


Abstract
RNA viruses encode an RNA-dependent RNA polymerase (RdRp) that catalyzes the synthesis of their RNA(s). In the case of positive-stranded RNA viruses belonging to the order Nidovirales, the RdRp resides in a replicase subunit that is unusually large. Bioinformatics analysis of this non-structural protein has now revealed a nidoviral signature domain (genetic marker) that is N-terminally adjacent to the RdRp and has no apparent homologs elsewhere. Based on its conservation profile, this domain is proposed to have nucleotidylation activity. We used recombinant non-structural protein 9 of the arterivirus equine arteritis virus (EAV) and different biochemical assays, including irreversible labeling with a GTP analog followed by a proteomics analysis, to demonstrate the manganese-dependent covalent binding of guanosine and uridine phosphates to a lysine/histidine residue. Most likely this was the invariant lysine of the newly identified domain, named nidovirus RdRp-associated nucleotidyltransferase (NiRAN), whose substitution with alanine severely diminished the described binding. Furthermore, this mutation crippled EAV and prevented the replication of severe acute respiratory syndrome coronavirus (SARS-CoV) in cell culture, indicating that NiRAN is essential for nidoviruses. Potential functions supported by NiRAN may include nucleic acid ligation, mRNA capping and protein-primed RNA synthesis, possibilities that remain to be explored in future studies.
INTRODUCTION
Positive-stranded (+) RNA viruses of the order Nidovirales can infect either vertebrate (families Arteriviridae and Coronaviridae) or invertebrate hosts (Mesoniviridae and Roniviridae) (1,2). Examples of nidoviruses with high economic and societal impact are the arterivirus porcine reproductive and respiratory syndrome virus (PRRSV) (3) and the zoonotic coronaviruses (CoVs) causing severe acute respiratory syndrome (SARS) and Middle East respiratory syndrome (MERS) in humans (4–6). While nidoviruses constitute a monophyletic group, their genome size differences are striking, with genomes ranging from 13–16 kb for arteriviruses to 20–21 kb for mesoniviruses and 25–34 kb for roniviruses and coronaviruses, which may reflect different stages of the largest genome expansion known to have occurred in RNA viruses (7).
Nidoviruses are characterized by their distinct polycistronic genome organization, the conservation of key replicative enzymes, and a common genome expression and replication strategy (8) (Figure 1). Their distinctive transcription mechanism involves the synthesis of a variable set of subgenomic (sg) mRNAs, which are 3′ co-terminal with the viral genome (reviewed in (9, 10)). In most nidoviruses, sg mRNAs and genome also share a common 5′ leader sequence. The synthesis of sg mRNAs (transcription) and genome RNA (replication) is performed by a poorly characterized replication-transcription complex (RTC) that is comprised of multiple protein subunits (reviewed in (11,12,13)) encoded in two large open reading frames (ORFs), ORF1a and ORF1b, which are translated from the nidoviral genomic RNA (Figure 1A). The two polyproteins (pp), pp1a and pp1ab, the latter resulting from ribosomal frameshifting during genome translation, are auto-catalytically processed by multiple cognate proteases, one of which (the 3C-like (3CLpro) or main (Mpro) protease) is responsible for the large majority of cleavages (14). Downstream of ORF1b, nidovirus genomes contain multiple smaller ORFs, known as the 3′ ORFs (7), which are expressed from the sg mRNAs described above.
Figure 1.
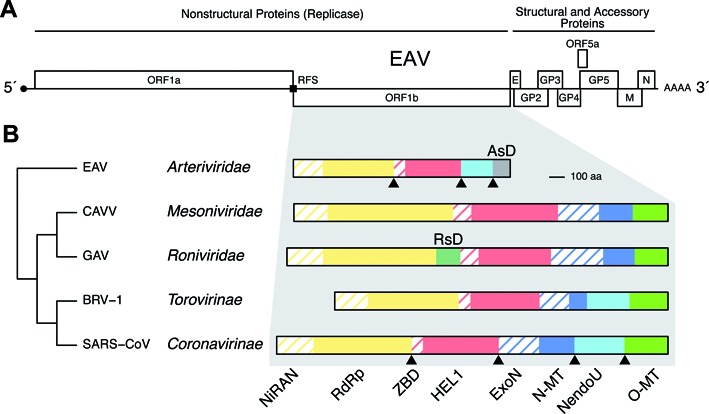
Genome organization and ORF1b-encoded enzymes and domains of nidoviruses. (A) The genome organization of Equine arteritis virus (EAV), including replicase open reading frames (ORFs) 1a and 1b, and 3′ ORFs encoding structural proteins, is shown. Genomes of other nidoviruses employ similar organizations while they may vary in respect to size of different regions and number of 3′ ORFs. RFS, ribosomal frameshift site. (B) ORF1b size and domain comparison between the five nidovirus (sub)families is shown for EAV (Arteriviridae), Cavally virus (CAVV, Mesoniviridae), Gill-associated virus (GAV, Roniviridae), Breda virus (BRV-1, Torovirinae) and Severe acute respiratory syndrome coronavirus (SARS-CoV, Coronavirinae); see Supplementary Table 1 for details regarding these viruses. NiRAN, nidovirus RdRp-associated nucleotidyltransferase; RdRp, RNA-dependent RNA polymerase; ZBD, Zn-binding domain; HEL1, helicase superfamily 1 core domain; ExoN, exoribonuclease; N-MT, N7-methyltransferase; NendoU, nidovirus uridylate-specific endoribonuclease; O-MT, 2′-O-methyltransferase; AsD, arterivirus-specific domain; RsD, ronivirus-specific domain. Depicted is a simplified domain organization since most enzymes are part of multidomain proteins. Note that viruses of the Torovirinae subfamily encode a truncated version of N-MT. Triangles, established cleavage sites by 3CLpro in two virus (sub)families; ORF1b-encoded proteins of other viruses may be proteolytically processed in a similar way. The order of emergence of different nidovirus (sub)families is presented by a simplified tree on the left.
During evolution, most conserved proteins of nidoviruses have diverged more extensively than those of organisms of the Tree of Life. In line with the principal function of each region, genome conservation increases from 3′ ORFs to ORF1a to ORF1b (7). Accordingly, the 3′ ORF region encodes virion proteins and, optionally, accessory proteins that are predominantly group- or family-specific and mediate virus–host interactions (15,16). ORF1a encodes a variable number of proteins that include co-factors of the RNA-dependent RNA polymerase (RdRp) and other ORF1b-encoded enzymes, three hydrophobic proteins mediating the association of the RTC with membranes and the viral proteases (13,17,18,19). The latter group includes the 3CLpro, which is the only ORF1a-encoded enzyme conserved in all nidoviruses. In contrast, ORF1b is highly conserved and encodes different RNA-processing enzymes that critically control viral RNA synthesis (Figure 1B). These invariantly include the RdRp and a superfamily 1 helicase domain (HEL1), which is fused with a multinuclear Zn-binding domain (ZBD). RdRp and HEL1 are expressed as part of two different cleavage products residing next to each other in pp1ab (8). The RdRp is believed to mediate the synthesis of all viral RNA molecules, while over the years the unwinding activity of the helicase was implicated in the control of replication, transcription, translation, virion biogenesis, and, most recently, post-transcriptional RNA quality control (reviewed in (20)). Among the lineage-specific proteins encoded in ORF1b are four enzymes. A 3′-5′ exoribonuclease (ExoN, in Coronaviridae, Mesoniviridae and Roniviridae) and an N7-methyltransferase (N-MT, in the Coronavirinae subfamily, Mesoniviridae and Roniviridae) constitute adjacent domains in the same pp1b cleavage product. They were implicated in RNA proofreading (19,21–22) and in 5′ end cap formation (23,24), respectively. Downstream of this subunit, nidoviruses encode an uridylate-specific endoribonuclease of unknown function (NendoU, in Arteriviridae and Coronaviridae) (25,26) and/or a 2′-O-methyltransferase (O-MT) (in Coronaviridae, Mesoniviridae and Roniviridae), which was implicated in 5′ end cap modification and immune evasion (23,27–29). All six ORF1b-encoded enzymes have distantly related viral and/or cellular homologs. Additionally, Roniviridae and Arteriviridae encode family-specific domains of unknown origin and function, RsD (30) and AsD (31,32), respectively.
The protein subunit containing the RdRp domain is known as non-structural protein (nsp) 9 in Arteriviridae and nsp12 in Coronaviridae (8). Its major ORF1b-encoded part varies in size from ∼700 to ∼900 amino acid residues and is N-terminally extended by a portion encoded in ORF1a. The RdRp-containing replicase subunit of nidoviruses thus seems to be larger than the characterized RdRps of other RNA viruses, which commonly comprise less than 500 amino acid residues (33). RdRps adopt variations of an α/β fold (reviewed in (34)) and have characteristic conserved sequences (motifs). In nidoviruses, these motifs were mapped to the C-terminal one-third of the RdRp-containing protein (35,36), whose tertiary structure is available only as a template-based model for SARS-CoV nsp12 (37,38).
With one notable exception (N-MT; 24), all ORF1b-encoded enzymes were initially identified by comparative genomic analysis involving viral and cellular proteins see (31,39,13,36) and references there. These assignments were fully corroborated by their subsequent biochemical characterization (25–26,29,40–45). Furthermore, the (in)tolerance to replacement of active site residues as tested in reverse genetics studies of coronaviruses and arteriviruses in general correlated well with the observed enzyme conservation. Accordingly, the replacement of conserved residues of the nidovirus-wide conserved RdRp, ZBD and HEL1 were lethal (46–48), while virus mutants were crippled upon inactivation of ExoN, NendoU or O-MT enzymes (49–51), which are conserved in only some of the nidovirus families (30). This correlation is noteworthy since it coherently links the results of the experimental characterization of a few nidoviruses in cell culture systems to evolutionary patterns that were shaped by natural selection in many hosts over an extremely large time frame. The fact that this correlation is evident for nidoviruses overall, rather than for separate families, indicates that nidovirus-wide comparative genomics provides sensible models to the functional characterization of the most conserved replicative proteins.
In the present study, we aimed to elucidate the domain organization, origin and function of the RdRp-containing proteins of nidoviruses by integrating bioinformatics, biochemistry and reverse genetics in a manner that was validated in many prior studies. Our extensive bioinformatics analysis revealed a novel domain, encoded upstream of the RdRp domain within the same cleavage product. It is conserved in all nidoviruses and has no apparent viral or cellular homologs, making it a second genetic marker for the order Nidovirales. Based on results obtained using EAV and SARS-CoV, this domain was concluded to have an essential nucleotidylation activity and was named nidovirus RdRp-associated nucleotidyltransferase (NiRAN). Its potential functions in nidovirus replication may include RNA ligation, protein-primed RNA synthesis, and the guanylyltransferase function that is necessary for mRNA capping.
MATERIALS AND METHODS
Virus genomes
Genomes of nidoviruses were retrieved from GenBank (52) and RefSeq (53) using Homology-Annotation hYbrid retrieval of GENetic Sequences (HAYGENS) tool http://veb.lumc.nl/HAYGENS. Genomes of all viruses were used to produce sequence alignments (see below), which were purged to retain only subsets of viruses representing the known diversity of each nidovirus family for downstream bioinformatics analyses. For the Arteriviridae and Coronaviridae families, one representative was drawn randomly from each evolutionary compact cluster corresponding to known and tentative species that were defined with the help of DEmARC1.3 (54). Twenty nine viruses of the family Mesoniviridae were clustered into six groups, whose intra- and inter-group evolutionary distance was below and above 0.075, respectively. One representative was chosen randomly from each of the six groups. For the Roniviridae family, two viruses, each prototyping a species, were used. To retrieve information about genomes, the SNAD program (55) was used. The final subsets include 30, 5, 10, 6 and 2 sequences representing all established and putative taxa of corona-, toro-, arteri-, mesoni- and roniviruses, respectively (Supplementary Table S1).
Multiple sequence alignments and secondary structure prediction
Multiple sequence alignments (MSAs) of proteins were generated using the Viralis platform (56) and assisted by HMMER 3.1 (57), Muscle 3.8.31 (58) and ClustalW 2.012 (59) programs in default modes. We have produced family-wide MSAs of nsp12 of coronaviruses, nsp9 of arteriviruses and their counterparts of mesoniviruses and roniviruses, whose borders have been tentatively mapped through limited similarity with known 3CLpro cleavage sites of these viruses (60,61). They included NiRAN and RdRp domains delineated as described separately. For simplicity, we will refer to the proteins of mesoni- and roniviruses as nsp12t, with ‘t’ standing for tentative, since the proteolytic cleavage of the replicase polyproteins of these viruses remains to be addressed in detail. Besides NiRAN and RdRp, we have also produced family-specific MSAs of three other nidovirus-wide conserved protein domains: 3CLpro, HEL1 and ZBD. Family-specific MSAs of the NiRAN domain were combined in a stepwise manner using the profile mode of ClustalW with subsequent manual local refinement, which was limited and guided by results obtained using HHalign of the HH-suite 2.0.15 software (62,63) when and if the two programs disagreed. The produced MSAs included one, two, three, four and five (sub)families, respectively, namely: Coronavirinae and Torovirinae (named CoTo), Coronaviridae and Mesoniviridae (CoToMe), Coronaviridae, Mesoniviridae and Roniviridae (CoToMeRo), Coronaviridae, Mesoniviridae, Roniviridae and Arteriviridae (CoToMeRoAr). The final MSA of NiRAN is presented in Supplementary Figure S1 in an annotated format and Supplementary Table S2 in FASTA format.
