

Abstract
In bacteria and archaea, short fragments of foreign DNA are integrated into Clustered Regularly Interspaced Short Palindromic Repeat (CRISPR) loci, providing a molecular memory of previous encounters with foreign genetic elements. In Escherichia coli, short CRISPR-derived RNAs are incorporated into a multi-subunit surveillance complex called Cascade (CRISPR-associated complex for antiviral defense). Recent structures of Cascade capture snapshots of this seahorse-shaped RNA-guided surveillance complex before and after binding to a DNA target. Here we determine a 3.2 Å x-ray crystal structure of Cascade in a new crystal form that provides insight into the mechanism of double-stranded DNA binding. Molecular dynamic simulations performed using available structures reveal functional roles for residues in the tail, backbone and belly subunits of Cascade that are critical for binding double-stranded DNA. Structural comparisons are used to make functional predictions and these predictions are tested in vivo and in vitro. Collectively, the results in this study reveal underlying mechanisms involved in target-induced conformational changes and highlight residues important in DNA binding and protospacer adjacent motif recognition.
INTRODUCTION
All immune systems must recognize and eliminate foreign invaders while avoiding self-antigens that would lead to an autoimmune reaction. In many bacteria (∼50%) and most archaea (∼90%), immunity to invading genetic parasites is achieved by integrating short fragments of invading DNA into Clustered Regularly Interspaced Short Palindromic Repeats (CRISPRs) (1–8). Transcripts from these loci are processed into short CRISPR-derived RNAs (crRNAs) that assemble with CRISPR-associated (Cas) proteins into large ribonucleoprotein surveillance complexes that use the crRNA as a guide to bind complementary DNA targets, called protospacers. While all CRISPR systems rely on crRNAs for sequence-specific detection of invading nucleic acids, phylogenetic analyses of CRISPR loci and cas genes have identified three distinct CRISPR system types (Type I, II and III) that are further separated into at least 12 subtypes (Type I-A to I-F, Type II-A to II-C and Type III-A to III-C) (9).
crRNA-guided surveillance complexes must distinguish ‘nonself’ (viral and plasmid DNA) from ‘self’ (chromosomal DNA) sequences that are complementary to the crRNA guide. Type I and Type II CRISPR systems identify ‘nonself’ targets through recognition of a protospacer adjacent motif (PAM) (10–17). In contrast, Type III complexes prevent self targeting by detecting complementary base pairing that extends beyond the crRNA guide and into the 5′-handle (7,18,19). Foreign DNA recognition results in conformational changes in the CRISPR surveillance machinery that activate cis- or trans-acting nucleases for target destruction.
Escherichia coli K12 contains a Type I-E system that consists of eight cas genes and a downstream CRISPR locus (Figure 1A). Five of these cas genes encode proteins that assemble (Cse11, Cse22, Cas76, Cas5e1 and Cas6e1) with a 61-nt crRNA into a large seahorse-shaped complex called Cascade (CRISPR-associated complex for antiviral defense) (Figure 1) (10,20,21). Recently, three x-ray crystal structures and an additional cryo-EM reconstruction of Cascade were determined at different stages of DNA target surveillance (22–25). The crystal structures explain how six Cas7 proteins assemble into a helical backbone with the 32-nt crRNA guide sequence presented in short helical segments, while head and tail proteins interact with conserved regions of the 3′ and 5′ repeat sequences, positioned at opposite ends of the complex (22–24). The unbound and ssDNA-bound crystal structures adopt different conformational states, and the cryo-EM reconstruction offers new insights into the mechanism of binding double-stranded DNA (dsDNA), which is the physiologically relevant target. Collectively, these structures offer snapshots of Cascade at different stages of DNA surveillance, however the contemporaneous nature of these publications has precluded a comparison of these structures.
Figure 1.
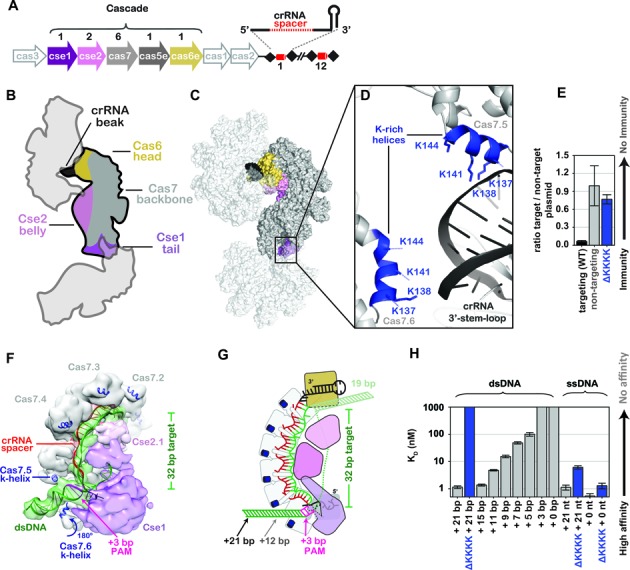
A lysine-rich vise is critical for binding double-stranded DNA (dsDNA). (A) The Clustered Regularly Interspaced Short Palindromic Repeat (CRISPR)-mediated adaptive immune system in Escherichia coli (Type I-E) consists of eight cas genes (arrows) and a CRISPR locus (black diamonds and red cylinders). Five of the cas genes (colored arrows) encode proteins that assemble with a 61-nt crRNA to from the seahorse-shaped complex composed of an unequal number of five different Cas proteins. The stoichiometry of Cas proteins in Cascade is indicated above the arrows. (B) Cartoon schematic of how three symmetry-related Cascade complexes pack together in the crystal in a head-to-tail and head-to-belly arrangement. Subunits are colored according to the scheme used in panel A. (C) Surface rendition of symmetry-related Cascade complexes. (D) The interface between the 3′ stem-loop of one Cascade assembly with lysine-rich (K-rich) helices of another. Positively charged K-rich helices are indicated in blue. (E) Plasmid curing results. The ΔKKKK mutation results in immunodeficiency. (F) Model of Cascade bound to dsDNA docked into the cryo-EM density EMDB 5929. The K-helices of Cas7.5 and Cas7.6 are indicated. (G) Schematic of Cascade-binding dsDNA. The protospacer adjacent motif and number of base pairs required to reach through the K-helices (+12) are indicated. (H) Equilibrium dissociation constants of Cascade and ΔKKKK Cascade with various dsDNA and ssDNA substrates containing 3′-ends with variable lengths.
Here we present a 3.2 Å x-ray crystal structure of Cascade in a new crystal form, which reveals interactions between protein and nucleic acid subunits. This structure, combined with insights from recent x-ray and cryo-EM structures of Cascade, offer a mechanistic explanation for target-induced conformational rearrangements and reveals new functions for specific residues involved in target recognition. These structure-guided insights are used to test function using a combination of in vivo and in vitro assays. Collectively, the data presented here support a DNA targeting mechanism where positively charged residues in the backbone make nonsequence-specific interactions with dsDNA substrates that are necessary to initiate RNA-guided strand invasion. Conserved residues along the belly stabilize directional hybridization with the crRNA, driving the conformational rearrangements that lock the complex on the DNA target and recruit the Cas3 nuclease for target destruction.
