


Abstract
The CRISPR-Cas9 system uses guide RNAs to direct the Cas9 endonuclease to cleave target sequences. It can, in theory, target essentially any sequence in a genome, but the efficiency of the predicted guide RNAs varies dramatically. If no targeted cells are obtained, it is also difficult to know why the experiment fails. We have developed a transient transfection based method to enrich successfully targeted cells by co-targeting the hypoxanthine phosphoribosyltransferase (HPRT) gene. Cells are transfected with two guide RNAs that target respectively HPRT and the gene of interest. HPRT targeted cells are selected by resistance to 6-thioguanine (6-TG) and then examined for potential alterations to the gene targeted by the co-transfected guide RNA. Alterations of many genes, such as AAVS1, Exo1 and Trex1, are highly enriched in the 6-TG resistant cells. This method works in both HCT116 cells and U2OS cells and can easily be scaled up to process multiple guide RNAs. When co-targeting fails, it is straightforward to determine whether the target gene is essential or the guide RNA is ineffective. HPRT co-targeting thus provides a simple, efficient and scalable way to enrich gene targeting events and to identify the cause of failure.
The CRISPR-Cas9 system is a revolutionary technology for gene targeting in cells (1–3). It consists of two components: a guide RNA and the Cas9 endonuclease that respectively pairs with the target sequence and then cleaves it (4–6). The guide RNA contains 19 nt that in theory can be custom designed to target almost any sequence in a genome (5–8). In practice, the effectiveness of the predicted guide RNAs varies dramatically (8). This problem, when compounded by other commonly encountered problems such as poor efficiency of DNA transfection or low titer of viruses, can make the isolation of successfully targeted cells a really laborious task (9). Low transfection efficiency can be improved by special techniques like nucleofection (6), but they are expensive and not readily available to all labs. A common method to enrich successfully targeted cells is to use drug resistance or green fluorescent protein (GFP) markers to select for cells that have integrated the guide RNA-expressing DNA into chromosomes (7,10). Fluorescence-activated cell sorting (FACS) sorting only enriches cells expressing high levels of GFP but not necessarily Cas9, while integration of foreign DNA, especially viral DNA, into the chromosome is itself an alteration to the genome and might cause oncogenic transformation. In some cases, Cas9 is also integrated into the genome and constitutively expressed (7). Persistent expression of the guide RNA in combination with Cas9 might lead to increasing probabilities of off-target cleavages over long-term culturing (11). In addition, if no targeted cells are obtained, it is difficult to track down the cause of failure. The guide RNA might be ineffective or the correctly targeted cells might have died off and only cells not expressing the guide RNA might have survived.
To overcome these shortcomings, we have developed a transient plasmid DNA transfection-based method to enrich successfully targeted cells without the need for DNA integration into chromosomes by co-targeting the cellular hypoxanthine phosphoribosyltransferase (HPRT) gene. This gene encodes a protein that catalyzes the conversion of hypoxanthine to inosinemonophosphate and guanine to guanosine monophosphate in the non-essential purine salvage pathway (12). HPRT+ cells are sensitive to 6-thioguanine (6-TG), which can be converted to the nucleotide form by HPRT and incorporated into DNA by DNA polymerase, killing cells by a process involving postreplicative mismatch repair (13–15). The strategy is to transfect cells with two plasmids that express respectively a HPRT guide RNA and a guide RNA for the gene of interest. Cas9 can be expressed from the gene on a separate plasmid, a plasmid carrying the HPRT gRNA or integrated into the chromosome (if such a cell line is already available). If a cell becomes resistant to 6-TG, it would suggest that this cell should also be competent to target the gene of interest as long as the gRNA is effective. Thus if the targeted gene is not altered in the resulting 6-TG resistant cells, it would suggest that the guide RNA is ineffective. On the other hand, if no 6-TG resistant cells can be obtained by co-targeting, it would suggest that the gene of interest might be essential.
We have tested this method with guide RNAs for HPRT and the non-essential AAVS1 locus in HCT116 cells, a colorectal cancer cell line with a near diploidic karyotype (16). The results showed a dramatic enrichment of AAVS1 targeting events from below detection without co-targeting to over 80% with co-targeting. Other non-essential genes such as Trex1 and Exo1 were also successfully enriched by HPRT co-targeting. The method also worked in U2OS cells, an osteosarcoma cell line with a complicated karyotype (17). Co-targeting with guide RNAs for DNA topoisomerase 2α (Top2α) gave rise to no 6-TG resistant cells, which is consistent with Top2α being essential for cell proliferation (18). On the other hand, co-targeting with some other guide RNAs gave rise to plenty of 6-TG resistant cells but no alteration to the intended sequences, suggesting that the guide RNAs were ineffective. Together, these results demonstrate that co-targeting the HPRT gene provides a simple and efficient method to enrich successfully targeted cells. It can also be easily used to evaluate the effectiveness of guide RNAs and the essentialness of target genes.
