens (human), P. troglodytes (chimp), M. musculus (mouse), G. gallus (chicken), C. intestinalis (squirt), M. domestica (apple) and Epstein barr virus (virus).
The need for training data which is appropriate for non-human species has been raised previously (31,37,48,50), and attempts have been made to address the issue. Gudys et al. proposed a methodology (HuntMi) for creating taxon-specific training data wherein sequences are pooled from many genomes within a broad taxon (37). They also examined a range of pattern classification approaches, concluding that random forests were most effective for the feature set proposed in their study (37). Wu et al. propose the use of similarly multi-species pooled positive datasets, however they note that this methodology does not perform well for all taxa, noting Mycetozoa as a taxon for which insufficient data are available for a pooled dataset generation approach (48). Lertampaiporn et al. utilize a positive training set which contains pre-miRNA sequences pooled from the genomes of H. sapiens, Arabidopsis thaliana and Oryza sativa in order to broaden the usefulness of their classifier beyond strictly human miRNA studies (31). While the above measures represent important steps towards increasing the accuracy of miRNA prediction in diverse species, we here demonstrate that a more advanced framework for species-specific training data selection has the potential to vastly improve miRNA prediction accuracy across a range of species. This approach is particularly useful for niche species that are of great scientific interest due to their genetic uniqueness, but are phylogenetically distant from model organisms such as H. sapiens which are typically used to create single-species or multi-species pooled training data.
While the issue of class imbalance (i.e. the large number of negative sequences versus true miRNAs in a genome) is widely acknowledged (31,37–38,45), and has been addressed during training using techniques such as SMOTE (38,31,51), it remains largely unaddressed in the testing and evaluation of miRNA prediction methods. Therefore, as adopted in (52), we introduce in this study precision–recall curves using real-world class imbalance levels as a means for comparing performance of miRNA prediction methods. Relative to the widely used metrics of geometric mean and AUC, precision–recall curves account for the large class imbalance observed in actual genomes (estimated at 1000:1 for most genomes; see below). This performance metric has been adopted in other relevant fields, e.g. protein–protein interaction prediction (53), as it quantifies the performance of a classifier in terms that are of direct interest to actual users of the method—those who will perform follow-up experimental validation of predictors.
In this study, we present a framework for the dynamic generation of species-specific training data, suitable for the creation of highly accurate Species-specific MIRna Predictors (SMIRP). Such a method is particularly needed for newly sequenced species of biological interest and for species for which high-quality miRNA data is not already available. This framework can be applied to generate training data for any miRNA classification method, including current leading methods. Our framework leverages sequence clustering techniques in order to produce positive training data representing diverse miRNAs. Selection techniques are applied to these clusters to tailor the dataset towards any species, with an emphasis on those miRNA that appear to be highly conserved. Negative datasets are built using miRNA-like hairpins from species that are closely related to the target species, providing negative training data more likely to resemble those found in the target species. We demonstrate a positive correlation between the use of training data from species which are closely related to a species of interest and classification performance on a hold-out species, providing clear evidence that our species-specific methods successfully leverage phylogenetic information for classification. We further demonstrate improved performance of two SVM-based classifiers and one random forest-based classifier for miRNA studies on reptile, insect, plant and virus genomes. Trained species-specific miRNA prediction systems and all training and test data are freely available at http://bioinf.sce.carleton.ca/SMIRP. As previously mentioned, this approach is particularly useful for niche species that are of important model organisms of study, but suffer from being phylogenetically distant from the model organisms that are typically used to create single-species or multi-species pooled training data.
MATERIALS AND METHODS
Framework overview
Figure 2 illustrates our proposed framework for constructing species-specific positive and negative datasets for training and evaluating miRNA prediction systems. Known miRNA sequences are first gathered from multiple species. These sequences are then clustered by identity or similarity such that redundant training data are grouped together. A single representative sequence is selected from each of the p largest clusters, such that the representative sequence derives from the species considered to be most closely related to the target species. For example, if studying Drosophila melanogaster, one would prefer training data from other insect species to data from human. The resulting sequences are used as the positive training data. Negative data are similarly taken from one or more species deemed to be closely related to the target species, for which annotated coding regions and ncRNA are available. Selection of source species for negative training data is performed manually based on phylogenetic information from resources such as the NCBI taxonomy browser (54) or that provided by miRBase (4). The SMIRP framework is robust with respect to this selection in that performance is generally consistent where negative training data are selected from various species within the same family. For example, when a D. melanogaster-specific classifier is retrained using negative data from D. simulans instead of D. pseudoobscura, no significant or consistent increase in performance is observed (see Supplementary Figure S2). For all experiments performed in this study, species selection for negative training datasets was performed manually based on the phylogenetic grouping of the miRBase database. Within the lowest ranking taxon containing the target species, the species with the highest number of known miRNA was chosen as the training species.
Figure 2.

Overview depiction of species-specific training dataset generation framework.
Generating species-specific positive training sets
Species-specific positive training datasets were built using the miRBase v.19 database (4). This database contains 20 982 miRNA sequences across 193 species. Redundant sequences and sequences containing non-AGCU characters were removed from the dataset, resulting in an initial training dataset containing 19 161 sequences. CD-hit (55) was then used to generate clusters of sequences within our initial dataset, using a threshold of 80% sequence identity. Default CD-hit parameters were used for clustering.
Using the miRBase sequence dataset and the clusters described above, we then developed positive training datasets for miRNA classification that were targeted towards the species of interest (referred to here as our target species). These datasets were developed as follows:
For a given positive integer p, the largest p clusters were chosen from the CD-hit clustering results. Larger values of p provide a larger number of positive data, however smaller values of p provide higher-quality data, representing larger families of well-conserved miRNA. Supplementary Figure S1 demonstrates that our method is largely insensitive to this tradeoff. Therefore, optimization of the parameter p is not a required step in the generation of positive training sets. For the experiments below, p was set to match the number of training data used to train either microPred (P = 692) or HeteroMirPred (P = 1000), however larger values of p may be used in general.
