


Abstract
Processing and post-transcriptional regulation of RNA often depend on binding of regulatory molecules to short motifs in RNA. The effects of such interactions are difficult to study, because most regulatory molecules recognize partially degenerate RNA motifs, embedded in a sequence context specific for each RNA. Here, we describe Library Sequencing (LibSeq), an accurate massively parallel reporter method for completely characterizing the regulatory potential of thousands of short RNA sequences in a specific context. By sequencing cDNA derived from a plasmid library expressing identical reporter genes except for a degenerate 7mer subsequence in the 3′UTR, the regulatory effects of each 7mer can be determined. We show that LibSeq identifies regulatory motifs used by RNA-binding proteins and microRNAs. We furthermore apply the method to cells transfected with RNase H recruiting oligonucleotides to obtain quantitative information for >15000 potential target sequences in parallel. These comprehensive datasets provide insights into the specificity requirements of RNase H and allow a specificity measure to be calculated for each tested oligonucleotide. Moreover, we show that inclusion of chemical modifications in the central part of an RNase H recruiting oligonucleotide can increase its sequence-specificity.
INTRODUCTION
In recent years there has been an increasing awareness of the importance of post-transcriptional regulation in gene expression (1). Most genes are regulated post-transcriptionally to fine-tune expression or provide rapid responses to external stimuli. In fact, global comparisons between mRNA and protein levels in different cell types show correlation for some genes, but also reveal thousands of genes where the protein level does not reflect the mRNA level (2). RNA processing, quality checkpoints, nuclear export, and translation of mRNA molecules provide many opportunities for regulation, and are in most cases dependent on the interaction between a short stretch of the RNA polymer and a regulatory molecule (3). The same is true when potentially therapeutic molecules such as oligonucleotides and short interfering RNAs (siRNAs) are used to manipulate gene expression. Thus, for both endogenous and exogenous regulatory molecules, characterisation of the regulatory consequence of interaction with different RNA sequences is the key to understanding how targeted regulation is obtained.
There are two major strategies for investigating the effects of regulatory molecules. First, cells can be treated with the regulator and the changes in gene expression monitored. The gene sequences can then be correlated to gene expression changes to pinpoint the particular motif responsible for the regulation. One example of this strategy is in the analysis of microRNA (miRNA) regulation, where the measurement of global expression changes clearly identified complementarity to the miRNA seed sequence in the 3′-UTR as the major determinant of miRNA regulation (4). However, this type of analysis is hampered by the fact that (i) expression levels may change because of secondary effects, and (ii) in every gene a potential regulatory sequence will be in a particular sequence context, which affects the regulatory impact. An alternative strategy is to identify the direct RNA binding sites. Systematic Evolution of Ligands by EXponential Enrichment (SELEX) experiments and more recently sequencing based methods allow binding motifs of RNA-binding proteins (RNA-BPs) to be identified (5,6). This information is helpful, but neglects the effects that the cellular environment can have on binding. Information about binding in a cellular context can be obtained with the ultraviolet cross-linking and immunoprecipitation (CLIP) method, which allows in vivo binding of RNA-BPs to be identified (7). The drawback for these methods is that they detect only binding, but not the regulatory consequence of the interaction.
RNase H recruiting oligonucleotides are an example of a type of regulator for which it remains challenging to determine regulatory effects and specificity. Upon binding of the oligonucleotides to their complementary RNA target sequence, RNase H is recruited and the RNA strand is cleaved (8,9). After RNA cleavage, the resulting unprotected RNA cleavage fragments are rapidly degraded by cellular exonucleases (10). A therapeutic oligonucleotide targeting and degrading apolipoprotein B (APOB) by an RNase H dependent mechanism was recently approved for the treatment of homozygous familial hypercholesterolemia (11) and many others are in clinical development (12). Besides the intended RNA target, it is well known that RNase H recruiting oligonucleotides may also cause degradation of unintended off-targets via binding to partially complementary target sites (13,14). Presumably, off targeting depends on whether the binding affinity between oligonucleotide and RNA target region is sufficient to allow appreciable amounts of duplex to form (14), and whether RNase H tolerates structural changes induced by duplex mismatches and bulges (15). In a therapeutic context, off targeting may potentially lead to unwanted effects or toxicities (16) and should therefore be minimized when RNase H recruiting oligonucleotides are designed. However, the mechanistic principles that determine which of the many possible imperfectly matched off-target regions in the transcriptome are targeted by RNase H remains unknown.
