


Abstract
The human reference assembly remains incomplete due to the underrepresentation of repeat-rich sequences that are found within centromeric regions and acrocentric short arms. Although these sequences are marginally represented in the assembly, they are often fully represented in whole-genome short-read datasets and contribute to inappropriate alignments and high read-depth signals that localize to a small number of assembled homologous regions. As a consequence, these regions often provide artifactual peak calls that confound hypothesis testing and large-scale genomic studies. To address this problem, we have constructed mapping targets that represent roughly 8% of the human genome generally omitted from the human reference assembly. By integrating these data into standard mapping and peak-calling pipelines we demonstrate a 10-fold reduction in signals in regions common to the blacklisted region and identify a comprehensive set of regions that exhibit mapping sensitivity with the presence of the repeat-rich targets.
INTRODUCTION
Short-read mapping and enrichment studies from functional whole-genome datasets are important for guiding our understanding of genome regulation (1). To ensure high-confidence peak calls many analyses focus exclusively on non-repetitive regions of the genome or only on mapped reads that have a single, best alignment score, thereby removing read alignments that map equally well to multiple repeat sequences (2–4). When enrichment profiles include repeat sequences with more than one optimal placement in the genome it is often assumed (i.e. when using the Burrows-Wheeler Alignment Tool, bwa mem, http://arxiv.org/abs/1303.3997) that the multiple mapping reads are assigned randomly, thereby promoting a uniform distribution of alignments to avoid mapping. This multi-mapping strategy is challenged by sequences that are dramatically underrepresented in the genome assembly, such as tandem repeats that occupy millions of bases within centromeric and acrocentric regions (5). In these cases, reads are forced to map to a small number of homologous regions in the genome, resulting in large, artifactual read pile-ups (as illustrated in Figure 1a). It is possible, given the limited representation of these sequences in the assembled genome, that a single region can provide a best alignment score challenging standard efforts to eliminate mapping to repeats. Such outliers confound large-scale genomic analyses, involving training sets in supervised learning methods or efforts to study Pearson correlations between datasets.
Figure 1.
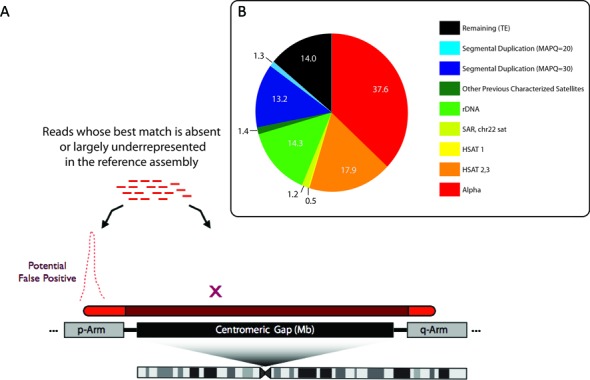
Large, multi-megabase sized regions of the human genome remain incomplete due to highly repetitive regions of the human genome, mapping to centromere/heterochromatin assigned gaps, and including sequences that remain missing from subtelomeric regions in the acrocentric short arms. As shown in (A), these regions are marked in the genome by gaps or space holders to indicate regions that are enriched for long arrays of tandemly repeated DNA. Often the edges of these gaps provide some representation of the sequences across the entirety of the array (shown as red if included in the assembly and shaded red if inferred to be present in the gap region). Sequence reads from the entire region are expected to be present in high-throughput, whole-genome datasets. When mapping to a partial reference, these reads find their best alignments on the regions represented in the assembly. As a result, a large number of reads (representing the multi-megabase arrays) align with high read depth, resulting in false positive sites in the genome. To account for these mapping errors we have designed mapping targets, collectively called a ‘sponge database’ with the various distribution of DNA families shown in (B) for the collection of 1.5 million remaining unassembled reads from the HuRef genome.
Standard analyses aimed to characterize high-throughput sequencing data are aware of these artifact alignments. Efforts to mitigate these mapping errors often involve providing an additional ‘decoy’ database, or a collection of sequences that are missing from the human genome that are useful to ensure proper alignment (6). Additionally, previous methods have been used to track sites of extensive read pile-up capable of generating artifact peak calls, known as ‘blacklisted’ regions (1) (http://www.broadinstitute.org/∼anshul/projects/encode/rawdata/blacklists/hg19-blacklist-README.pdf). These blacklisted sites in the genome enable researchers to filter these robust, non-biological signals of enrichment from downstream studies and hypotheses testing. Often, determining the location of these artifact signals is an active, and in some cases manual, process that must be repeated with each reference genome update, and released datasets vary considerably based on the method of prediction. Furthermore, blacklist coordinates typically emphasize sites that are observed across a collection of genomic data, thereby omitting artifact alignments that are specific to one or a small proportion of samples.