A representative sequence was chosen from each of the p clusters. For each cluster, the representative sequence of the cluster is the sequence which is found in the species nearest to the representative species in terms of phylogenetic classification. The phylogenetic classifications given within the miRBase database were used to classify species for the purpose of representative sequence selection.
In the event of multiple candidate representative sequences within a cluster whose species are equally close (phylogenetically) to our target species, the candidate sequence whose length is closest to the mean length of sequences within the cluster is chosen as the representative sequence for the cluster.
The resulting positive training dataset contains miRNA sequences that are highly conserved across species. Because homologs of these sequences have been verified in many species, the positive training dataset also represents miRNA whose functional annotation is well studied. Importantly, no two miRNA sequences are alike with >80% identity; therefore the dataset does not contain redundant sequences. In addition, miRNA sequences within the dataset are phylogenetically similar to the target species, increasing the likelihood of conservation between training data and miRNA to be predicted in unannotated target species.
Generating species-specific negative training datasets
Negative training datasets for a target species were generated from coding region (exonic) sequences and ncRNA sequences of species that are closely related to the target species, based on phylogenetic distance. Coding region and ncRNA sequences were retrieved from the European Nucleotide Archive (56). Once data was retrieved, the following steps were carried out:
Coding and ncRNA sequence data were combined and formatted into a FASTA format which is compatible with the ViennaRNA package (57).
Pseudo-miRNA sequences were extracted from the coding and ncRNA sequence data. Pseudo-miRNA sequences are defined as those that fold into a hairpin structure containing at least 18 stem pairs and a minimum free energy of at most −15 kCal/mol. These folding criteria are commonly used for the determination of miRNA hairpin candidates, and are the criteria with which the microPred negative training dataset was built (38,39).
Redundant pseudo-miRNA sequences were removed from the negative training set. A pseudo-miRNA sequence was considered redundant if it was a substring of another pseudo-miRNA sequence in the negative training set. The user can optionally specify that clustering should be also applied to the negative training data, as is done to the positive training data. Applying this optional step may affect prediction performance depending on the test species; all results below correspond to datasets built without using this option. A subset of candidate hairpin sequences of size n was chosen at random from the full list of sequences.
The resulting negative training sets contain n pseudo-miRNA sequences, which are derived from coding regions and ncRNA of a close relative to the target species.
RESULTS
Demonstrating effectiveness of framework
To demonstrate the effectiveness of our proposed species-specific dataset generation framework, it was applied to four diverse target species representing four distinct phyla: Anolis carolinensis, D. melanogaster, A. thaliana and Rhesus lymphocryptovirus. We refer to these four target species as ‘hold-out test species’. For each hold-out test species, we first generated species-specific positive and negative training sets for which data from the hold-out test species is withheld (i.e. we pretend that no sequence annotation is available for the target species). Testing datasets for each of the hold-out test species were extracted based on the withheld (known) annotations (i.e. true miRNA and exonic hairpin regions). In order to demonstrate the broad applicability of our framework, we have applied it to both the widely cited microPred classification pipeline (38) and the newer HeteroMirPred classification pipeline (31). Training sets generated by our framework are compared against equivalent datasets using human-only data (as used in (38)) and multi-species pooled data (as used in (31,37,48)). In all cases, the species-specific training data generated by our framework leads to substantial and consistent performance gains. Each step of this evaluation procedure is described in detail in the following sections.
Hold-out test species datasets
Positive and negative hold-out species datasets were generated for each of our four hold-out test species. The four positive test sets consist of all pre-miRNA sequences present in miRBase v.19 for the given hold-out test species. Corresponding negative test sets were built using the negative set generation method described in the Materials and Methods section; data was retrieved from the hold-out species genome. For species where an abundance of negative test data was present, the dataset size parameter n was set to 500. The number of sequences in each of the positive and negative hold-out sets can be seen in Table 1.
Table 1. Hold-out datasets used for testing of species-specific dataset generation.
| Species | Size of positive hold-out dataset | Size of negative hold-out dataset |
|---|---|---|
| A. carolinensis | 282 | 500 |
| D. melanogaster | 237 | 443 |
| A. thaliana | 298 | 500 |
| R. lymphocryptovirus | 35 | 86 |
Reference training datasets
We compared our species-specific training datasets with the training datasets used in the microPred and HeteroMirPred studies. These two datasets represent human-only training data and pooled multi-species training data, respectively.
MicroPred training dataset
The microPred training dataset, available at http://www.cs.ox.ac.uk/people/manohara.rukshan.batuwita/microPred.htm, has become a de-facto standard for the training of miRNA prediction methods, having been used in numerous studies since it was introduced in 2009 (40,29,31–32,37–38). This dataset contains 691 human pre-miRNA sequences as well as 9248 pseudo-miRNA hairpin candidates that appear in human coding regions and human ncRNA regions.
HeteroMirPred training dataset
The HeteroMirPred training dataset was not made publicly available, therefore we have re-created the dataset using the parameters described by Lertampaiporn et al. (31). The original positive training dataset consists of 1000 randomly selected non-redundant pre-miRNAs—600 from the H. sapiens genome, 200 from the O. sativa genome and 200 from the A. thaliana genome. Because A. thaliana is one of the species used in our hold-out test sets, our re-creation of the HeteroMirPred dataset uses Glycine max pre-miRNA in place of A. thaliana pre-miRNA.