In this study, we describe LibSeq (short for Library Sequencing), which is an accurate massive parallel-sequencing-based method for completely characterizing the regulatory potential of thousands of short RNA sequences in a invariant sequence context. These putative regulatory RNA sequences occur as 7mer motifs in the 3′UTR of a reporter mRNA. We show that LibSeq detects functional motifs for endogenous regulators such as RNA-BPs and miRNAs in HeLa cells, and identifies the regulatory impact of exogenous factors such as chemically modified DNA oligonucleotides designed to recruit RNase H. The LibSeq strategy has many potential applications. Here, we use LibSeq to investigate the sequence and chemical modification pattern required for RNase H recruiting oligonucleotides and devise a measure that can be used to compare the specificity of candidate oligonucleotides. Using this measure, we demonstrate that shortening of the central stretch of unmodified DNA nucleotides in a RNase H recruiting oligonucleotide can increase its specificity This finding has important implications for the design of RNase H recruiting oligonucleotides.
MATERIALS AND METHODS
Luciferase measurements
HeLa cells were maintained in Dulbecco DMEM growth medium with 10% Fetal Bovine serum (FBS), 1% penicillin/streptomycin (5000 units/ml penicillin and 5000 μg/ml streptomycin) and 1% l-glutamine (200 mM). The day before transfection, 70 000 HeLa cells/well were plated in 24-well culture plates. For each well, 11 ng Renilla plasmids, 209 ng perfect match 7mer clone plasmid and oligonucleotide corresponding to a final concentration of 25 nM were added to 50 μl optiMEM (Life Technologies) and transfections were performed in triplicate with Lipofectamine 2000 (Life Technologies) according to manufacturer's protocol. After transfection, cells were incubated in a CO2 incubator for 4 h, after which the growth medium was changed to medium containing antibiotics. Twenty-four hours after transfection the cells were harvested and the luciferase expression was quantified with the Dual-Luciferase Reporter Assay System (Promega). Cells were lysed with 100 μl of 1XPassiv Lysis Buffer pr. 24-well and mixed gently for 20–25 min. Fifteen microliters of the cell-lysates were then used to measure renilla and firefly luciferase fluorescence intensity using a GloMax 20/20 luminometer.
LibSeq gene expression vector library synthesis
The plasmid LibSeq library was synthesised by Eurofins-MWG, Germany, as a custom Gene Evolution library with seven degenerate positions using the Ligation Chain Reaction method (17). The linear library had the following sequence: 5′-AGATCTCATAGCTGGATGTGTGGAAAAAGTGGATATTTGAAGAAAANNNNNNNATATTTTAATACGAAGAGGACACTCCTCGAG-3′ and was cloned into the 3′ UTR of the pGL4.12-TK-MCS+ vector, which has previously been described (18) using the BglII/XhoI restriction sites. Part of the linear library sequence is identical to the region on the human chromosome 18, between positions 174684746 and 174684800 and contains a miRNA-1 target site in the human HAND2 gene.