MATERIALS AND METHODS
Cell culture and reagents
The human HCT116 and U2OS cells, Dulbecco's Modified Eagle Medium (DMEM), fetal bovine serum, penicillin/streptomycin (P/S), L-glutamine, non-essential amino acids (NEAA) and G418 were obtained from the Tissue Culture Facility at the Fox Chase Cancer Center. Cas9-expressing HCT116 cells were constructed by infecting HCT116 cells with a lentivirus containing Cas9 under the control of a doxycycline-inducible promoter (Addgene, MA, USA) (8). A cell clone with high and stringently-controlled expression of Cas9 was selected for this study. Cells were grown in DMEM supplemented with 10% FBS, 2 mM L-glutamine, NEAA and P/S at 37°C under a 5% CO2 humidified atmosphere. 6-TG and Crystal violet were purchased from Sigma-Aldrich (MO, USA).
Guide RNA construction
Plasmids expressing Cas9 (hCas9-pcDNA3.3-TOPO) and AAVS1 guide RNA T2 were obtained from Addgene (MA, USA) (6). The 400 bp backbone for guide RNA expression was modified to add two Sap sites to allow the insertion of 19 nt custom designed targeting sequences. The 19 nt guide RNA sequences used in this study are ((N)19NGG strand; 5′->3′): AAGTAATTCACTTACAGTC (HPRT gRNA), TCCCCTCCAGACTCGCACA (Trex1 gRNA), GCCATAATTACAGAGGACT (Exo1 gRNA), ATCTTTACAATGCTCAGGT (Top2α gRNA) and GTTTTATCCTGATGCCGAC (BLM gRNA4).
Guide RNA targeting in cells
Plasmids expressing guide RNAs and Cas9 were introduced into cells with Lipofectamine 2000 (Invitrogen, CA, USA) following the manufacturer's protocol. Cells were seeded in 24-well plates and grown to a confluency of 90–99%. A typical transfection reaction contains: 0.1 μg plasmid of the HPRT gRNA, 0.3 μg plasmid of the guide RNA for the gene of interest and 0.4 μg of Cas9 plasmid. In the case of Cas9-expressing cells, the Cas9 plasmid was replaced with more of the plasmid of guide RNA for the gene of interest. Cells were re-seeded 16–18 h post-transfection at different densities and in two sets of plates. For selection of HPRT− cells, 6-TG was added to the media (10 μg/ml final concentration) 3 days later and cells were grown for 10 days with two changes of 6-TG containing media. One set of plates were stained with Crystal Violet to visualize colonies. 6-TG resistant cells from the other set of plates were pooled and grown up for genomic DNA preparation.
Amplification of the targeted regions
Genomic DNA was extracted from cells using the following protocol. Confluent cells grown in 24-well plates were first extracted with 1 ml of 100 mM PIPES (pH6.9), 1 mM ethylenediaminetetraacetic acid (EDTA), 0.25% Triton X-100, 4M glycerol at room temperature for 1 min. The remaining nuclei were incubated with 10 mM Tris (pH7.5), 1 mM EDTA, 500 mM NaCl, 0.5% Triton X-100 and 100 μg/ml proteinase K at 37°C for 2–4 h. The genomic DNA was transferred to eppendorf tubes and heated at 90°C for 30 min. The targeted regions were amplified from genomic DNA with Hot Start Taq DNA polymerase (NEB, MA, USA). The following primers were used (all in 5′->3′): GATGCTCACCTCTCCCACAC and ACATCCATGGGACTTCTGCC (for HPRT); ACAGGAGGTGGGGGTTAGAC and TATATTCCCAGGGCCGGTTA (for AAVS1); GCAGACCCTCATCTTTTTCG and TACTGGGCTCAGATAGTTGAC (for Trex1); TCCAGTTCCAGCTGCCTAGA and GTCTGCACATTCCTAGCCGA (for Exo1); GAGGTGTAGGCATTTGCTGC and GCCACTCTGTGAACCAGGTA (for BLM). All oligonucleotides were synthesized by Integrated DNA technologies (IA, USA).