MATERIALS AND METHODS
Cascade expression and purification
Protein expression and purification was performed using previously described methods (10,21). Briefly, cas genes and CRISPR RNAs were coexpressed in E. coli Bl21 (DE3) cells using three or four different expression vectors, where either Cse2 or Cas7 is fused to an N-terminal Strep tag (Supplementary Table S1). Cells were grown in LB media under antibiotic selection and induced at an OD600nm of 0.5 using 0.2 mM isopropyl-β-D-1-thiogalactopyranoside (IPTG). Cells were cultured overnight at 16°C, pelleted by centrifugation (5000 g for 10 min), suspended in lysis buffer (100 mM Tris–HCl pH 8.0, 150 mM NaCl, 1 mM Ethylenediaminetetraaceticacid (EDTA), 1 mM tris(2-carboxyethyl)phosphine (TCEP) and 5% glycerol), and frozen at −80°C. Cells were lysed by sonication and lysates were clarified by centrifugation (22 000 g for 30 min). Cascade self-assembles in vivo and the complex was affinity purified on StrepTrap HP resin (GE) using N-terminal Strep-II tags on either Cse2 or Cas7. Elution of Cascade was performed using the lysis buffer supplemented with 2.5 mM desthiobiotin. The Strep-II tag was removed with HRV-3C protease, followed by a second purification using StrepTrap HP resin. Cascade was concentrated prior to gel filtration chromatography using either a 10/300 Superose6 column (GE Healthcare) equilibrated with 50 mM Tris–HCl pH 7.5, 100mM NaCl, 1 mM TCEP and 5% glycerol, or a 26/60 Superdex 200 (GE Healthcare) equilibrated with 50 mM Tris–HCl pH 7.5.
Cascade crystallization and structure determination
Cascade crystals were grown using hanging-drop vapor diffusion at 4, 12, 18 and 24°C from equal volumes (2 + 2 μl) of concentrated Cascade protein (A280 = 30–45) and mother liquor (0.1 M HEPES pH 7.0, 0–0.1 M KCl and 8–14% (w/v) PEG 8000). The crystal used in this study grew in the presence of an 11-nt ssDNA containing a 5′-CTT-3′ PAM and 8-nt (5′-AATACCGT-3′) complementary to the crRNA-guide sequence in a 2:1 oligonucleotide:protein ratio. However, the target DNA is not observed in the electron density. Hanging-drop experiments were carried out in EasyXtal 15-Well DG-Tool plates (Qiagen). Crystals generally appear after 3 weeks of incubation. Crystals were harvested and cryoprotected in mother liquor supplemented with 20% (v/v) PEG 400 before flash cooling in liquid nitrogen. Diffraction data were collected on beamline 23-ID at the Advanced Photon Source at the Argonne National Lab (Supplementary Table S2). Data processing was carried out using XDS and Aimless (26,27). A previously determined structure of Cascade was used as an initial search ensemble to determine the structure by molecular replacement using Phaser (28). Model building was performed using COOT (29), the model was refined using Phenix.refine (30), and validated using Molprobity (Supplementary Table S2) (31).
Structural analysis and graphical rendering
Structures were analyzed and figures were rendered using PyMol or Chimera (32,33). Movies were made using Chimera. Molecular dynamics simulations were performed using Cascade structures (PDB ID: 4TVX, 4QYZ and EMDB ID: 5929) (22,23,25). Two different molecular dynamic simulations we preformed. In the first simulation we used atomic coordinates from the unbound structure of Cascade (4TVX), whereas the second simulation used atomic coordinates from Cascade (4QYZ) bound to ssDNA. However, parts of the ssDNA bound structure are disordered and not present in the x-ray density. To create a model based on the ssDNA-bound structure, the unbound (4TVX) and ssDNA bound (4QYZ) structures of Cascade were superimposed using the Cas7 backbone (RMSD 0.71 Å). The aligned unbound structure was used to model amino acid positions not observed in the ssDNA-bound structure. For both simulations, the models were refined using Molecular Dynamics Flexible Fitting (MDFF) by generating force potentials from a simulated 5-Å resolution map of the unbound Cascade structure or the ssDNA-bound Cascade structure, while simultaneously enforcing harmonic restraints to maintain secondary structure (34,35). This simulation ran for 100 picoseconds (ps) followed by a 2-ps energy minimization in Nanoscale Molecular Dynamics (36). This atomic model was then fit into the ∼9 Å resolution cryo-EM map of Cascade bound to dsDNA (25), and B-form DNA was modeled into density at the tail of the complex using Chimera. The 9 Å resolution cryo-EM density accommodates two distinct starting models, each positioning the globular domain of Cse1 (Cas8) in a different conformational state. These differences are derived from whether we used the ssDNA bound or unbound x-ray crystal structure as the basis to model the conformational state of the Cse1 subunit. Atomic coordinates of these models were refined using MDFF for 150 ps, followed by a 2-ps energy minimization. All simulations were performed at 300°K with 2-femtosecond time steps. Correlation coefficients were determined using Chimera's Fit in Map function.
Electrophoretic mobility shift assay (EMSA)
Oligonucleotides (Operon) listed in Supplementary Table S3 were 5′-end labeled with γ32P-ATP (PerkinElmer) using T4 polynucleotide kinase (NEB). Labeled oligonucleotides were purified by phenol/chloroform extraction followed by MicroSpin G-25 columns (GE Healthcare). dsDNA was prepared by mixing labeled oligonucleotides with more than five-fold molar excess of the complementary oligonucleotide, in hybridization buffer (20 mM HEPES pH 7.5, 75 mM NaCl, 2mM EDTA, 10% glycerol and 0.01% bromophenol blue). The mixture was incubated at 95°C for 5 min, and gradually cooled to 25°C in a thermocycler. Oligonucleotide duplexes were gel purified, ethanol precipitated and recovered in hybridization buffer.