To reveal all local similarities between two MSAs, their profiles were compared using an align routine in HH-suite 2.0.15, whose results were visualized in a dot-plot fashion with the -dthr = 0.25 and -dwin = 10. Statistical significance of similarity was measured using % of confidence and expectation value (E). HH-suite calculates those for the best local hit in an MSA, regardless whether the latter was produced using the local or global mode of the program. Consequently, similarity of global MSAs may be underestimated. Based on family-specific MSAs of NiRAN and RdRp, the secondary structure of these domains was predicted using software Jpred 3 (64) and PSIPRED (65). In both cases, the sequence with the least gaps was selected from the sequences forming the MSA. The prediction was made only for columns of the MSA in which the selected sequence does not contain gaps. The MSAs were converted into the final figure using ESPript (66).
Homology detection in protein databases
The obtained MSAs were converted into Hidden Markov Model (HMM) profiles or position-specific scoring matrices (PSSM) and used as queries to search for homologs in three different types of databases composed of: individual sequences (nr database, including GenBank CDS translations, RefSeq proteins, SwissProt, PIR and PRF (67)), profiles (PFAM A (68)), and protein 3D structures (PDB (69)). For GenBank scanning, HMMER 3.1 software (57) was used with the E-value cutoff of 10. To search for homologs among protein profiles and 3D structures, HHsearch of HH-suite 2.0.15 software (62,63) and pGenTHREADER 8.9 software (70–72) were used, respectively.
In comparisons with the PDB (www.rcsb.org, (69)) using pGenTHREADER, RdRps of different viruses dominated the hit list for the best sampled nidoviruses, corona- and arteriviruses, and they were consistently present among the top hits for the two other families. Typically the similarity between a nidovirus query and a target encompassed the entire target and was limited to the C-terminal part of the query, with the N-terminal ∼250 and 350 amino acid residues remaining unmatched in arteriviruses and other nidoviruses, respectively (Figure 2A). Likewise, the C-terminal part of nsp9/nsp12/nsp12t matched the RdRp profiles of different virus families in PFAM (68) and an in-house database although this analysis was complicated by the presence of nidovirus sequences in the top-hit PFAM profile (see below). Based on these results we concluded that nsp9, nsp12 and nsp12t contain N-terminal domains that are not part of canonical RdRps. This domain is referred to as NiRAN in this manuscript.
Figure 2.
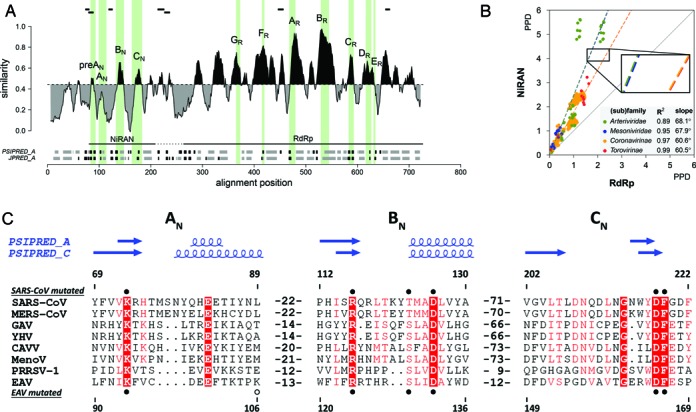
Delineation and divergence of the NiRAN domain in the RdRp-containing proteins of nidoviruses. (A) Sequence variation, domain organization and secondary structure of the RdRp-containing protein of arteriviruses, and location of peptides identified by mass spectrometry after FSBG-labeling of arterivirus nsp9. Shown is the similarity density plot obtained for the multiple sequence alignment (MSA) of proteins including NiRAN and RdRp domains of arteriviruses. To highlight the regional deviation of conservation from that of the MSA average, areas above and below the mean similarity are shaded in black and grey, respectively. Uncertainty in respect to the domain boundary between NiRAN and RdRp is indicated by a dashed horizontal line. Positions of conserved sequence motifs of NiRAN and RdRp are indicated by vertical shading areas; motifs are labeled. Below the similarity density plot, secondary structure elements, predicted based on the arterivirus MSA using PSIPRED (PSIPRED_A) and Jpred 3 (JPRED_A), are presented in grey for α-helices, black for β-strands. (B) Relative scale of divergence of NiRAN versus RdRp in four different nidovirus (sub)families. Shown is scatter plot of PPDs of the NiRAN (y-axis) versus PPDs of RdRp (x-axis), which were calculated from the respective four PhyML trees. Dashed lines depict linear regressions fit in four differently highlighted PPD distributions, with its detail being magnified in the zoom-in; R2 and slope values of the regressions are listed in the inset panel. The solid diagonal line corresponds to the matching rate of PPDs for the two domains and is provided for comparison. (C) MSA of the three conserved NiRAN motifs of eight representative nidoviruses and their predicted secondary structures. Absolutely conserved residues are in white font, while partially conserved residues are highlighted. Secondary structure predictions were made with PSIPRED (64) based on arterivirus (PSIPRED_A) or coronavirus (PSIPRED_C) MSAs. Residues mutated in recombinant SARS-CoV (Coronaviridae) non-structural protein (nsp) 12 and recombinant EAV (Arteriviridae) nsp9 are indicated by filled (conserved) and empty (control) circles, above and below the alignment respectively. Mutated residues D445A in EAV and K103A, D618A in SARS-CoV are not shown. Amino acid numbers above and below the alignment refer to SARS-CoV nsp12 and EAV nsp9, respectively. MERS-CoV, Middle East respiratory syndrome coronavirus (Coronaviridae); GAV (Roniviridae); YHV, yellow head virus (Roniviridae); CAVV (Mesoniviridae); MenoV, Meno virus (Mesoniviridae); PRRSV-1, porcine reproductive and respiratory syndrome virus, European genotype (Arteriviridae). For other abbreviations, see Figure 1.
Evolutionary analyses
To estimate the divergence of NiRAN and RdRp, two analyses were conducted. Distribution of similarity density in MSAs of NiRAN and RdRp was plotted using R package Bio3D (73) under the conservation assessment method ‘similarity’, substitution matrix Blosum62 (74) and a sliding window of 11 MSA columns. Peaks of similarity were attributed to the known RdRp motifs G, F, A, B, C, D, E (35), or named and assigned to the newly recognized motifs of NiRAN, preA, A, B and C. Suffix R and N were added to motif labels of the RdRp and NiRAN domain, respectively. Reconstruction of phylogenetic trees of NiRAN and RdRp of different (sub)families was performed using PhyML 3.0, with the WAG amino acid substitution matrix, allowing substitution rate heterogeneity among sites (eight categories) and 1000 iterations of non-parametric bootstrapping (75). Pairwise patristic distances (PPDs) between viruses were calculated from these trees using R package ‘ape’ (76). They were used to assess relative rates of evolution of NiRAN and RdRp domains through the comparison of linear regressions, which were fit into the respective PPD distributions as implemented in R package ‘stats’ (77).
Protein expression and purification
Nucleotides 5256 to 7333 of the genome of the EAV Bucyrus strain were cloned into a pASK3 (IBA) vector essentially as described (47) to yield a construct that expresses nsp9 that is N-terminally fused to ubiquitin and tagged with hexahistidine at its C-terminus. Mutations were introduced according to the QuikChange protocol and verified by sequencing. Plasmids were transformed into Escherichia coli C2523/pCG1, which constitutively express the Ubp1 protease to remove the ubiquitin tag during expression and thereby generate the native nsp9 N-terminus. Cells were cultured in Luria Broth in the presence of ampicillin (100 μg/ml) and chloramphenicol (34 μg/ml) at 37°C until an OD600 >0.7. At this point protein expression was induced by the addition of anhydrotetracycline to a final concentration of 200 ng/ml and incubation was continued at 20°C overnight. Cell pellets were harvested by centrifugation and stored at −20°C until further use.
Proteins were batch purified by immobilized metal ion affinity chromatography using Co2+ Talon beads. In short, cell pellets were resuspended in lysis buffer (20 mM HEPES, pH 7.5, 10% glycerol (v/v), 10 mM imidazole, 5 mM β-mercaptoethanol) supplemented with 500 mM NaCl. Lysis was achieved by a 30-min incubation with 0.1 mg/ml lysozyme and five subsequent cycles of 10-s sonication to shear genomic DNA. Cellular debris was removed by centrifugation at 20 000 g for 20 min. The cleared supernatant was recovered and equilibrated Talon-beads were added. After 1 h of binding under agitation, beads were washed four times for 15 min with a 25-times bigger volume of lysis buffer containing first 500 mM, than 250 mM, and finally twice 100 mM NaCl. In the end, proteins were eluted twice with lysis buffer containing 100 mM NaCl and 150 mM imidazole. Both fractions were pooled and dialyzed twice for 6 h or longer against an at least 100-fold bigger volume of 20 mM HEPES, pH 7.5, 50% glycerol (v/v), 100 mM NaCl, 2 mM DTT. All steps of the purification were performed at 4°C or on ice. All mutant proteins were expressed and purified in parallel with the wild-type protein used as reference in nucleotidylation assays. Protein concentrations were measured by absorbance at 280 nm using a calculated extinction coefficient of 93 170 M−1cm−1 and a molecular mass of 77 885 Da for wild-type nsp9. Typical protein yields were 5 mg/l culture and nucleotidylation activity was observed for at least 4 months if stored at −20°C at a concentration below 15 μM. Finally, the absence of the N-terminal ubiquitin tag was confirmed by mass spectrometry.
Nucleotidylation assay
Nucleotidylation assays were performed in a total volume of 10 μl containing, unless specified otherwise, 50 mM Tris, pH 8.5, 6 mM MnCl2, 5 mM DTT, up to 2.5 μM nsp9 and 0.17 μM [α-32P]NTP (Perkin Elmer, 3000 Ci/mmol). Furthermore, 12.5% glycerol (v/v), 25 mM NaCl, 5 mM HEPES, pH 7.5, and 0.5 mM DTT were carried over from the protein storage buffer. In preliminary experiments magnesium (1–20 mM) did not support nucleotidylation activity and was consequently not pursued further. Samples were incubated for 30 min at 30°C. Reactions were stopped by addition of 5 μl gel loading buffer (62.5 mM Tris, pH 6.8, 100 mM dithiothreitol (DTT), 2.5% sodium dodecyl sulphate (SDS), 10% glycerol, 0.005% bromophenol blue) and denaturing of the proteins by heating at 95°C for 5 min. 12% sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) gels were run, stained with Coomassie G-250, and destained overnight. After drying, phosphorimager screens were exposed to gels for 5 h and scanned on a Typhoon variable mode scanner (GE healthcare), after which band intensities were analyzed with ImageQuant TL software (GE healthcare). The buffers used to find the pH optimum of the nucleotidylation reaction were MES (pH 5.5–6.5), MOPS (pH 7.0), Tris (pH 7.5–8.5) and CHES (pH 9.0–9.5) (20 mM).
To assess the chemical nature of the nucleotide-protein bond, the pH was temporarily shifted after product formation. To this end, 1 μl HCl or NaOH (both 1 M) was added before incubation at 95°C for 4 min. Afterward the original pH was restored by addition of the complementary base or acid, and samples were separated and analyzed as described.
FSBG labeling and mass spectrometry
Reaction mixtures were the same as described for the nucleotidylation assay with two modifications: radioactive nucleotides were replaced by up to 2 mM of the reactive guanosine-5'-triphosphate (GTP) analog 5′-(4-fluorosulfonylbenzoyl)guanosine (FSBG) (78), of which the synthesis is described in supplementary Materials and Methods, and samples were incubated for 1 h at 30°C to increase the ratio between labeled and unlabeled protein. Subsequently, the protein (20 μg) was reduced by addition of 5 mM DTT and denatured in 1% SDS for 10 min at 70°C. Next, the samples were alkylated by addition of 15 mM iodoacetamide and incubation for 20 min at RT. Next, the protein was applied to a centrifugal filter (Millipore Microcon, MWCO 30 kDa) and washed three times with NH4HCO3 (25 mM) before a protease digestion was performed with 2 μg trypsin in 100 μl NH4HCO3 overnight at RT. Recovered peptides were treated with 50 mM NaOH for 25 min, desalted using Oasis spin columns (Waters) and finally analyzed by on-line nano-liquid chromatography tandem mass spectrometry on an LTQ-FT Ultra (Thermo, Bremen, Germany). Tandem mass spectra were searched against the Uniprot database, using mascot version 2.2.04, with a precursor accuracy of 2 ppm and product ion accuracy of 0.5 Da. Carbamidomethyl was set as a fixed modification, and oxidation, N-acetylation (protein N-terminus) and FSBG were set as variable modifications.
Label release
For analysis of the released nucleotides, 350 pmol of nsp9 were nucleotidylated with [α-32P]nucleoside-5'-triphosphates ([α-32P]NTPs) as described above for 1 h at 30°C. After the reaction free NTPs were removed by buffer exchange and extensive washing with the help of a centrifugal filter (Millipore ultrafree-0.5, MWCO 10 kDa). Protein was precipitated with a five times greater volume of acetone overnight at −20°C. The resulting pellet was resuspended in 20 mM Tris, pH 8.5, 100 mM NaCl. Equal amounts of the solutions were incubated at 95°C for 4 min after addition of HCl or NaOH (1 M). Samples were adjusted to their original pH and spotted onto polyethylenimine cellulose thin layer chromatography plates, which were developed in 80% acetic acid (1 M), 20% ethanol (v/v), 0.5 M LiCl. Plates were dried and phosphorimaging was performed as described above. Non-radioactive nucleotide standards were run on each plate and visualized by UV-shadowing to allow the identification of the radioactive products.