RESULTS
Exon8 of the HPRT gene can be efficiently targeted by the CRISPR-Cas9 system in HCT116 cells
To test HPRT co-targeting, we first attempted to identify a guide RNA that could efficiently disrupt the HPRT gene. One guide RNA was found to be highly effective at targeting exon8, which encodes an active site for the HPRT protein and is frequently mutated in 6-TG resistant cells (19,20). The plasmid expressing the HPRT guide RNA was co-transfected with the Cas9-expressing plasmid into HCT116 cells. The transfected cells were reseeded in 6-well plates and selected with 6-TG. The resulting 6-TG resistant colonies were stained with crystal violet. As shown in Figure 1A, compared to the seeding control (with no 6-TG selection), ca. 1% of the cells transfected with the two plasmids became resistant to 6-TG. To ascertain that 6-TG resistance was the result of targeted disruption of the HPRT gene, we amplified the fragment spanning the guide RNA sequence from the genomic DNA of the pooled 6-TG resistant cells. Sequence analysis indeed showed expected alterations to the sequence adjacent to the PAM motif (CCA) (Figure 1B). The major alteration was an extra ‘T’ peak after the third nucleotide from PAM and the peaks afterward became mixtures of nucleotides. To rule out the possibility of bad sequencing reactions, the pooled polymerase chain reaction (PCR) products were subcloned into the pGEM-T vector and sequenced individually. Out of 15 HPRT products, 8 carried a ‘T’ insertion, 2 carried a ‘TG’ insertion and the remaining 5 carried deletions of 2–15 nt (Figure 1C and D). Overall, the data were consistent with an initial cleavage between the third and fourth nucleotide away from PAM followed by further deletions/insertions before religation (4).
Figure 1.
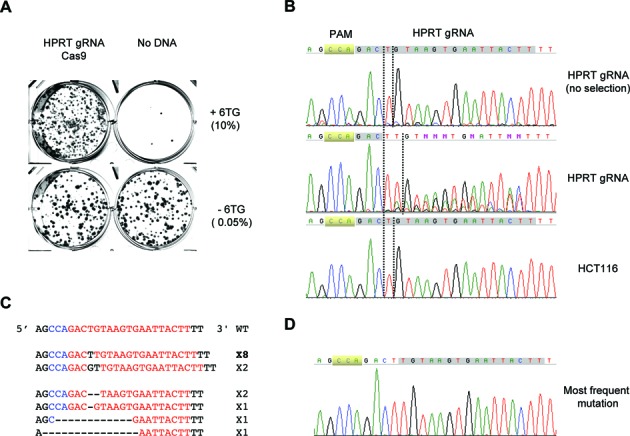
The HPRT gene can be efficiently disrupted by a guide RNA targeting exon8 in HCT116 cells. (A) Staining of colonies formed after HCT116 cells were transfected with plasmids expressing HPRT guide RNA or Cas9 and selected for 6-TG (10μg/ml). Cells were seeded at 0.05% (no 6-TG) and 5% (+6-TG). (B) Sequences of PCR products of the HPRT region in the genomic DNA isolated from the pooled HPRT targeted but unselected cells, 6-TG resistant HPRT targeted cells, or parental HCT116 cells. (C) Sequences of individual inserts of the cloned PCR products from the genomic DNA of the 6-TG resistant cells. (D) Sequencing chromatogram of the most frequent mutation.
Targeting of the AAVS1 locus is dramatically enriched in HPRT co-targeted cells
To test the co-targeting strategy, we first chose the AAVS1 locus, which can be efficiently disrupted by the T2 guide RNA (6). HCT116 cells were transfected with the Cas9 plasmid and the AAVS1 plasmid or with these two and the HPRT plasmid. The trasnfected cells were reseeded and selected for 6-TG resistance. As shown in Figure 2A, co-transfection with Cas9, AAVS1 and HPRT resulted in many 6-TG resistant colonies, but that with Cas9 and AAVS1 produced only background level of colonies. The AAVS1 fragment was then amplified from the genomic DNA isolated from either the 6-TG resistant co-targeted cells or the unselected AAVS1 singly targeted cells. Sequence analysis showed that the AAVS1 locus was very different in the two populations of cells. While the AAVS1 singly targeted cells showed only wild-type sequence, the AAVS1 and HPRT co-targeted cells showed dramatic alterations. The major change was consistent with a ‘G’ deletion after the third nucleotide away from PAM (TGG) (Figure 2B). This was confirmed by the sequencing of individual fragments after the pooled PCR products were subcloned into the pGEM-T vector. Eleven out of thirteen AAVS1 products were mutants: 6 carrying a ‘G’ insertion and 5 carrying deletions of 2–12 nt (Figure 2C and D). The data were again consistent with an initial cleavage between the third and fourth nucleotides from PAM followed by small deletions/insertions before religation.
Figure 2.
