


Abstract
Structural biology experiments and structure prediction tools have provided many high-resolution three-dimensional structures of nucleic acids. Also, molecular dynamics force field parameters have been adapted to simulating charged and flexible nucleic acid structures on microsecond time scales. Therefore, we can generate the dynamics of DNA or RNA molecules, but we still lack adequate tools for the analysis of the resulting huge amounts of data. We present MINT (Motif Identifier for Nucleic acids Trajectory) — an automatic tool for analyzing three-dimensional structures of RNA and DNA, and their full-atom molecular dynamics trajectories or other conformation sets (e.g. X-ray or nuclear magnetic resonance-derived structures). For each RNA or DNA conformation MINT determines the hydrogen bonding network resolving the base pairing patterns, identifies secondary structure motifs (helices, junctions, loops, etc.) and pseudoknots. MINT also estimates the energy of stacking and phosphate anion-base interactions. For many conformations, as in a molecular dynamics trajectory, MINT provides averages of the above structural and energetic features and their evolution. We show MINT functionality based on all-atom explicit solvent molecular dynamics trajectory of the 30S ribosomal subunit.
INTRODUCTION
Nucleic acids, especially RNA, acquire many complicated tertiary structures to perform cellular functions (1). Provided that this tertiary structure is known, one of the common tools to investigate the structural and dynamical properties of nucleic acids and their complexes on atomic scale is molecular dynamics (MD) (2). With this technique riboswitches (3), protein–RNA complexes (4,5) and even the entire ribosome (6) have been studied. Other methods to sample the conformational space are the stochastic-based Monte Carlo techniques. Their applications to RNA molecules include the investigation of folding kinetics (7,8). Most importantly, all these simulation methods generate large data sets, i.e. many molecule conformations, which have to be post-processed.
Many computational tools have been designed to analyze single RNA conformations (9). One of the most comprehensive is Assemble2 (10), which reads the RNA secondary structure, constructs structural alignments of several RNAs, and overall facilitates RNA structure prediction and modeling. For detailed geometric analyses of RNA, especially its helical fragments, the Curves+ (11) or 3DNA (12) can be used. The programs apply standard reference frame (13) and describe the mutual position of two nucleotides and the conformation of the backbone using torsional angles. Many tools can analyze RNA structures based on the contacts and interactions between nucleotides. For basic identification of base pairs, stacking interactions and structural elements, such as helices, bulges and pseudoknots the MC-Annotate (14) can be used. More detailed description, together with the two-dimensional (2D) representations of RNA, can be obtained with RNAView (15) or RNAmap2D. The latter can also analyze complexes of nucleic acids and other molecules such as proteins or ligands and metal ions (16). The ClaRNA program additionally classifies each contact based on the similarity to a reference obtained from a large number of experimentally determined RNA structures (17). Recently announced, DSSR, a new component of the 3DNA, can define the secondary structures of RNA from three-dimensional (3D) coordinates, recognize motifs and non-pairing interactions (18). However, it does not analyze energetics of the recognized interactions.
On the other hand, there are only few programs for the analysis of RNA conformation sets
obtained from full-atom MD simulations. One is Canal, which
applies Curves+ (11) to
every trajectory frame and computes statistics, histograms and correlations for various
measures such as groove widths and depths, backbone dihedrals and base pairing
parameters. 3DNA includes scripts facilitating the analysis of MD
data for nucleic acids (12), and
do extends
3DNA applications to GROMACS trajectories (19).
extends
3DNA applications to GROMACS trajectories (19).
In fact, there is no complex tool to help analyze the dynamics of both the secondary and tertiary RNA structures.Therefore, we have designed Motif Identifier for Nucleic acids Trajectory (MINT) to characterize multiple RNA structures and the changes in hydrogen bonds, base pairing patterns, stacking and secondary structure motifs. The conformation sets can be from MD trajectories, Monte Carlo simulations, X-ray or nuclear magnetic resonance-derived conformations of the same molecule. MINT works for both RNA and DNA. However, since it is mainly RNA that acquires complicated 3D folds, we describe the software based on the RNA example.
MATERIALS AND METHODS
MINT works in a single and multiple conformation mode. For a single RNA or DNA conformation MINT outputs:
nucleotides forming helices, hairpin loops, internal loops, junctions, pseudoknots and other motifs, together with their classification,
all Watson–Crick (WC) edge and non-Watson–Crick (non-WC) edge pairs, along with their cis or trans configuration and edge-to-edge classification (20).
the number of WC-edge and non-WC-edge hydrogen bonds (and their sum) per nucleotide,
the stacking energy: van der Waals (VDW) and electrostatic interaction terms (and their sum) per nucleotide,
all phosphate anion–π interacting nucleotides,
files necessary for the visualization of the above properties.
The multiple conformation mode works as a standalone package to analyze many conformations of one molecule, e.g. from a trajectory. MINT computes the above listed properties for every frame/conformation and, in addition, outputs the statistics:
nucleotides forming helices, loops, pseudoknots and other motifs together with their occurrence (i.e. the frame numbers in which these motifs were detected and the percentage of trajectory time they lasted),
clusters of secondary structure motifs and average motifs along with 2D and 3D contacts,
all WC-edge, non-WC-edge pairs and triples, stacking and anion–π interacting nucleotides with their occurrence,
for each nucleotide MINT lists the nucleotides with which it formed hydrogen bonds (giving the number of hydrogen bonds and their occurrence),
the average secondary structure,
correlations in the breaking and forming of the WC-edge pairs,
the average number of WC-edge and non-WC-edge hydrogen bonds (and their sum) per nucleotide,
the average stacking energy – VDW and electrostatic terms (and their sum) per nucleotide,
visualizations of the outputs and files that can be used in VARNA (21), Visual Molecular Dynamics (VMD) (22), Chimera (23) and RNAMovies (24).
MINT is written in Python language. BioPython (25) is used to read files in the Protein Data Bank format (.pdb) and MDAnalysis (26) to process trajectories. The analysis of many frames/conformations can be run in parallel on any number of CPUs and is limited only by the amount of memory. MINT splits the trajectory into pieces of equal lengths and analyzes each sub-trajectory on a separate core but at the same time. Finally, the program computes statistics for all frames. The software, manual and server are available at http://mint.cent.uw.edu.pl.