Libseq plasmid pool sequencing
Sequencing libraries of the LibSeq plasmid pool were prepared by amplification of plasmids using Phusion High-Fidelity DNA Polymerase (New England Biolabs) following the manufacturer's recommendations, using 1 ng plasmid as template, with the forward primer 5′-AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCTNNATCTCATAGCTGGATGTGTGG-3′, NN indicating a sample specific barcode, and the reverse primer 5′-CAAGCAGAAGACGGCATACGAGATCATTCTAGTTGTGGTTTGTCCA-3′. The PCR program was 98°C, 30 s; (98°C, 10 s; 61°C, 20 s; 72°C, 20 s) x 4; (98°C, 10 s; 72°C, 20 s) x 12. The PCR product was then separated on a 0.5% agarose gel and the resulting 246 nucleotide amplicon was cut from the gel and extracted with the QIAquick Gel Extraction Kit (Qiagen) and purified using Agencourt AMPure XP beads (Beckman Coulter). Single read sequencing with read length 100 was performed on an Illumina HiSeq 2000 sequencing system.
Libseq transfection and RNA preparation
One day before transfection 1.6 × 106 HeLa cells were plated in 15 ml of Dulbecco DMEM growth medium supplemented with 10% FBS and 1% l-glutamine (200 mM), but no antibiotics, in a 10 cm petri dish so that cells were 90–95% confluent at the time of transfection. Transfection was performed using Lipofectamine 2000 (Life Technologies) as described by the manufacturer. In all transfections, 6.3 μg LibSeq library plasmid DNA was diluted in 1.5 ml of Opti-MEM I Reduced Serum Medium (Life Technologies). For oligonucleotide transfections, 450 pmol of oligonucleotide was also added per sample (final concentration 25 nM). Twenty-four hours after transfection cells were washed with PBS and harvested with 2 ml of Tri Reagent (Sigma–Aldrich). Subsequently, RNA was extracted as described by the Tri Reagent manufacturer's protocol. The purified total RNA was then enriched for mRNA using the Poly(A)Purist MAG kit (Ambion) following the kit protocol and DNase treated using Turbo DNase (Life Technologies). The manufacturer's Rigid Treatment protocol was used with the addition of an extra round of DNase treatment, followed by a further 30 min of incubation. After DNase treatment, two rounds of ethanol precipitation were performed.
cDNA and sequencing library synthesis
cDNA was synthesized with the SuperScript III First-Strand Synthesis System (Life Technologies), using 0.5 μg enriched mRNA as input. A LibSeq mRNA specific primer containing an overhang with Illumina flowcell adapter binding sequence (5′-CAAGCAGAAGACGGCATACGAGATCATTCTAGTTGTGGTTTGTCCA-3′) was used in the first strand reaction, generating LibSeq cDNA transcripts. RT-PCR was performed as described in manufacturer's protocol. In all experiments, a minus-RT control reaction was run in parallel, where no reverse transcriptase was added and cDNA therefore not synthesized. Subsequently, the library was PCR amplified with forward primer (5′-AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCTNNATCTCATAGCTGGATGTGTGG-3′) and reverse primer (5′-CAAGCAGAAGACGGCATACGAGAT-3′). NN in the forward primer denotes the sample specific barcode. Both primers contain 5′ overhangs with Illumina adapter sequences. The reverse primer binds to the 5′ overhang of the reverse transcription primer and will therefore support amplification of LibSeq cDNA, but not residual LibSeq plasmid DNA. To ensure that no plasmid DNA was amplified in the PCR, no amplification product was allowed in the minus-RT control sample. Twenty-four cycles of amplification was performed using Phusion High-Fidelity DNA Polymerase following manufacturer's recommendations. The remaining steps in the library preparation and sequencing were performed in an identical manner to the sequencing of the LibSeq plasmid pool, as described above.