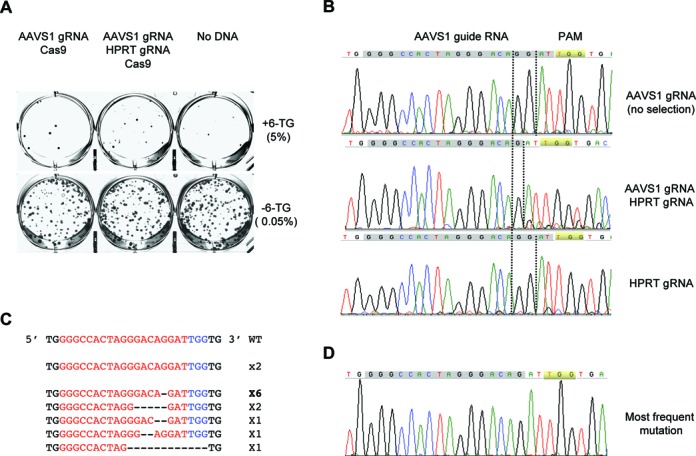
The AAVS1 locus is efficiently disrupted in the HPRT co-targeted cells. (A) Staining of colonies formed after HCT116 cells were transfected with plasmids expressing HPRT guide RNA or Cas9 and selected with 6-TG (10μg/ml). Cells were seeded at 0.05% (no 6-TG) and 5% (+6-TG). (B) Sequences of PCR products of the AAVS1 region in the genomic DNA isolated from the pooled AAVS1 targeted but unselected cells, 6-TG resistant HPRT and AAVS1 co-targeted cells, or HPRT only targeted cells. (C) Sequences of individual inserts of the cloned PCR products from the genomic DNA of the co-targeted 6-TG resistant cells. (D) Sequencing chromatogram of the most frequent mutation.
HPRT co-targeting works for other genes
We then used the co-targeting method to target other genes and found it to work as efficiently as for the AAVS1 locus. One of the genes that was efficiently co-targeted was Trex1 (21). The genomic DNA showed clear alterations to the region targeted by the Trex1 guide RNA in the 6-TG resistant co-targeted cells but not in the unselected cells (Figure 3A). The major change was consistent with an insertion of ‘G’ between the third and fourth nucleotide from PAM (CCG). This was confirmed by the sequencing of the cloned individual PCR fragments, with 7 out of 14 containing an extra ‘G’. Another gene that was efficiently co-targeted was Exo1 (22,23). As shown in Figure 3B, the Exo1 gene was extensively altered in the region targeted by the Exo1 guide RNA in the 6-TG resistant cells but not in the unselected cells. Sequencing of the cloned individual PCR fragments showed that 20 out of 25 contained deletions and the other 5 contained insertions. Consistent with the more complicated chromatogram of the pooled PCR, 9 of the deletions were long, ranging from 286 to 604 nt in length, suggesting that the repair of this DNA double-strand break (DSB) is prone to resection in HCT116 cells.
Figure 3.
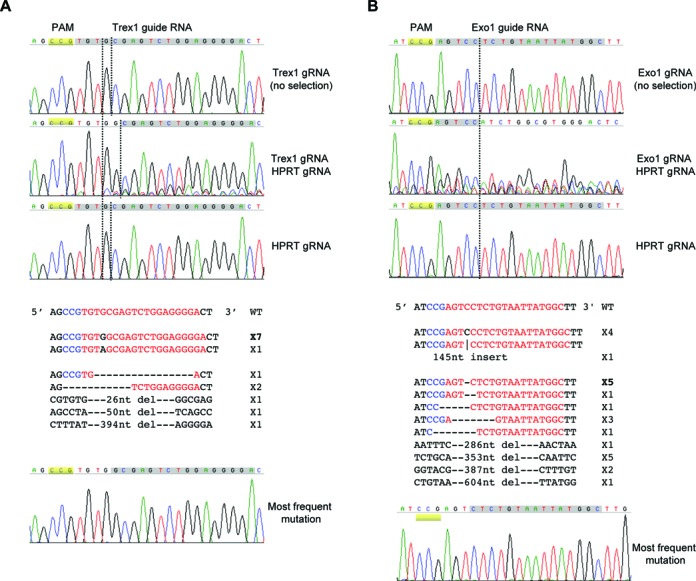
Other genes can also be efficiently co-targeted. (A) Sequence data of the Trex1 guide RNA targeted region. Top: sequencing chromatograms of the PCR products from the Trex1 targeted but unselected cells, 6-TG resistant HPRT and Trex1 co-targeted cells, or HPRT only targeted cells. Middle: sequences of individual Trex1 inserts of the cloned PCR products from the genomic DNA of the co-targeted 6-TG resistant cells. Bottom: sequencing chromatogram of the most frequent mutation. (B) Sequence data of the Exo1 guide RNA targeted region. Top: sequencing chromatograms of the PCR products from the Exo1 targeted but unselected cells, 6-TG resistant HPRT and Exo1 co-targeted cells, or HPRT only targeted cells. Middle: sequences of individual Exo1 inserts of the cloned PCR products from the genomic DNA of the co-targeted 6-TG resistant cells. Bottom: sequencing chromatogram of the most frequent mutation. The HCT116 cells in these experiments carried an integrated Cas9 gene under the control of the TET promoter.