Implementation
First, MINT reads a .pdb file with coordinates and then a file with many conformations of the same molecule. The supported formats for the latter are .dcd, .xyz, .trr and .crd. For every inputted frame MINT maps the hydrogen bonds, recognizes base pairs and writes the dot-bracket representation of the secondary structure. Next, it runs the algorithm to classify structural motifs and at the same time searches for stacking and anion–π interactions (Figure 1).
Figure 1.
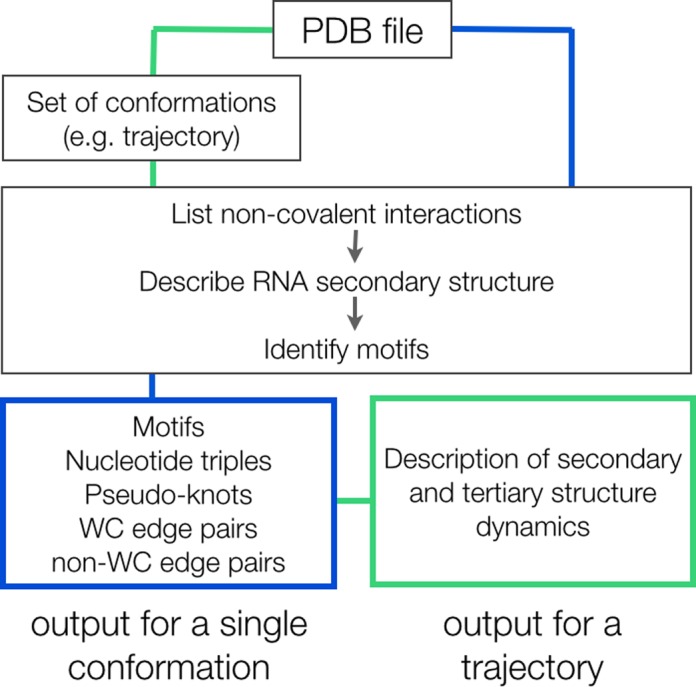
MINT workflow. The main function implements the analysis of a single frame. For a trajectory, the function first creates a table of all atoms from a .pdb file with their coordinates from the entire trajectory. While analyzing trajectory frames, the coordinates are read from the created table.
Hydrogen bond definition
A hydrogen bond is the basic term of the program. It is defined as a non-covalent interaction in which a hydrogen atom of a donor is placed close to the acceptor. The hydrogen bond criteria are defined by an angle between the acceptor, hydrogen atom and the donor (default minimal angle is 140 degrees) and a distance between the donor and acceptor (default: 3.25 Å) or a distance between the acceptor and hydrogen atom (default: 2.8 Å).
Donors and acceptors
To analyze nucleic acids we defined a list of possible acceptors and donors for all standard nucleotides (A, U, T, G, C). Following the classification by Leontis and Westhof (20) the acceptors and donors are assigned to the nucleotide edges shown in Figure 2.
Figure 2.

Nucleotide edges with hydrogen donors in green, acceptors in blue and atoms that serve as both hydrogen donor and acceptor in orange (27).
MINT checks if there is a hydrogen bond created by any of the donors or acceptors of all possible pairs placed within a defined distance cutoff. Knowing the atoms participating in hydrogen bonds, the program determines the interacting edges and using the edge information classifies a pair. Note that we use the edge-to-edge classification (20,27), instead of the concept of the canonical base pairs, to unambiguously and consistently describe all possible geometric pairs that may, even transiently, occur in a trajectory.
Base pair geometric isomerism
For detected nucleotide pairs geometric isomerism of their glycosidic bonds is computed. The program measures the torsion angle formed by four atoms (C1’, N1 in pyrimidines and C1’, N9 in purines) and depending on its value a cis or trans conformation is denoted.
Aromatic stacking
For almost 300 geometries of stacked base dimers, Šponer et al. compared ab initio stacking energies with energies obtained using simple pairwise-additive empirical potentials (28–31). They found that calculations applying the Lennard-Jones potential and Coulombic terms with atom-centered point charges reproduce the ab initio stacking energies of base dimers within +/−1.5 kcal/mol (30). This agreement suggests that calculations based on empirical potentials approximate well the stacking interaction energy between nucleobases. Therefore, we estimate the energy of stacking between two nucleobases as the sum of electrostatic (Uel) and van der Waals (UVDW) interaction terms:
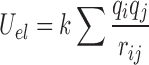 |
(1) |
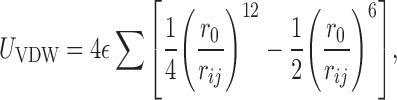 |
(2) |
where k is the Coulomb constant (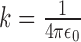 ),
qi and
qj are atom-centered point
charges, rij is the distance between
the atoms i, j,
),
qi and
qj are atom-centered point
charges, rij is the distance between
the atoms i, j, 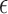 is the depth of the
Lennard-Jones potential well for atoms i, j, and
r0 is the sum of VDW radii of atoms
i and j. The sum runs over all atoms of both
interacting nucleobases.
is the depth of the
Lennard-Jones potential well for atoms i, j, and
r0 is the sum of VDW radii of atoms
i and j. The sum runs over all atoms of both
interacting nucleobases.
The planar shape of nucleobases ensures that the lowest VDW energies are obtained for parallel orientations of bases with the largest geometric overlap. With this in mind, we assume that two nucleobases are stacked, if their VDW energy is lower than a given threshold. By default it is set to −0.5 kcal/mol, and was estimated by trial and error but seems appropriate for non-modified nucleobases. MINT provides the VDW parameters and partial atomic charges for RNA and DNA nucleotides from the Amber (32) and Charmm (33,34) force fields. Users may also supply their own parameters.
Anion–π contacts
Following recent study on the types of non-covalent contacts in RNA, MINT also analyzes the phosphate oxygen contacts with nucleobases, the so-called anion–π contacts (P. Auffinger 2013, personal communication). An example of such contact is shown in Figure 3. These contacts are detected based on the distance between the oxygen atom of a phosphate group of one nucleotide and the center of mass of another nucleobase ring. The energy of interaction for these non-covalent contacts is estimated in the same way as for aromatic stacking. Only systems with the distance lower than a given threshold (default is 5 Å) are considered as anion–π interacting. Since there is no guarantee that the current force fields, based on empirical potentials, describe well these types of contacts, the user has to verify the energetics of the recognized complexes.