To address this challenge, we have generated mapping targets (referred to here as the ‘sponge’ sequence database) that represent roughly 8% of the human genome that are missing or underrepresented in the human reference assembly. Similar to the ‘decoy genome’ strategy, the sponge database improves read mapping by allowing best scored alignments to a collection of sequences that are not typically present in the reference chromosome assemblies. However, unlike previous methods, the sponge database is also useful in reducing artifacts when allowing for multi-mapping read alignments. By representing a stoichiometric version of repeat-rich sequences missing from the human reference genome, exact multi-mapping read alignments are randomly distributed across all possible sites. As a result, it is possible to greatly reduce large, artifactual read pile-ups in the human reference genome. When including the sponge sequence database in short-read mapping and peak-calling sequence protocols we observe a sharp decrease in read alignments within blacklisted regions. We show that this reduction, while useful in targeting characterized artifact regions, does not alter enrichment profiles that benefit from multiple lines of biological support. By applying these analyses to DNase short-read datasets we remove aberrant mapping, thereby improving large-scale correlation values. As an extension of this work, we are able to identify additional sites that demonstrate mapping sensitivity in the presence of the sponge mapping targets and are common across datasets and variable among individuals. Here we demonstrate the utility of this approach on the human reference genome using available functional and genomic datasets, with the expectation that this method will be easily extendable to additional datasets and genome assemblies that lack prior annotation of artifact regions.
MATERIALS AND METHODS
Creating the sponge database: characterizing unmapped read libraries
The sponge database is defined by a collection of HuRef whole-genome shotgun (WGS) reads that are not included in human chromosome assemblies (i.e. HuRef: GCA_000002125.2, GRCh37: GCA_000001405.1 and/or GRCh38: GCA_000001305.2) (7). HuRef read fasta files and quality information were downloaded directly from the NCBI Trace Archive using the following query: CENTER_NAME = ‘JCVI’ and SPECIES_CODE = ‘HOMO SAPIENS’ and center_project = ‘GENOMIC-SEQUENCING-DIPLOID-HUMAN-REFERENCE-GENOME’. HuRef assembled contigs used in this study are provided through GenBank ABBA00000000.1; BioProject: PRJNA19621, BioSample: SAMN02981236. HuRef WGS reads were considered ‘unmapped’ and included in the sponge database if they remain unassembled, i.e. excluded from all assembled contigs, or if they are present in an assembled contig that lacks assignment to a particular chromosome reference assembly. To identify the reads that passed the criteria to be included in the sponge database we used available ‘posmap’ data tables that reference each WGS read assignment to published contig assemblies: ftp://ftp.jcvi.org/pub/data/huref/h6.posmap.frgctg.gz. In total, our initial HuRef WGS sponge database represents 246 Mb of unmapped sequences, or roughly 8.2% of the genome (7), described below:
Satellite DNAs
Initial characterization of read annotation for tandem repeats was performed using RepeatMasker (‘s’ crossmatch– sensitive, default ‘human’ parameters/Library release: 20140131; http://wwwrepeatmaskerorg) (8). Uncharacterized sequences were studied for the presence of a de novo tandem repeat (tandem repeat finder, using parameters Match = 2 Mismatch = 7 Delta = 7 PM = 80 PI = 10 Minscore = 50 MaxPeriod = 1000) (9) or homology using RepeatMasker software with a specified library of previously characterized human satellite DNAs in GenBank, with the following accessions: M25748.1,M25749.1, AF020783.1, AF020782.1, X87951.1, AJ245409.1, X68546.1, X68545.1, M25748.1, M25749.1. Satellite sequences representing alpha satellite and HSat2,3 were collected from previous published databases using the HuRef genome (10–12). Alpha satellite reference models are only included in the GRCh37 version of the sponge database, as these sequences are present in the GRCh38 chromosome reference assemblies. In an effort to present stoichiometric estimates in the sponge database, individual satellite read libraries were normalized relative to genome-wide sequence coverage (with read estimates provided in Supplementary Table S1). Normalized alpha satellite sequences were included as reference models (method previously described in (11)), where the length of the linear sequence is determined by normalized read depth.