Co-targeting is also effective in U2OS cells
We also tested if co-targeting works in other types of cells. U2OS is a commonly used cell line for cell biology experiments owing to its large and flat nucleus. It has a complicated karyotype with many genes present in multiple copies. U2OS cells were first transfected with plasmids expressing Cas9 and HPRT guide RNA and then selected for 6-TG resistance. As shown in Figure 4A, 6-TG resistant cells were readily obtained, demonstrating that the HPRT gene could be targeted in this cell line. We then repeated the experiment with Cas9 and two guide RNAs, one for HPRT and one for AAVS1. 6-TG resistant cells were selected, genomic DNA isolated and the AAVS1 locus amplified. Sequence analysis showed clearly that the AAVS1 locus in the co-targeted cells was altered (Figure 4B). In contrast, cells transfected but not selected by 6-TG showed a sequence chromatogram indistinguishable from that of the wild-type. Notably, the sequence change in the co-targeted U2OS cells was more extensive than in the similarly co-targeted HCT116 cells. Only half of the guide RNA sequence was still retained, presumably due to different DSB repair activities in the two cell lines. This was confirmed by the sequencing of individual AAVS1 PCR products after they were subcloned into the pGEM-T vector. Sixteen out of seventeen products carried small deletions or insertions that were consistent with an initial cleavage between the third and fourth nucleotide from PAM followed by end polishing before religation (Figure 4C and D).
Figure 4.
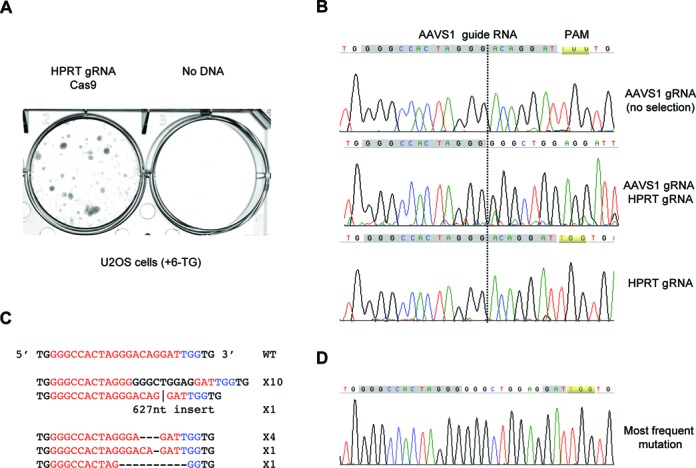
Co-targeting also works in U2OS cells. (A) Staining of colonies formed after U2OS cells were transfected with different plasmids and selected with 10μg/ml 6-TG. (B) Sequences of PCR products of the AAVS1 region in the AAVS1 targeted but unselected cells, 6-TG resistant HPRT and AAVS1 co-targeted cells or untargeted U2OS cells. (C) Sequences of individual inserts of the cloned PCR products from the genomic DNA of the co-targeted 6-TG resistant cells. (D) Sequencing chromatogram of the most frequent mutation.
Co-targeting can easily evaluate the effectiveness of guide RNAs and the essentialness of target genes
Of the many guide RNAs we tested for co-targeting, three types of results were obtained. The first type is demonstrated by AAVS1, Trex1 and Exo1, showing efficient targeting. The second type is that only background level 6-TG colonies survived selection. Figure 5A shows the result of HPRT co-targeting with the Top2α guide RNA. Only a few colonies emerged after the first round of 6-TG selection and they all died off upon passaging. This is consistent with Top2α being an essential gene for DNA replication, transcription, chromosome condensation and segregation (18). Thus the failure to obtain HPRT co-targeted cells is a strong indication that the guide RNA is effective but the target gene might be essential. The third type of result is illustrated by co-targeting with a guide RNA for the Bloom syndrome gene (BLM). Many 6-TG resistant colonies were obtained (Figure 5A), but sequence analysis showed no alteration to the targeted region (Figure 5B). Because these cells were clearly competent for targeting the HPRT gene, the conclusion has to be that this particular guide RNA for BLM is ineffective in HCT116 cells.
Figure 5.