LibSeq data processing
LibSeq sequencing reads were processed in Galaxy (19). First, 20 nts of the reads’ 3′ ends were trimmed away and any reads with more than 10% of positions having quality score <20 were discarded. The remaining reads were clipped immediately downstream of the seven degenerate positions (using the clipping sequence ATATTTTAAT). Reads not containing the clipping sequence, having one or more failed base calls or being shorter than 15 nts were then discarded. The remaining reads were grouped according to barcode (and thus sample) and reverse complemented. Again, clipping was performed, this time with the sequence TTTTCTTCAAAT, corresponding to the reverse complement of the sequence immediately upstream of the 7mers, and reads not carrying the sequence were discarded. Next, reads were filtered by length to ensure that all reads consisted of a 7mer sequence. Finally, reads were once again reverse complemented and converted to tabular format, so that the 7mers could be grouped and counted using the Galaxy grouping tool. The counts for the different experiments are available in Supplementary Table S1.
Endogenous signal, effects of miRNA and protein binding 7mers
The analysis of the impact of the endogenous regulatory environment is based on two plasmid sequencing experiments and three LibSeq transfection experiments. Two 7mer subsequences (‘GGGCCAA’ and ‘ACATACG’) with abnormally high abundances in some samples, most likely due to contamination with unique clonings of these 7mers, were excluded from the analysis. Human miRNA seed sequences were retrieved from the TargetScan v6.0 database (20). The miRNAs expressed in HeLa cells were identified according to the miRNA cloning data performed by Tuschl and coworkers (21). Proteins binding strongly to 7mers were identified from the cisBP database using the criteria applied by the cisBP authors (7mer motifs with an E score >0.45) (22). RNA-BPs expressed in HeLa cells were defined as the subset of RNA-BPs being among the 2000 most expressed in HeLa cells according to the quantitative proteomics data for the HeLa cell line produced by Nagaraj et al. (23).
Oligonucleotide synthesis and purification
LNA-modified DNA oligonucleotides were synthesized with complete phosphorothioate backbones using standard phosphoramidite protocols on an ÄKTA Oligopilot (GE Healthcare, Denmark). After synthesis, the oligonucleotides were deprotected and cleaved from the solid support using aqueous ammonia at 65°C overnight. Three buffers were applied: A: 10 mM NaOH, B: 10 mM NaOH, 2 M NaCl, C: B/EtOH 4/1. The oligonucleotides were purified by ion-exchange high-performance liquid chromatography by applying a gradient of buffer B: 0–80% in 38 column volumes (CV) followed by a washout of the column with 100% buffer B (4 CV) and 100% buffer C (4 CV) before re-equilibration with 100% buffer A (4 CV). The collected fractions from the purification were analyzed by ultra-performance liquid chromatography and fractions with purity >85% were pooled and adjusted to pH 7–8 with HCl (1 M). Subsequently, the pooled fractions were desalted on an Äkta CrossFlow using a Millipore-membrane with a 1 kDa cut-off (Pellicon 2 Mini Ultrafiltration Module PLAC C 0.1 m2) and lyophilized. Finally, liquid chromatography-mass spectrometry (reverse phase and electrospray ionization-mass spectrometry) was used to verify compound identity and purity.
Calculation of binding score
Hybridization between two complementary nucleic acid strands is mainly governed by hydrogen bonding between base pairs on opposite strands and base stacking. In addition, nucleic acid polymers are flexible and can form bulges or loops that also needs to be considered when evaluating the thermodynamically most favored hybridization (24–26). Energy parameters for the binding of LNA-modified phosphorothioate oligonucleotides to RNA have not been published, but the LNA-modification is known to induce an RNA-like conformation of the DNA (27). We therefore make a rough estimate of the standard free energy of binding, ΔG, using parameters for RNA–RNA interactions (26). In addition, we calculate a simple binding score, using the Needleman-Wunsch algorithm (28), between oligonucleotide and RNA. The oligonucleotide sequences are listed in Table 1, and the RNA sequence is 3′-ATTTTATANNNNNNNAAAAGAAG-5′, where N indicates the positions of the 7mer. The Needleman-Wunsch alignment was performed without end gap and gap opening penalties, with a gap extension penalty of –1 and with substitution scores of 1 for each Watson–Crick base pair and zero otherwise. The maximal score with this scheme equals the length of the oligonucleotide. Binding energies and binding scores for all 7mers are available in Supplementary Table S2.