While not explicitly stated in the study by Lertampaiporn et al., the negative set used to train the HeteroMirPred classifier is likely to be the same negative set used to train the microPred classifier. We believe this to be true since the two datasets have the same number of coding region sequences and ncRNA sequences, and the negative set generation methodology described by the two studies implies that this is the case. Therefore, we have used the microPred negative training set in our re-creation of the HeteroMirPred training dataset.
Species-specific training datasets
For each hold-out test species, the framework described above was applied to create species-specific training datasets. Positive data were selected with preference to samples from species which are phylogenetically similar to each of the hold-out species. Negative data were selected from species that were closely related to the hold-out test species, as follows: Xenopus tropicalis for the hold-out species A. carolinensis, D. pseudoobscura for D. melanogaster, Arabidopsis lyrata for A. thaliana and Epstein Barr virus for R. lymphocryptovirus. In order to ensure a fair comparison between existing training datasets and species-specific training datasets, the numbers of positive and negative patterns used in the species-specific datasets (p and n, respectively) match the number of positive and negative patterns used in the respective existing training set. Species-specific training sets based on the microPred classifier use P = 692 and n = 10 000, while species-specific training sets based on the heteroMirPred classifier use P = 1000 and n = 10 000. Minority class datasets were oversampled using SMOTE (58) such that positive and negative training datasets contained the same number of samples.
Model classifiers
We demonstrate the applicability of our dataset generation method using local implementations of the microPred and HeteroMirPred classifiers as published in (38) and (31), respectively. The microPred and HeteroMirPred classifiers had to be implemented locally since the original implementations were unsuitable for our experiments because they do not produce prediction confidence results (only binary predictions), and therefore cannot be analysed using precision–recall or ROC curves. We have therefore generated SVM classifiers, following the feature set and training protocol used in the original reports, using the LibSVM library (59). SVM hyperparameters found to be optimal over the reference datasets were used for all species-specific classifiers.
Classifier test protocol
For each hold-out test species, species-specific training datasets were compared with their respective human-specific and pooled training datasets on microPred and HeteroMirPred-like classifiers using the following protocol.
Training datasets were stratified into 10 equal subsets, as in an outer 10-fold cross-validation experiment. A classifier was then produced for each training set, such that each classifier was trained on 90% of the total training data. Each classifier was then used to predict the complete hold-out species test dataset, thereby providing 10 estimates of classification performance. The total performance of the 10 classifiers on the hold-out dataset was used as a measure of the effectiveness of the training set on which the 10 classifiers were trained. The same test procedure was used for both species-specific training datasets and reference training datasets.
Measuring performance of targeted species-specific models on hold-out datasets
We compare the performance of our species-specific training data generation approach with the approaches of the microPred and HeteroMirPred classification studies using the metrics of precision and recall. Since the class imbalance in our test data is not necessarily reflective of the actual degree of imbalance expected when the classifier is applied to a complete genome, we use the prevalence-corrected precision defined as:
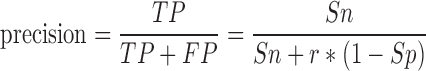 |
Where TP is the estimated true positive rate of the classifier, FP is the estimated false positive rate of the classifier, r is the expected ratio of negative to positive samples in the real-world data, and Sn and Sp are the estimated sensitivity and specificity of the classifier, respectively. Precision can be interpreted as the portion of predictions that are actually true miRNAs. Recall is synonymous with the sensitivity measure used in previous miRNA prediction studies and is defined as:
 |
Relative to the geometric mean and AUC metrics commonly used in the field of miRNA prediction, precision and recall better elucidate real-world performance for de novo miRNA experiments, where large class imbalances are expected, as these metrics operate at the expected class imbalance for a given classification problem.
Actual class imbalance in genome-wide miRNA prediction experiments varies based on the genome used. Within eukaryotic genomes we estimate the real-world class imbalance in miRNA prediction experiments to be ∼1000:1 in favour of the negative class. This is considered to be a conservative estimate, as the relatively compact D. melanogaster genome contains ∼800 000 non-redundant hairpin structures which satisfy conditions for miRNA candidacy, while the number of miRNA in this genome is only 466 as of miRBase v.20 (4) resulting in a ratio of 1716:1. Similarly, the H. sapiens genome contains ∼11 000 000 hairpin structures (60) and 2578 miRNA as of miRBase v.20 (ratio of 4267:1). Therefore, in the calculation of precision and recall within eukaryotic genomes, we set r to 1000, representing a 1:1000 class imbalance. Similarly, we estimate the class imbalance of de novo miRNA prediction within smaller viral genomes to be 1:100 and set the r value to 100 in accordance with this estimate.
Experimental results
Figure 3 presents the precision–recall curves for microPred-like classifiers trained on species-specific training data and human-specific training data as the classifier decision threshold varies from permissive to conservative. Figure 4 presents equivalent precision–recall curves for HeteroMirPred-like classifiers trained on species-specific training data and multi-species pooled training data. As can clearly be seen in these figures, our species-specific approach provides a consistent and substantial boost in recall for a wide range of precision values for all four test species.
Figure 3.

Comparison of species-specific training data with human-specific data on microPred-like model. In all precision–recall curves, the dashed blue curve indicates microPred-like prediction using human-trained model, while the solid red curve indicates the tailored species-specific approach developed in this study. MiRNA predictions were carried out for (A) A. carolinensis, (B) A. thaliana, (C) D. melanogaster and (D) R. lymphocryptovirus.
Figure 4.

Comparison of species-specific training data with human-specific data on HeteroMirPred-like model. All other information as mentioned in Figure 3.