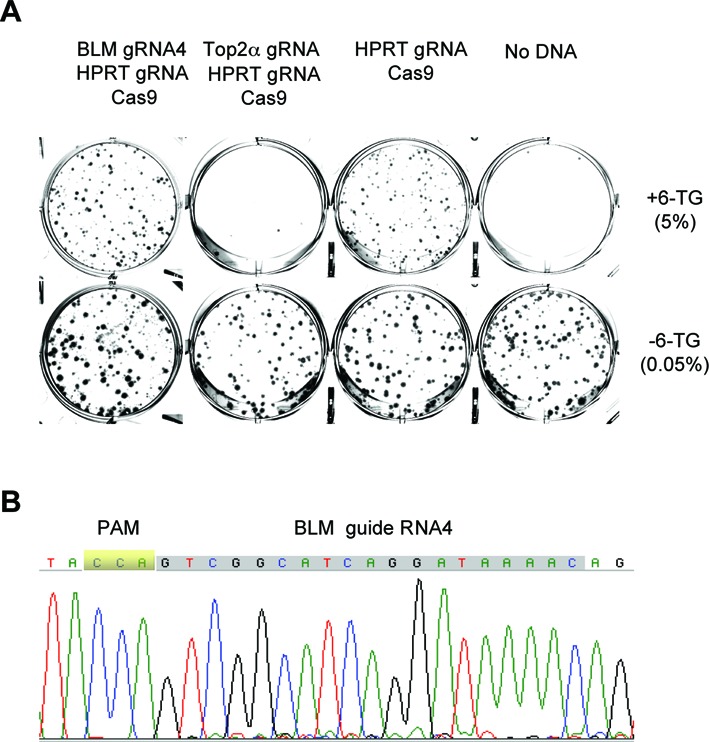
Co-targeting allows quick evaluation of gene essentiality and guide RNA effectiveness. (A) Staining of colonies formed after HCT116 cells were transfected with different plasmids and selected with 10μg/ml 6-TG. Cells were seeded at 0.05% (no 6-TG) and 5% (+6-TG). The HCT116 cells in these experiments carried an integrated Cas9 gene under the control of the TET promoter. (B) Sequencing chromatogram of the BLM region of the genomic DNA isolated from the pooled HPRT and BLM co-targeted 6-TG resistant cells.
DISCUSSION
We have developed a simple co-targeting method for enriching CRISPR-Cas9 mediated gene targeting in cultured cells. It involves only conventional transfections with plasmid DNA expressing guide RNAs targeting the HPRT gene and the gene of interest. The resulting HPRT− cells are selected by the nucleotide analogue 6-TG. For many guide RNAs tested, the target genes are efficiently disrupted in 6-TG resistant cells. One can then proceed to easily isolate individual cell clones from the greatly enriched mutant populations for phenotypic characterizations. If the target gene is not altered in 6-TG resistant cells, one can confidently conclude that the guide RNA is ineffective. On the other hand, if no 6-TG resistant cells survive, it would strongly suggest that the target gene might be essential. This method can thus be adapted to confirm the leads from whole genome screens of essential genes (7,8). However, to definitively rule out any potential off-target effects, one has to show that the phenotype can be complemented by re-expressing a gRNA-resistant copy of the target gene. In a single round of experiments, one can either obtain cells with the target gene disrupted, conclude that the target gene might be essential or that the guide RNA is ineffective.
This method is extremely easy and safe to perform. It only uses basic tissue culture setup and standard transfection reagents. No sophisticated equipment, expensive reagents or complicated protocols are required. It is thus very easy to scale up simply by increasing the number of transfections. We routinely do 12 transfections at a time. It does not involve viruses, the most commonly used way to deliver guide RNA-expressing DNA into cells. Viruses can be difficult to prepare with consistent quality and take extra time to characterize. They also raise concerns about laboratory safety and secondary effects of viral DNA integration into chromosomes. In particular, many guide RNAs can cause various degrees of off-target cleavages (11). As such, if cells also have an unregulated Cas9 gene integrated into chromosomes, constitutive expressions of the guide RNA and Cas9 over long-term poses a risk to the integrity of the genome. The HPRT co-targeting method involves only transient transfections with normal plasmid DNA. The frequency of plasmid DNA integration into chromosomes is extremely low. Genomic loci cleaved by Cas9 appear to be quickly repaired by NHEJ rather than become hot spots of plasmid DNA integration (Yan, H.; unpublished data). If plasmids also carry a gene conferring resistance to a drug such as G418, the risk of their integration into chromosomes can be further eliminated by checking the HPRT− cell clones for G418 sensitivity.