Ribosomal DNA sequences
Reads containing ribosomal DNA sequence were identified using RepeatMasker (‘s’ crossmatch– sensitive) using a complete repeating unit (GenBank Accession: U13369.1). In support of the published ribosomal DNA repeat copy number estimates (13,14), we predict roughly 450 copies of the ribosomal DNA repeat based on read depth estimates. Within the sponge database we used 3469 sampled rDNA reads (total bp: 17218973) to provide 400x copies of the 42 999 repeat.
Remaining unmapped and previously uncharacterized sequences
Remaining, high quality unmapped HuRef WGS reads (defined as containing at least 100 bases with as least a phred score of 30) were included in the sponge database if they did not align to the human reference chromosome assemblies (GRCh37: GCA_000001405.1 and/or GRCh38: GCA_000001305.2) with greater than 95% identity with at least 60% read alignment (mapping performed using bwasw, default parameters). Reads removed due to shared high sequence homology with chromosome reference assemblies were assumed to represent misassembled sequences specific to the HuRef assembly that are correctly represented in the reference assemblies. Remaining WGS reads were reduced to an estimated stoichiometric amount (or the randomly selected 0.125 proportion of the 8x sequence coverage in the HuRef WGS genome, representing roughly 1x coverage (7)).
Mitochondria sequences
In addition to HuRef WGS reads, we included a separate sequence database containing human mitochondrial DNAs (Human mtDB: http://www.mtdb.igp.uu.se/ (15)). In an attempt to represent a rough stoichiometric estimate of mitochondrial genomes we included 1000 mitochondria genomes from a diverse collection of human populations (15) (Supplementary Figure S1) representing an estimate of non-nuclear genomes expected from an average human cell (16). A total of 500 published mitochondrial genomes were organized in tandem to avoid edge effects introduced when mapping reads to either the start or the end of the circular genome.
Final stoichiometric sponge sequence libraries used in this study are available in Supplementary Datasets 1 and 2 (sponge_GRCh37 and sponge_GRCh38, respectively). As sequence abundance of repeat families is suspected to vary, we also generated mapping target read databases of increasing coverage (1x, 2x, 4x and 8x sequence coverage).
Alignments and peak-calling protocol with the general sponge against short-read datasets
The sponge database functions in two ways to eliminate artifact read alignments: first, the sequence database provides an opportunity to generate exact match alignments to sequences included in the sponge database, thereby removing inexact matches from the reference assembly. Second, the sponge database is useful in identifying and eliminating signals due to multiple exact, or ‘best scored’ matches that are shared between the sponge and the reference database (with randomly distributed read alignments using bwa mem, http://bio-bwa.sourceforge.net/bwa.shtml - 13). This strategy relies on random distribution of read alignments over possible sites of multi-mapping sites and benefits from rough stoichiometric representation of sequences in the human genome.
We used the sponge database as a mapping target to study read alignment profiles across diverse ENCODE ChIP-seq datasets (1), as well as low-coverage genomic datasets from two individuals from diverse populations (HuRef (10), Western European and GM12939, Yoruba). Links to datasets are provided in Supplementary Table S2. Reads were mapped against a reference database containing GRCh37 assembly (GCA_000001405.1; chromosomes and mitochondrial genome) with or without the sponge sequence database. Comparisons with the decoy sequences used in the standard 1000 genome analysis pipeline (6) used the reference database: hs37d5 (ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/technical/reference/phase2_reference_assembly_sequence/hs37d5.fa.gz).