Reverse genetics of EAV
Alanine-coding mutations for conserved and control residues were introduced into full-length cDNA clone pEAV211 (79) using appropriate shuttle vectors and restriction enzymes. The presence of the mutations was confirmed by sequencing. pEAV plasmid DNA was in vitro transcribed with the mMessage-mMachine T7 kit (Ambion), and the synthesized RNA was transfected into BHK-21 cells after LiCl precipitation as described previously (80). Virus replication was monitored by immunofluorescence microscopy until 72 h post transfection (p.t.) using antibodies directed against nsp3 and N protein as described (81) and by plaque assays (80) using transfected cell culture supernatants, to monitor the production of viral progeny.
Sequence analysis of the nsp9-coding region was performed to either verify the presence of the introduced mutations or to monitor the presence of (second site) reversions. For this purpose, fresh BHK-21 cells were infected with virus-containing cell culture supernatants and total RNA was extracted with Tripure Isolation Reagent (Roche Applied Science) after appearance of cytopathic effect (CPE) (typically at 18 h post infection (p.i.)). EAV-specific primers were used to reverse transcribe RNA and PCR amplify the nsp9-coding region (nt 5256–7333). RT-PCR fragments of the EAV genome were sequenced after gel purification and sequences compared to those of the respective RNA used for transfection.
Reverse genetics of SARS-CoV
Mutations in the SARS-CoV nsp12-coding region were engineered in prSCV, a pBeloBac11 derivative containing a full-length cDNA copy of the SARS-CoV Frankfurt-1 sequence (82) by using ‘en passant recombineering’ as described in Tischer et al. (83). The (mutated) BAC DNA was linearized with NotI, extracted with phenol–chloroform, and transcribed with T7 RNA Polymerase (mMessage-mMachine T7 kit; Ambion) using an input of 2 μg of BAC DNA per 20-μl reaction. Viral RNA transcripts were precipitated with LiCl according to the manufacturer's protocol. Subsequently, 6 μg of RNA were electroporated into 5 × 106 BHK-Tet-SARS-N cells, which expressed the SARS-CoV N protein following 4 h induction with 2 μM doxycycline as described previously (84). Electroporated BHK-Tet-SARS-N cells were seeded in a 1:1 ratio with Vero-E6 cells. Viral protein expression and the production of viral progeny was followed until 72 h p.t. by immunofluorescence microscopy using antibodies directed against nsp4 and N protein and by plaque assays of cell culture supernatants, respectively (both methods were described previously in Subissi et al. (84)). All work with live SARS-CoV was performed inside biosafety cabinets in a biosafety level 3 facility at Leiden University Medical Center.
For sequence analysis of viral progeny, fresh Vero-E6 cells were infected with harvests from viable mutants taken at 72 h p.t., and SARS-CoV RNA was isolated 18 h p.i. using TriPure Isolation Reagent (Roche Applied Science) as described in the manufacturer's instructions. Random hexamers were used to prime the RT reaction, which was followed by amplification of the nsp12-coding region (nt 13398–16166) by using SARS-CoV-specific primers. RT-PCR products were sequenced to verify the presence of the introduced mutations.
RESULTS
Delineation of a novel, unique domain that is conserved upstream of the RdRp in polyproteins of all nidoviruses
Inspection of the intra-family sequence conservation for (sub)family-specific MSAs of nsp9, nsp12 and nsp12t (see ‘Materials and Methods’ section for technical details) using density similarity plots (Supplementary Figure S2) confirmed the association of characteristic RdRp motifs with some of the most prominent conservation peaks, located in the C-terminal half of nsp9 and nsp12 (RdRp domain). For nsp12t, similar conclusions could be drawn although the conservation profiles of these viruses, especially roniviruses, were of lesser resolution due to the overall higher similarity that was the result of the limited virus sampling and divergence. Importantly, also the N-terminal half of nsp9 and nsp12 (NiRAN domain) included a few above-average conservation peaks although the overall conservation was evidently highest around the established RdRp motifs (Figure 2A; Supplementary Figure S2). Likewise, NiRAN compared to RdRp accepted two–to-three times more substitutions in four nidovirus (sub)families (Figure 2B). In this comparison, slopes of the four PPD distributions were strikingly similar, particularly in the pairs of the Coronavirinae and Torovirinae (60.6 and 60.5, respectively) and the Mesoniviridae and Arteriviridae (67.9 and 68.1). Thus, NiRAN must have evolved under similar constraints in different lineages of nidoviruses, which is compatible with a common function of this domain.
Next, we investigated the relation of the NiRAN domains of the four different families by pairwise profile–profile comparisons using HHalign in local mode (see Supplementary Figure S3 and Figure 3 for all results and a selection of thereof, respectively). This analysis revealed strong support (∼98% confidence and E = 7.7e-09–1.7e-08) for the similarity between NiRANs of coronavirus/torovirus nsp12 and mesonivirus nsp12t, and moderate support (∼21–30% confidence and E = 0.00051–0.00091) for the similarity between the respective domains of mesoni- and roniviruses. Based on these observations, we have aligned the NiRAN domain of coronavirus nsp12 and mesonivirus nsp12t using the profile mode of ClustalW, with the MSA being slightly adjusted taking into account the HHalign-mediated results. This MSA of two families was superior compared to each of the two family-specific MSAs with respect to its similarity to the MSA of roniviruses (∼54–75% confidence and E = 0.00011–0.00049). Consequently, the ronivirus MSA was added to the MSA of corona/toro- and mesoniviruses to generate an MSA of the NiRAN of the three families, hereafter called ExoN-encoding nidoviruses with reference to the domain that distinguishes this group from arteriviruses (Figure 1B). In the above HHsearch local alignments, almost the full-length NiRAN domains were aligned.
Figure 3.
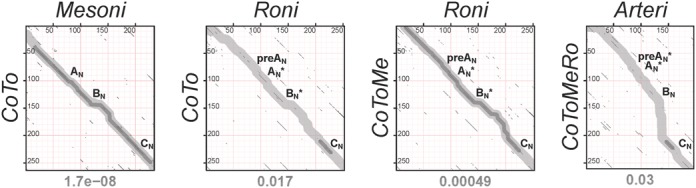
Establishing sequence conservation between NiRAN domains of different (sub)families. Shown are four pairwise dot-plots that compare HMM profiles of NiRAN domains of different origins using HHalign. For the entire set of dot-plots generated, please see Supplementary Figure S3. The bottom three plots correspond to steps used to produce the nidovirus-wide NiRAN MSA (Supplementary Figure S1), while the top plot is shown for comparison. Coordinates of query and target HMMs are presented on y-axis and x-axis, respectively. All local similarities between two profiles are depicted as black dots. Transparent fat dark and light gray lines on the dot-plot show paths of HHalign alignments, obtained in local and global modes, respectively. The E-value of the top local alignment is specified below each dot-plot. In the profile–profile alignment produced in global mode, conserved amino acids of NiRAN motifs may have been properly aligned or not. If conserved residues of a motif were aligned, the corresponding region of the alignment path is labeled with the respective motif name without an asterisk. If the misalignment of conserved residues was limited to a shift of one or two residues (HMM–HMM alignment columns), the corresponding region of the alignment path is labeled with the respective motif name plus an asterisk.
In contrast to the above observations, the support for similarity between the NiRAN MSAs of arteriviruses and ExoN-encoding nidoviruses, separately or combined, in our HHalign-based analysis was relatively weak (E = 0.03–0.4), particularly with respect to confidence (1.5% or worse). This could be due to the similarity being recognized only in a small C-terminal region. This experience prompted us to compare conserved motifs and predicted secondary structures of the domains of these families (Supplementary Figures S1 and S2). Ten residues were found to be invariant in the conserved NiRAN of the ExoN-encoding nidoviruses. They map to three motifs designated AN (with a K-x[6–9]-E pattern in ExoN-encoding nidoviruses), BN (R-x[8–9]-D) and CN (T-x-DN-x4-G-x[2,4]-DF), respectively, with motifs BN and CN representing the most prominent conservation peaks of this domain in coronaviruses (Supplementary Figure S2). Remarkably, similar conserved motifs are present in the NiRAN of arteriviruses (Figure 2A and C), where BN and CN again occupy the two most prominent peaks (Supplementary Figure S2). The three motifs are similarly positioned relative to the ORF1a/ORF1b frameshift signal in all nidoviruses, and, importantly, they were aligned in arteriviruses and the ExoN-encoding nidoviruses using HHalign in global mode (Figure 3, rightmost plot). Specifically, all four invariant residues of motifs AN and BN of ExoN-encoding nidoviruses are also conserved in arteriviruses although with slightly smaller distances separating the two residues of each pair (Supplementary Figure S1 and Figure 2C). In the most highly conserved motif CN, the aspartate-phenylalanine dipeptide and likely glycine (the only deviating arginine at this position in the lactate dehydrogenase-elevating virus isolate U15146 may result from a sequencing error) are absolutely conserved among nidoviruses while the other invariant residues of ExoN-encoding nidoviruses appear to have been replaced by similar residues in arteriviruses. Additionally, there is a good agreement between the predicted secondary structure for the domains of arteriviruses and ExoN-encoding nidoviruses, particularly in the area encompassing the three sequence motifs as well as regions immediately upstream of motif AN (named preAN motif) and downstream of motif CN (Supplementary Figure S1). In ExoN-encoding nidoviruses, motifs BN and CN are separated by a variable region of 40–60 amino acid residues that does not include absolutely conserved residues, while in arteriviruses motifs BN and CN are adjacent. Based on these observations, we concluded that nsp9, nsp12 and nsp12t contain the NiRAN domain, which is conserved in all nidoviruses, although we acknowledge that the support for the conservation of different motifs between different nidovirus (sub)families is not equally strong. Also, we noted that, at this stage, it was not possible to precisely define the C-terminal border of the NiRAN domain. NiIRAN and RdRp may thus, be adjacent or separated by another small domain of variable size in different nidoviruses (Supplementary Figure S2).
To gain insight into the origin and function of the NiRAN domain, we compared MSA-based profiles of this domain and its individual motifs of different nidovirus families and the entire order with the PFAM, GenBank, Viralis DB and PDB databases. As a control, we used the HMM profiles of four other domains that are conserved in all nidoviruses, 3CLpro, RdRp, ZBD and HEL1. We expected to find hits to either other nidovirus proteins, if NiRAN would have emerged by duplication or non-nidovirus proteins, if the NiRAN ancestor would have been acquired from an external source. None of the database scans involving the NiRAN retrieved a non-nidovirus hit whose E-value was better than 0.065 for HMMER and 1.3 for the HHsearch program from HH-suite (Figure 4) and none of these hits had sequences similar to the motifs of the NiRAN. In contrast, statistically significant hits with virus and/or host proteins were identified for the nidoviral control proteins either in both or one of the scans; according to annotation, at least some of these hits were true positives in the functional and/or structural sense. Likewise, in scans of the PDB using pGenTHREADER, all top hits for the NiRAN of the four virus families had low support (P = 0.014 or worse) with no match of the conserved motifs. In contrast, top hits for four RdRp queries were supported with P-values of 0.0003 or better and targeted RdRps of other viruses, at least for arteri- and coronavirus queries.
Figure 4.

Comparison of nidovirus-wide conserved domains with sequence databases. Shown are histograms depicting E-values of the best non-nidovirus hits obtained during HMMER-mediated profile-sequence (A) and HHsearch-mediated profile–profile (B) searches of the GenBank and PFAM A databases, respectively, using MSA profiles of five nidovirus-wide conserved domains encoded by four nidovirus families. The identity of the non-nidoviral top-hit in the respective databases is specified. Stars indicate hits whose homologous relationship with the respective query is also supported by the functional and/or structural annotation of the respective targets.
EAV nsp9 has Mn2+-dependent nucleotidylation activity with UTP/GTP preference
Since we could not identify any homologs of the NiRAN domain whose prior characterization would facilitate the formulation of a hypothesis about its function, we have reviewed the available information about nidovirus genome organization and replicative enzymes, and the results described above. The data were most compatible with the hypothesis that this domain is an RNA processing enzyme, in view of (i) the abundance of RNA processing enzymes in the ORF1b-encoded polyprotein (Figure 1B); and (ii) the profile of invariant residues, composed of aspartate, glutamate, lysine, arginine and phenylalanine (and possibly glycine) (Figure 2C), the first four of which are among the most frequently employed catalytic residues (85). Since the domain is uniquely conserved in nidoviruses, we hypothesized that its activity might work in concert with that of another, similarly unique RNA processing enzyme. At the time of this consideration, the NendoU endoribonuclease of nidoviruses was believed to be such an enzyme (25) (assessment revised in 2011, (30)). Consequently, we reasoned that a ligase function would be a natural counterpart for the endoribonuclease (NendoU), as observed in many biological processes, and would fit in the functional cooperation framework outlined in our previous analysis of the SARS-CoV proteome (39). This hypothesis was also compatible with the predicted α/β structural organization of NiRAN (Supplementary Figure S1) and the lack of detectable similarity between NiRAN and the highly diverse nucleotidyltransferase superfamily, to which nucleic acid ligases belong. This superfamily is known to include groups that differ even in the most conserved sequence motifs, especially in proteins of viral origin (86,87). Based on mechanistic insights obtained with other ligases, we expected that the conserved lysine might be the principal catalytic residue of the NiRAN domain.