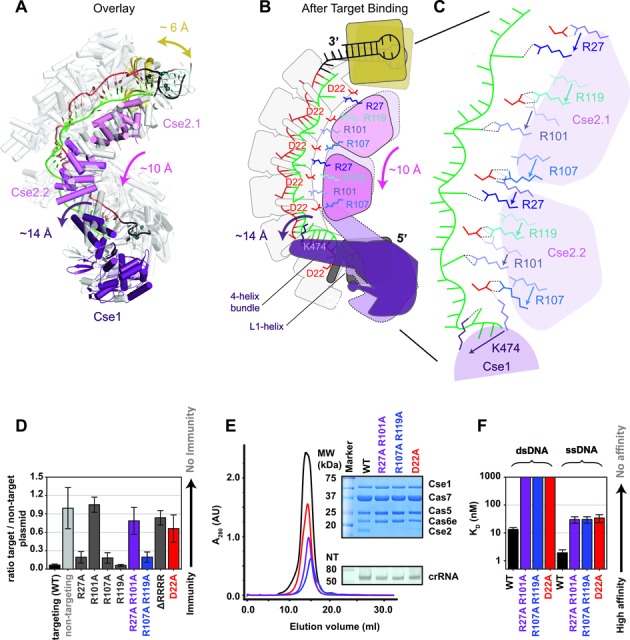
Increasing concentrations of Cascade were incubated with oligonucleotides in hybridization buffer plus 1 mM TCEP. Samples were incubated for 15 min at 37°C, loaded onto a 6% native polyacrylamide gel and run for 3 h at 150 volts at 4°C. Gels were dried, exposed to phosphor storage screens and scanned with a Typhoon (GE Healthcare) phosphorimager. Bound and unbound DNA fractions were quantified using ImageQuant software (GE Healthcare) or GelQuant.NET software (biochemlabsolutions.com). After background subtraction, the fractions of bound oligonucleotides were plotted against total Cascade concentration. The data were fit by nonlinear regression analysis using the equation:
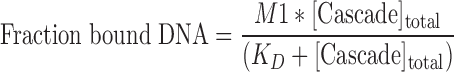 |
Where M1 is the amplitude of the binding curve. Reported KDs are the average of three independent experiments and error bars represent standard deviations.
Plasmid curing assays
Plasmid curing assays were preformed according to previously described methods (37). In brief, E. coli BL21 (AI) cells were transformed with pWUR547 (CRISPR 7xP7), pWUR397 (Cas3) and pWUR400 (Cse1, Cse2, Cas7, Cas5e and Cas6e) plasmids. These cells were made electrocompetent using standard methods and transformed with an equal molar mixture of pUC19 (nontarget) and pUC19-P7-CAT (target) containing a 350-bp insert of the phage P7 genome flanked by a 5′-CAT-3′ PAM (Supplementary Table S1). Cells used for a negative control (immunodefective) were identical except pWUR547 (CRISPR 7xP7) was replaced with a nontargeting CRISPR (pWUR630), which targets a protospacer that is not present on either pUC19 or pUC19-P7-CAT plasmids. After transformation the cells were grown for 2 h at 37°C in 1 ml of LB-media supplemented with 100 μg/ml ampicillin, 0.2% L-arabinose and 0.2 mM IPTG. Cells were plated on LB-agar plates supplemented with 100 μg/ml ampicillin, 0.2% L-arabinose and 0.2 mM IPTG at 37°C overnight. The next day, 40 colonies were randomly screened for the presence of pUC19 or pUC-P7-CAT by colony PCR using forward (5′-CAGTGAGCGCAACGCAATTA) and reverse (5′-AACTATGCGGCATCAGAGCA) primers that amplify a 454-bp region of pUC19 or a 768-bp region in pUC-P7-CAT. PCR reactions were separated by agarose gel electrophoresis and stained with sybr safe (Supplementary Figure S1). The error bars represent standard error of the mean calculated from three independent experiments. Mutants of Cascade were made by site directed mutagenesis using the pWUR400 plasmid as a template. Primers for mutagenesis are listed in Supplementary Table S4.
RESULTS
A lysine-rich vise is essential for dsDNA binding and unwinding
We determined a 3.2 Å x-ray structure of Cascade in a new crystal form using molecular replacement methods (Figure 1 and Supplementary Table S2). The asymmetric unit contains two structurally similar seahorse-shaped Cascade assemblies that superimpose on two previously determined structures of Cascade with a root-mean-square deviation of <1.1 Å for equivalently positioned Cα atoms (Supplementary Figure S2 and Supplementary Table S2) (22,24). However, the arrangement of Cascade molecules in the new crystal form provides new insights into the mechanism of dsDNA binding. Symmetrically related Cascade complexes are packed in a head-to-tail and head-to-belly arrangement (Figure 1B and C), with the 3′-stem-loop of one Cascade placed between two positively charged lysine-rich helices (K137, K138, K141 and K144) on Cas7.5 and Cas7.6 subunits. The backbone of Cascade is composed of six Cas7 subunits that share a ‘right-hand’ morphology consisting of fingers, palm and a thumb (Figure 1). The lysine-rich helix (K-rich helix) is located on the ‘pinky’ of the fingers domain of Cas7.1 to Cas7.5, but the fingers domain of Cas7.6 is rotated ∼180 degrees. Previously we speculated that the lysine-rich helix on Cas7.5 and Cas7.6 may play a role in stabilizing dsDNA during target recognition and our new crystal structure reveals nonsequence-specific interactions between the lysines and the negatively charged phosphate backbone (Figure 1D) (22). To test the importance of these lysines in Cascade-mediated immunity, we made a quadruple mutant of Cas7 (K137A, K138A, K141A and K144A) denoted ΔKKKK, and tested the mutant using an in vivo plasmid curing assay. Plasmid curing assays indicate that the ΔKKKK mutation results in severe immunodeficiency (Figure 1E).
To model potential interactions between the K-rich helices and dsDNA, we docked the atomic coordinates of Cascade and B-form dsDNA into the 9 Å cryo-EM reconstruction of Cascade and then used MDFF to fit the atomic model into the EM density while maintaining bond angles and distances (34,35,38). This model indicates that the 3′-end of a bound dsDNA target is positioned between the K-rich helices of Cas7.5 and Cas7.6. We hypothesized that the K-rich helices of Cas7.5 and Cas7.6 function as a molecular vise grip that binds the phosphate backbone of dsDNA and positions the target for PAM recognition and crRNA-guided strand invasion (Figure 1G and Supplementary Movie S1). To test this hypothesis, we purified ΔKKKK Cascade (Supplementary Figure S3A), and performed electrophoretic mobility shift assays (EMSAs) using dsDNA containing a protospacer and a 5′-CAT-3′ PAM. Wild type Cascade bound to a 72-bp dsDNA target containing a protospacer and PAM with high affinity (KD = 1.14 nM), while the ΔKKKK mutation resulted in a severe binding defect (KD > 1000 nM) (Figure 1H and Supplementary Figure S4B). In contrast to dsDNA targets, the ΔKKKK mutation results in modest binding defects for ssDNA targets (Figure 1H). These biochemical data indicate the immunodeficiency of the ΔKKKK mutant is a result of a dsDNA-binding defect.