By including these mapping targets in standard alignment (Burrows-Wheeler Alignment Tool, BWA-MEM, allowing for multi-mapping, with default alignment parameters) and peak-calling protocols (Model-based Analysis of ChIP-Seq (MACS, version macs14)) (17) we were able to evaluate improvement in read assignment within blacklisted regions, study gain and/or loss of enrichment profiles and characterize both common and cell-line specific sites not previously identified to present artifact alignments. Primary analyses were performed using the stoichiometric version of the sponge database (estimated to represent sequences in roughly 1x coverage). Additionally, we included increasing proportions of the database (1x, 2x, 4x and 8x coverage) to evaluate mapping efficiencies that are sensitive to sequence abundance of a given mapping target. Alignment files and significant peaks were initially evaluated by intersecting with BED files provided for three separate blacklists (Description available http://www.broadinstitute.org/∼anshul/projects/encode/rawdata/blacklists/hg19-blacklist-README.pdf; Blacklist based on RepeatMasker annotation: ftp://encodeftp.cse.ucsc.edu/users/akundaje/rawdata/blacklists/hg19/Duke_Hg19SignalRepeatArtifactRegions.bed.gz; Merged Consensus blacklist: ftp://encodeftp.cse.ucsc.edu/users/akundaje/rawdata/blacklists/hg19/wgEncodeHg19ConsensusSignalArtifactRegions.bed.gz and Ultra-high signal artifacts: ftp://encodeftp.cse.ucsc.edu/users/akundaje/rawdata/blacklists/hg19/wgEncodeHg19ConsensusSignalArtifactRegions.bed.gz). Previously characterized CTCF peaks served as controls in our study (TCF Binding Sites by ChIP-seq from ENCODE/University of Washington; obtained from UCSC Genome Table Browser GRCh37/hg19: Track = ‘UW CTCF Binding’; Table = ‘GM78 CTCF Ht 1 (wgEncodeUwTfbsGm12878CtcfStdHotspotsRep1)’). Transcript locations were defined by RefSeq (18) (RefSeq assembly accession: GCF_000001405.13; obtained from UCSC Genome Table Browser for GRCh37/hg19: Track = ‘RefSeq Genes’; Table = ‘refGene’), and control promoter regions were defined as 1 kb upstream of a defined RefSeq gene. Intersection between bedfiles was made with bedtools intersect (v2.18.2) (19).
Correlation plots and generation of dendogram of DNAse I datasets
Read mapping protocols were performed as previously described (20) and in line with ENCODE 3 established alignment protocols (notably, differs from previous mapping protocols used in this study in that multi-mapping reads are ignored), with or without the addition of sponge mapping targets (with stoichiometric estimates, 1x coverage). Comparisons between bigWig alignment files used a base-by-base Pearson correlation with bigWigCorrelation software (version 2), developed in house to enable correlation values between full genome bigWig files (commonly involving up to 3 billion data points each) and also convert to a bedgraph format necessary for downstream analyses. This tool is publically available through the UCSC applications/software code release (v316; with procedure to obtain the user application tool set and how to build the tools is in this README: http://genome-source.cse.ucsc.edu/gitweb/?p=kent.git;a=blob;f=src/userApps/README). Pairwise comparisons were performed using bigWigCorrelation and scored based on similarity or ranking by summed Pearson correlation values. The resulting outputs were converted to a square matrix and clusters were visualized using imagesc in MatLab (MATLAB, The MathWorks Inc. 2000, Natick, MA, USA). Subtracting correlation values generated when using the sponge mapping targets from those values without the sponge mapping targets (or GRCh37 chromosome assemblies: GCA_000001405.1) provided a second matrix to identify regions of increased correlation.
The dendrogram was generated with bigWig alignment files from DNAse datasets from a variety of cell types and individuals (with mapping protocol described above). Clustering was performed in a hierarchical manner, using in-house utility bigWigCluster, where linkage was determined by merging individual DNase datasets in a pairwise manner based on ranked similarity. Merging was performed with an in-house utility, bigWigMerge, used to merge together multiple bigWigs into a single output bedGraph necessary when using the bigWigCorrelation software. These tools are also available through the UCSC applications/software code release (v316; with procedure to obtain the user application tool set and how to build the tools is in this README: http://genome-source.cse.ucsc.edu/gitweb/?p=kent.git;a=blob;f=src/userApps/README). The resulting binary tree (.json format) was visualized using the cluster function from Javascript library d3 to generate a radial dendrogram. The root node, representing the merged result of bigWig files, was placed at the center of the graph. Smaller node size corresponds to similar bigWig children, and larger node corresponds to diverging bigWig children.
RESULTS
Description of sponge database
The human reference genome remains incomplete; yet, roughly 8% of sequences that occupy unfinished regions (typically associated with centromeric and constitutive heterochromatin regions) are present in most short-read datasets. These sequences are typically misaligned to a small number of sites that are present in the reference assembly that share sufficient sequence similarity, often resulting in large artifactual read pile-ups (Figure 1a). To address this problem we have generated a database containing those sequences missing from the human genome, representing 246 Mb or roughly 8.2% of the HuRef genome (7). The majority (80%) of the unmapped sequences are characterized as large tandem repeat families, including ribosomal repeats and satellite DNAs, previously observed to be enriched in centromeric regions and acrocentric short arms (Figure 1b).