In order to provide useful summary metrics for miRNA classification, we also report recall rates at the 90% and 50% precision levels, representing the number of near-guaranteed predictions made by the classifier (90% precision), and the portion of true miRNA expected to be recovered when operating at a typical acceptable experimental validation rate (50% precision). Table 2A and B summarize the recall rates of microPred-like classifiers trained on species-specific training data and the default microPred human training data, at precision rates of 90% and 50%, respectively. On average, application of species-specific training sets increases the recall rate at 90% precision by 152.3% and the recall rate at 50% precision by 199.9%. Tables 3A and B summarize analogous data using a HeteroMirPred-like classifier. Consistent with the results for the microPred-like classifier, substantial increases are observed for our proposed species-specific approach; on average, the recall rate at 90% precision is increased by 533.2% and recall rate at 50% precision is increased by 396.1%.
Table 2A. Recall at 90% precision, human-specific and our tailored species-specific training data using the microPred-like classifier.
| Hold-out test species | Human-specific training data | Species-specific training data | Increase in recall (%) |
|---|---|---|---|
| A. carolinensis | 0.254 | 0.583 | 130 |
| D. melanogaster | 0.094 | 0.311 | 231 |
| A. thaliana | 0.360 | 0.583 | 61.9 |
| R. lymphocryptovirus | 0.158 | 0.453 | 187 |
Table 3A. Recall at 90% precision, pooled training data and our tailored species-specific training data using the HeteroMirPred-like classifier.
| Hold-out test species | Pooled training data | Species-specific training data | Increase in recall (%) |
|---|---|---|---|
| A. carolinensis | 0.215 | 0.611 | 184 |
| D. melanogaster | 0.036 | 0.634 | 1660 |
| A. thaliana | 0.260 | 0.767 | 195 |
| R. lymphocryptovirus | 0.325 | 0.625 | 92.3 |
A test of significance was applied to each ‘Increase in recall’ result in Tables 2A, B, 3A and B. For each pair of SMIRP and reference classifiers, a distribution of increases in recall expected under the null hypothesis (H0: there is no significant difference in achievable recall between the two methods) was computed at both the 50% and 90% precision levels. These distributions were formed by repeatedly (N = 10 000) pooling the prediction scores from the two methods and drawing pseudo-samples labelled as SMIRP and reference. Row ordering was preserved resulting in a paired (matched) experiment design. For each pair of pseudosamples, the increase in recall was recorded. P-values were then computed for the actually observed increases in recall. All results in all four Tables 2A–3B were found to be significant at the P < 0.01 level except for the recall@precision = 50% results for R. lymphocryptovirus in Tables 2B and 3B.
Table 3B. Recall at 50% precision, pooled training data and our tailored species-specific training data using the HeteroMirPred-like classifier.
| Hold-out test species | Pooled training data | Species-specific training data | Increase in recall (%) |
|---|---|---|---|
| A. carolinensis | 0.232 | 0.620 | 167 |
| D. melanogaster | 0.051 | 0.639 | 1150 |
| A. thaliana | 0.285 | 0.781 | 174 |
| R. lymphocryptovirus | 0.325 | 0.629 | 93.5 |
Table 2B. Recall at 50% precision, human-specific and our tailored species-specific training data using the microPred-like classifier.
| Hold-out test species | Human-specific training data | Species-specific training data | Increase in recall (%) |
|---|---|---|---|
| A. carolinensis | 0.262 | 0.695 | 165 |
| D. melanogaster | 0.094 | 0.497 | 429 |
| A. thaliana | 0.370 | 0.588 | 58.9 |
| R. lymphocryptovirus | 0.208 | 0.514 | 147 |
Tables 4A and B present the number of miRNA recovered using our pipeline which do and do not share sequence similarity (80% or greater) with training data. Of the hold-out miRNA recovered by our microPred-like and HeteroMirPred-like classifiers, 60.6% and 59.6% do not share significant similarity with any of the training data. Therefore, SMIRP is capable of predicting miRNA which are not homologous to existing miRNA.
Table 4A. Average number of miRNA recovered at 50% precision which are and are not homologous to training data (MicroPred-like classifier).
| Hold-out test species | Number of miRNA recovered | Homologous to training data | Not homologous to training data |
|---|---|---|---|
| A. carolinensis | 196 | 87 | 109 |
| D. melanogaster | 118 | 73 | 45 |
| A. thaliana | 175 | 39 | 136 |
| R. lymphocryptovirus | 16 | 0 | 16 |
Here, homology is defined as 80% sequence identity or higher.
Table 4B. Average number of miRNA recovered at 50% precision which are, and are not, homologous to training data (HeteroMirPred-like classifier).
| Hold-out test species | Number of miRNA recovered | Homologous to training data | Not homologous to training data |
|---|---|---|---|
| A. carolinensis | 175 | 94 | 81 |
| D. melanogaster | 152 | 76 | 76 |
| A. thaliana | 232 | 65 | 167 |
| R. lymphocryptovirus | 22 | 0 | 22 |
Effect of phylogenetic distance on classification performance
In order to elucidate the effect of phylogenetic similarity within positive and negative datasets, we have performed additional classification experiments on the A. thaliana hold-out set. In each of these experiments, we varied the phylogenetic similarity between the hold-out species and our positive and negative training sets. Seven training datasets were generated, for which the following phylogenetic groups (clades) were removed: genus A. thaliana, family Arabidopsis, order Brassicaceae, clade Eurosids II, clade Rosids, clade Eudicots, kingdom Plantae. Negative training datasets were built using the following representative species, respectively: A. lyrata, Brassica napus, Theobroma cacao, Cucumis melo, O. sativa, Physcomitrella patens and H. sapiens. Figure 5 demonstrates a clear inverse correlation between classification performance and phylogenetic distance between training data and hold-out test data. This serves to validate SMIRP's underpinning hypothesis, in that training data should be taken preferentially from species as closely related to the target species as possible.