Co-targeting works in both HCT116 and U2OS cells, two cell lines with drastically different karyotypes. In principle, it should also work in any in vitro cultured cell line that can be transfected and is sensitive to 6-TG. The concentration of 6-TG for selection might need to be determined experimentally for each cell line. For cell lines that have high efficiency of transfection, such as 293 cells, co-targeting is not necessary but might still help (5,6). One potential problem of the co-targeting method is the induction of chromosomal translocations, which can occur at a low frequency when two DSBs are simultaneously present in close spatial proximity (24,25). The individual cell clones with the disrupted targeted gene should thus be checked for potential translocations between the HPRT gene and the target gene. Another potential downside of the co-targeting method is the disruption of the HPRT gene, which is involved in the salvage of purines from degraded DNA. Under normal growth conditions purine salvage is unimportant and HPRT is completely dispensable for cell growth and proliferation. However, it does affect neurodevelopment in mice and causes Lesch-Nyhan Disease disease in humans carrying the mutations (26). Should it be a concern, the HPRT gene can easily be put back into cells by standard transfection or corrected by CRISPR-Cas9 mediated homologous recombinational repair. Finally, the very use of 6-TG to kill non-targeted cells also excludes its application to gene targeting in whole organisms. For this purpose one might have to use genetic methods instead of drug selection, such as those developed in C. elegans (27–29). Overall, HPRT co-targeting provides an easy, safe, scalable and efficient non-viral method to enrich the successfully targeted cells, determine if a gene might be essential and evaluate if a guide RNA is effective.
Acknowledgments
The authors would like to thank Dr Christopher Seeger for helpful discussions during the preparation of the manuscript.
FUNDING
National Institute of Health [R01 GM57962 to H.Y.]. Funding for open access charge: National Institutes of Health [R01 GM57962 to H.Y.].
Conflict of interest statement. None declared.
REFERENCES
- 1.Mali P., Esvelt K.M., Church G.M. Cas9 as a versatile tool for engineering biology. Nat. Methods. 2013;10:957–963. doi: 10.1038/nmeth.2649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cong L., Zhang F. Genome engineering using CRISPR-Cas9 system. Methods Mol. Biol. 2014;1239:197–217. doi: 10.1007/978-1-4939-1862-1_10. [DOI] [PubMed] [Google Scholar]
- 3.Doudna J.A., Charpentier E. Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science. 2014;346:1258096. doi: 10.1126/science.1258096. [DOI] [PubMed] [Google Scholar]
- 4.Jinek M., East A., Cheng A., Lin S., Ma E., Doudna J. RNA-programmed genome editing in human cells. Elife. 2013;2:e00471. doi: 10.7554/eLife.00471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cong L., Ran F.A., Cox D., Lin S., Barretto R., Habib N., Hsu P.D., Wu X., Jiang W., Marraffini L.A., et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mali P., Yang L., Esvelt K.M., Aach J., Guell M., DiCarlo J.E., Norville J.E., Church G.M. RNA-guided human genome engineering via Cas9. Science. 2013;339:823–826. doi: 10.1126/science.1232033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shalem O., Sanjana N.E., Hartenian E., Shi X., Scott D.A., Mikkelsen T.S., Heckl D., Ebert B.L., Root D.E., Doench J.G., et al. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science. 2014;343:84–87. doi: 10.1126/science.1247005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang T., Wei J.J., Sabatini D.M., Lander E.S. Genetic screens in human cells using the CRISPR-Cas9 system. Science. 2014;343:80–84. doi: 10.1126/science.1246981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Miyaoka Y., Chan A.H., Judge L.M., Yoo J., Huang M., Nguyen T.D., Lizarraga P.P., So P.L., Conklin B.R. Isolation of single-base genome-edited human iPS cells without antibiotic selection. Nat. Methods. 2014;11:291–293. doi: 10.1038/nmeth.2840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yang L., Guell M., Byrne S., Yang J.L., De Los Angeles A., Mali P., Aach J., Kim-Kiselak C., Briggs A.W., Rios X., et al. Optimization of scarless human stem cell genome editing. Nucleic Acids Res. 2013;41:9049–9061. doi: 10.1093/nar/gkt555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fu Y., Foden J.A., Khayter C., Maeder M.L., Reyon D., Joung J.K., Sander J.D. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat. Biotechnol. 2013;31:822–826. doi: 10.1038/nbt.2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Silverman L.J., Kelley W.N., Palella T.D. Genetic analysis of human hypoxanthine-guanine phosphoribosyltransferase deficiency. Enzyme. 1987;38:36–44. doi: 10.1159/000469188. [DOI] [PubMed] [Google Scholar]