Figure 3.
The phosphate group oxygen atom ‘stacking’ over the guanine base. The oxygen and base atoms are shown as spheres of sizes corresponding to their VDW radii. A fragment is from the 4GD2.pdb file.
Modified nucleotides
Modified nucleotides are found in many functional non-coding RNAs, e.g. tRNAs (35,36). However, the standard Amber (32) and Charmm (33,34) force fields provide parameters only for the non-modified nucleotides. Also, the edge-to-edge classification of hydrogen bonds between RNA bases was proposed only for non-modified nucleotides A, U, G and C (20).
For modified nucleotides we provide the VDW parameters and partial atomic charges developed by Aduri et al. (37) for 107 naturally occurring modifications. For the edge-to-edge classification of base pairs formed by these nucleotides, we assign their atoms to four distinct edges: the WC edge, the Hoogsteen edge, the sugar edge and to the edge termed modification. Atoms common for the modified and non-modified nucleobase are assigned as in the non-modified one (Figure 2). If an atom existing in a non-modified nucleotide is substituted by one other atom or if one atom is added, we assign such atoms to the same edge as in the non-modified case. The 2'O methyl carbon is classified into the sugar edge. All other atoms of the modified nucleotide are classified into the modification edge.
If MINT detects a modified nucleotide, for which there are no parameters, it automatically assigns its atoms to edges. For atoms assigned to the modification edge the stacking interaction energy is set to 0. However, the user may add parameters for modified nucleotides. The force field parameters prepared for all-atom MD simulation of modified nucleotides can be further adopted to MINT parameter format.
Representation of RNA secondary structure
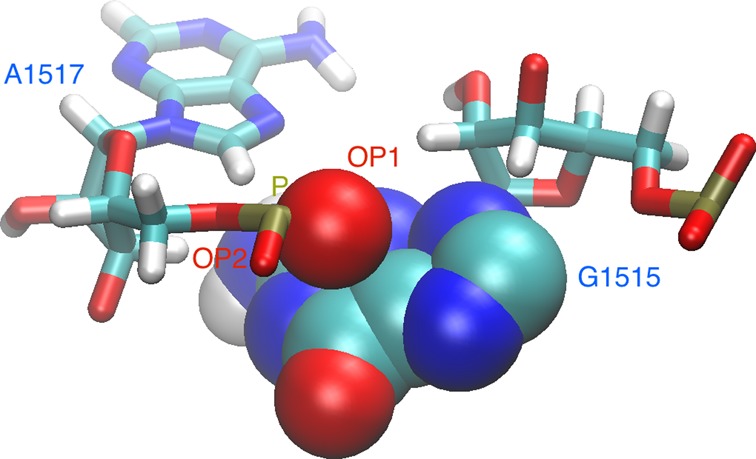
After detecting the WC-edge pairs, we create a list representation of the RNA secondary structure. Most nucleotides have only one WC-edge partner but in MD trajectory it may transiently happen that a second WC-edge partner is encountered, and such a triple is not considered in the secondary structure analysis. The index in the list represents the nucleotide number; the stored value is the index of its WC-edge partner. The list is easy interpretable if the arcs connecting the pairs are drawn as in Figure 4.
Figure 4.
An example of a list-representation of RNA secondary structure. Every cell of the matrix contains the nucleotide number that is WC-edge paired with the nucleotide indicated by the matrix index (above the cell). Therefore, nucleotide no 1 pairs with nucleotide no 25, nucleotide no 2 with 24 and so on. Base pairing is marked by black curved lines and the WC-edge interactions creating a pseudoknot by red curves.
Pseudoknots
The list-representation contains also pseudoknots–non-secondary motifs formed by WC-edge pairing. MINT detects a pseudoknot fold if the arcs intersect. A pseudoknot is a symmetric structure so in Figure 4 both the three pairs 6–17, 7–16 and 8–15 and the two pairs 12–21, 13–20 form a pseudoknot.
To erase pseudoknots from the list representation of the secondary structure, so they do not disturb the motif-search algorithm, we use a conflict elimination method (38) leading to a nested structure containing the maximum number of base pairs. In this case the pairs 12–21 and 13–20 are removed and classified as a pseudoknot.
RNA motif description
MINT describes motifs by numbering unpaired nucleotides detected between base pairs on the edges of the motif. For examples of the codes describing motifs see Supplementary Figure S1. A four nucleotide loop of a hairpin is assigned a single number 4. An asymmetric internal loop, with three unpaired nucleotides in one strand, is represented by two numbers 0–3. A symmetric internal loop, with three unpaired nucleotides in each strand, is coded as 3–3, and a three-way junction, without unpaired nucleotides: 0–0–0.
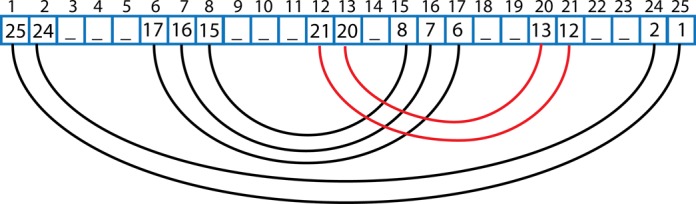
Motif-search algorithm
The algorithm uses a list representation of the RNA secondary structure. To detect helices and other motifs it walks through the list and stores the information about the structure (Figure 5).
Figure 5.
A scheme of the algorithm traveling around the secondary structure of RNA in the graph and list representation. The arrows and their numbers indicate sequential steps of the algorithm. Blue arrows mark helices and green arrows other structural motifs. Orange lines show the jumps that the algorithm takes after distinguishing a motif. The STOP sign indicates the position where the algorithm terminates.
For two sequential WC-edge paired nucleotides, the algorithm stores that the nucleotide is a part of a helix (step 1 in Figure 5). If a helix ends, i.e. an unpaired nucleotide is encountered ahead of a pair, the algorithm starts to travel around the motif; it remembers the first pair, the border of the motif, and goes to the index stored in the list (step 2). The algorithm moves back until it encounters an unpaired nucleotide—with decreasing indexes (steps 3–9); if a pair is found, the algorithm goes to the indicated position in the list (step 10) and again moves back (steps 11–13); the motif ends once the algorithm finds itself one step ahead of the previously remembered starting index of the motif (step 13). After a motif is found and classified, the algorithm jumps one step further from the last seen pair (step 14), and behaves identically: stores the helix (step 15), jumps to a pair of the last nucleotides (step 16), travels around the motif (steps 17–22), finds itself one step ahead of the starting motif (step 22) and jumps to the last seen pair plus one index (step 23). The algorithm stops searching for the motifs once the index is larger than the value stored in the list (step 23).