Table 1. Sequence and LNA-modification pattern for oligonucleotides evaluated by LibSeq.
| Name | Sequence (5′ to 3′)a |
|---|---|
| A | TATcagctacTTT |
| B | TATtgaacgtTTT |
| B-4DNA | TATtgaAcgttTT |
| B-3DNA | TAttgAacgTtTT |
| B-1DNA | TATtGaAcGtTtT |
aUpper case plus bold font indicate LNA and lower case DNA. All nucleotides are phosphorothioate-linked.
Calculation of sequence-specificity
We define the sequence-specificity of an RNase H recruiting oligonucleotide as the ratio of the rate of degradation of the intended target to the sum of the rates of degradation of the unintended targets (29)
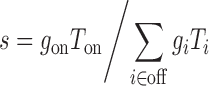 |
(1) |
where in a LibSeq experiment Ti is the abundance of mRNA reporter i, and gi is the degradation constant for the oligonucleotide on that reporter. The reporters are divided into the one with perfectly matching 7mer, which is the intended target (i = on), and those with imperfectly matched 7mers but which are still degraded to some extent (reduced by at least 50% in sample containing the regulatory oligo relative to control) (i = off).
To estimate gi from LibSeq data we assume a basic model of the mRNA reporter life cycle, in which the rate of transcription is a constant, Vi, but unique to each reporter, and the endogenous degradation rate as well as the exogenous, oligonucleotide-induced, degradation rate are additive and described by first-order kinetics. The derivative of the abundance Ti with respect to time can then be written
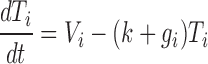 |
The endogenous degradation constant k is here assumed the same for all mRNA reporters. At steady state Ti does not change, so (dTi /dt) is equal to zero, and Ti = Vi /(k + gi). When no oligonucleotide is present, Tino = Vi /k. Measuring mRNA reporter abundances both in the absence and presence of oligonucleotide, where 0 < Ti ≤ Tino, we can therefore write the ratio as
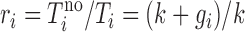 |
which rearranges to gi = k(ri − 1). Inserting this in (Equation 1), the sequence-specificity can now be written as
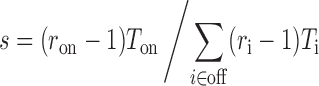 |
(2) |
We furthermore define a relative sequence-specificity, s’, where only the ratios ri are included, as
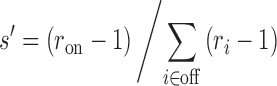 |
(3) |
Using this formulation of the sequence-specificity (Equation 3), it is only the relative fold change in abundances that influence sequence-specificity, and not the absolute abundances. Since RNA levels for the same gene may differ widely between different biological systems, but oligonucleotide-induced degradation constants do not, the sequence-specificity as defined in (Equation 3) is more oligonucleotide-centric and system-independent, and is therefore the measure used for comparing oligonucleotides with different modification designs in this study.
RESULTS
Reproducible detection of RNA motifs with LibSeq
To investigate the regulatory potential of thousands of short sequences in a constant context, we prepared a library of expression plasmids that contains a degenerate sequence of seven nucleotides (referred to as a 7mer) positioned in the 3′UTR of a luciferase reporter gene (Figure 1). Each 7mer can affect expression of the reporter and serve as a barcode after sequencing of the plasmid library or cDNA isolated from cells transfected with the library (Figure 1). We wanted to minimise effects of the 3′ UTR sequence flanking the 7mer and therefore embedded the degenerate sequence in an AU rich region from the 3′ UTR of the human HAND2 gene, which encompasses a highly efficient miR-1 target site (14). Using computational prediction, we find that 7mer sequences embedded in this context generally have higher accessibility than 7mer sequences embedded in sequences randomly picked from the transcriptome (Supplementary Figure S1A).