To detect this putative NTP-dependent RNA ligase activity, we took advantage of the universal ligase mechanism, which can be separated into three steps (88). First, an NTP molecule, typically adenosine triphosphate (ATP), is bound to the enzyme's binding pocket, and a covalent bond is established between the nucleotide's α-phosphate, nucleoside-5'-monophosphate (NMP) and the side chain of either lysine or histidine, while pyrophosphate is released. Since this protein–NMP is a true, temporarily stable intermediate, it can be readily detected by biochemical methods. In contrast, demonstration of the following two steps, NMP transfer to the 5′ phosphate of an RNA substrate and subsequent ligation of a second RNA molecule under release of the NMP, depends on the availability of target RNA sequences whose identification is often not as straightforward. Thus, we first assessed our hypothesis by testing the covalent binding of a nucleotide, known as nucleotidylation.
To this end, recombinant EAV nsp9 was expressed in E. coli, purified, and incubated with each of the four NTPs, which were 32P-labeled at the α-position. Samples were analyzed using denaturing SDS-PAGE to discriminate between covalent and affinity-based nucleotide binding. As can be seen in Figure 5A, we could indeed detect a radioactively labeled product with a mobility comparable to that of nsp9 in the presence of GTP and uridine-5'-triphosphate (UTP). To verify that this labeled band corresponded to a protein and did not result from 3′ end labeling of co-purified E. coli RNA or polyG synthesis by the RNA polymerase residing in the C-terminal domain of nsp9, guanylylation was followed by the addition of either proteinase K or RNase T1, which cleaves single-stranded RNA after G residues. As expected, only protease treatment removed the band while incubation with RNase T1 had no effect on the product (Figure 5B). The same result was obtained after uridylylation using RNase A, which cleaves after pyrimidines in single-stranded RNA (data not shown). Furthermore, as the use of GTP labeled in the γ-position did not result in a radioactive product, we conclude that this phosphate, in agreement with the general nucleotidylation mechanism, is released during the reaction (Figure 5B).
Figure 5.
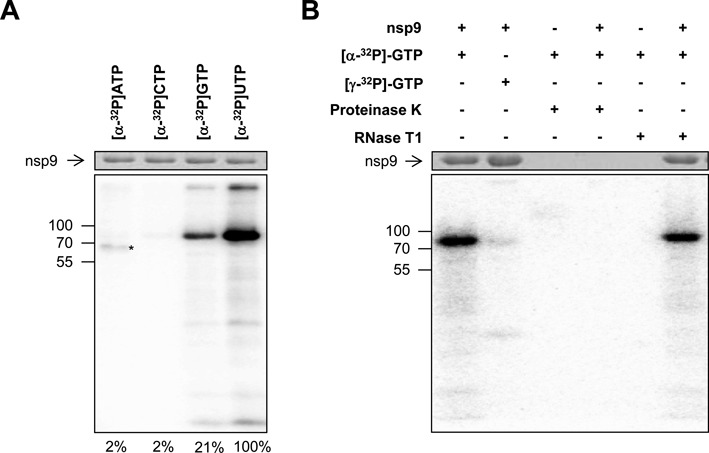
EAV nsp9 has nucleotidylation activity. Purified recombinant EAV nsp9 (78 kDa) was incubated with the indicated 32P-labeled NTP in the presence of MnCl2. After denaturing SDS-PAGE, reaction products were visualized by Coomassie brilliant blue staining (top panels) and phosphor imaging (bottom panels). Positions of molecular weight markers are depicted on the left in kDa. (A) Uridylylation and guanylylation activity as revealed by covalent binding of the respective radioactive nucleotide to nsp9. Note that the protein indicated with an asterisk likely is an Escherichia coli-derived impurity reacting with ATP. Relative band intensities are shown at the bottom. (B) Guanylylation was distinguished from RNA polymerization by incubating the products generated during the nucleotidylation assay with proteinase K (1 mg/ml) or with RNase T1 (0.5 U), which cleaves single-stranded RNA after G residues, for 30 min at 37°C.
Unexpectedly, we observed a marked substrate specificity of nsp9 for UTP, which resulted in the accumulation of five times more enzyme–nucleotide complex than observed with GTP. In contrast, we observed no covalent binding with ATP or cytidine-5'-triphosphate (CTP) as substrates (Figure 5A). The observed substrate preferences are remarkable for two reasons. First, since both UTP and GTP are present in significantly lower concentrations under physiological conditions than ATP (89) and are in general not used as primary energy source, it suggests that the identity of the base, rather than the energy stored within the phosphodiester bonds, may be critical for a subsequent step in the reaction pathway. This implies that reaction pathways other than RNA ligation, which predominantly utilizes ATP, must be considered. Second, the selective utilization of only one pyrimidine and one purine substrate raised questions about the nature and number of active sites involved, for instance, whether both nucleotides bind to separate binding sites or utilize different catalytic residues within the same binding site. Unfortunately, there are no crystal structures for any of the nidovirus nsp9/nsp12/nsp12t subunits available to date, which might have been used to resolve this matter in docking studies.
To characterize the NTP binding further, we compared the pH dependence of both activities. Interestingly, while the relative activities below pH 8.5 were identical with both substrates, the relative guanylylation activity was exceedingly higher than uridylylation at a pH above 8.5 (Figure 6A). To exclude that the observed pattern is due to a difference in the metal ion requirement, we determined the optimal manganese concentration for nucleotidylation with both substrates. As is apparent from Figure 6B, both activities share the same broad optimum between 6 and 10 mM MnCl2. This result made it unlikely that manganese oxidation and a concomitant decrease of available Mn2+ ions, as we observed at a pH above 9.0, would selectively favour the utilization of one of the two substrates. The observed difference between guanylylation and uridylylation with regard to its pH optimum may thus be genuine. For instance, this slightly broadened or—more likely—shifted pH optimum of guanylylation may be the result of a GTP-induced spatial reorientation of amino acid side chains in the vicinity of the catalytic residue and a concomitant alteration of its pKa. Alternatively, it may also be explained by the two substrates using different binding sites. These possibilities were partially addressed in the experiments described in the subsequent sections.
Figure 6.
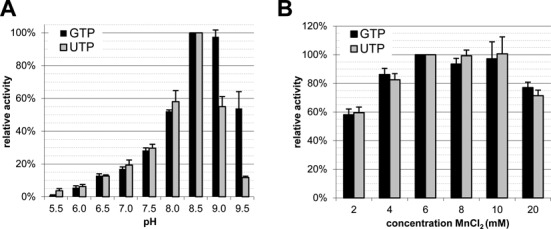
EAV nsp9 guanylylation has a slightly broader or shifted pH optimum compared to uridylylation while the metal ion requirement is identical. (A) The pH optimum in the range from 5.5 to 9.5 was determined using the buffers listed in ‘Materials and Methods’ section. (B) Assessment of the optimal MnCl2 concentration for nucleotidylation. Error bars represent the standard deviation of the mean based on three independent experiments.
FSBG labeling of nsp9 suggests the presence of a nucleotide binding site in the NiRAN domain
To verify that the newly discovered nucleotidylation activity is associated with the NiRAN domain, we first sought to establish the presence of the expected nucleotide binding site. To this end, we replaced the substrate in the nucleotidylation assay with the reactive guanosine analog 5′-(4-fluorosulfonylbenzoyl)guanosine (FSBG) (Supplementary Figure S4A) (78). Depending on the exact shape of the nucleotide binding pocket this compound may be suitable for binding and reacting with any nucleophile within the pocket, leaving behind a stable sulfonylbenzoyl tag that can be readily detected by mass spectrometry. In this way, residues that are lining the binding site can be identified. However, because the points of attack of FSBG (sulfonyl group sulfur) and GTP (α-phosphorus) are spatially separated (∼4Å, Supplementary Figure S4A and B), these residues are not necessarily of biological relevance to nucleotidylation but rather are indicative of the local neighborhood of the nucleotidylation reaction.
After analysis of the nucleotidylation reaction mixture by mass spectrometry, seven modified peptides representing five distinct nsp9 regions could be assigned: three in (the vicinity of) the NiRAN domain and two in the RdRp domain (Figure 2A and Supplementary Figure S4C). In agreement with previously published results (78), we found only lysine and tyrosine residues to be modified, as these are thought to provide the chemically most stable bonds. The selectivity of the modification was evident from the fact that only seven lysine and tyrosine residues served as nucleophile for the reaction. Furthermore, we identified all these peptides in independent experiments using FSBG concentrations ranging from 25 μM to 2 mM. Within this range, a concentration of 100 μM was sufficient to detect all seven peptides. Together this strongly suggests that the reaction with FSBG only occurred after binding to a specific site(s) and did not originate from random collisions. Furthermore, the two modified residues in the EAV RdRp are located in either a predicted α-helix or in a loop not far upstream and downstream of the AR and ER motifs, respectively, which are involved in NTP binding in other, better characterized RdRps. The five modified residues in the EAV NiRAN domain are poorly conserved in related arteriviruses and are located in the vicinity of one of the three major motifs in either a predicted loop region (1 residue) or a β-strand (4 residues). These findings are compatible with the expected properties of the FSBG modification that may label any nucleophile within a 4 Å distance from the NTP-binding site(s). We therefore conclude that the peptides identified in this experiment reflect the presence of a nucleotide binding site within the RdRp required for RNA synthesis and a second binding site that is located in the NiRAN domain, which could serve for nucleotidylation.
Conserved residues of the NiRAN domain but not of the RdRp domain are required for nucleotidylation activity
In a next step, we examined the importance of conserved NiRAN residues for the guanylylation and uridylylation activities by characterization of alanine substitution mutants of several residues, including five invariant residues, in recombinant EAV nsp9. Notably, none of these mutations significantly reduced expression or stability (data not shown), indicating that they are most likely compatible with the protein's structure. Subsequent characterization demonstrated that all conserved NiRAN residues that were probed (Figure 2, Table 1) are important for nucleotidylation activity, as their replacement with alanine led, with the exception of S129A, to a drop to below 10% of wild-type protein activity. In contrast, alanine substitution of a non-conserved N-terminal residue (K106A) as well as of a conserved residue in the RdRp domain (D445A of motif AR), which is known to be essential for the polymerase activity in other RNA viruses (34), had only a mild effect, preserving at least 75% of the activity (Figure 7). Thus, we concluded that the identified sequence motifs in the EAV nsp9 NiRAN domain are functionally connected to the nucleotidylation activity. Whether the decrease in activity was due to a loss of affinity, impairment of catalysis or both remains to be established. In addition, as the level of remaining activity (again with exception of the S129A mutant) did not depend on the substrate used, both guanylylation and uridylylation are likely catalyzed by the same active site.
Table 1. Reverse genetics analysis of EAV nsp9 and SARS-CoV nsp12 mutants.
| Virus | Motif | Mutant | Mutation | Virus titers (PFU/ml at 16–18 h p.t.) | nsp9/nsp12 sequence of P1 virusa |
|---|---|---|---|---|---|
| wt | 1·107, 2·108 | n.d. | |||
| AN | K94A | AAA→GCA | <20, <20 | Reversion | |
| Non-conserved | K106A | AAA→GCA | 3·105, 2·106 | GCA | |
| BN | R124A | CGU→GCU | <20, <20 | Reversion | |
| EAV | BN | S129A | UCG→GCG | 1·104, 5·103 | Reversion |
| BN | D132A | GAU→GCU | 3·104, 6·103 | Reversion | |
| CN | D165A | GAU→GCU | 3·103, 1·104 | Reversion | |
| CN | F166A | UUU→GCU | <20, <20 | n.a. | |
| AR | D445A | GAC→GCC | <20, 1·104 | Reversion | |
| wt | 4·106, 3·105 | n.d. | |||
| AN | K73A | AAG→GCC | <20, <20 | n.a. | |
| Non-conserved | K103A | AAG→GCA | <20, <20 | GCA | |
| BN | R116A | CGU→GCU | <20, <20 | n.a. | |
| SARS-CoV | BN | T123A | ACA→GCU | 1·105, 4·105 | GCU |
| BN | D126A | GAU→GCG | <20, <20 | n.a. | |
| CN | D218A | GAU→GCU | <20, <20 | n.a. | |
| CN | F219A | UUC→GCG | 2·104, 8·102 | GCG | |
| AR | D618A | GAU→GCG | <20, <20 | n.a. |
aVirus-containing supernatants were collected at 72 h p.t. and subsequently used for re-infection of fresh BHK-21 (EAV) or Vero-E6 (SARS-CoV) cells. Total RNA was isolated after appearance of CPE and nsp9/nsp12 coding regions were sequenced. All results were confirmed in a second independent experiment. n.d., not done; n.a., not applicable (non-viable phenotype).
Figure 7.
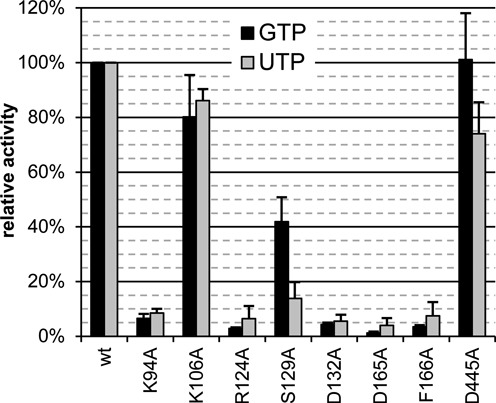
Conserved NiRAN residues are essential for the nucleotidylation activity. Alanine substitution of conserved NiRAN residues dramatically decreased the nucleotidylation activity of nsp9. In contrast, mutation of the non-conserved K106 in the NiRAN domain or the conserved D445 in the RdRp domain had only a mild effect on activity. Error bars represent the standard deviation of the mean based on three independent experiments.