Structural models of Cascade bound to dsDNA suggest that at least 12 additional bps on the 3′-side of the protospacer sequence are required to extend through the K-rich helices. Accordingly, the model predicts that 3′-truncations of the dsDNA will disrupt high-affinity binding to Cascade. To test this model, we performed EMSAs using a series of dsDNA substrates with identical protospacers, but incrementally shorter 3′-ends. We observed a direct correlation between the length of the 3′-end and binding affinity. dsDNA substrates with 3′-ends that extend 8 bps beyond the PAM bind with high affinity (Kd < 5 nM), while substrates with shorter 3′-ends display significant binding defects (Figure 1H and Supplementary Figure S4B). Binding affinities directly correlated with the length of the 3′-end and dsDNA substrates that do not reach the lysine vise (+3 and +0) result in binding affinities comparable to nontarget DNA (KD > 1000 nM). However, these binding defects are primarily limited to dsDNA substrates. The length of the 3′-end had no significant impact on binding affinities for short ssDNA substrates, though longer ssDNA did bind with lower affinity to the ΔKKKK mutant (Figure 1H and Supplementary Figure S4C). Together, these results suggest the K-rich vise positions dsDNA for subsequent PAM scanning and duplex unwinding.
PAM recognition and strand invasion
Cascade distinguishes self from nonself sequences through recognition of a trinucleotide PAM sequence located adjacent to the protospacer (12,39). Previous structural, biochemical and genetic work suggested that residues 125 to 131 of Cse1 form a loop (L1) that participates in PAM recognition (40). The crystal structure of Cascade reveals that L1 includes a short α-helix that extends through a pore formed by Cas5e (22,24), and that the L1-helix interacts with nucleobases in the 5′-handle of the crRNA (Figure 2A) (25,40). However, DNA binding induces a conformational change in Cascade, and L1 is disordered in the ssDNA bound structure of Cascade (23). This suggests that conformational changes induced by DNA binding may extract the L1-helix from the Cas5e pore. To clarify the functional role of the L1-helix, we compared unbound structures of Cascade to atomic models of Cascade-bound dsDNA that we generated using MDFF (Figure 2A-B). MDFF models generated using atomic coordinates from the ssDNA bound structure indicate that L1 moves out of the Cas5e pore and repositions itself next to the PAM. However, a similar simulation performed using atomic coordinates from the unbound structure of Cascade reveals only modest changes in the position of residues in the L1-helix (Supplementary Figure S5). The different position of L1 between these models reflects bias introduced by the position of L1 in the starting model. Regardless of the starting model, both simulations indicate that two residues in the L1-helix (T125 and N126) become more accessible for PAM interactions. Furthermore, both models indicate that a β-hairpin on Cse1 (residues 343 to 366) positions itself between the complementary and displaced strands of the protospacer.
Figure 2.
The L1-helix and a long β-hairpin in the Cse1 subunit are involved in protospacer adjacent motif (PAM) recognition and duplex destabilization. (A) In the structure of Cascade prior to DNA binding, L1 sits inside the Cas5e pore. F129, V130 and N131 make contact with nucleobases in the 5′-handle of the crRNA. T125 and N126 are solvent accessible. (B) Molecular Dynamic Flexible Fitting was used to model atomic coordinates of Cascade prior to target binding, into the cryo-EM density of Cascade bound to a dsDNA target with a PAM. The model is colored according to changes in distance relative to the PAM. Motion toward the PAM is colored orange and motion away from the PAM is blue. In the simulation, the L1-helix is positioned proximal to the PAM and the β-hairpin is positioned between single-strand regions of the DNA target. (C) Mutations in the L1-helix (T125N/N126K) or the β-hairpin (R351G/N353P/A355S/S356R) result in immunodeficiency. (D) Elution profile of the L1-helix and β-hairpin mutants. The insert shows a Commassie blue-stained SDS-PAGE gel (top) and a denaturing polyacrylamide gel of phenol-extracted crRNA isolated from each of the Cascade complexes (bottom). (E) Equilibrium dissociation constants of Cascade, the L1-helix and β-hairpin mutants for dsDNA substrates containing either 5′-CAT-3′ or 5′-AAT-3′ PAM and for ssDNA substrates containing a 5′-CAT-3′ PAM.
To test the importance of the L1-helix and the β-hairpin in dsDNA target recognition, we mutated L1 and the β-hairpin of Cse1 to mimic the Cse1 sequence from Streptococcus thermophilus (Supplementary Figure S6), which recognizes a 5′-AAT-3′ PAM rather than a 5′-CAT-3′ PAM recognized by E. coli Cascade (41). Mutations in L1 were restricted to solvent accessible residues (i.e., T125N and N126K) (Figure 2A-B). The β-hairpin is surface exposed in both MDFF models and six residues (343–366) were replaced with the corresponding amino acids from the S. thermophilus Cse1 sequence (Supplementary Figure S6). Mutations in either the L1-helix or the β-hairpin result in immunodeficiencies comparable to controls performed using a nontargeting CRISPR (Figure 2C). To determine if the immunodeficiencies were due to assembly defects, we purified Cascade complexes containing either the L1-helix or the β-hairpin mutations. The L1-helix and β-hairpin mutants express and purify like wild type, suggesting that these mutations do not perturb assembly of the complex (Figure 2D). Next, we used the purified complexes to perform EMSAs to determine if these mutations perturb DNA binding. The L1-helix and β-hairpin mutations result in only minor binding defects for ssDNA, but these same mutants result in significant binding defects for dsDNA substrates that contain a 5′-CAT-3′ or a 5′-AAT-3′ PAM, and a protospacer (Figure 2E and Supplementary Figure S7). These results indicate that residues in the L1-helix or the β-hairpin are not necessary for recognition of ssDNA targets and that the L1-helix or the β-hairpin are not individually responsible for PAM recognition, but they are critical for binding dsDNA.
An arginine relay facilitates conformational rearrangements during target binding
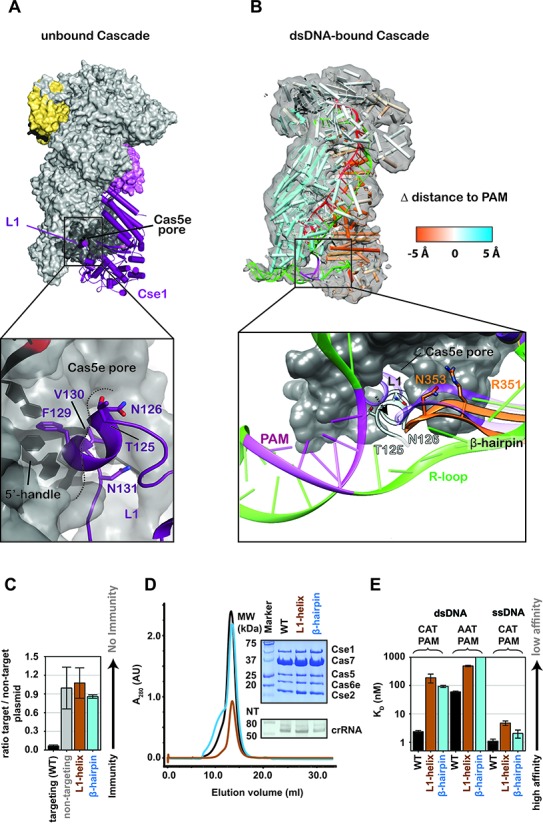
Comparison of unbound and DNA-bound x-ray structures reveals that the helical Cas7 backbone of Cascade is a rigid structure that does not move upon target binding (Figure 3) (22–24). In fact, the hexameric Cas7 backbones from these two structures superimpose with an RMSD of 0.71 Å over all equivalently positioned Cα atoms. In contrast to the structurally rigid Cas7 backbone, target binding triggers a dramatic conformational rearrangement in the belly (Cse2) and tail (Cse1) subunits. The two Cse2 subunits slide ∼10 Å toward the tail upon target binding, and the four-helix bundle of Cse1 tilts ∼14 Å closer to the bound ssDNA (Figure 3A and Supplementary Movie S1). To understand the functional implications of this conformational rearrangement, we examined interactions between the Cas7 and Cse2 subunits in the unbound and ssDNA-bound crystal structures. In the unbound state, two conserved arginines (R27 and R101) on each Cse2 subunit form salt bridges with conserved aspartic acid residues (D22) displayed on the Cas7 subunits. Target binding induces a conformational change that moves the Cse2 subunits down the Cas7 backbone. The R27 and R101 residues form new contacts with kinked-out bases on the bound DNA target, and compensatory salt bridges are formed between additional arginines on Cse2 (R107 and R119) and D22 residues further down the backbone (Figure 3B-C).