Figure 5.

Recall @ 50% Precision on A. thaliana hold-out test set, as phylogenetic distance between training data and A. thaliana is systematically increased. X-axis labels describe the phylogenetic group which was withheld during training dataset generation. Classification performance clearly decreases as the phylogenetic distance between training and testing species is increased.
Application of SMIRP to random forest classifiers
In order to demonstrate the applicability of SMIRP across multiple classifier types, we have compared the SMIRP dataset generation technique with the taxon-specific dataset generation approach of HuntMi (37) for the training of random forest classifiers. The HuntMi feature set, which contains the 21 microPred features and seven additional features, was used for this experiment. Random (decision) forest classifiers were trained using the scikit-learn (61) python library. Classifier hyperparameters were set to default values, with the exception of the number of trees which was set to n = 500. This high number of trees allows for more fine-grained classification confidence results relative to the lower default value of n = 10 since confidence is derived from the voting results among n individual trees. Classifiers were trained using the SMIRP species-specific datasets described above, and also using taxon-specific datasets representing animals, plants and viruses (as appropriate to each test hold-out species). Taxon-specific positive datasets contain all experimentally validated miRNA from miRBase version 19 for the respective taxon, excluding that of the hold-out species. Taxon-specific negative datasets are those provided by HuntMi (37). Each of these classifiers was then tested on the hold-out species test sets described above. Note that the random forest classification models used during these experiments are not identical to those used in the original HuntMi study (for example, the specific form of class probability thresholding differs here). As demonstrated in Supplementary Figure S3, species-specific datasets outperform the taxon-specific datasets by a large margin for all four hold-out species across all three major taxa. Importantly, since these experiments involve random forests, as opposed to SVMs used elsewhere in this manuscript, these results also demonstrate the broad applicability of the SMIRP framework to existing and future classifiers, regardless of machine learning approach.
Results of genome-wide Biomphalaria glabrata miRNA prediction
We applied the SMIRP dataset generation framework along with the HeteroMirPred-like classifier to the unannotated Biomphalaria glabrata (snail) genome with the goal of predicting novel miRNA. SMIRP was used as described above, using P = 600 and n = 10000. L. gigantean was used as the negative reference species. Using the SMIRP framework, 202 miRNA were discovered within the B. glabrata genome. Of these, 107 miRNAs are novel, and a significant portion of these novel miRNA were found to target genes involved in cellular processes specific to snail, such as secretory mucal proteins and shell formation. These results, which are detailed in Supplementary Table S1, demonstrate the ability of SMIRP to predict a large number of novel miRNAs within the unannotated genome of a non-model species.
DISCUSSION
Currently, state of the art methods for miRNA prediction do not provide adequate specificity for the efficient discovery of novel miRNA during genome-scale experiments on unannotated genomes. These novel miRNA discovery experiments are performed in the presence of very high-class imbalance (typically on the order of 1000 negative hairpin regions per one true positive miRNA), and experimental validation of positives is costly and time-consuming. As a result, very high precision and specificity are demanded of classifiers, and current efforts that are often tuned to maximize the geometric mean of sensitivity and specificity do not meet this demand. Furthermore, we have demonstrated that specificity of miRNA prediction decreases substantially when classifiers are asked to make miRNA predictions on species dissimilar to the species on which they were trained.
In order to provide precise classification of miRNA in unannotated genomes potentially distant from model species, we have introduced a framework for species-specific miRNA classification, which increases prediction performance for arbitrary species. This framework dynamically produces classification models for the test species under study. Positive training sets are produced through a two-step filtering process on the set of all available miRNA sequences from multiple species:
Generate clusters of miRNA based on sequence identity or similarity. The largest such clusters are representative of a large number of highly confident miRNAs that are conserved across species.
Select a representative miRNA from the largest clusters. Selection is based on phylogenetic similarity to the target species, increasing likelihood of conservation between representative miRNA and target species. In addition, selection of a single miRNA from each cluster ensures that the positive training set contains no redundant miRNA.
Negative training sets are built using coding regions and ncRNAs from annotated genomes of species that are closely related to the target species. As with the positive training sets, selection of closely related species here increases the likelihood of sequence conservation between the negative training set and the target genome.
We have demonstrated that SMIRP, our species-specific dataset generation framework, provides a dramatic increase in classifier performance relative to the human-specific dataset generation method of the microPred study (38), as well as the multi-species pooled dataset generation method of the HeteroMirPred study (31) and the taxon-specific method of HuntMi (37). This increase in performance holds across four distinct hold-out species representing four distinct phyla. By reporting precision at realistic class imbalance levels, our tests reflect the high-specificity operating points which are required during genome-wide miRNA prediction studies. Relative to pooled (HeteroMirPred) or human-specific (microPred) dataset generation methods, SMIRP results in a 4x increase in recall when demanding a precision of 90% (i.e. 4x more true miRNAs are identified while expecting 90% of predicted miRNAs to be true). Consistent increases in classification performance were observed when using both SVM and random forest classifiers indicating the broad applicability of the SMIRP framework. We have demonstrated that SMIRP-based classifiers are able to predict novel miRNA, without homology to training data. Applying this method to the unannotated genome of B. glabrata (snail), 202 miRNA were discovered, of which 107 were novel and many of these are snail-specific. SMIRP can be applied to any existing or future miRNA prediction method, providing increased classification performance for all experiments on unannotated genomes.