In contrast to these results, the mutation at position S129, the only targeted residue that is fully conserved in arteriviruses but may be replaced by threonine in other nidoviruses, exhibited a slightly different effect on guanylylation and uridylylation. Mutant S129A displayed an intermediate activity when using GTP but was almost as deficient as mutants of the nidovirus-wide conserved residues when UTP was used as substrate (Figure 7). This finding may indicate that S129 is specifically involved in the hydrogen bond network between protein and UTP. Alternatively, as the covalent binding of the nucleotide occurs via a nucleophilic attack on the α-phosphate, this serine may in principle be suitable to play this role. Although to our knowledge nucleic acid ligases typically employ lysine and rarely histidine as catalytic residues (88,90), we cannot exclude that uridylylation occurs via this S129 while guanylylation utilizes another amino acid.
Nucleotidylation occurs via the formation of a phosphoamide bond
In order to identify which type of amino acid is the catalytic residue involved in nucleotidylation, we probed the chemical stability of the bond formed between enzyme and nucleotide. To this end, we subjected the nucleotidylation product to either a higher or a lower pH for 4 min, while the protein was heat denatured. The loss of the radioactive label under acidic or alkaline conditions is an indicator for the type of bond that is formed (Figure 8A) (91). As evident from Figure 8B, the bond between guanosine phosphate and nsp9 was acid-labile but stable under alkaline conditions, which is indicative of a phosphoamide bond originating from either a lysine or histidine. This result was also confirmed for uridylylation (data not shown), excluding a direct role for S129 in the attachment of the uridine phosphate. Since there is no conserved histidine present in the NiRAN domain, K94 is the most likely candidate within this domain to fulfill the role of catalytic residue.
Figure 8.
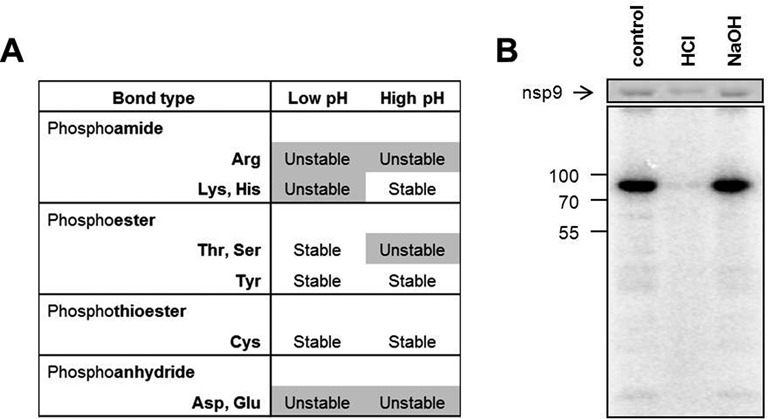
A phosphoamide bond is formed between nsp9 and the guanosine phosphate. (A) Chemical stability of different phosphoamino acid bonds. Adapted from (91). (B) The protein was labeled with [α-32P]GTP and subsequently incubated at pH 8.5 (control) or under acidic or alkaline conditions. Reaction products were visualized after denaturing SDS-PAGE by Coomassie brilliant blue staining (top panel) and phosphor imaging (bottom panel). Size markers are depicted on the left in kDa.
Guanosine and uridine phosphates may be attached via different phosphate groups
So far we have demonstrated that guanylylation and uridylylation are essentially equally sensitive to replacement of NiRAN residues, share the same metal ion requirements, and both rely on the formation of a phosphoamide bond. We therefore concluded that there is only one active site responsible for nucleotidylation, which allows utilization of both substrates. Interestingly, if this were true, discrimination of GTP and UTP against ATP and CTP would be solely based on the presence of an oxygen at C6 of GTP and C4 of UTP. However, given the pronounced size difference between UTP and GTP, the positions of both substrates within the binding site are unlikely to be equivalent. In principle, two binding scenarios are possible. First, the ribose and phosphate moieties of both nucleotides could occupy the same position within the binding site, for example by forming hydrogen bonds via the ribose's 2′ and 3′ hydroxyl groups and charge interactions between the protein and the phosphates. Yet, due to the size difference of the bases (pyrimidine vs. purine), any additional interactions between protein and bases would involve different hydrogen bond networks, potentially involving water molecules in the case of the smaller UTP. Alternatively, due to stacking interactions between an aromatic residue of the protein and the bases, uracil and the pyrimidine ring of guanine might occupy equivalent positions. As this would inevitably lead to the relative misplacement of the ribose and phosphates of UTP compared to GTP, the catalytic residue may compensate for the size difference by re-adjusting and attacking the β- instead of the α-phosphate of UTP.
To explore the above possibility, nsp9 was nucleotidylated as before and non-bound label was removed by extensive washing until no residual radioactivity was detected in the wash buffer. The nucleotide-protein bond was subsequently broken by lowering of the pH and the released nucleotide was analyzed by thin layer chromatography. While nsp9 incubated with GTP clearly released significantly more of the expected guanosine-5'-monophosphate (GMP) in an acidic environment than under alkaline conditions, the results after uridylylation were not as conclusive. Although also in this case the monophosphate was released after HCl treatment, the intensity did not match that of GMP and a second product was present in higher quantities (Figure 9A). This may indicate that uridine-5'-monophosphate (UMP) is either further hydrolyzed under these conditions or that in fact a UMP–protein adduct is only the minor product after uridylylation. Therefore, it remains unclear whether the binding of UTP indeed forces an attack of the β-phosphate. To exclude that the observed GMP release is caused by the treatment with HCl, control samples lacking nsp9 were also investigated. As expected this did not result in a product with equivalent mobility to GMP (Figure 9B).
Figure 9.
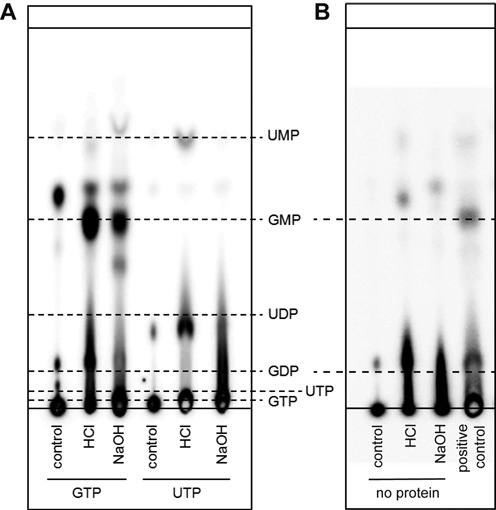
GMP is released from labeled EAV nsp9 under acidic conditions. (A) nsp9 was labeled with [α-32P]GTP or [α-32P]UTP and was incubated at pH 8.5 (control) or under acidic or alkaline conditions after removal of non-incorporated nucleotides. Resulting products were separated with PEI-cellulose TLC. Solid lines represent the position where samples have been spotted (bottom) and the running front (top). Dashed lines represent the respective mobilities of the indicated nucleotides. (B) [α-32P]GTP was incubated under the same conditions as in (A) but omitting nsp9. An nsp9-containing sample treated with HCl served as positive control.
NiRAN nucleotidylation is essential for EAV and SARS-CoV replication in cell culture
To establish the importance of the NiRAN domain for nidoviral replication, we used reverse genetics to engineer both EAV and SARS-CoV mutants in which conserved NiRAN residues were substituted with alanine. Following transfection of in vitro-transcribed full-length RNA into permissive cells, viral protein expression and progeny release were monitored (Table 1). As expected for such conserved residues, most alanine substitutions were either lethal for the virus or resulted in a severely crippled virus that reverted, thus confirming the essential role of the nucleotidylation activity during the viral replication cycle. Similarly, also replacement of a conserved aspartate in motif A of the downstream RdRp domain, which is known to be required for the activity of polymerases in other (+) RNA viruses (34), was tolerated in neither EAV nor SARS-CoV. Notable exceptions to this general pattern, in addition to the replacements of non-conserved lysine residues included as controls, were the T123A and F219A mutations in SARS-CoV nsp12. These mutations were stably maintained although they produced a mixed plaque phenotype comprising wild-type-sized and smaller plaques, with F219A also demonstrating a markedly lower progeny titer (at least two logs reduced) than the wild-type control (Figure 10). The reason for this differential behavior of these two SARS-CoV mutants in comparison to those of EAV is unclear at the moment.
Figure 10.
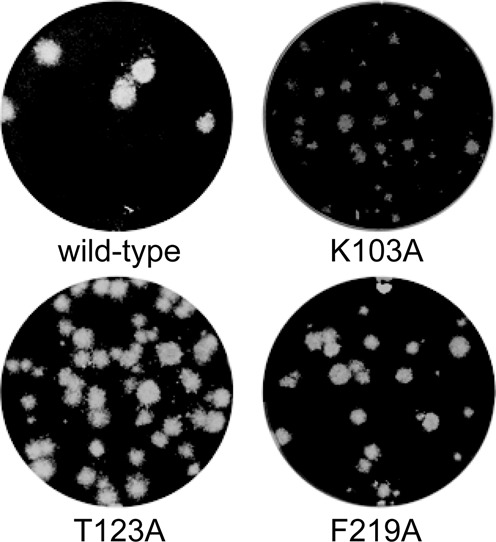
Plaque phenotypes of viable SARS-CoV NiRAN mutants. Progeny virus harvested at 3 days post transfection was used for plaque assays (see ‘Materials and Methods’ section) on Vero-E6 cell monolayers, which were fixed and stained after 3 days to visualize virus-induced plaques.
DISCUSSION
NiRAN is the first enzymatic genetic marker of the order Nidovirales
The NiRAN domain described in this study is the fourth ORF1b-encoded enzyme involved in RNA-dependent processes identified in arteriviruses and the seventh in coronaviruses. As in most prior studies of nidoviral replicative proteins, this identification was initiated by comparative genomics analysis. Unlike all other nidovirus enzymes, however, NiRAN was found to have no appreciable sequence similarity with proteins outside the order Nidovirales. Even the similarity between the arteriviral NiRAN and that of other nidoviruses was found to be marginal. These results suggested that NiRAN either is a unique enzyme specific to nidoviruses or has diverged from its paralogs beyond recognition, i. e. to an extent that cannot be ascertain by even the most powerful HMM-based tools currently available. The latter possibility is not merely hypothetical given that five out of the seven amino acid residues that are evolutionary invariant in the NiRAN domain belong to the most common residues found in proteins. We expect this uncertainty to be resolved in the future when the sampling of nidoviruses will be expanded, sequence profile techniques will be further advanced, and tertiary structures of the proteins analyzed in this study may become available.
Besides technical challenges in the identification of NiRAN, this domain also stands out for its properties that are indicative of an unknown but critical role in nidovirus replication (see below). NiRAN is the only ORF1b-encoded domain that is located upstream of the RdRp and resides within the same non-structural protein. This implies that NiRAN may influence the folding of the downstream RdRp domain. It would be reasonable to expect cross-talk between these domains, potentially coupling the reactions and processes they catalyze. Thus, NiRAN is a prime candidate regulator and/or co-factor of the RdRp, a property that should be taken into account in future experiments aiming at the characterization of the RdRp or reconstitution of RTC activity in vitro.
The exclusive conservation of NiRAN in nidoviruses is indicative of its acquisition by a nidovirus ancestor before the currently known nidovirus families diverged. This makes the domain a genetic marker of this virus order, only the second after the previously identified ZBD and the first with enzymatic activity. It may not be a coincidence that each of these markers is associated with a key enzyme in (+) RNA virus replication, RdRp and HEL1, respectively. The HEL1-modulating role of the ZBD and its involvement in all major processes of the nidovirus replicative cycle have been documented (reviewed in (20)). Similar studies could be performed to probe the function(s) of NiRAN.
Possible functions of conserved NiRAN residues
We here demonstrated that NiRAN is essential for EAV and SARS-CoV replication in cell culture by testing mutants in which conserved residues had been replaced. The mutated viruses were either crippled (and in most cases reverted to wt) or dead, depending on the targeted residue and the virus studied. Importantly the magnitude of the observed effect paralleled that caused by replacement of an RdRp active site residue in the same virus. This parallel is most notable because of the much higher divergence of the NiRAN sequence compared to the RdRp. Also, the significance of NiRAN for virus replication must be different from that of NendoU, the only other ORF1b-encoded enzyme that has been probed extensively by mutagenesis in reverse genetics in both corona- and arteriviruses (25,50,92). Two of those studies revealed that EAV and mouse hepatitis virus (MHV) NendoU mutants with replacements in the active site were stable and in the latter case even displayed similar plaque phenotypes as the wild-type virus while being only slightly delayed in growth (50,92).
In our biochemical assays of the nidovirus RdRp subunit (40,42,93), we detected the new nucleotidylation activity that was associated with the NiRAN of EAV nsp9, as demonstrated by mass spectrometry analysis (Figure 2A and Supplementary Figure S4) and the importance of conserved NiRAN residues for this activity (Figure 7). Nucleotidylation was most pronounced with UTP as substrate but was also observed with GTP (Figure 5A). Despite their size difference, both substrates appeared to be utilized by the same NiRAN binding site since uridylylation as well as guanylylation depended on the same conserved residues. To our knowledge such dual specificity has never been reported for a protein of an RNA virus and (likely) a host. Our results strongly suggested the nucleotidylated residue to be either a lysine or a histidine (Figure 8) located in the N-terminal part of nsp9. Since NiRAN lacks a conserved histidine, the conserved lysine of motif A (K94 in EAV nsp9) is the most likely target for nucleotidylation.