Figure 3.
Conserved arginines on the Cse2 subunits participate in a relay that stabilizes target binding induced conformational changes. (A) Overlay of Cascade crystal structures before (gray) and after (colored) target binding. (B-C) Schematic of the conformational change induced by target binding. The two Cse2 subunits move ∼10-Å down the backbone of Cascade. Salt bridges between the belly (Cse2 R27 and R101) and the backbone (Cas7 D22) are broken. R27 and R101 are repositioned to stabilize the flipped-out bases on the DNA target strand and compensatory salt bridges are formed by Cse2 R107 and R119 with D22 residues on Cas7 subunits further down the backbone. (D) Plasmid-curing assays reveal that R101A mutations result in strong immune system defects, while individual mutation of the other Cse2 arginines shows little or no measurable defect. The D22A mutation also results in immunodeficiency. (E) Elution profile of the WT Cascade and different mutants. The insert shows a Commassie blue-stained SDS-PAGE gel (top) and a denaturing polyacrylamide gel of phenol extracted crRNA isolated for the Cascade complexes (bottom). The Cse2 R27A/R101A, Cse2 R107A/R119A and Cas7 D22A mutants lack Cse2. (F) Equilibrium dissociation constants of Cascade and Cascade mutants for a 72-bp dsDNA target and a 72-nt ssDNA target containing a 5′-CAT-3′ PAM.
This ‘molecular relay’ of arginines anchors the Cse2 subunits to the Cascade complex during target binding, and results in a net gain of four ionic interactions with the kinked-out bases. We hypothesized that newly formed interactions between flipped-out bases and the arginines on Cse2 would contribute to the stability of the target-bound conformational state. To test this hypothesis we performed EMSAs using dsDNA and ssDNA targets that are missing a nucleobase at every 6th position of the target-strand protospacer (Supplementary Table S3). We observed a five-fold binding defect in abasic ssDNA substrates (Supplementary Figure S8), suggesting that interactions between the Cse2 arginines R27 and R101, and kinked-out bases contribute to target binding. However, no significant difference was observed for binding abasic dsDNA substrates, which may not be surprising given that the relatively modest contribution gained by stabilizing the flipped-out bases may be masked in dsDNA substrates where abasic substitutions compensate for the binding defect observed in ssDNA by destabilizing the dsDNA duplex.
Salt bridges between D22 residues on Cas7 and conserved arginines on Cse2 (R27, R101, R107 and R119), appear to be critical for anchoring the Cse2 subunits to the Cas7 backbone (Figure 3 B-C). To determine the importance of the salt bridges in Cascade-mediated immunity we made point mutants of the salt-bridging arginines on Cse2 (R27A, R101A, R107A and R119A), and performed in vivo plasmid-curing assays (Figure 3D). Individual arginine mutations of R27A, R107A and R119A had no detectable effect on immunity, while R101A resulted in an immunodeficiency. Next, we made double mutants of the arginine residues that participate in salt bridging before (R27A/R101A) or after (R107A/R119A) target binding. The double-mutant R27A/R101A was immunocompromised, while R107A/R119A showed no significant immune defect, indicating that salt bridges formed before target binding are critical for immunity while compensatory interactions made after target binding are not essential in an overexpression system. Consistent with these results, the quadruple arginine mutant (R27A, R101A, R107A and R119A), denoted ΔRRRR, resulted in an immune defect comparable to R101A or the double R27A/R101A mutant. To confirm that the immune defect was a result of disrupting the observed salt bridges we mutated the aspartic acid residue (D22A) on Cas7. The D22A mutation resulted in a plasmid-curing defect similar to R101A, R27A/R101A and ΔRRRR, suggesting that salt-bridge formation between Cse2 R101 and Cas7 D22 is important for immunity.
To determine if the immunodeficiencies observed for the Cse2 double mutants R27A/R101A and R107A/R119A and the Cas7 D22A mutant are a consequence of perturbing Cascade assembly, target binding or Cas3 recruitment, we expressed and purified mutant Cascade complexes using a strep-tag on the N-terminus of Cas7. These mutations all result in Cascade complexes lacking the Cse2 subunits (Figure 3E). A faint Cse2 band is observed in D22A Cascade preparations. One explanation for this observation is that Cas7 has a second aspartic acid residue at position 21 that may partially compensate for the D22A mutations during purification. To determine the consequence of Cse2 destabilization on target binding we performed EMSAs using ssDNA and dsDNA targets. Cascade lacking Cse2 is unable to bind dsDNA tightly and has a 15-fold decrease in affinity for ssDNA targets (Figure 3F and Supplementary Figure S9). These data indicate that Cse2 facilitates binding to ssDNA, and that Cse2 is critical for efficient binding to dsDNA substrates (17,21,42,43).
DISCUSSION
Cascade is a crRNA-guided surveillance complex that efficiently binds foreign DNA and recruits a trans-acting nuclease for target degradation (41,44–46). Here we determined the x-ray structure of Cascade in a new crystal form that reveals nonsequence-specific interactions between K-rich helices located on the Cas7.5 and Cas7.6 backbone subunits and the negatively charged crRNA from a symmetry-related molecule (Figure 1D). Collectively, we show that the positive charge on the K-rich helix is necessary for dsDNA, but not ssDNA binding, and that the K-rich vise plays a fundamental role in dsDNA binding that is equivalent to the importance of the protospacer and the PAM. dsDNA substrates that contain a PAM and a protospacer, but do not have 3′-ends long enough to reach the K-rich vise are bound with affinities similar to dsDNAs lacking a PAM or protospacer (Figure 1H) (10–12,40). Our structural models indicate that 12 bps of duplex DNA stretch from the 3′-end of the protospacer through the K-rich vise, and we show that substrate with 3′-ends that extend across the K-rich vise are bound with high affinity. This distance is similar to the 9 bps protected by Cascade in nuclease protection assays (10).