In the sponge database, sequence families are included in rough stoichiometric amounts (as estimated by read depth coverage), to function in distributing multiple mapping reads when using standard alignment software (bwa mem). Large, artifact read pile-ups are often due to misalignment of reads containing repeat sequences that are vastly underrepresented in the genome. By presenting a more accurate estimate of repeat family abundance and sequence variation in the genome, read alignments are expected to assign randomly across all possible sites, thereby distributing read mappings equally and reducing mapping biases. In line with this database utility, we have included 1000 diverse mitochondrial genomes in the sponge database (15) (Supplementary Figure S1), representing a rough estimate of non-nuclear genomes expected from an average human cell (16).
The sponge database is similar to the ‘decoy genome’ database (hs37d5ss) (6), as it presents an alternative set of missing human sequences that is useful in reducing mapping errors. By including unassembled WGS reads, the sponge database (1x, stoichiometric estimate database, Supplementary Data 1 and 2) provides a larger representation of sequences (128 636 fasta entries, 201 Mb) compared to the decoy genome (4715, 35 Mb). Although both databases provide considerable alpha satellite annotation, the sponge database provides more robust representation of other abundant pericentromeric repeat families (Supplementary Figure S2). For example, 34% of the sponge database provides sequence mapping for Human Satellites II and III, whereas these sequences make up only 2.8% of the decoy genome. The sponge database also presents roughly twice the sequence diversity (930 million unique 24-mers compared to 445 million unique 24-mers in the decoy database). Like the decoy genome, the entire sponge database is relatively small (196M), ensuring that it is amendable to standard alignment protocols.
Including sponge mapping targets reduce read alignments in blacklisted regions
To test the utility of these mapping targets in reducing read enrichment profiles within characterized blacklisted regions, we initially evaluated read alignments using two low-coverage human genomic datasets (HuRef and GM19239, Supplementary Table S2). By doing so, we detected a 10-fold reduction in bases aligned to blacklisted regions provided across four public blacklist datasets available for GRCh37 (Figure 2a), with a reduction in read alignments across all previously characterized repeat annotation attributed to blacklist regions (Supplementary Figure S3). To further explore reduction in mapping to blacklist regions we surveyed read alignments across a diverse collection of short-read ENCODE experimental datasets (1) (Supplementary Table S2). Evaluation of both read alignments and peak calling across all datasets reveals a reduction in mapping to blacklisted regions (Figure 2b, Supplementary Figure S4). Similarly, we observed a reduction in read mapping within annotated blacklisted regions in the GRCh38 reference, which includes the alpha satellite reference models within the chromosome assemblies (Supplementary Figure S5). Further, we demonstrate genome-wide decreases in read mapping and peak calling within blacklisted regions were not sensitive to sequence coverage of the sponge database, as increasing sequence coverage from 1x to 8x did not present notable improvement. For each dataset we were able to assess reduction in peak prediction across blacklisted regions or regions with little experimental support (as indicated on a region on 1p11.2 that has been observed to be misassembled in the GRCh37 assembly (21), Figure 2c as a false positive alignments), while maintaining true positive alignments, or peaks with multiple experimental lines of support for enrichment. Comparison between the DNase I mapping data with either the sponge or decoy dataset revealed a general decrease in read mapping across blacklisted sites, however, the sponge database provided a larger reduction in false mapping attributed to blacklisted sites annotated as satellite/centromeric repeats (Supplementary Figure S6).
Figure 2.