Single-conformation analysis mode
To analyze a single .pdb file, we also provide the MINT web server at http://mint.cent.uw.edu.pl. Either an all-atom .pdb file or a PDB code can be submitted. In the latter case, the file with a specified PDB code will be automatically downloaded and protonated using Reduce (39). The user can further download the output files and visualize the secondary and tertiary structures colored by the computed descriptors analogous to the ones shown in Figure 6.
Figure 6.
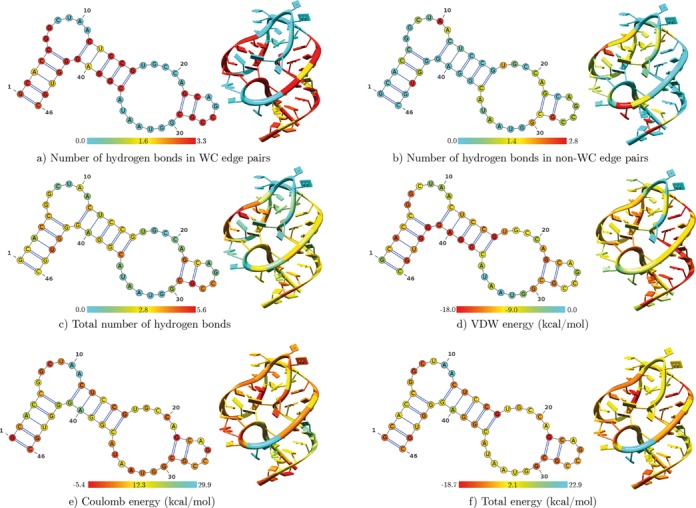
Secondary and tertiary structures of a 16S rRNA fragment (nucleotides 500–545) colored based on various descriptors calculated per nucleotide and averaged over the trajectory.
Trajectory analysis mode
For many RNA or DNA conformations, e.g. from the trajectory files, every frame is characterized as previously described. The main output is an .xls file listing all base pairs, helices, loops, junctions, nucleotide triples, pseudoknots, etc., with their topologies and participating nucleotides, as well as the frame numbers in which these motifs were detected.
Clustering
To describe the changes in the RNA secondary structure during dynamics, the detected motifs are clustered. Clustering is parameterized with two user-defined parameters: the minimal percentage of frames in which the motif has to be present to be classified and the minimal percentage of similarities between the two motifs to belong to the common cluster.
In the first step rare motifs are removed by filtering them according to the frequency of occurrence. Second, the motifs’ distance matrix is computed. The distance between motifs is defined as the number of their common nucleotides. The order of the nucleotides is not taken into account. Third, the motif with the longest list of partners is incorporated to the first cluster. Next, the second longest is chosen and so on. The motifs used in the first cluster and the sequential created clusters are deleted from the list—a motif can be present only in one cluster.
Dynamical propagation of the secondary structure
To detect correlations in the propagation of the secondary structure for every nucleotide, we compute a ϕ correlation coefficient defined as
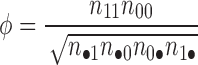 |
(3) |
where
n11 is the number of frames in which both nucleotides form a WC-edge pair, analogously n00 is the number of frames in which none of the nucleotides forms a WC-edge pair.
in the denominator n•1 = n11 + n01, n1• = n11 + n10, n•0 = n00 + n10, n0• = n00 + n01.
n01 is the number of frames in which the first nucleotide forms a WC-edge pair and the second nucleotide does not, analogously n10 is the number of frames in which the first nucleotide is WC-edge-paired and the second one is not. Note, the numbering in Python starts from 0 index.
The ϕ coefficient ranges from −1 to 1 so ϕ close to 0 suggests no correlation. A symmetric matrix with ϕ values is outputted both as a text file and heat map.
MD simulations of small ribosomal subunit
System preparation
The crystal structure of the Escherichia coli 30S subunit resolved with the 3.0 Å resolution (PDB code 4V9D) was taken as the starting conformation (40). This structure had the best resolution and longest 16S rRNA among the ribosome structures deposited in PDB (as of June 2012). The tRNAs were removed but the crystal waters and divalent ions were kept. Hydrogen atoms were added and the system was solvated with explicit waters extending at least 15 Å from any atom of the solute. Counterions and excess ions were added to achieve 0.15 M concentration of NaCl. These preparatory steps were performed with the VMD (22) program and CHARMM36 force field (41). The simulated system consisted of almost 866 000 atoms and trajectories were generated using NAMD (42).
Particle Mesh Ewald method, SHAKE algorithm with a time step of 2 fs and periodic boundary conditions were used. The cutoff parameter for the VDW and electrostatic interactions was set to 12 Å, the switching distance to 15 Å and pair list distance to 18 Å.
Simulation protocol
First, the solvent was energy minimized, second it was gradually heated, with the solute constrained. The temperature was increased from 30 K, increasing 10 K every 100 steps up to 300 K. Third, the system was equilibrated at 300 K in two phases. The constraints on solute atoms were gradually decreased in the following steps of 0.1 ns runs using the force constants: (i) 25 → 1 kcal/mol, (ii) 1 → 0.0076 kcal/mol, (iii) 0.0075 → 0.0042 kcal/mol, (iv) 0.0042 → 0.00167 kcal/mol. Next, unconstrained equilibration was performed for ∼35 ns at 300 K. It followed by 30 ns of the production, which was analyzed with MINT.
In the production phase the average root-mean-square deviation from the starting structure for heavy atoms was 4.2 ± 0.8 Å and the radius of gyration increased from the starting value of 66.2 Å to an average of 67.7 ± 0.1 Å (Supplementary Figure S2).
Results
We present MINT functionality based on the analysis of a 16S rRNA fragment (nucleotides 500–545, encompassing helix 18) from the 30 ns MD production trajectory of the 30S subunit (Supplementary Figure S3). The MINT output files comprehensively describe RNA conformations at both secondary and tertiary levels.