PAM recognition by Cascade is promiscuous, and different PAMs elicit distinct immune responses (39). Recognition of a protospacer flanked by one of the five different ‘interference’ PAMs elicits a response that involves the recruitment of Cas3 for degradation of the DNA target (39,47). However, protospacers flanked by one of 22 different ‘priming’ PAMs are still recognized by Cascade, but rather than eliciting Cas3 to degrade the target, these interactions appear to elicit a ‘priming’ response that promotes the rapid acquisition of adjacent protospacers (25,39,48). While the mechanism of PAM sensing remains unknown, we propose that PAM-dependent and PAM-priming modes of protospacer recognition will require the K-rich vise on Cas7.5 and Cas7.6, the L1-helix on Cse1, the β-hairpin on Cse1, and residues involved in the arginine relay that reposition the two Cse2 subunits during target binding (Figure 4). Residues in the L1-helix on Cse1 have previously been implicated in PAM detection (15,25), but structures of Cascade prior to engaging a DNA target suggested that these residues (F129, V130 and N131) are buried and unavailable for PAM interactions. However, target binding introduces a conformational change that may liberate the L1-helix for participation in PAM scanning. Our structural models and biochemical studies also identified a β-hairpin (residues 343–366) that is important for dsDNA target binding and may be involved in PAM sensing. Our attempts to change PAM detection from 5′-CAT-3′ to 5′-AAT-3′ by simply swapping solvent-accessible residues in the L1-helix, or β-hairpin from E. coli for residues found at equivalent positions in the Cse1 subunit from S. thermophilus failed. We think that this failure reflects our incomplete understanding of PAM recognition and we anticipate that higher resolution structures of Cascade bound to a dsDNA target containing a PAM will provide more insight into the mechanism of PAM sensing. While the resolution of our models precludes an atomic-resolution understanding of PAM recognition, we anticipate that PAM detection momentarily stalls Cascade, and that protracted lifetimes at PAM motifs may explain the proficiency of recognizing protospacers flanked by PAMs. This model is supported by recent evidence showing that the Csy complex (Type 1-F) preferentially interacts with dsDNA substrates containing PAMs, and that Cas9, a Type II-A crRNA-guided surveillance complex, preferentially samples PAM-rich regions of the lambda phage genome (14,16).
Figure 4.
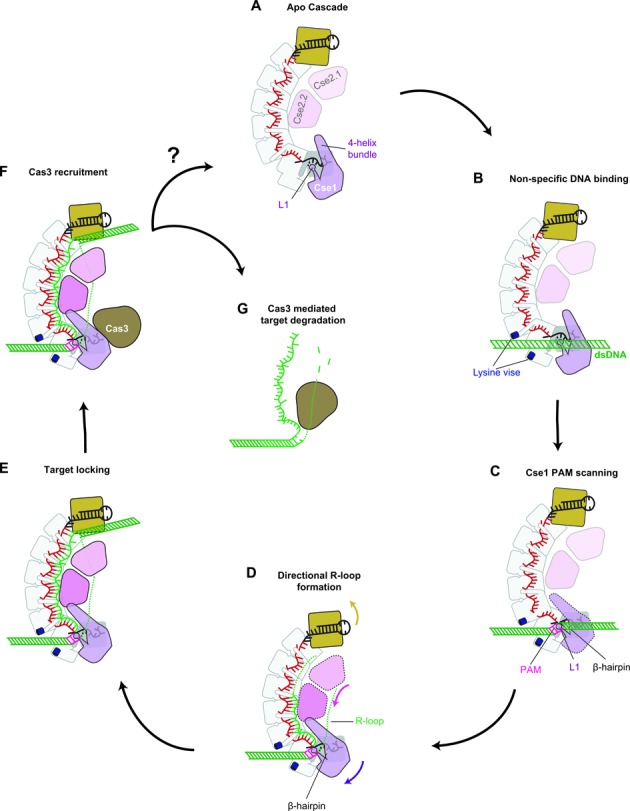
DNA target recognition and binding by Cascade. (A - B) A lysine-rich vise makes nonsequence-specific interactions with dsDNA. (C) DNA binding induces a conformational change and the L1-helix and β-hairpin are positioned for PAM scanning. (D) Directional hybridization between the DNA target and complementary crRNA drives an arginine relay along the belly subunits, in which arginines interact with kinked-out bases from the bound target strand and conserved aspartates on the Cas7 backbone. (E) The conformational rearrangement locks Cascade on the target and signal recruitment of the Cas3 helicase-nuclease. (F-G) Cas3 is recruited to the Cascade-DNA complex and the DNA is degraded by Cas3. Degradation of the DNA may release Cascade for another round of target detection, however turnover of Cascade has not been demonstrated.
PAM recognition initiates directional hybridization between the dsDNA target and complementary crRNA, displacing the noncomplementary strand of the DNA target and creating an R-loop that is predicted to be stabilized by Cse1 and Cse2 subunits (17,43). Complete R-loop formation results in a locking mechanism that increases the grip of Cascade on DNA substrates and Rutkauskas et al. demonstrated that the locking mechanism is required for Cas3 recruitment to bound targets (43). The structural analysis presented here reveals a series of conserved arginine resides on the Cse2 subunits that may contribute to locking by stabilizing flipped-out bases on the target strand and forming salt bridges with conserved acidic residues (i.e., D22) displayed on the Cas7 backbone (Supplementary Figure S10). Interestingly, the D22 residue is also strictly conserved in the Cas7 proteins of the Type III Cmr and Csm complexes, where it is required for endonucleolytic cleavage of RNA substrates (Supplementary Figure S10) (22). Although Cascade has no detectable RNase activity (data not shown), we show that D22 still plays a critical role in Cascade assembly and function. It is likely that this strictly conserved residue is derived from a common ancestor of Type I and Type III systems, but its function has evolved to match the needs of these diverse surveillance systems.
ACCESSION NUMBER
Atomic coordinates have been deposited into the Protein Data Bank with accession code 5CD4.
Supplementary Material
Acknowledgments
We thank Stan J.J. Brouns for providing plasmids used in this study and Brandon Smart and Maya Tsidulko for their help with statistical analysis. We are grateful to the members of the Wiedenheft lab for technical assistance and helpful discussions. X-ray diffraction data were collected using beamline 23-ID-B at the Advance Photon Source and beamline 12–2 at the Stanford Synchrotron Radiation Lightsource.