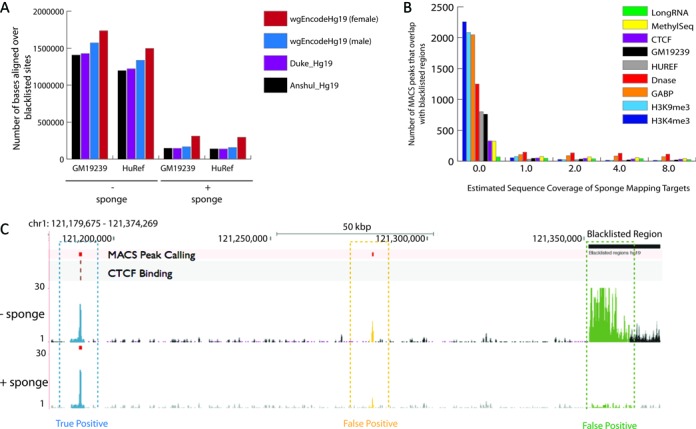
Reduction in artifact read alignments was observed in the presence of the sponge mapping targets when surveyed across blacklisted regions in figure (A) for four previously characterized datasets providing lists of annotated sites in hg19. When evaluating read mapping results with and without the sponge across low-coverage whole genomic datasets from two individual (HuRef, Western European and GM19239, Yoruba), we observe a 10-fold decrease. In panel (B) we observe a similar 10-fold or greater reduction in peaks called within blacklisted regions (shown here for the Anshul hg19 blacklisted data), including nine additional ENCODE functional datasets. Further, as one increases the abundance of the sponge database from 1x to 8x, we observe little improvement. Results for CTCF mapping in regions hg19 chr1:121,179,675–121,374,269 are shown with or without the sponge database in panel (C). MACS peak calls are indicated in red, and locations of CTCF binding are shown in the track highlighted in light browns. In the presence of sponge, mapping targets read alignment depth is decreased in regions that span a previously characterized blacklisted regions (shown in green) and labeled as a false positive. Alignments are reduced in regions that are not indicated as a blacklisted region, which appear to be novel (shown in orange), offering new sites of false positive alignments. Regions, as indicated in blue, that benefit from multiple lines of biological support still provide peak calls in the presence of the sponge mapping targets.
Use of sponge mapping targets improves large-scale analyses by reducing aberrant read alignments
To ensure sites of enrichment that benefit from multiple lines of biological evidence are not lost in the presence of the sponge mapping targets, we monitored changes in read depth and peak calls across select ENCODE datasets (1) (CTCF, promoter regions and RefSeq coding regions). By doing so, we observed that including the mapping targets had a very marginal effect on sites previously characterized by more than one dataset (e.g. CTCF ChIP-seq and CTCF transcription binding sites), with 99.3% of all peaks maintained (Figure 3a). Of those sites lost, the majority (89%, 179/201) are peaks that are associated with blacklisted regions. Likewise, we observed this trend for RNA sequence mapping with respect to RefSeq coordinates (98% peaks remaining) and promoter regions (97.8% using datasets from DNaseI, GABP and H3K4me3) (Figure 3b and c).
Figure 3.
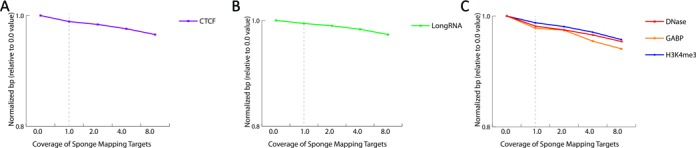
Sites of enrichment that benefit from multiple lines of biological evidence are not lost in the presence of the sponge mapping targets, as shown monitor changes in read depth for (A) CTCF with increasing abundance of the sponge database (1x–8x coverage), (B) long RNA datasets that overlap with characterized RefSeq gene locations and (C) within promoter regions, defined here as 1 kb upstream of a RefSeq gene and evaluated using DNase, GABP and H3K4me3 datasets.
Cell lines derived from similar tissues are expected to share similar sites of transcriptional regulation. This has been observed by correlations in genome-wide DNase I hypersensitive profiles that predict sites of open chromatin, identifying regions accessible to transcription factors. Artifact read alignments are expected to confound these large-scale correlations of pair-wise enrichment profiles by introducing noise. To evaluate improvement in genome-wide correlation datasets we selected 12 DNase I datasets (including seven fibroblast cell lines, three lymphoblast and two embryonic stem cells) to observe improvement in Pearson correlations when introducing the sponge mapping targets. Standard mapping protocols for ENCODE 3 filter multi-mapping reads, allowing us to specifically investigate the influence of the single, best read mapping with the addition of the sponge database. As a result, we observed an increase in correlation between related cell lines while removing spurious inter-correlations between cell-line groupings (Supplementary Figure S7). After the resulting small-scale run, we utilized the mapping targets pipeline to modestly improve correlations across a larger datasets representing different tissue and cell types, while maintaining biological meaningful correlations between cell lines as previously supported in the literature (20) (Supplementary Figure S8). Therefore, including the sponge mapping targets, even when filtering multi-mapping read alignments, is expected to provide some improvement to large-scale analyses of genome-wide read alignment data in addition to adjusting inference based on reducing local artifact peak calls.