The four pre-trained microPred-like and HeteroMirPred-like species-specific miRNA prediction systems evaluated in this study are available as SMIRP, at http://bioinf.sce.carleton.ca/SMIRP. We expect that these four classifiers will be useful for other closely related species. For example, the A. thaliana classifier is likely to be more effective for predicting miRNA in other plant species than would be the default human-only or multi-species pooled classifiers. Furthermore, all software for preparing species-specific datasets is available in open source at http://bioinf.sce.carleton.ca/SMIRP, as well as the training and testing data used in this study.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Natural Sciences and Engineering Research Council of Canada [RGPIN/327498-2009 to J.G.]. K.B.S. holds the Canada Research Chair in Molecular Physiology, while R.P. and K.K.B. both held NSERC postgraduate fellowships. Funding for open access charge: Research [RGPIN/327498-2009].
Conflict of interest statement. None declared.
REFERENCES
- 1.Humphreys D.T., Westman B.J., Martin D.I.K., Preiss T. MicroRNAs control translation initiation by inhibiting eukaryotic initiation factor 4E/cap and poly(A) tail function. Proc. Natl. Acad. Sci. U. S. A. 2005;102:16961–16966. doi: 10.1073/pnas.0506482102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bartel D. MicroRNAsGenomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 3.Miranda K.C., Huynh T., Tay Y., Ang Y.-S., Tam W.-L., Thomson A.M., Lim B., Rigoutsos I. A pattern-based method for the identification of MicroRNA binding sites and their corresponding heteroduplexes. Cell. 2006;126:1203–1217. doi: 10.1016/j.cell.2006.07.031. [DOI] [PubMed] [Google Scholar]
- 4.Kozomara A., Griffiths-Jones S. MiRBase: integrating microRNA annotation and deep-sequencing data. Nucleic Acids Res. 2011;39:152–157. doi: 10.1093/nar/gkq1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.La Torre A., Georgi S., Reh T.A. Conserved microRNA pathway regulates developmental timing of retinal neurogenesis. Proc. Natl. Acad. Sci. U. S. A. 2013;110:E2362–E2370. doi: 10.1073/pnas.1301837110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Le M.T.N., Xie H., Zhou B., Chia P.H., Rizk P., Um M., Udolph G., Yang H., Lim B., Lodish H.F. MicroRNA-125b promotes neuronal differentiation in human cells by repressing multiple targets. Mol. Cell. Biol. 2009;29:5290–5305. doi: 10.1128/MCB.01694-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Körner C., Keklikoglou I., Bender C., Wörner A., Münstermann E., Wiemann S. MicroRNA-31 sensitizes human breast cells to apoptosis by direct targeting of protein kinase C epsilon (PKCepsilon) J. Biol. Chem. 2013;288:8750–8761. doi: 10.1074/jbc.M112.414128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Iwasaki Y.W., Kiga K., Kayo H., Fukuda-Yuzawa Y., Weise J., Inada T., Tomita M., Ishihama Y., Fukao T. Global microRNA elevation by inducible Exportin 5 regulates cell cycle entry. RNA. 2013;19:490–497. doi: 10.1261/rna.036608.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Maistrovski Y., Biggar K.K., Storey K.B. HIF-1α regulation in mammalian hibernators: role of non-coding RNA in HIF-1α control during torpor in ground squirrels and bats. J. Comp. Physiol. B. 2012;182:849–859. doi: 10.1007/s00360-012-0662-y. [DOI] [PubMed] [Google Scholar]
- 10.Kowarsch A., Marr C., Schmidl D., Ruepp A., Theis F.J. Tissue-specific target analysis of disease-associated microRNAs in human signaling pathways. PLoS One. 2010;5:e11154. doi: 10.1371/journal.pone.0011154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Biggar K.K., Kornfeld S.F., Maistrovski Y., Storey K.B. MicroRNA regulation in extreme environments: differential expression of microRNAs in the intertidal snail Littorina littorea during extended periods of freezing and anoxia. Genomics Proteomics Bioinformatics. 2012;10:302–309. doi: 10.1016/j.gpb.2012.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Biggar K.K., Storey K.B. Evidence for cell cycle suppression and microRNA regulation of cyclin D1 during anoxia exposure in turtles. Cell Cycle. 2012;11:1705–1713. doi: 10.4161/cc.19790. [DOI] [PubMed] [Google Scholar]
- 13.Wu C.-W., Biggar K.K., Storey K.B. Dehydration mediated microRNA response in the African clawed frog Xenopus laevis. Gene. 2013;529:269–275. doi: 10.1016/j.gene.2013.07.064. [DOI] [PubMed] [Google Scholar]
- 14.Sebastian B., Aggrey S.E. MiR-Explore: predicting microRNA precursors by class grouping and secondary structure positional alignment. Bioinform. Biol. Insights. 2013;7:133–142. doi: 10.4137/BBI.S10758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lim L.P., Lau N.C., Weinstein E.G., Abdelhakim A., Yekta S., Rhoades M.W., Burge C.B., Bartel D.P. The microRNAs of Caenorhabditis elegans. Genes Dev. 2003;17:991–1008. doi: 10.1101/gad.1074403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang X., Zhang J., Li F., Gu J., He T., Zhang X., Li Y. MicroRNA identification based on sequence and structure alignment. Bioinformatics. 2005;21:3610–3614. doi: 10.1093/bioinformatics/bti562. [DOI] [PubMed] [Google Scholar]
- 17.Legendre M., Lambert A., Gautheret D. Profile-based detection of microRNA precursors in animal genomes. Bioinformatics. 2005;21:841–845. doi: 10.1093/bioinformatics/bti073. [DOI] [PubMed] [Google Scholar]