Author contributions: P.B.G.v.E., R.N.J., J.C. and B.W. designed the research; P.B.G.v.E., R.N.J., J.C., S.M.G. performed experiments; P.B.G.v.E., R.N.J., J.C., S.M.G., S.B. and B.W. analyzed the data and P.B.G.v.E., R.N.J. and B.W. wrote the paper.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
MSU's Graduate School and the Office of the Vice President for Research and Economic Development (to P.B.G.v.E.); Ruth L. Kirschstein National Research Service Award from the National Institutes of Health [F32 GM108436 to R.N.J.]; Undergraduate research from the Howard Hughes Medical Institute [#52006931 to J.C.], Montana IDeA Network of Biomedical Research Excellence from the National Institutes of Health [P20GM103474 to J.C.], Irving L. Weissman Undergraduate research (to J.C.); National Institutes of Health [R01GM097330 to S.B.]; National Institutes of Health [P20GM103500, R01GM108888 to B.W.], National Science Foundation EPSCoR [EPS-110134 to B.W.], M.J. Murdock Charitable Trust (to B.W.); Montana State University Agricultural Experimental Station; Advance Photon Source [ACB-12002, AGM-12006, DE-AC02-06CH11357]; Stanford Synchrotron Radiation Lightsource [DE-AC02-76SF00515, P41GM103393].
Conflict of interest statement. None declared.
REFERENCES
- 1.Barrangou R., Marraffini L.A. CRISPR-Cas Systems: prokaryotes upgrade to adaptive immunity. Mol. Cell. 2014;54:234–244. doi: 10.1016/j.molcel.2014.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jiang F., Doudna J.A. The structural biology of CRISPR-Cas systems. Curr. Opin. Struct. Biol. 2015;30:100–111. doi: 10.1016/j.sbi.2015.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Reeks J., Naismith J.H., White M.F. CRISPR interference: a structural perspective. Biochem. J. 2013;453:155–166. doi: 10.1042/BJ20130316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sorek R., Lawrence C.M., Wiedenheft B. CRISPR-mediated adaptive immune systems in bacteria and archaea. Annu. Rev. Biochem. 2013;82:237–266. doi: 10.1146/annurev-biochem-072911-172315. [DOI] [PubMed] [Google Scholar]
- 5.van der Oost J., Westra E.R., Jackson R.N., Wiedenheft B. Unravelling the structural and mechanistic basis of CRISPR-Cas systems. Nat. Rev. Microbiol. 2014;12:479–492. doi: 10.1038/nrmicro3279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gasiunas G., Sinkunas T., Siksnys V. Molecular mechanisms of CRISPR-mediated microbial immunity. Cell. Mol. Life Sci. 2014;71:449–465. doi: 10.1007/s00018-013-1438-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jackson R.N., Wiedenheft B. A conserved structural chassis for mounting versatile CRISPR RNA-guided immune responses. Mol. Cell. 2015;58:722–728. doi: 10.1016/j.molcel.2015.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tsui T.K., Li H. Structure principles of CRISPR-Cas surveillance and effector complexes. Annu. Rev. Biophys. 2015;44:255–229. doi: 10.1146/annurev-biophys-060414-033939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Makarova K.S., Haft D.H., Barrangou R., Brouns S.J., Charpentier E., Horvath P., Moineau S., Mojica F.J., Wolf Y.I., Yakunin A.F., et al. Evolution and classification of the CRISPR-Cas systems. Nat. Rev. Microbiol. 2011;9:467–477. doi: 10.1038/nrmicro2577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jore M.M., Lundgren M., van Duijn E., Bultema J.B., Westra E.R., Waghmare S.P., Wiedenheft B., Pul U., Wurm R., Wagner R., et al. Structural basis for CRISPR RNA-guided DNA recognition by Cascade. Nat. Struct. Mol. Biol. 2011;18:529–536. doi: 10.1038/nsmb.2019. [DOI] [PubMed] [Google Scholar]
- 11.Semenova E., Jore M.M., Datsenko K.A., Semenova A., Westra E.R., Wanner B., van der Oost J., Brouns S.J., Severinov K. Interference by clustered regularly interspaced short palindromic repeat (CRISPR) RNA is governed by a seed sequence. Proc. Natl. Acad. Sci. U.S.A. 2011;108:10098–10103. doi: 10.1073/pnas.1104144108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Westra E.R., Semenova E., Datsenko K.A., Jackson R.N., Wiedenheft B., Severinov K., Brouns S.J. Type I-E CRISPR-Cas systems discriminate target from non-target DNA through base pairing-independent PAM recognition. PLoS Genet. 2013;9:e1003742. doi: 10.1371/journal.pgen.1003742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Anders C., Niewoehner O., Duerst A., Jinek M. Structural basis of PAM-dependent target DNA recognition by the Cas9 endonuclease. Nature. 2014;513:569–573. doi: 10.1038/nature13579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rollins M.F., Schuman J.T., Paulus K., Bukhari H.S., Wiedenheft B. Mechanism of foreign DNA recognition by a CRISPR RNA-guided surveillance complex from Pseudomonas aeruginosa. Nucleic Acids Res. 2015;43:2216–2222. doi: 10.1093/nar/gkv094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sashital D.G., Jinek M., Doudna J.A. An RNA induced conformational change required for CRISPR RNA cleavage by the endonuclease Cse3. Nat. Struct. Mol. Biol. 2011;18:680–687. doi: 10.1038/nsmb.2043. [DOI] [PubMed] [Google Scholar]
- 16.Sternberg S.H., Redding S., Jinek M., Greene E.C., Doudna J.A. DNA interrogation by the CRISPR RNA-guided endonuclease Cas9. Nature. 2014;507:62–67. doi: 10.1038/nature13011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Szczelkun M.D., Tikhomirova M.S., Sinkunas T., Gasiunas G., Karvelis T., Pschera P., Siksnys V., Seidel R. Direct observation of R-loop formation by single RNA-guided Cas9 and Cascade effector complexes. Proc. Natl. Acad. Sci. U.S.A. 2014;111:9798–9803. doi: 10.1073/pnas.1402597111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Marraffini L.A., Sontheimer E.J. Self versus non-self discrimination during CRISPR RNA-directed immunity. Nature. 2010;463:568–571. doi: 10.1038/nature08703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Samai P., Pyenson N., Jiang W., Goldberg G.W., Hatoum-Aslan A., Marraffini L.A. Co-transcriptional DNA and RNA cleavage during type III CRISPR-Cas immunity. Cell. 2015;161:1164–1174. doi: 10.1016/j.cell.2015.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brouns S.J., Jore M.M., Lundgren M., Westra E.R., Slijkhuis R.J., Snijders A.P., Dickman M.J., Makarova K.S., Koonin E.V., van der Oost J., et al. Small CRISPR RNAs guide antiviral defense in prokaryotes. Science. 2008;321:960–964. doi: 10.1126/science.1159689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wiedenheft B., Lander G.C., Zhou K., Jore M.M., Brouns S.J.J., van der Oost J., Doudna J.A., Nogales E. Structures of the RNA-guided surveillance complex from a bacterial immune system. Nature. 2011;477:486–489. doi: 10.1038/nature10402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jackson R.N., Golden S.M., van Erp P.B., Carter J., Westra E.R., Brouns S.J., Van Der Oost J., Terwilliger T.C., Read R.J., Wiedenheft B., et al. Crystal structure of the CRISPR RNA-guided surveillance complex from Escherichia coli. Science. 2014;345:1473–1479. doi: 10.1126/science.1256328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mulepati S., Heroux A., Bailey S. Crystal structure of a CRISPR RNA-guided surveillance complex bound to a ssDNA target. Science. 2014;345:1479–1484. doi: 10.1126/science.1256996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhao H., Sheng G., Wang J., Wang M., Bunkoczi G., Gong W., Wei Z., Wang Y. Crystal structure of the RNA-guided immune surveillance Cascade complex in Escherichia coli. Nature. 2014;515:147–150. doi: 10.1038/nature13733. [DOI] [PubMed] [Google Scholar]