Characterization of novel artifact sites and regions that demonstrate peak-calling sensitivity to increased abundance of mapping targets
Artifact enrichment sites, which are not currently included as blacklist regions, were defined by the reduction of read alignments and loss of peak enrichment when studied in the presence of the sponge mapping targets. It is likely these novel sites will be classified as commonly associated sites, or read enrichments that were shared between two or more cell lines, and cell-line specific sites (Supplementary Figure S9). To address the opportunity for stoichiometric variation between individuals, we increased our study to monitor read depth and peak calling while increasing the size of the sponge database (2x, 4x and 8x) to track roughly 3% of sites that demonstrate mapping sensitivity in the presence of the sponge database (Supplementary Figure S10). In doing so, we were able to provide a summarized listing of regions in GRCh37/hg19 that appear to be sensitive across included datasets, as a method of filtering regions which are suspected to provide variable results in mapping studies using collection of cell lines from different individuals (Supplementary Table S3). Evaluation of these sites reveals dosage-dependent mapping targets show a sharp decrease in sensitivity, or reduction in read alignments, once past 2x stoichiometric estimates. Regions affected were associated with the larger pericentromeric satellite families (HSat2,3).
DISCUSSION
Ensuring correct read alignments is critical to high-throughput sequencing studies aimed at identifying sites of genome regulation and functional sequence annotation. Mapping of short reads generated from whole-genome datasets has demonstrated that particular sites in the reference genome are prone to artifactual increased alignments pile-ups, marked as blacklisted regions, which confound biological interpretation in these regions. These alignment errors are due to improper mapping of underrepresented repeated sequences to a limited number of assembled sites that share short stretches of sufficient sequence homology. Here we demonstrate that adding these sequences as mapping targets in an expected stoichiometric abundance, in effect presenting a more complete version of the human genome, is effective in correcting these mapping errors in standard mapping and analysis pipelines. To identify a precise set of regions of the human genome that are likely enriched due to false positive mapping we have evaluated sites with reduced read depth and peak calls in the presence of these additional mapping targets. We demonstrate the utility of this approach in reducing known artifacts in previously identified blacklisted regions. Further, we show that by eliminating this form of experimental noise we improve large-scale correlations between functional DNase datasets. We propose that the use of this strategy is useful in advancing de novo characterization of blacklisted regions in genomes that lack prior characterization.
Although repeat families included in our mapping dataset are known to vary between individuals, we observe that a single database representing roughly 1x coverage provides a sufficient stoichiometric representation to be useful in reducing false positive mapping. However, we do identify a small number of sites (∼3% observed in this study) that are indeed sensitive to the underlying target sequence abundance. We suspect that these sites may represent less abundant sequence variants that are represented in the assembly as well as in a lower proportion in the 1x database. These repeat variants may be present in tens of copies in one individual and tens of thousands of copies in another. Use of a single ‘sponge database’ from the HuRef genome may not be sufficient to capture these smaller signals of sequence evolution entirely. Rather, future studies may benefit from increasing the sponge database to annotate these regions for each cell line or alternatively use the database to generate cell-line specific databases by mapping genomic data to the sponge sequence library. By doing so one is more likely to overcome this limitation and have a better donor-matched definition of repeat abundance and variation in these regions. We expect this method to be extendable to other non-human genomes, by which species-specific sponge databases could provide a general mechanism to reduce artifact alignments.
Supplementary Material
Acknowledgments
We would like to acknowledge the ENCODE Analysis Working Group for providing blacklisted regions and feedback on the manuscript. Thanks to Donna Karolchik for editing the manuscript.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
NHGRI (5U41HG002371 and 3U41HG004568–09S1). WJK is also supported by Kent Informatics Inc.
Conflict of interest statement. None declared.