- 18.Dezulian T., Remmert M., Palatnik J.F., Weigel D., Huson D.H. Identification of plant microRNA homologs. Bioinformatics. 2006;22:359–360. doi: 10.1093/bioinformatics/bti802. [DOI] [PubMed] [Google Scholar]
- 19.Sewer A., Paul N., Landgraf P., Aravin A., Pfeffer S., Brownstein M.J., Tuschl T., van Nimwegen E., Zavolan M. Identification of clustered microRNAs using an ab initio prediction method. BMC Bioinformatics. 2005;6:267. doi: 10.1186/1471-2105-6-267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gao D., Middleton R., Rasko J.E.J., Ritchie W. MiREval 2.0: a web tool for simple microRNA prediction in genome sequences. Bioinformatics. 2013;29:3225–3226. doi: 10.1093/bioinformatics/btt545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang L., Li J., Zhu R., Xu L., He Y., Zhang R. A novel stepwise support vector machine (SVM) method based on optimal feature combination for predicting miRNA precursors. African J. Biotechnol. 2011;10:16720–16731. [Google Scholar]
- 22.Kadri S., Hinman V., Benos P. V. HHMMiR: efficient de novo prediction of microRNAs using hierarchical hidden Markov models. BMC Bioinformatics. 2009;10(Suppl. 1):S35. doi: 10.1186/1471-2105-10-S1-S35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhong L., Wang J.T.L., Wen D., Aris V., Soteropoulos P., Shapiro B.A. Effective classification of microRNA precursors using feature mining and AdaBoost algorithms. OMICS. 2013;17:486–493. doi: 10.1089/omi.2013.0011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lopes I., Schliep A., de Carvalho A. The discriminant power of RNA features for pre-miRNA recognition. BMC Bioinformatics. 2013;15:1–21. doi: 10.1186/1471-2105-15-124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shakiba N., Rueda L. MicroRNA identification using linear dimensionality reduction with explicit feature mapping. BMC Proc. 2013;7:S8. doi: 10.1186/1753-6561-7-S7-S8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu X., He S., Skogerbø G., Gong F., Chen R. Integrated sequence-structure motifs suffice to identify microRNA precursors. PLoS One. 2012;7:e32797. doi: 10.1371/journal.pone.0032797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wei L., Liao M., Gao Y., Ji R., He Z., Zou Q. Improved and promising identification of human microRNAs by incorporating a high-quality negative set. IEEE/ACM Trans. Comput. Biol. Bioinform. 2013;11:192–201. doi: 10.1109/TCBB.2013.146. [DOI] [PubMed] [Google Scholar]
- 28.Titov I.I., Vorozheykin P.S. Ab initio human miRNA and pre-miRNA prediction. J. Bioinform. Comput. Biol. 2013;11:1343009. doi: 10.1142/S0219720013430099. [DOI] [PubMed] [Google Scholar]
- 29.Xiao J., Tang X., Li Y., Fang Z., Ma D., He Y., Li M. Identification of microRNA precursors based on random forest with network-level representation method of stem-loop structure. BMC Bioinformatics. 2011;12:165. doi: 10.1186/1471-2105-12-165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ng K.K.L.S., Mishra S.K.S., Loong K., Ng S. De novo SVM classification of precursor microRNAs from genomic pseudo hairpins using global and intrinsic folding measures. Bioinformatics. 2007;23:1321–1330. doi: 10.1093/bioinformatics/btm026. [DOI] [PubMed] [Google Scholar]
- 31.Lertampaiporn S., Thammarongtham C., Nukoolkit C., Kaewkamnerdpong B., Ruengjitchatchawalya M. Heterogeneous ensemble approach with discriminative features and modified-SMOTEbagging for pre-miRNA classification. Nucleic Acids Res. 2013;41:e21. doi: 10.1093/nar/gks878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xuan P., Guo M.Z., Wang J., Wang C.Y., Liu X.Y., Liu Y. Genetic algorithm-based efficient feature selection for classification of pre-miRNAs. Genet. Mol. Res. 2011;10:588–603. doi: 10.4238/vol10-2gmr969. [DOI] [PubMed] [Google Scholar]
- 33.Yousef M., Jung S., Showe L.C., Showe M.K. Learning from positive examples when the negative class is undetermined–microRNA gene identification. Algorithms Mol. Biol. 2008;3:2. doi: 10.1186/1748-7188-3-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yousef M., Nebozhyn M., Shatkay H., Kanterakis S., Showe L.C., Showe M.K. Combining multi-species genomic data for microRNA identification using a Naive Bayes classifier. Bioinformatics. 2006;22:1325–1334. doi: 10.1093/bioinformatics/btl094. [DOI] [PubMed] [Google Scholar]
- 35.Jiang P., Wu H., Wang W., Ma W., Sun X., Lu Z. MiPred: classification of real and pseudo microRNA precursors using random forest prediction model with combined features. Nucleic Acids Res. 2007;35:W339–W344. doi: 10.1093/nar/gkm368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Loong K., Ng S., Mishra S.K., Ng K.L.S. De novo SVM classification of precursor microRNAs from genomic pseudo hairpins using global and intrinsic folding measures. Bioinformatics. 2007;23:1321–1330. doi: 10.1093/bioinformatics/btm026. [DOI] [PubMed] [Google Scholar]
- 37.Gudyś A., Szcześniak M.W., Sikora M., Makalowska I. HuntMi: an efficient and taxon-specific approach in pre-miRNA identification. BMC Bioinformatics. 2013;14:83. doi: 10.1186/1471-2105-14-83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Batuwita R., Palade V. MicroPred: effective classification of pre-miRNAs for human miRNA gene prediction. Bioinformatics. 2009;25:989–995. doi: 10.1093/bioinformatics/btp107. [DOI] [PubMed] [Google Scholar]