- 25.Hochstrasser M.L., Taylor D.W., Bhat P., Guegler C.K., Sternberg S.H., Nogales E., Doudna J.A. CasA mediates Cas3-catalyzed target degradation during CRISPR RNA-guided interference. Proc. Natl. Acad. Sci. U.S.A. 2014;111:6618–6623. doi: 10.1073/pnas.1405079111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Evans P.R., Murshudov G.N. How good are my data and what is the resolution. Acta Crystallogr. D. 2013;69:1204–1214. doi: 10.1107/S0907444913000061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kabsch W. Xds. Acta Crystallogr. D Biol. Crystallogr. 2010;66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mccoy A.J., Grosse-Kunstleve R.W., Adams P.D., Winn M.D., Storoni L.C., Read R.J. Phaser crystallographic software. J Appl. Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Emsley P., Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr. D. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 30.Afonine P.V., Grosse-Kunstleve R.W., Echols N., Headd J.J., Moriarty N.W., Mustyakimov M., Terwilliger T.C., Urzhumtsev A., Zwart P.H., Adams P.D., et al. Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. D Biol. Crystallogr. 2012;68:352–367. doi: 10.1107/S0907444912001308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen V.B., Arendall W.B. 3rd, Headd J.J., Keedy D.A., Immormino R.M., Kapral G.J., Murray L.W., Richardson J.S., Richardson D.C. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.DeLano W.L. The PyMOL Molecular Graphics System. Version 1.7.4 Schrödinger, LLC; 2002. [Google Scholar]
- 33.Goddard T.D., Huang C.C., Ferrin T.E. Software extensions to UCSF chimera for interactive visualization of large molecular assemblies. Structure. 2005;13:473–482. doi: 10.1016/j.str.2005.01.006. [DOI] [PubMed] [Google Scholar]
- 34.Chan K.Y., Trabuco L.G., Schreiner E., Schulten K. Cryo-electron microscopy modeling by the molecular dynamics flexible fitting method. Biopolymers. 2012;97:678–686. doi: 10.1002/bip.22042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Trabuco L.G., Villa E., Mitra K., Frank J., Schulten K. Flexible fitting of atomic structures into electron microscopy maps using molecular dynamics. Structure. 2008;16:673–683. doi: 10.1016/j.str.2008.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kale L., Skeel R., Bhandarkar M., Brunner R., Gursoy A., Krawetz N., Phillips J., Shinozaki A., Varadarajan K., Schulten K., et al. NAMD2: greater scalability for parallel molecular dynamics. J. Comput. Phys. 1999;151:283–312. [Google Scholar]
- 37.Almendros C., Guzman N.M., Diez-Villasenor C., Garcia-Martinez J., Mojica F.J. Target motifs affecting natural immunity by a constitutive CRISPR-Cas system in Escherichia coli. PLoS One. 2012;7:e50797. doi: 10.1371/journal.pone.0050797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Trabuco L.G., Villa E., Schreiner E., Harrison C.B., Schulten K. Molecular dynamics flexible fitting: a practical guide to combine cryo-electron microscopy and X-ray crystallography. Methods. 2009;49:174–180. doi: 10.1016/j.ymeth.2009.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fineran P.C., Gerritzen M.J., Suarez-Diez M., Kunne T., Boekhorst J., van Hijum S.A., Staals R.H., Brouns S.J. Degenerate target sites mediate rapid primed CRISPR adaptation. Proc. Natl. Acad. Sci. U.S.A. 2014;111:1629–1638. doi: 10.1073/pnas.1400071111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sashital D.G., Wiedenheft B., Doudna J.A. Mechanism of foreign DNA selection in a bacterial adaptive immune system. Mol. Cell. 2012;48:606–615. doi: 10.1016/j.molcel.2012.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sinkunas T., Gasiunas G., Waghmare S.P., Dickman M.J., Barrangou R., Horvath P., Siksnys V. In vitro reconstitution of Cascade-mediated CRISPR immunity in Streptococcus thermophilus. Embo Journal. 2013;32:385–394. doi: 10.1038/emboj.2012.352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Westra E.R., Nilges B., van Erp P.B., van der Oost J., Dame R.T., Brouns S.J. Cascade-mediated binding and bending of negatively supercoiled DNA. RNA Biol. 2012;9:1134–1138. doi: 10.4161/rna.21410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rutkauskas M., Sinkunas T., Songailiene I., Tikhomirova M.S., Siksnys V., Seidel R. Directional R-Loop formation by the CRISPR-Cas Surveillance Complex Cascade provides efficient off-target site rejection. Cell Rep. 2015;10:1534–1543. doi: 10.1016/j.celrep.2015.01.067. [DOI] [PubMed] [Google Scholar]
- 44.Westra E.R., Swarts D., Staals R., Jore M.M., Brouns S.J.J., Oost J.v.d. The CRISPRs, They Are A-Changin’: how prokaryotes generate adaptive immunity. Annu. Rev. Genet. 2012;46:311–339. doi: 10.1146/annurev-genet-110711-155447. [DOI] [PubMed] [Google Scholar]
- 45.Jackson R.N., Lavin M., Wiedenheft B. Fitting CRISPR-associated Cas3 into the Helicase Family Tree. Curr. Opin. Struct. Biol. 2014;24:106–114. doi: 10.1016/j.sbi.2014.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mulepati S., Bailey S. In vitro reconstitution of an Escherichia coli RNA-guided immune system reveals unidirectional, ATP-dependent degradation of DNA target. J. Biol. Chem. 2013;288:22184–22192. doi: 10.1074/jbc.M113.472233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Westra E.R., van Erp P.B.G., Künne T., Wong S.P., Staals R.H.J., Seegers C.L.C., Bollen S., Jore M.M., Semenova E., Severinov K., et al. CRISPR immunity relies on the consecutive binding and degradation of negatively supercoiled invader DNA by Cascade and Cas3. Mol. Cell. 2012;46:595–605. doi: 10.1016/j.molcel.2012.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Blosser T.R., Loeff L., Westra E.R., Vlot M., Kunne T., Sobota M., Dekker C., Brouns S.J., Joo C. Two distinct DNA binding modes guide dual roles of a CRISPR-Cas protein complex. Mol. Cell. 2015;58:60–70. doi: 10.1016/j.molcel.2015.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.