REFERENCES
- 1.Encode Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Koehler R., Issac H., Cloonan N., Grimmond S.M. The uniqueome: a mappability resource for short-tag sequencing. Bioinformatics. 2011;27:272–274. doi: 10.1093/bioinformatics/btq640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Derrien T., Estelle J., Marco Sola S., Knowles D.G., Raineri E., Guigo R., Ribeca P. Fast computation and applications of genome mappability. PLoS One. 2012;7:e30377. doi: 10.1371/journal.pone.0030377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lee H., Schatz M.C. Genomic dark matter: the reliability of short read mapping illustrated by the genome mappability score. Bioinformatics. 2012;28:2097–2105. doi: 10.1093/bioinformatics/bts330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Eichler E.E., Clark R.A., She X. An assessment of the sequence gaps: unfinished business in a finished human genome. Nat. Rev. Genet. 2004;5:345–354. doi: 10.1038/nrg1322. [DOI] [PubMed] [Google Scholar]
- 6.Li H. Towards better understanding of artifacts in variant calling from high-coverage samples. Bioinformatics. 2014;30:2843–2851. doi: 10.1093/bioinformatics/btu356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Levy S., Sutton G., Ng P.C., Feuk L., Halpern A.L., Walenz B.P., Axelrod N., Huang J., Kirkness E.F., Denisov G., et al. The diploid genome sequence of an individual human. PLoS Biol. 2007;5:e254. doi: 10.1371/journal.pbio.0050254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jurka J., Kapitonov V.V., Pavlicek A., Klonowski P., Kohany O., Walichiewicz J. Repbase Update, a database of eukaryotic repetitive elements. Cytogenet. Genome Res. 2005;110:462–467. doi: 10.1159/000084979. [DOI] [PubMed] [Google Scholar]
- 9.Benson G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 1999;27:573–580. doi: 10.1093/nar/27.2.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hayden K.E., Strome E.D., Merrett S.L., Lee H.R., Rudd M.K., Willard H.F. Sequences associated with centromere competency in the human genome. Mol. Cell Biol. 2013;33:763–772. doi: 10.1128/MCB.01198-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Miga K.H., Newton Y., Jain M., Altemose N., Willard H.F., Kent W.J. Centromere reference models for human chromosomes X and Y satellite arrays. Genome Res. 2014;24:697–707. doi: 10.1101/gr.159624.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Altemose N., Miga K.H., Maggioni M., Willard H.F. Genomic characterization of large heterochromatic gaps in the human genome assembly. PLoS Comput. Biol. 2014;10:e1003628. doi: 10.1371/journal.pcbi.1003628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ganley A.R., Kobayashi T. Highly efficient concerted evolution in the ribosomal DNA repeats: total rDNA repeat variation revealed by whole-genome shotgun sequence data. Genome Res. 2007;17:184–191. doi: 10.1101/gr.5457707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gonzalez I.L., Sylvester J.E. Complete sequence of the 43-kb human ribosomal DNA repeat: analysis of the intergenic spacer. Genomics. 1995;27:320–328. doi: 10.1006/geno.1995.1049. [DOI] [PubMed] [Google Scholar]
- 15.Ingman M., Gyllensten U. mtDB: Human Mitochondrial Genome Database, a resource for population genetics and medical sciences. Nucleic Acids Res. 2006;34:D749–D751. doi: 10.1093/nar/gkj010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Robin E.D., Wong R. Mitochondrial DNA molecules and virtual number of mitochondria per cell in mammalian cells. J. Cell. Physiol. 1988;136:507–513. doi: 10.1002/jcp.1041360316. [DOI] [PubMed] [Google Scholar]
- 17.Zhang Y., Liu T., Meyer C.A., Eeckhoute J., Johnson D.S., Bernstein B.E., Nusbaum C., Myers R.M., Brown M., Li W., et al. Model-based analysis of ChIP-Seq (MACS) Genome Biol. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pruitt K.D., Tatusova T., Maglott D.R. NCBI reference sequences (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res. 2007;35:D61–D65. doi: 10.1093/nar/gkl842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Quinlan A.R., Hall I.M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stergachis A.B., Neph S., Reynolds A., Humbert R., Miller B., Paige S.L., Vernot B., Cheng J.B., Thurman R.E., Sandstrom R., et al. Developmental fate and cellular maturity encoded in human regulatory DNA landscapes. Cell. 2013;154:888–903. doi: 10.1016/j.cell.2013.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dennis M.Y., Nuttle X., Sudmant P.H., Antonacci F., Graves T.A., Nefedov M., Rosenfeld J.A., Sajjadian S., Malig M., Kotkiewicz H., et al. Evolution of human-specific neural SRGAP2 genes by incomplete segmental duplication. Cell. 2012;149:912–922. doi: 10.1016/j.cell.2012.03.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.