Given the non-radioactive endogenous NTP pool present in E. coli, these results imply that during its expression a part of the recombinant nsp9 may have already been converted to the described nucleoside adducts. Consequently, only the free nsp9 must have been available for nucleotidylation by its NiRAN domain using radioactive GTP/UTP. The nucleotidylated fraction of the total protein pool depends on many factors, including the adduct's stability, and remains unknown. However, this uncertainty does not undermine the validity of the established nucleotidylation activity of nsp9, given the specificity and selectivity documented here, which were determined using different techniques and various controls to arrive at a consistent set of properties of the enzyme. Combined, the results of our biochemical and bioinformatics analyses assigned nucleotidylation activity to the NiRAN domain beyond a reasonable doubt. To rationalize the protein's ability to bind nucleoside phosphates covalently, future studies may focus on the role of protein–nucleoside adducts as reaction intermediates for possible downstream processes, three of which are discussed below.
Next to K94 and/or conserved R124 of motif BN, which may mediate NTP binding via interactions with the negatively charged phosphates, a third conserved residue which may contribute to NTP binding is the motif CN phenylalanine (F166 in EAV). Since phenylalanine would most likely interact with the nucleotide substrate by base stacking, its contribution in terms of binding energy would be one order of magnitude lower than that of electrostatic interactions of lysine/arginine with the phosphates (94). Based solely on this consideration, F166 could be expected to be of ‘lesser’ importance than the basic residues. However, this was apparently not the case since the replacement of the aromatic residue with alanine was lethal for EAV while substitution of either of the basic residues led to a low level of replication that eventually facilitated reversion (Table 1). All these substitutions require two nucleotide point mutations to revert back to wild-type, which should be an extremely rare event during a single round of replication. Consequently, the non-viable phenotype of the F166A mutant may hint at a lower tolerance of single-nucleotide partial revertants (F166V or F166S) in comparison to those originating from K94A (K94T or K94E) and R124A (R124P or R124G). Alternatively, the observed non-viable F166A phenotype may be explained by a vital interaction between NiRAN and RdRp or other proteins involving F166. In contrast to EAV, the homologous residue in SARS-CoV nsp12, F219, appeared to be less essential since its replacement merely reduced progeny titers and altered the plaque phenotype, while the nucleotide changes were maintained. At present, the exact reason for this difference between EAV and SARS-CoV is unclear, but it suggests that the role and/or regulation of this conserved phenylalanine may have evolved in these distantly related nidoviruses, whose NiRAN domains are of strikingly different sizes; such evolution has parallels in other enzymes (95).
Since neither binding of phosphates nor base stacking would enable the enzyme to discriminate between the four bases, it is likely that some of the conserved residues are involved in the formation of a hydrogen bond network that is specific for GTP or UTP. The conserved serine/threonine of motif BN could be a candidate as substitution of this serine in EAV nsp9 (S129) was the only mutation that had a differential effect on guanylylation and uridylylation (Figure 7). Finally, in agreement with observations for other nucleotidylate-forming enzymes (96–98), also nsp9 nucleotidylation is metal-dependent (Figure 5B), potentially due to an important role for metal ions in coordination of the triphosphate or charge neutralization of the pyrophosphate leaving group. In our in vitro system it was Mn2+ rather than the most common divalent cation Mg2+ that supported nucleotidylation activity when tested over a wide concentration range. We propose that at least one of the three acidic conserved residues (E100, D132 and D165 in EAV nsp9) is directly involved in the binding of this essential manganese ion(s). Since the concentration of this cation in cells is lower than that required to observe nucleotidylation in vitro, we cannot exclude the possibility that another co-factor or substrate modulates this property of the enzyme in vivo, and/or that another metal ion is used.
Possible roles of nucleotidylation in the context of viral replication
The identification of the nucleotidylation activity raises the question which role it may play in the nidovirus replicative cycle. In the discussion that follows, we will consider the pros and cons of the involvement of NiRAN's nucleotidylation activity in three previously described functions that are not involved in energy-dependent metabolic processes: nucleic acid ligation, mRNA capping and protein-primed RNA synthesis.
Ligase function
We initially considered NiRAN to be a non-canonical ATP-dependent RNA ligase. It was reasoned that, in the context of nidovirus replication, such an activity could be the functional complement of the NendoU endoribonuclease (7). Moreover, at that time both enzymes were considered to have been conserved across all taxa during evolution of the nidovirus lineage. However, it recently became clear that NendoU is conserved only in nidoviruses infecting vertebrate hosts. Consequently, our original hypothesis would not explain why this putative ligase would be conserved in roni- and mesoniviruses, which do not encode the endoribonuclease. Another complication regarding that original hypothesis has emerged from the present study, which identified NiRAN as being UTP/GTP-specific. Although the hydrolysis of all NTPs results in the release of the same amount of energy, ATP-dependent RNA ligases dominate the ligase family. It would therefore be surprising, if nidoviruses encoded a ligase that strongly discriminates against ATP. To our knowledge the GTP-specific tRNA-splicing ligase RtcB is the only currently known example of a protein involved in nucleic acid strand joining exhibiting this kind of substrate specificity (90). Furthermore, thus far no substrates that would require a ligase function were identified in nidovirus replication, which however remains poorly characterized in general.
5′ end cap guanylyltransferase function
Besides RNA ligases, also guanylyltransferases (GTases) employ a very similar mechanism of nucleotidylation and are used to permanently modify the 5′ end of RNA with the bound GMP in a process called RNA capping (reviewed in (99)). Intriguingly, three of the four enzyme activities required for cap formation and modification, namely an RNA-triphosphatase and two methyltransferases, have been identified in coronaviruses (44,100), with the missing activity being the GTase. Furthermore, recent characterization of EAV nsp10 in our lab (unpublished data) showed that it resembles its coronavirus homolog in terms of possessing RNA-triphosphatase activity, which is required prior to GTase activity in the conventional capping pathway. In line with these findings, experimental evidence supporting the presence of a cap structure on genomic RNA was reported for three very distantly related viruses of the Nidovirales order, namely for MHV (Coronavirinae) (101), Equine torovirus (Torovirinae) (102) and Simian hemorrhagic fever virus (Arteriviridae) (103). Importantly, the known GTases of (+) RNA viruses, flavivirus NS5 (104), alphavirus nsP1 and orthologous proteins (97,105), do neither share conserved features nor do they resemble host GTases. Thus, the possibility of NiRAN being a cap-synthesizing GTase could be reconciled with our current knowledge of the structural and sequence diversity of this class of enzymes. This cannot be said, however, about NiRAN's substrate preference for UTP over GTP, which has not been reported for GTases mediating cap formation.
Protein-priming function
If UTP binding by NiRAN reflects a genuine property of the enzyme, another mechanism that might utilize its nucleotidylation activity may be protein-primed RNA synthesis. This strategy is used by many viruses including the large group of picornavirus-like viruses, which notably have evolutionary affinity to nidoviruses (35,36). In these viruses, a nucleotide is covalently attached to a protein that is commonly known as VPg (viral protein genome-linked), which may then be extended to a dinucleotide. This dinucleotide is subsequently base-paired to the 3′ end of the viral RNA where it serves as primer for the synthesis of the complementary RNA strand (106). Interestingly, the first nucleotide of the EAV genome is a G while the 3′ end is equipped with a poly(A) tail. Thus, the dual specificity of nsp9 for GTP and UTP would be compatible with the different requirements for the initiation of the synthesis of genomic and subgenomic RNAs of positive and negative polarity, respectively. To which extent this property is conserved across nidoviruses remains to be established.
While considering this mechanism, it is instructive to take into account observations that distinguish nidoviruses from VPg-utilizing viruses. First, to our knowledge, all currently described nucleotide-VPg bonds are realized via the hydroxyl group of either a tyrosine or a serine/threonine (107–111), while NiRAN most likely uses the invariant lysine residue (Figure 8). This problem could be resolved if NiRAN assumes the role of the RdRp of VPg-encoding viruses and transfers the bound nucleotide to another protein that subsequently serves as VPg. Second, at least for coronaviruses, the VPg-based mechanism would not be compatible with the previously proposed primase-based mechanism (112) for the initiation of RNA synthesis. However, the latter mechanism remains tentative since it assigns primase activity to a protein complex that, according to a recent study (84), may merely be a processivity co-factor for the nsp12 RdRp. Finally, as mentioned before, the mRNAs of several nidoviruses were concluded to be capped at their 5′ end, a modification that is not observed in known VPg-utilizing viruses. To use both VPg priming and capping, it would be necessary to actively or passively remove the attached protein in order to allow mRNA capping to commence. This sequence of events would constitute a novel, and perhaps unlikely, variant of the capping pathway, as the RNA's 5′ end would not be di- or triphosphorylated after VPg removal, a requirement for entering any of the known viral capping pathways (99). Thus, if NiRAN would be part of a VPg-utilizing mechanism, this might differ considerably from those currently described and could possibly also vary among nidoviruses.
In view of the considerations outlined for each of the three possible scenarios employing nucleotidylation activity, it is evident that presently none of these can be reconciled with the evolutionary, structural and functional characteristics of NiRAN described in this study without additional assumptions. This may reflect yet-to-be revealed specifics of the nidovirus RTC and its unparalleled complexity.
Supplementary Material
Acknowledgments
The authors thank Bruno Canard, Etienne Decroly, Isabelle Imbert, Barbara Selisko, Lorenzo Subissi and Aartjan te Velthuis for helpful discussions; Chris Lauber and Erik Hoogendoorn for help with the DEmARC-based analysis, and Daniel Cupac and Linda Boomaars for technical assistance. A.E.G. is a member of the Netherlands Bioinformatics Center (NBIC) Faculty.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
European Union Seventh Framework program through the EUVIRNA project (European Training Network on (+) RNA virus replication and Antiviral Drug Development [264286]; SILVER project [260644]; Netherlands Organization for Scientific Research (NWO) through TOP-GO [700.10.352]; Leiden University Fund; Collaborative Agreement in Bioinformatics between Leiden University Medical Center and Moscow State University (MoBiLe). Funding for open access charge: Leiden University Medical Center and Netherlands Organisation for Scientific Research.
Conflict of interest statement. None declared.