Nucleotide surrounding and interactions
The main MINT output provides a general overview of the contacts in the conformation set. It lists all nucleotides with the number of WC- and/or non-WC-edge hydrogen bond pairs formed by a particular nucleotide, as well as stacking interactions: Coulomb, VDW, their sum and averages over all frames. This output, whose fragment is listed in Table 1, helps detect unusual hydrogen bonding patterns, e.g. with the high average number of non-WC-edge bonds. Whole output listing also the average VDW and Coulomb energies is shown in Supplementary Table S1.
Table 1. Average numbers of hydrogen bonds (hbonds) with standard deviations observed in a trajectory for WC-edge and non-WC-edge pairs.
| Nucleotide no. | WC hbonds | Non-WC hbonds | Nucleotide no. | WC hbonds | Non-WC hbonds |
|---|---|---|---|---|---|
| G527 | 3.2±0.6 | 2.4±0.7 | A532 | 0.0±0.0 | 0.5±0.5 |
| C528 | 3.1±0.5 | 1.5±0.5 | A533 | 0.2±0.7 | 1.1±0.2 |
| G529 | 0.0±0.0 | 1.8±0.4 | U534 | 0.0±0.0 | 0.4±0.5 |
| G530 | 0.0±0.1 | 0.0±0.1 | A535 | 0.0±0.0 | 1.2±0.4 |
| U531 | 0.0±0.0 | 0.2±0.5 | C536 | 3.1±0.5 | 1.2±0.5 |
MINT allows projecting the computed descriptors on tertiary and secondary structures. It creates six .pdb files in which the temperature factor column for each nucleotide is replaced with, respectively, the number of WC-edge, non-WC-edge hydrogen bonds, and their sum, Coulomb, VDW energy and stacking energy. Therefore, one may view the structure in, e.g. VMD (22) or Chimera (23) and produce images analogous to the ones shown in Figure 6. To annotate average secondary structures and create images, MINT uses the VARNA (21) visualization applet. Examples of secondary structure graphics for helix 18 are shown in Figure 6 and for the entire 3′ major domain of 16S RNA in Supplementary Figure S4.
MINT also lists statistics of nucleotide contacts giving the percentage of frames in which a given pair, triple or other pattern occurred. An example is listed in Table 2 and shown in Figure 7.
Table 2. Nucleotide hydrogen contacts observed in a trajectory.
| Nucleotide | Hydrogen bond contacts | |
|---|---|---|
| G527 | C522 A535: 87% | |
| C528 | G521 A535: 74% | |
| G529 | G517 C519 A520: 39% | C519 A520: 37% |
| G530 | no contatcs: 100% | |
| U531 | no contatcs: 83% | |
| A532 | no contatcs: 51% | U516 A520: 15% |
| A533 | U516 A520 A535: 22% | |
| U534 | no contacts: 57% | C511: 20% U512: 16% |
| A535 | G527 C528 A533: 48% | G527 C528: 18% |
| C536 | G515 G521: 43% | |
Each row lists: the nucleotide type with its number, the nucleotides it hydrogen bonds with, the percentage of trajectory time these contacts were formed. The ‘no contacts’ denote the percentage of frames the nucleotide did not create any hydrogen bonds. Only the contacts present in more than 10% of the trajectory are listed so the contacts in a row do not have to sum up to 100%. For example, A535 for 48% of trajectory time contacts G527, C528 and A533, but 18% of time only G527 and C528 (Figure 7). This dynamic interaction is revealed in MD—in the crystal A535 pairs only with U516. For full output which includes also the number of hydrogen bonds see Supplementary Table S2.
Figure 7.
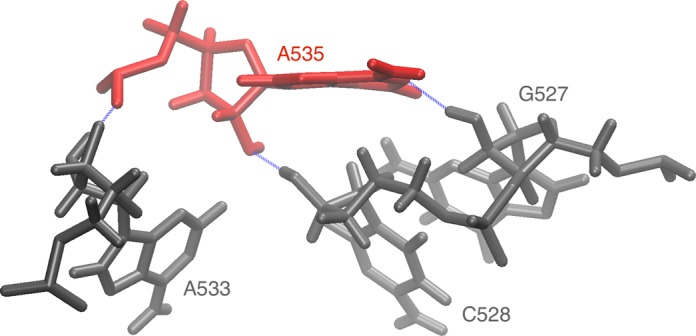
Prevalent hydrogen bonding pattern of A535. The conformation is from the 9.49 ns frame with hydrogen bonds marked as blue dashed lines. Nucleotides are listed in Table 2. The structural context of this arrangement is shown and explained in Supplementary Figure S5.
Classification of pairs
The nucleic acid structure is characterized with a list of pairs described according to the edge-to-edge (Figure 2) classification (20), geometric isomerism and percentage of frames a certain pair occurred in the trajectory along with frame numbers. Table 3 shows a fragment of the output listing nucleotide pairs.
Table 3. The MINT output listing nucleotide pairs.
| Nucleotide pair | Interacting edges | Configuration | % of frames |
|---|---|---|---|
| G527/A535 | Sugar/WC | trans | 81% |
| G527/A535 | Sugar/Sugar | trans | 15% |
| C528/A535 | Sugar/Sugar | trans | 97% |
| C528/A533 | Sugar/Sugar | cis | 13% |
| U531/G1207 | Sugar/Sugar | cis | 16% |
| A532/U1056 | WC*Hoogsteen/WC*Sugar | cis | 20% |
| A532/A1055 | WC*Hoogsteen/Sugar | trans | 17% |
| A532/A1055 | WC*Hoogsteen/Sugar | cis | 16% |
| A533/A535 | Sugar/Sugar | trans | 55% |
| A533/C536 | Sugar/WC*Hoogsteen | trans | 22% |
Each pair is classified by interacting nucleotide edges, configuration, and the percentage of trajectory frames in which the pair was detected. The * notation is used if only one hydrogen bond between nucleotides is found, involving corner atoms, and it is impossible to assign the edge uniquely.
Stacking interactions
The stacking energy is presented as the sum of the Coulomb and VDW energies between the bases of two nucleotides with an energy threshold for stacked bases on the VDW energy (see Materials and Methods). An example of the output is shown in Table 4.