- 13.Gallagher R.E., Ferrari A.C., Zulich A.W., Yen R.W., Testa J.R. Cytotoxic and cytodifferentiative components of 6-thioguanine resistance in HL-60 cells containing acquired double minute chromosomes. Cancer Res. 1984;44:2642–2653. [PubMed] [Google Scholar]
- 14.Pan B.F., Nelson J.A. Characterization of the DNA damage in 6-thioguanine-treated cells. Biochem. Pharmacol. 1990;40:1063–1069. doi: 10.1016/0006-2952(90)90494-6. [DOI] [PubMed] [Google Scholar]
- 15.Swann P.F., Waters T.R., Moulton D.C., Xu Y.Z., Zheng Q., Edwards M., Mace R. Role of postreplicative DNA mismatch repair in the cytotoxic action of thioguanine. Science. 1996;273:1109–1111. doi: 10.1126/science.273.5278.1109. [DOI] [PubMed] [Google Scholar]
- 16.Roschke A.V., Tonon G., Gehlhaus K.S., McTyre N., Bussey K.J., Lababidi S., Scudiero D.A., Weinstein J.N., Kirsch I.R. Karyotypic complexity of the NCI-60 drug-screening panel. Cancer Res. 2003;63:8634–8647. [PubMed] [Google Scholar]
- 17.Janssen A., Medema R.H. Genetic instability: tipping the balance. Oncogene. 2013;32:4459–4470. doi: 10.1038/onc.2012.576. [DOI] [PubMed] [Google Scholar]
- 18.Wang J.C. Cellular roles of DNA topoisomerases: a molecular perspective. Nat. Rev. Mol. Cell. Biol. 2002;3:430–440. doi: 10.1038/nrm831. [DOI] [PubMed] [Google Scholar]
- 19.Mittelstaedt R.A., Heflich R.H. Analysis of in vivo mutation in exon 8 of the rat hprt gene. Mutat. Res. 1994;311:139–148. doi: 10.1016/0027-5107(94)90082-5. [DOI] [PubMed] [Google Scholar]
- 20.Wilson J.M., Young A.B., Kelley W.N. Hypoxanthine-guanine phosphoribosyltransferase deficiency. The molecular basis of the clinical syndromes. N. Engl. J. Med. 1983;309:900–910. doi: 10.1056/NEJM198310133091507. [DOI] [PubMed] [Google Scholar]
- 21.Mazur D.J., Perrino F.W. Identification and expression of the TREX1 and TREX2 cDNA sequences encoding mammalian 3′–>5′ exonucleases. J. Biol. Chem. 1999;274:19655–19660. doi: 10.1074/jbc.274.28.19655. [DOI] [PubMed] [Google Scholar]
- 22.Schmutte C., Marinescu R.C., Sadoff M.M., Guerrette S., Overhauser J., Fishel R. Human exonuclease I interacts with the mismatch repair protein hMSH2. Cancer Res. 1998;58:4537–4542. [PubMed] [Google Scholar]
- 23.Wilson D.M. 3rd, Carney J.P., Coleman M.A., Adamson A.W., Christensen M., Lamerdin J.E. Hex1: a new human Rad2 nuclease family member with homology to yeast exonuclease 1. Nucleic Acids Res. 1998;26:3762–3768. doi: 10.1093/nar/26.16.3762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Richardson C., Jasin M. Frequent chromosomal translocations induced by DNA double-strand breaks. Nature. 2000;405:697–700. doi: 10.1038/35015097. [DOI] [PubMed] [Google Scholar]
- 25.Zhang Y., McCord R.P., Ho Y.J., Lajoie B.R., Hildebrand D.G., Simon A.C., Becker M.S., Alt F.W., Dekker J. Spatial organization of the mouse genome and its role in recurrent chromosomal translocations. Cell. 2012;148:908–921. doi: 10.1016/j.cell.2012.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kang T.H., Park Y., Bader J.S., Friedmann T. The housekeeping gene hypoxanthine guanine phosphoribosyltransferase (HPRT) regulates multiple developmental and metabolic pathways of murine embryonic stem cell neuronal differentiation. PLoS One. 2013;8:e74967. doi: 10.1371/journal.pone.0074967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kim H., Ishidate T., Ghanta K.S., Seth M., Conte D., Jr., Shirayama M., Mello C.C. A co-CRISPR strategy for efficient genome editing in Caenorhabditis elegans. Genetics. 2014;197:1069–1080. doi: 10.1534/genetics.114.166389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Arribere J.A., Bell R.T., Fu B.X., Artiles K.L., Hartman P.S., Fire A.Z. Efficient marker-free recovery of custom genetic modifications with CRISPR/Cas9 in Caenorhabditis elegans. Genetics. 2014;198:837–846. doi: 10.1534/genetics.114.169730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ward J.D. Rapid and precise engineering of the Caenorhabditis elegans genome with lethal mutation co-conversion and inactivation of NHEJ repair. Genetics. 2015;199:363–377. doi: 10.1534/genetics.114.172361. [DOI] [PMC free article] [PubMed] [Google Scholar]