- 39.Xue C., Li F., He T., Liu G.-P., Li Y., Zhang X. Classification of real and pseudo microRNA precursors using local structure-sequence features and support vector machine. BMC Bioinformatics. 2005;6:310. doi: 10.1186/1471-2105-6-310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Han K. Effective sample selection for classification of pre-miRNAs. Genet. Mol. Res. 2011;10:506–518. doi: 10.4238/vol10-1gmr1054. [DOI] [PubMed] [Google Scholar]
- 41.Ding J., Zhou S., Guan J. MiRenSVM: towards better prediction of microRNA precursors using an ensemble SVM classifier with multi-loop features. BMC Bioinformatics. 2010;11(Suppl. 1):S11. doi: 10.1186/1471-2105-11-S11-S11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Titov I.I., Vorozheykin P.S. Ab initio human miRNA and pre-miRNA prediction. J. Bioinform. Comput. Biol. 2013;11:1343009. doi: 10.1142/S0219720013430099. [DOI] [PubMed] [Google Scholar]
- 43.Friedländer M.R., Chen W., Adamidi C., Maaskola J., Einspanier R., Knespel S., Rajewsky N. Discovering microRNAs from deep sequencing data using miRDeep. Nat. Biotechnol. 2008;26:407–415. doi: 10.1038/nbt1394. [DOI] [PubMed] [Google Scholar]
- 44.Friedländer M.R., Mackowiak S.D., Li N., Chen W., Rajewsky N. miRDeep2 accurately identifies known and hundreds of novel microRNA genes in seven animal clades. Nucleic Acids Res. 2012;40:37–52. doi: 10.1093/nar/gkr688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Saçar M.D., Hamzeiy H., Allmer J. Can MiRBase provide positive data for machine learning for the detection of MiRNA hairpins. J. Integr. Bioinform. 2013;10:215. doi: 10.2390/biecoll-jib-2013-215. [DOI] [PubMed] [Google Scholar]
- 46.Burnside J., Ouyang M., Anderson A., Bernberg E., Lu C., Meyers B.C., Green P.J., Markis M., Isaacs G., Huang E., et al. Deep sequencing of chicken microRNAs. BMC Genomics. 2008;9:185. doi: 10.1186/1471-2164-9-185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Szittya G., Moxon S., Santos D.M., Jing R., Fevereiro M.P.S., Moulton V., Dalmay T. High-throughput sequencing of Medicago truncatula short RNAs identifies eight new miRNA families. BMC Genomics. 2008;9:593. doi: 10.1186/1471-2164-9-593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wu Y., Wei B., Liu H., Li T., Rayner S. MiRPara: a SVM-based software tool for prediction of most probable microRNA coding regions in genome scale sequences. BMC Bioinformatics. 2011;12:107. doi: 10.1186/1471-2105-12-107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hsu S.-D., Tseng Y.-T., Shrestha S., Lin Y.-L., Khaleel A., Chou C.-H., Chu C.-F., Huang H.-Y., Lin C.-M., Ho S.-Y., et al. miRTarBase update 2014: an information resource for experimentally validated miRNA-target interactions. Nucleic Acids Res. 2014;42:D78–D85. doi: 10.1093/nar/gkt1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mathelier A., Carbone A. MIReNA: finding microRNAs with high accuracy and no learning at genome scale and from deep sequencing data. Bioinformatics. 2010;26:2226–2234. doi: 10.1093/bioinformatics/btq329. [DOI] [PubMed] [Google Scholar]
- 51.Dang X.T. A novel over-sampling method and its application to miRNA prediction. J. Biomed. Sci. Eng. 2013;06:236–248. [Google Scholar]
- 52.Xu Y., Zhou X., Zhang W. MicroRNA prediction with a novel ranking algorithm based on random walks. Bioinformatics. 2008;24:i50–i58. doi: 10.1093/bioinformatics/btn175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pitre S., Hooshyar M., Schoenrock A., Samanfar B., Jessulat M., Green J.R., Dehne F., Golshani A. Short co-occurring polypeptide regions can predict global protein interaction maps. Sci. Rep. 2012;2:239. doi: 10.1038/srep00239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sayers E.W., Barrett T., Benson D.A., Bryant S.H., Canese K., Chetvernin V., Church D.M., DiCuccio M., Edgar R., Federhen S., et al. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2009;37:D5–D15. doi: 10.1093/nar/gkn741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Huang Y., Niu B., Gao Y., Fu L., Li W. CD-HIT suite: a web server for clustering and comparing biological sequences. Bioinformatics. 2010;26:680–682. doi: 10.1093/bioinformatics/btq003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Leinonen R., Akhtar R., Birney E., Bower L., Cerdeno-Tárraga A., Cheng Y., Cleland I., Faruque N., Goodgame N., Gibson R., et al. The European Nucleotide Archive. Nucleic Acids Res. 2011;39:D28–D31. doi: 10.1093/nar/gkq967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hofacker I., Fontana W. Fast folding and comparison of RNA secondary structures. Chem. Mon. 1994;125:167–188. [Google Scholar]
- 58.Chawla N., Bowyer K. SMOTE: synthetic minority over-sampling technique. J. Artificial Intell. Res. 2011;16:321–357. [Google Scholar]
- 59.Chang C., Lin C. LIBSVM: a library for support vector machines. ACM Trans. Intell. Syst. Technol. 2011;2:1–27. [Google Scholar]
- 60.Bentwich I. Prediction and validation of microRNAs and their targets. FEBS Lett. 2005;579:5904–5910. doi: 10.1016/j.febslet.2005.09.040. [DOI] [PubMed] [Google Scholar]
- 61.Pedregosa F., Varoquaux G. Scikit-learn: machine learning in Python. J. Mach. Learn. Res. 2011;12:2825–2830. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.