Table 4. List of stacked bases along with their trajectory averaged stacking energy and the percentage of frames the interaction was present.
| Stacked bases | Average Coulomb energy (kcal/mol) | Average VDW energy (kcal/mol) | Average stacking energy (kcal/mol) | % of frames |
|---|---|---|---|---|
| G527/C528 | −6.8±1.3 | −5.5±0.5 | −12.2±1.4 | 100% |
| C528/G529 | −2.8±1.1 | −3.9±0.5 | −6.7±1.2 | 99% |
| C528/A533 | 6.8±0.9 | −0.6±0.1 | 6.2±0.9 | 28% |
| G529/A533 | 2.3±0.3 | −0.6±0.1 | 1.8±0.3 | 22% |
| A532/G1206 | 1.9 ±0.8 | −1.1±0.5 | 0.8±0.9 | 85% |
| A532/A1055 | 15.1±2.6 | −1.1±0.4 | 14.1±2.6 | 40% |
| A532/U1056 | 5.5±2.5 | −0.9±0.3 | 4.7±2.5 | 22% |
| A533/C536 | 6.7±1.9 | −5.7±0.6 | 0.9±2.0 | 77% |
A nucleotide can show up several times because it may create alternative stacking interactions with different nucleotides.
An analogous table is constructed for the anion–π contacts — their energy is calculated as the sum of the Coulomb and VDW energies between a base and oxygen atom (Supplementary Table S3).
Dynamical correlations within hydrogen bonding patterns
Based on the contacts that each nucleotide forms in each frame, we computed the ϕ coefficient. Figure 8 shows its illustration as a heat map. The uncorrelated regions (ϕ close to 0) characterize nucleotides which either do not form pairs or form a strong pair that never opens in the course of the dynamics. Therefore, the heat map does not characterize the secondary structure (43) but rather the mobile parts of the structure. The synchronous movement of two nucleotides is indicated by positive ϕ (the same pair opens and closes). This heat map should be analyzed taking into account the pseudoknot in this fragment (not seen in a simple 2D representation) whose 3D structure is described in Supplementary Figure S6. A negative correlation occurs for an asynchronous movement, i.e. if a nucleotide is paired (‘closed’) while the other is open and conversely. A trajectory-based example for nucleotides 504 and 542, in which the G542 base ‘jumps’ between two others, is shown in Supplementary Figure S7. Overall, the heat map is useful while searching for the non-obvious structural blocks.
Figure 8.
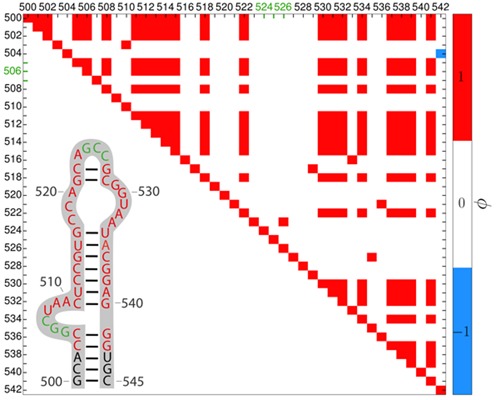
Heat map of the ϕ correlation coefficient for the 503–542 16S rRNA region from the MD simulation. The inset shows the secondary structure (43) with nucleotides creating a pseudoknot in green. Axes labels stand for nucleotide numbers. The ϕ coefficients larger than +0.4 (the cutoff for the color scale is defined by the user) are in red, lower than −0.4 in blue and the rest is in white. Nucleotides correlate with themselves so the diagonal is red. Every paired nucleotide and its neighboring pairs have ϕ above 0.4, indicating positive correlations in accord with their synchronous movement.
Dynamics of structural motifs
For every frame MINT creates a dot-bracket representation of the secondary structure. Next, it generates an .xml file, which can be further imported in RNAMovies (24) to visualize the evolution of the secondary structure in a trajectory (Supplementary Figure S8).
All detected motifs and their classification are written to a separate sheet whose fragment is listed in Table 5. The motifs are further clustered. A cluster contains various secondary structure motifs appearing in the same RNA region so it describes the structural flexibility of the given fragment (Figure 9).
Table 5. A fragment of the MINT output with a list of secondary structure motifs and their occurrence.
| Cluster | Motif | Motif forming nucleotides | % of frames | |
|---|---|---|---|---|
| No. | No. | Code | ||
| 0 | 0 | 4 | C522-G527-C526-C525-G524-A523-C522- | 100% |
| 1 | 1 | 0–6 | C504-G541-G540-C511-A510-A509-U508-C507-G506-G505-C504- | 99% |
| 2 | 2 | 7–5 | G515-C536-A535-U534-A533-A532-U531-G530-G529-C528-G521-A520-C519-C518-G517-U516-G515- | 88% |
| 3 | 4–0 | A520-A533-A532-U531-G530-G529-C528-G521-A520- | 12% | |
| 4 | 2–4 | G515-C536-A535-U534-A533-A520-C519-C518-G517-U516-G515- | 12% | |
A motif is labeled according to classification shown in Supplementary Figure S1.
Figure 9.
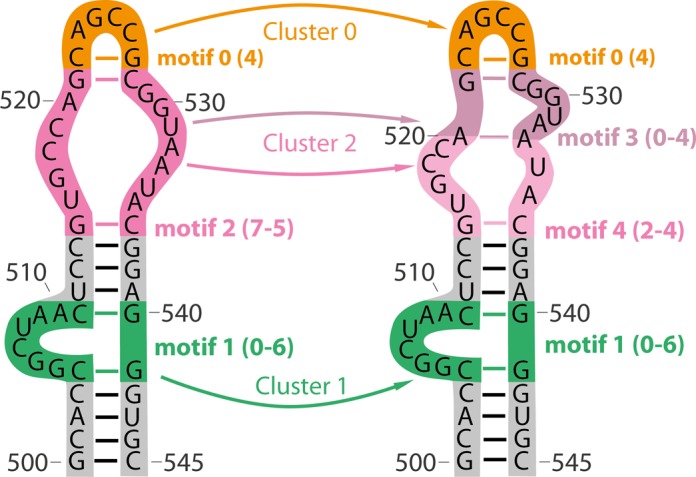
Schematic representation of clustering of motifs listed in Table 5. Cluster 0 consists of motif 0 with code 4, cluster 1 of motif 1 with codes 0–6, and cluster 2 of motifs no. 2, 3 and 4. For motif codes see Supplementary Figure S1. The scheme presents two secondary structures of the same RNA fragment that was simulated. Motifs engaging the same nucleotides fall in the same clusters.