REFERENCES
- 1.de Groot R.J., Cowley J.A., Enjuanes L., Faaberg K.S., Perlman S., Rottier P.J., Snijder E.J., Ziebuhr J., Gorbalenya A.E. Order Nidovirales. In: King AMQ, Adams MJ, Carstens EB, Lefkowitz EJ, editors. Virus Taxonomy. Ninth Report of the International Committee on Taxonomy of Viruses. Amsterdam: Elsevier Academic Press; 2012. pp. 785–795. [Google Scholar]
- 2.Lauber C., Ziebuhr J., Junglen S., Drosten C., Zirkel F., Nga P.T., Morita K., Snijder E.J., Gorbalenya A.E. Mesoniviridae: a proposed new family in the order Nidovirales formed by a single species of mosquito-borne viruses. Arch. Virol. 2012;157:1623–1628. doi: 10.1007/s00705-012-1295-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Neumann E.J., Kliebenstein J.B., Johnson C.D., Mabry J.W., Bush E.J., Seitzinger A.H., Green A.L., Zimmerman J.J. Assessment of the economic impact of porcine reproductive and respiratory syndrome on swine production in the United States. J. Am. Vet. Med. Assoc. 2005;227:385–392. doi: 10.2460/javma.2005.227.385. [DOI] [PubMed] [Google Scholar]
- 4.Coleman C.M., Frieman M.B. Coronaviruses: important emerging human pathogens. J. Virol. 2014;88:5209–5212. doi: 10.1128/JVI.03488-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hilgenfeld R., Peiris M. From SARS to MERS: 10 years of research on highly pathogenic human coronaviruses. Antiviral Res. 2013;100:286–295. doi: 10.1016/j.antiviral.2013.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zaki A.M., van Boheemen S., Bestebroer T.M., Osterhaus A.D.M.E., Fouchier R.A.M. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N Engl J Med. 2012;367:1814–1820. doi: 10.1056/NEJMoa1211721. [DOI] [PubMed] [Google Scholar]
- 7.Lauber C., Goeman J.J., Parquet M.C., Nga P.T., Snijder E.J., Morita K., Gorbalenya A.E. The footprint of genome architecture in the largest genome expansion in RNA viruses. PLoS Pathog. 2013;9:e1003500. doi: 10.1371/journal.ppat.1003500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Snijder E.J., Siddell S.G., Gorbalenya A.E. The order Nidovirales. In: Mahy BW, ter Meulen V, editors. Topley and Wilson's Microbiology and Microbial Infections: Virology Volume. London: Hodder Arnold; 2005. pp. 390–404. [Google Scholar]
- 9.Pasternak A.O., Spaan W.J., Snijder E.J. Nidovirus transcription: how to make sense…. J. Gen. Virol. 2006;87:1403–1421. doi: 10.1099/vir.0.81611-0. [DOI] [PubMed] [Google Scholar]
- 10.Sawicki S. G., Sawicki D. L., Siddell S. G. A contemporary view of coronavirus transcription. J. Virol. 2007;81:20–29. doi: 10.1128/JVI.01358-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brian D. A., Baric R. S. Coronavirus genome structure and replication. Curr. Top. Microbiol. Immunol. 2005;287:1–30. doi: 10.1007/3-540-26765-4_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Masters P.S. The molecular biology of coronaviruses. Adv. Virus Res. 2006;66:193–292. doi: 10.1016/S0065-3527(06)66005-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gorbalenya A.E., Enjuanes L., Ziebuhr J., Snijder E.J. Nidovirales: evolving the largest RNA virus genome. Virus Res. 2006;117:17–37. doi: 10.1016/j.virusres.2006.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ziebuhr J., Snijder E.J., Gorbalenya A.E. Virus-encoded proteinases and proteolytic processing in the Nidovirales. J. Gen. Virol. 2000;81:853–879. doi: 10.1099/0022-1317-81-4-853. [DOI] [PubMed] [Google Scholar]
- 15.Weiss S.R., Leibowitz J.L. Coronavirus pathogenesis. Adv Virus Res. 2011;81:85–164. doi: 10.1016/B978-0-12-385885-6.00009-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Perlman S., Netland J. Coronaviruses post-SARS: update on replication and pathogenesis. Nat Rev Microbiol. 2009;7:439–450. doi: 10.1038/nrmicro2147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Subissi L., Imbert I., Ferron F., Collet A., Coutard B., Decroly E., Canard B. SARS-CoV ORF1b-encoded nonstructural proteins 12–16: replicative enzymes as antiviral targets. Antiviral Res. 2014;101:122–130. doi: 10.1016/j.antiviral.2013.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Neuman B.W., Angelini M.M., Buchmeier M.J. Does form meet function in the coronavirus replicative organelle. Trends Microbiol. 2014;22:642–647. doi: 10.1016/j.tim.2014.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Denison M.R., Graham R.L., Donaldson E.F., Eckerle L.D., Baric R.S. Coronaviruses: an RNA proofreading machine regulates replication fidelity and diversity. RNA Biol. 2011;8:270–279. doi: 10.4161/rna.8.2.15013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lehmann K.C., Snijder E.J., Posthuma C.C., Gorbalenya A.E. What we know but do not understand about nidovirus helicases. Virus Res. 2014;202:12–32. doi: 10.1016/j.virusres.2014.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bouvet M., Imbert I., Subissi L., Gluais L., Canard B., Decroly E. RNA 3′-end mismatch excision by the severe acute respiratory syndrome coronavirus nonstructural protein nsp10/nsp14 exoribonuclease complex. Proc. Natl. Acad. Sci. U.S.A. 2012;109:9372–9377. doi: 10.1073/pnas.1201130109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Minskaia E., Hertzig T., Gorbalenya A.E., Campanacci V., Cambillau C., Canard B., Ziebuhr J. Discovery of an RNA virus 3′->5′ exoribonuclease that is critically involved in coronavirus RNA synthesis. Proc. Natl. Acad. Sci. U.S.A. 2006;103:5108–5113. doi: 10.1073/pnas.0508200103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bouvet M., Debarnot C., Imbert I., Selisko B., Snijder E.J., Canard B., Decroly E. In vitro reconstitution of SARS-coronavirus mRNA cap methylation. PLoS Pathog. 2010;6:e1000863. doi: 10.1371/journal.ppat.1000863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen Y., Cai H., Pan J., Xiang N., Tien P., Ahola T., Guo D. Functional screen reveals SARS coronavirus nonstructural protein nsp14 as a novel cap N7 methyltransferase. Proc. Natl. Acad. Sci. U.S.A. 2009;106:3484–3489. doi: 10.1073/pnas.0808790106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ivanov K.A., Hertzig T., Rozanov M., Bayer S., Thiel V., Gorbalenya A.E., Ziebuhr J. Major genetic marker of nidoviruses encodes a replicative endoribonuclease. Proc. Natl. Acad. Sci. U.S.A. 2004;101:12694–12699. doi: 10.1073/pnas.0403127101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nedialkova D.D., Ulferts R., van den Born E., Lauber C., Gorbalenya A.E., Ziebuhr J., Snijder E.J. Biochemical characterization of arterivirus nonstructural protein 11 reveals the nidovirus-wide conservation of a replicative endoribonuclease. J. Virol. 2009;83:5671–5682. doi: 10.1128/JVI.00261-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen Y., Su C., Ke M., Jin X., Xu L., Zhang Z., Wu A., Sun Y., Yang Z., Tien P., et al. Biochemical and structural insights into the mechanisms of SARS coronavirus RNA ribose 2′-O-methylation by nsp16/nsp10 protein complex. PLoS Pathog. 2011;7:e1002294. doi: 10.1371/journal.ppat.1002294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Daffis S., Szretter K.J., Schriewer J., Li J., Youn S., Errett J., Lin T.Y., Schneller S., Zust R., Dong H., et al. 2′-O methylation of the viral mRNA cap evades host restriction by IFIT family members. Nature. 2010;468:452–456. doi: 10.1038/nature09489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Decroly E., Imbert I., Coutard B., Bouvet M., Selisko B., Alvarez K., Gorbalenya A.E., Snijder E.J., Canard B. Coronavirus nonstructural protein 16 is a cap-0 binding enzyme possessing (nucleoside-2′O)-methyltransferase activity. J. Virol. 2008;82:8071–8084. doi: 10.1128/JVI.00407-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nga P.T., Parquet M.C., Lauber C., Parida M., Nabeshima T., Yu F., Thuy N.T., Inoue S., Ito T., Okamoto K., et al. Discovery of the first insect nidovirus, a missing evolutionary link in the emergence of the largest RNA virus genomes. PLoS Pathog. 2011;7:e1002215. doi: 10.1371/journal.ppat.1002215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gorbalenya A.E. Big nidovirus genome. When count and order of domains matter. Adv. Exp. Med. Biol. 2001;494:1–17. [PubMed] [Google Scholar]
- 32.Lehmann K.C., Hooghiemstra L., Gulyaeva A., Samborskiy D., Zevenhoven-Dobbe J., Snijder E., Gorbalenya A.E., Posthuma C.C. Arterivirus nsp12 versus the coronavirus nsp16 2′-O-methyltransferase: comparison of the C-terminal cleavage products of two nidovirus pp1ab polyproteins. J. Gen. Virol. 2015 doi: 10.1099/vir.0.000209. doi:10.1099/vir.0.000209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lang D.M., Zemla A.T., Zhou C.L. Highly similar structural frames link the template tunnel and NTP entry tunnel to the exterior surface in RNA-dependent RNA polymerases. Nucleic Acids Res. 2013;41:1464–1482. doi: 10.1093/nar/gks1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ng K.K., Arnold J.J., Cameron C.E. Structure-function relationships among RNA-dependent RNA polymerases. Curr. Top. Microbiol. Immunol. 2008;320:137–156. doi: 10.1007/978-3-540-75157-1_7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gorbalenya A.E., Pringle F.M., Zeddam J.L., Luke B.T., Cameron C.E., Kalmakoff J., Hanzlik T.N., Gordon K.H., Ward V.K. The palm subdomain-based active site is internally permuted in viral RNA-dependent RNA polymerases of an ancient lineage. J. Mol. Biol. 2002;324:47–62. doi: 10.1016/S0022-2836(02)01033-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gorbalenya A.E., Koonin E.V., Donchenko A.P., Blinov V.M. Coronavirus genome: prediction of putative functional domains in the non-structural polyprotein by comparative amino acid sequence analysis. Nucleic Acids Res. 1989;17:4847–4861. doi: 10.1093/nar/17.12.4847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Azzi A., Lin S.X. Human SARS-coronavirus RNA-dependent RNA polymerase: activity determinants and nucleoside analogue inhibitors. Proteins. 2004;57:12–14. doi: 10.1002/prot.20194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xu X., Liu Y., Weiss S., Arnold E., Sarafianos S.G., Ding J. Molecular model of SARS coronavirus polymerase: implications for biochemical functions and drug design. Nucleic Acids Res. 2003;31:7117–7130. doi: 10.1093/nar/gkg916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Snijder E.J., Bredenbeek P.J., Dobbe J.C., Thiel V., Ziebuhr J., Poon L.L., Guan Y., Rozanov M., Spaan W.J., Gorbalenya A.E. Unique and conserved features of genome and proteome of SARS-coronavirus, an early split-off from the coronavirus group 2 lineage. J. Mol. Biol. 2003;331:991–1004. doi: 10.1016/S0022-2836(03)00865-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ahn D.G., Choi J.K., Taylor D.R., Oh J.W. Biochemical characterization of a recombinant SARS coronavirus nsp12 RNA-dependent RNA polymerase capable of copying viral RNA templates. Arch. Virol. 2012;157:2095–2104. doi: 10.1007/s00705-012-1404-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bautista E.M., Faaberg K.S., Mickelson D., McGruder E.D. Functional properties of the predicted helicase of porcine reproductive and respiratory syndrome virus. Virology. 2002;298:258–270. doi: 10.1006/viro.2002.1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Beerens N., Selisko B., Ricagno S., Imbert I., van der Zanden L., Snijder E.J., Canard B. De novo initiation of RNA synthesis by the arterivirus RNA-dependent RNA polymerase. J. Virol. 2007;81:8384–8395. doi: 10.1128/JVI.00564-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ivanov K.A., Thiel V., Dobbe J.C., van der Meer Y., Snijder E.J., Ziebuhr J. Multiple enzymatic activities associated with severe acute respiratory syndrome coronavirus helicase. J. Virol. 2004;78:5619–5632. doi: 10.1128/JVI.78.11.5619-5632.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ivanov K.A., Ziebuhr J. Human coronavirus 229E nonstructural protein 13: characterization of duplex-unwinding, nucleoside triphosphatase, and RNA 5′-triphosphatase activities. J. Virol. 2004;78:7833–7838. doi: 10.1128/JVI.78.14.7833-7838.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Seybert A., van Dinten L.C., Snijder E.J., Ziebuhr J. Biochemical characterization of the equine arteritis virus helicase suggests a close functional relationship between arterivirus and coronavirus helicases. J. Virol. 2000;74:9586–9593. doi: 10.1128/jvi.74.20.9586-9593.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Seybert A., Posthuma C.C., van Dinten L.C., Snijder E.J., Gorbalenya A.E., Ziebuhr J. A complex zinc finger controls the enzymatic activities of nidovirus helicases. J. Virol. 2005;79:696–704. doi: 10.1128/JVI.79.2.696-704.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.te Velthuis A.J., van den Worm S.H., Sims A.C., Baric R.S., Snijder E.J., van Hemert M.J. Zn(2+) inhibits coronavirus and arterivirus RNA polymerase activity in vitro and zinc ionophores block the replication of these viruses in cell culture. PLoS Pathog. 2010;6:e1001176. doi: 10.1371/journal.ppat.1001176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.van Dinten L.C., van Tol H., Gorbalenya A.E., Snijder E.J. The predicted metal-binding region of the arterivirus helicase protein is involved in subgenomic mRNA synthesis, genome replication, and virion biogenesis. J. Virol. 2000;74:5213–5223. doi: 10.1128/jvi.74.11.5213-5223.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Eckerle L.D., Lu X., Sperry S.M., Choi L., Denison M.R. High fidelity of murine hepatitis virus replication is decreased in nsp14 exoribonuclease mutants. J. Virol. 2007;81:12135–12144. doi: 10.1128/JVI.01296-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Posthuma C.C., Nedialkova D.D., Zevenhoven-Dobbe J.C., Blokhuis J.H., Gorbalenya A.E., Snijder E.J. Site-directed mutagenesis of the Nidovirus replicative endoribonuclease NendoU exerts pleiotropic effects on the arterivirus life cycle. J. Virol. 2006;80:1653–1661. doi: 10.1128/JVI.80.4.1653-1661.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zust R., Cervantes-Barragan L., Habjan M., Maier R., Neuman B.W., Ziebuhr J., Szretter K.J., Baker S.C., Barchet W., Diamond M.S., et al. Ribose 2′-O-methylation provides a molecular signature for the distinction of self and non-self mRNA dependent on the RNA sensor Mda5. Nat. Immunol. 2011;12:137–143. doi: 10.1038/ni.