Next, for a given cluster MINT computes an average motif by taking the longest motif from the cluster and annotating it based on the average WC- and non-WC-edge hydrogen bonds. Such a list is used to identify active nucleotides (which form and break hydrogen bonds during the simulation) as presented in Supplementary Table S4.
In summary, MINT output consists of an .xls file with separate sheets (and, if requested, the .csv files), .pdb files for structure visualization, .png files with secondary structures colored by the hydrogen bonding and stacking energy, an .xml input file to be readily used in RNAMovies and a simple text file with a detailed description of the RNA or DNA structure in each conformation frame. Supplementary Figure S9 presents MINT benchmarks and performance depending on the RNA chain length and CPUs.
CONCLUSION
We characterized a tool for post-processing MD-derived trajectories or other large conformation sets of nucleic acid molecules. MINT calculates various descriptors for nucleic acid structures, provides their time evolution and facilitates their visualization. MINT lists the statistics of these descriptors to enable relating them with function, e.g. experimental mutational analyses listed in Supplementary Table S5 for ribosomal RNA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
University of Warsaw [CeNT/BST and ICM/KDM/G31-4]; National Science Centre [DEC-2012/05/B/NZ1/00035 to J.T., M.J. and DEC-2011/03/N/NZ2/02482 to M.J.]; European Social Fund [contract no UDA-POKL.04.01.01-00-072/09-00 to M.J.]; Foundation for Polish Science (Team project co-financed by European Regional Development Fund operated within Innovative Economy Operational Programme).
Conflict of interest statement. None declared.
REFERENCES
- 1.Bloomfield V.A., Crothres D.M., Tinoco I.J. Nucleic acids: structures, properties, and functions. Sausalito, CA: University Science Books; 1999. [Google Scholar]
- 2.Šponer J., Lankas F. Computational studies of RNA and DNA. Dordrecht: Springer; 2006. [Google Scholar]
- 3.Onuchic J.N., Sanbonmatsu K.Y. Magnesium fluctuations modulate RNA dynamics in the SAM-I riboswitch. J. Am. Chem. Soc. 2012;134:12043–12053. doi: 10.1021/ja301454u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Krepl M., Réblová K., Koča J., Šponer J. Bioinformatics and molecular dynamics simulation study of L1 stalk non-canonical rRNA elements: kink-turns, loops, and tetraloops. J. Phys. Chem. B. 2013;117:5540–5555. doi: 10.1021/jp401482m. [DOI] [PubMed] [Google Scholar]
- 5.Panecka J., Mura C., Trylska J. Interplay of the bacterial ribosomal A-site, S12 protein mutations and paromomycin binding: a molecular dynamics study. PLoS ONE. 2014;9:e111811. doi: 10.1371/journal.pone.0111811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Whitford P.C., Onuchic J.N., Sanbonmatsu K.Y. Connecting energy landscapes with experimental rates for aminoacyl-tRNA accommodation in the ribosome. J. Am. Chem. Soc. 2010;132:13170–13171. doi: 10.1021/ja1061399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nivón L.G., Shakhnovich E.I. All-atom Monte Carlo simulation of GCAA RNA folding. J. Mol. Biol. 2004;344:29–45. doi: 10.1016/j.jmb.2004.09.041. [DOI] [PubMed] [Google Scholar]
- 8.Tang X., Thomas S., Tapia L., Giedroc D.P., Amato N.M. Simulating RNA folding kinetics on approximated energy landscapes. J. Mol. Biol. 2008;381:1055–1067. doi: 10.1016/j.jmb.2008.02.007. [DOI] [PubMed] [Google Scholar]
- 9.Laing C., Schlick T. Computational approaches to 3D modeling of RNA. J. Phys. Condens. Matter. 2010;22:283–101. doi: 10.1088/0953-8984/22/28/283101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jossinet F., Ludwig T.E., Westhof E. Assemble: an interactive graphical tool to analyze and build RNA architectures at the 2D and 3D levels. Bioinformatics. 2010;26:2057–2059. doi: 10.1093/bioinformatics/btq321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lavery R., Moakher M., Maddocks J.H., Petkeviciute D., Zakrzewska K. Conformational analysis of nucleic acids revisited: Curves+ Nucleic Acids Res. 2009;37:5917–5929. doi: 10.1093/nar/gkp608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lu X.J., Olson W.K. 3DNA: a versatile, integrated software system for the analysis, rebuilding, and visualization of three-dimensional nucleic-acid structures. Nat. Protoc. 2008;3:1213–1227. doi: 10.1038/nprot.2008.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Olson W.K., Bansal M., Burley S.K., Dickerson R.E., Gerstein M., Harvey S.C., Heinemann U., Lu X.J., Neidle S., Shakked Z., et al. A standard reference frame for the description of nucleic acid base-pair geometry. J. Mol. Biol. 2001;313:229–237. doi: 10.1006/jmbi.2001.4987. [DOI] [PubMed] [Google Scholar]
- 14.Gendron P., Lemieux S., Major F. Quantitative analysis of nucleic acid three-dimensional structures. J. Mol. Biol. 2001;308:919–936. doi: 10.1006/jmbi.2001.4626. [DOI] [PubMed] [Google Scholar]
- 15.Yang H. Tools for the automatic identification and classification of RNA base pairs. Nucleic Acids Res. 2003;31:3450–3460. doi: 10.1093/nar/gkg529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pietal M.J., Szostak N., Rother K.M., Bujnicki J.M. RNAmap2D - calculation, visualization and analysis of contact and distance maps for RNA and protein-RNA complex structures. BMC Bioinformatics. 2012;13:333. doi: 10.1186/1471-2105-13-333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wale T., Chojnowski G., Gierski P., Bujnicki J.M. ClaRNA: a classifier of contacts in RNA 3D structures based on a comparative analysis of various classification schemes. Nucleic Acids Res. 2014;42:e151. doi: 10.1093/nar/gku765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lu X.J., Olson W.K., Bussemaker H.J. The RNA backbone plays a crucial role in mediating the intrinsic stability of the GpU dinucleotide platform and the GpUpA/GpA miniduplex. Nucleic Acids Res. 2010;38:4868–4876. doi: 10.1093/nar/gkq155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kumar R., Grubmüller H. do_x3dna: a tool to analyze structural fluctuations of dsDNA or dsRNA from molecular dynamics simulations. Bioinformatics. 2015 doi: 10.1093/bioinformatics/btv190. doi:10.1093/bioinformatics/btv190. [DOI] [PubMed] [Google Scholar]