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Benson D.A., Cavanaugh M., Clark K., Karsch-Mizrachi I., Lipman D.J., Ostell J., Sayers E.W. GenBank. Nucleic Acids Res. 2013;41:D36–D42. doi: 10.1093/nar/gks1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pruitt K.D., Brown G.R., Hiatt S.M., Thibaud-Nissen F., Astashyn A., Ermolaeva O., Farrell C.M., Hart J., Landrum M.J., McGarvey K.M., et al. RefSeq: an update on mammalian reference sequences. Nucleic Acids Res. 2014;42:D756–D763. doi: 10.1093/nar/gkt1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lauber C., Gorbalenya A.E. Partitioning the genetic diversity of a virus family: approach and evaluation through a case study of picornaviruses. J. Virol. 2012;86:3890–3904. doi: 10.1128/JVI.07173-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sidorov I.A., Reshetov D.A., Gorbalenya A.E. SNAD: sequence name annotation-based designer. BMC Bioinformatics. 2009;10:251. doi: 10.1186/1471-2105-10-251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gorbalenya A.E., Lieutaud P., Harris M.R., Coutard B., Canard B., Kleywegt G.J., Kravchenko A.A., Samborskiy D.V., Sidorov I.A., Leontovich A.M., et al. Practical application of bioinformatics by the multidisciplinary VIZIER consortium. Antiviral Res. 2010;87:95–110. doi: 10.1016/j.antiviral.2010.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Finn R.D., Clements J., Eddy S.R. HMMER web server: interactive sequence similarity searching. Nucleic Acids Res. 2011;39:W29–W37. doi: 10.1093/nar/gkr367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Edgar R.C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Larkin M.A., Blackshields G., Brown N.P., Chenna R., McGettigan P.A., McWilliam H., Valentin F., Wallace I.M., Wilm A., Lopez R., et al. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- 60.Ziebuhr J., Bayer S., Cowley J.A., Gorbalenya A.E. The 3C-like proteinase of an invertebrate nidovirus links coronavirus and potyvirus homologs. J. Virol. 2003;77:1415–1426. doi: 10.1128/JVI.77.2.1415-1426.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Blanck S., Stinn A., Tsiklauri L., Zirkel F., Junglen S., Ziebuhr J. Characterization of an alphamesonivirus 3C-like protease defines a special group of nidovirus main proteases. J. Virol. 2014;88:13747–13758. doi: 10.1128/JVI.02040-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Soding J. Protein homology detection by HMM-HMM comparison. Bioinformatics. 2005;21:951–960. doi: 10.1093/bioinformatics/bti125. [DOI] [PubMed] [Google Scholar]
- 63.Remmert M., Biegert A., Hauser A., Soding J. HHblits: lightning-fast iterative protein sequence searching by HMM-HMM alignment. Nat. Methods. 2012;9:173–175. doi: 10.1038/nmeth.1818. [DOI] [PubMed] [Google Scholar]
- 64.Cole C., Barber J.D., Barton G.J. The Jpred 3 secondary structure prediction server. Nucleic Acids Res. 2008;36:W197–W201. doi: 10.1093/nar/gkn238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jones D.T. Protein secondary structure prediction based on position-specific scoring matrices. J. Mol. Biol. 1999;292:195–202. doi: 10.1006/jmbi.1999.3091. [DOI] [PubMed] [Google Scholar]
- 66.Robert X., Gouet P. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 2014;42:W320–W324. doi: 10.1093/nar/gku316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.NCBI Resource Coordinators. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2015;43:D6–D17. doi: 10.1093/nar/gku1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Finn R.D., Bateman A., Clements J., Coggill P., Eberhardt R.Y., Eddy S.R., Heger A., Hetherington K., Holm L., Mistry J., et al. Pfam: the protein families database. Nucleic Acids Res. 2014;42:D222–D230. doi: 10.1093/nar/gkt1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Berman H.M., Westbrook J., Feng Z., Gilliland G., Bhat T.N., Weissig H., Shindyalov I.N., Bourne P.E. The Protein Data Bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Jones D.T. GenTHREADER: an efficient and reliable protein fold recognition method for genomic sequences. J. Mol. Biol. 1999;287:797–815. doi: 10.1006/jmbi.1999.2583. [DOI] [PubMed] [Google Scholar]
- 71.McGuffin L.J., Jones D.T. Improvement of the GenTHREADER method for genomic fold recognition. Bioinformatics. 2003;19:874–881. doi: 10.1093/bioinformatics/btg097. [DOI] [PubMed] [Google Scholar]
- 72.Lobley A., Sadowski M.I., Jones D.T. pGenTHREADER and pDomTHREADER: new methods for improved protein fold recognition and superfamily discrimination. Bioinformatics. 2009;25:1761–1767. doi: 10.1093/bioinformatics/btp302. [DOI] [PubMed] [Google Scholar]
- 73.Grant B.J., Rodrigues A.P., ElSawy K.M., McCammon J.A., Caves L.S. Bio3d: an R package for the comparative analysis of protein structures. Bioinformatics. 2006;22:2695–2696. doi: 10.1093/bioinformatics/btl461. [DOI] [PubMed] [Google Scholar]
- 74.Henikoff S., Henikoff J.G. Amino acid substitution matrices from protein blocks. Proc. Natl. Acad. Sci. U.S.A. 1992;89:10915–10919. doi: 10.1073/pnas.89.22.10915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Guindon S., Dufayard J.F., Lefort V., Anisimova M., Hordijk W., Gascuel O. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst. Biol. 2010;59:307–321. doi: 10.1093/sysbio/syq010. [DOI] [PubMed] [Google Scholar]
- 76.Paradis E., Claude J., Strimmer K. APE: analyses of phylogenetics and evolution in R language. Bioinformatics. 2004;20:289–290. doi: 10.1093/bioinformatics/btg412. [DOI] [PubMed] [Google Scholar]
- 77.R Development Core Team. R: a Language and Environment for Statistical Computing. Vienna: R Foundation for statistical computing; 2011. [Google Scholar]
- 78.Hanoulle X., Van D.J., Staes A., Martens L., Goethals M., Vandekerckhove J., Gevaert K. A new functional, chemical proteomics technology to identify purine nucleotide binding sites in complex proteomes. J. Proteome. Res. 2006;5:3438–3445. doi: 10.1021/pr060313e. [DOI] [PubMed] [Google Scholar]
- 79.van den Born E., Gultyaev A.P., Snijder E.J. Secondary structure and function of the 5′-proximal region of the equine arteritis virus RNA genome. RNA. 2004;10:424–437. doi: 10.1261/rna.5174804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Nedialkova D.D., Gorbalenya A.E., Snijder E.J. Arterivirus Nsp1 modulates the accumulation of minus-strand templates to control the relative abundance of viral mRNAs. PLoS Pathog. 2010;6:e1000772. doi: 10.1371/journal.ppat.1000772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.van der Meer Y., van T.H., Locker J.K., Snijder E.J. ORF1a-encoded replicase subunits are involved in the membrane association of the arterivirus replication complex. J. Virol. 1998;72:6689–6698. doi: 10.1128/jvi.72.8.6689-6698.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Pfefferle S., Krahling V., Ditt V., Grywna K., Muhlberger E., Drosten C. Reverse genetic characterization of the natural genomic deletion in SARS-Coronavirus strain Frankfurt-1 open reading frame 7b reveals an attenuating function of the 7b protein in-vitro and in-vivo. Virol. J. 2009;6:131. doi: 10.1186/1743-422X-6-131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Tischer B.K., Smith G.A., Osterrieder N. En passant mutagenesis: a two step markerless red recombination system. Methods Mol. Biol. 2010;634:421–430. doi: 10.1007/978-1-60761-652-8_30. [DOI] [PubMed] [Google Scholar]
- 84.Subissi L., Posthuma C.C., Collet A., Zevenhoven-Dobbe J.C., Gorbalenya A.E., Decroly E., Snijder E.J., Canard B., Imbert I. One severe acute respiratory syndrome coronavirus protein complex integrates processive RNA polymerase and exonuclease activities. Proc. Natl. Acad. Sci. U.S.A. 2014;111:e3900. doi: 10.1073/pnas.1323705111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Bartlett G.J., Porter C.T., Borkakoti N., Thornton J.M. Analysis of catalytic residues in enzyme active sites. J. Mol. Biol. 2002;324:105–121. doi: 10.1016/s0022-2836(02)01036-7. [DOI] [PubMed] [Google Scholar]
- 86.Henderson B.R., Saeedi B.J., Campagnola G., Geiss B.J. Analysis of RNA binding by the dengue virus NS5 RNA capping enzyme. PLoS One. 2011;6:e25795. doi: 10.1371/journal.pone.0025795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Shuman S., Schwer B. RNA capping enzyme and DNA ligase: a superfamily of covalent nucleotidyl transferases. Mol. Microbiol. 1995;17:405–410. doi: 10.1111/j.1365-2958.1995.mmi_17030405.x. [DOI] [PubMed] [Google Scholar]
- 88.Shuman S., Lima C.D. The polynucleotide ligase and RNA capping enzyme superfamily of covalent nucleotidyltransferases. Curr. Opin. Struct. Biol. 2004;14:757–764. doi: 10.1016/j.sbi.2004.10.006. [DOI] [PubMed] [Google Scholar]
- 89.Traut T.W. Physiological concentrations of purines and pyrimidines. Mol. Cell Biochem. 1994;140:1–22. doi: 10.1007/BF00928361. [DOI] [PubMed] [Google Scholar]
- 90.Chakravarty A.K., Subbotin R., Chait B.T., Shuman S. RNA ligase RtcB splices 3′-phosphate and 5′-OH ends via covalent RtcB-(histidinyl)-GMP and polynucleotide-(3′)pp(5′)G intermediates. Proc. Natl. Acad. Sci. U.S.A. 2012;109:6072–6077. doi: 10.1073/pnas.1201207109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Duclos B., Marcandier S., Cozzone A.J. Chemical properties and separation of phosphoamino acids by thin-layer chromatography and/or electrophoresis. Methods Enzymol. 1991;201:10–21. doi: 10.1016/0076-6879(91)01004-l. [DOI] [PubMed] [Google Scholar]
- 92.Kang H., Bhardwaj K., Li Y., Palaninathan S., Sacchettini J., Guarino L., Leibowitz J.L., Kao C.C. Biochemical and genetic analyses of murine hepatitis virus Nsp15 endoribonuclease. J. Virol. 2007;81:13587–13597. doi: 10.1128/JVI.00547-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.te Velthuis A.J., Arnold J.J., Cameron C.E., van den Worm S.H., Snijder E.J. The RNA polymerase activity of SARS-coronavirus nsp12 is primer dependent. Nucleic Acids Res. 2010;38:203–214. doi: 10.1093/nar/gkp904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kumar N.V., Govil G. Theoretical studies on protein-nucleic acid interactions. III. Stacking of aromatic amino acids with bases and base pairs of nucleic acids. Biopolymers. 1984;23:2009–2024. doi: 10.1002/bip.360231015. [DOI] [PubMed] [Google Scholar]
- 95.Bartlett G.J., Borkakoti N., Thornton J.M. Catalysing new reactions during evolution: economy of residues and mechanism. J. Mol. Biol. 2003;331:829–860. doi: 10.1016/s0022-2836(03)00734-4. [DOI] [PubMed] [Google Scholar]
- 96.Schmelz S., Naismith J.H. Adenylate-forming enzymes. Curr. Opin. Struct. Biol. 2009;19:666–671. doi: 10.1016/j.sbi.2009.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ahola T., Laakkonen P., Vihinen H., Kaariainen L. Critical residues of Semliki Forest virus RNA capping enzyme involved in methyltransferase and guanylyltransferase-like activities. J. Virol. 1997;71:392–397. doi: 10.1128/jvi.71.1.392-397.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Nandakumar J., Shuman S., Lima C.D. RNA ligase structures reveal the basis for RNA specificity and conformational changes that drive ligation forward. Cell. 2006;127:71–84. doi: 10.1016/j.cell.2006.08.038. [DOI] [PubMed] [Google Scholar]
- 99.Decroly E., Ferron F., Lescar J., Canard B. Conventional and unconventional mechanisms for capping viral mRNA. Nat. Rev. Microbiol. 2012;10:51–65. doi: 10.1038/nrmicro2675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Bouvet M., Debarnot C., Imbert I., Selisko B., Snijder E.J., Canard B., Decroly E. In vitro reconstitution of SARS-coronavirus mRNA cap methylation. PLoS Pathog. 2010;6:e1000863. doi: 10.1371/journal.ppat.1000863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Lai M.M., Patton C.D., Stohlman S.A. Further characterization of mRNA's of mouse hepatitis virus: presence of common 5′-end nucleotides. J. Virol. 1982;41:557–565. doi: 10.1128/jvi.41.2.557-565.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.van Vliet A.L., Smits S.L., Rottier P.J., de Groot R.J. Discontinuous and non-discontinuous subgenomic RNA transcription in a nidovirus. EMBO J. 2002;21:6571–6580. doi: 10.1093/emboj/cdf635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Sagripanti J.L., Zandomeni R.O., Weinmann R. The cap structure of simian hemorrhagic fever virion RNA. Virology. 1986;151:146–150. doi: 10.1016/0042-6822(86)90113-3. [DOI] [PubMed] [Google Scholar]
- 104.Issur M., Geiss B.J., Bougie I., Picard-Jean F., Despins S., Mayette J., Hobdey S.E., Bisaillon M. The flavivirus NS5 protein is a true RNA guanylyltransferase that catalyzes a two-step reaction to form the RNA cap structure. RNA. 2009;15:2340–2350. doi: 10.1261/rna.1609709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Ahola T., Ahlquist P. Putative RNA capping activities encoded by brome mosaic virus: methylation and covalent binding of guanylate by replicase protein 1a. J. Virol. 1999;73:10061–10069. doi: 10.1128/jvi.73.12.10061-10069.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Paul A.V., Rieder E., Kim D.W., van Boom J.H., Wimmer E. Identification of an RNA hairpin in poliovirus RNA that serves as the primary template in the in vitro uridylylation of VPg. J. Virol. 2000;74:10359–10370. doi: 10.1128/jvi.74.22.10359-10370.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Ambros V., Baltimore D. Protein is linked to the 5′ end of poliovirus RNA by a phosphodiester linkage to tyrosine. J. Biol. Chem. 1978;253:5263–5266. [PubMed] [Google Scholar]
- 108.Pan J., Lin L., Tao Y.J. Self-guanylylation of birnavirus VP1 does not require an intact polymerase activity site. Virology. 2009;395:87–96. doi: 10.1016/j.virol.2009.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Mitra T., Sosnovtsev S.V., Green K.Y. Mutagenesis of tyrosine 24 in the VPg protein is lethal for feline calicivirus. J. Virol. 2004;78:4931–4935. doi: 10.1128/JVI.78.9.4931-4935.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Jiang J., Laliberte J.F. The genome-linked protein VPg of plant viruses-a protein with many partners. Curr. Opin. Virol. 2011;1:347–354. doi: 10.1016/j.coviro.2011.09.010. [DOI] [PubMed] [Google Scholar]
- 111.Zeddam J.L., Gordon K.H., Lauber C., Alves C.A., Luke B.T., Hanzlik T.N., Ward V.K., Gorbalenya A.E. Euprosterna elaeasa virus genome sequence and evolution of the Tetraviridae family: emergence of bipartite genomes and conservation of the VPg signal with the dsRNA Birnaviridae family. Virology. 2010;397:145–154. doi: 10.1016/j.virol.2009.10.042. [DOI] [PubMed] [Google Scholar]
- 112.Imbert I., Guillemot J.C., Bourhis J.M., Bussetta C., Coutard B., Egloff M.P., Ferron F., Gorbalenya A.E., Canard B. A second, non-canonical RNA-dependent RNA polymerase in SARS coronavirus. EMBO J. 2006;25:4933–4942. doi: 10.1038/sj.emboj.7601368. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.