- 20.Leontis N.B., Stombaugh J., Westhof E. The non-Watson-Crick base pairs and their associated isostericity matrices. Nucleic Acids Res. 2002;30:3497–3531. doi: 10.1093/nar/gkf481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Darty K., Denise A., Ponty Y. VARNA: interactive drawing and editing of the secondary structure. Bioinformatics. 2009;25:1974–1975. doi: 10.1093/bioinformatics/btp250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Humphrey W., Dalke A., Schulten K. VMD: visual molecular dynamics. J. Mol. Graphics. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 23.Pettersen E.F., Goddard T.D., Huang C.C., Couch G.S., Greenblatt D.M., Meng E.C., Ferrin T.E. UCSF Chimera–a visualization system for exploratory research and analysis. J. Comput. Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 24.Evers D., Giegerich R. RNA Movies: visualizing RNA secondary structure spaces. Bioinformatics. 1999;15:32–37. doi: 10.1093/bioinformatics/15.1.32. [DOI] [PubMed] [Google Scholar]
- 25.Cock P.J., Antao T., Chang J.T., Chapman B.A., Cox C.J., Dalke A., Friedberg I., Hamelryck T., Kauff F., Wilczynski B.E.A. Biopython: freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics. 2009;25:1422–1423. doi: 10.1093/bioinformatics/btp163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Michaud-Agrawal N., Denning E.J., Woolf T.B., Beckstein O. MDAnalysis: a toolkit for the analysis of molecular dynamics simulations. J. Comput. Chem. 2011;32:2319–2327. doi: 10.1002/jcc.21787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lescoute A., Westhof E. The interaction networks of structured RNAs. Nucleic Acids Res. 2006;34:6587–6604. doi: 10.1093/nar/gkl963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Carter A.P., Clemons W.M., Brodersen D.E., Morgan-Warren R.J., Wimberly B.T., Ramakrishnan V. Functional insights from the structure of the 30S ribosomal subunit and its interactions with antibiotics. Nature. 2000;407:340–348. doi: 10.1038/35030019. [DOI] [PubMed] [Google Scholar]
- 29.Šponer J., Gabb H.A., Leszczynski J., Hobza P. Base-base and deoxyribose-base stacking interactions in B-DNA and Z-DNA: a quantum-chemical study. Biophys. J. 1997;73:76–87. doi: 10.1016/S0006-3495(97)78049-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Šponer J., Leszczynski J., Hobza P. Nature of nucleic acid–base stacking: nonempirical ab initio and empirical potential characterization of 10 stacked base dimers. Comparison of stacked and H-bonded base pairs. J. Phys. Chem. 1996;100:5590–5596. [Google Scholar]
- 31.Šponer J., Leszczynski J., Hobza P. Base stacking in cytosine dimer. A comparison of correlated ab initio calculations with three empirical potential models and density functional theory calculations. J. Comput. Chem. 1995;17:841–850. [Google Scholar]
- 32.Wang J., Cieplak P., Kollman P.A. How well does a restrained electrostatic potential (RESP) model perform in calculating conformational energies of organic and biological molecules? J. Comput. Chem. 2000;21:1049–1074. [Google Scholar]
- 33.Mackerell A.D., Banavali N.K. All-atom empirical force field for nucleic acids: II. Application to molecular dynamics simulations of DNA and RNA in solution. J. Comput. Chem. 2000;21:105–120. [Google Scholar]
- 34.Foloppe N., Mackerell A.D. All-atom empirical force field for nucleic acids: I. Parameter optimization based on small molecule and condensed phase macromolecular target data. J. Comput. Chem. 2000;21:86–104. [Google Scholar]
- 35.Machnicka M.A., Milanowska K., Osman Oglou O., Purta E., Kurkowska M., Olchowik A., Januszewski W., Kalinowski S., Dunin-Horkawicz S., Rother K.M.E.A. MODOMICS: a database of RNA modification pathways–2013 update. Nucleic Acids Res. 2013;41:D262–D267. doi: 10.1093/nar/gks1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cantara W.A., Crain P.F., Rozenski J., McCloskey J.A., Harris K.A., Zhang X., Vendeix F.A., Fabris D., Agris P.F. The RNA modification database, RNAMDB: 2011 update. Nucleic Acids Res. 2011;39:195–201. doi: 10.1093/nar/gkq1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Aduri R., Psciuk B.T., Saro P., Taniga H., Schlegel H.B., SantaLucia J. AMBER force field parameters for the naturally occurring modified nucleosides in RNA. J. Chem. Theory Comput. 2007;3:1464–1475. doi: 10.1021/ct600329w. [DOI] [PubMed] [Google Scholar]
- 38.Smit S., Rother K., Heringa J., Knight R. From knotted to nested RNA structures: a variety of computational methods for pseudoknot removal. RNA. 2008;14:410–416. doi: 10.1261/rna.881308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Word J.M., Lovell S.C., Richardson J.S., Richardson D.C. Asparagine and glutamine: using hydrogen atom contacts in the choice of side-chain amide orientation. J. Mol. Biol. 1999;285:1735–1747. doi: 10.1006/jmbi.1998.2401. [DOI] [PubMed] [Google Scholar]
- 40.Dunkle J.A., Wang L., Feldman M.B., Pulk A., Chen V.B., Kapral G.J., Noeske J., Richardson J.S., Blanchard S.C., Cate J.H. Structures of the bacterial ribosome in classical and hybrid states of tRNA binding. Science. 2011;332:981–984. doi: 10.1126/science.1202692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Denning E.J., Priyakumar U.D., Nilsson L., MacKerell A.D. Jr. Impact of 2-hydroxyl sampling on the conformational properties of RNA: update of the CHARMM all-atom additive force field for RNA. J. Comput. Chem. 2011;32:1929–1943. doi: 10.1002/jcc.21777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Phillips J.C., Braun R., Wang W., Gumbart J., Tajkhorshid E., Villa E., Chipot C., Skeel R.D., Kalé L., Schulten K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Noller H.F., Woese C.R. Secondary structure of 16S ribosomal RNA. Science. 1981;212:403–411. doi: 10.1126/science.6163215. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.