Figure 1.
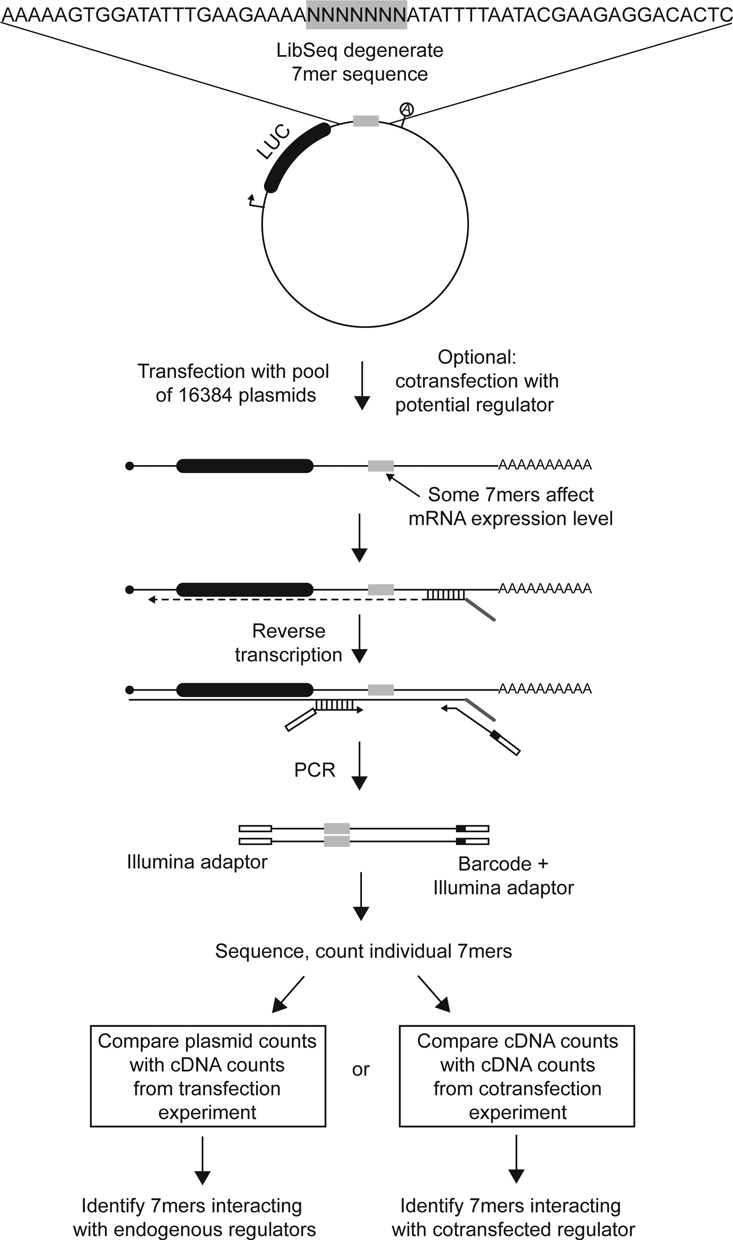
Schematic representation of the LibSeq strategy. LibSeq experiments are performed with a pool of reporter plasmids that are identical except for a seven nucleotide sequence located in the 3′ UTR of the reporter gene. Each individual 7mer sequence can affect the expression of the reporter mRNA through interactions with endogenous or exogenous regulator molecules and at the same time serve as a barcode allowing the expression to be evaluated using massive parallel sequencing.
To characterize the LibSeq library, we amplified and sequenced the LibSeq plasmid pool and found that the composition of the plasmid library was highly reproducible (Figure 2A). The frequency of each of the four nucleotides at the first six positions in the 7mers, counting from the 5′-end was close to the expected 0.25 (Supplementary Figure S1B). At the last position, T was enriched in low abundance 7mers, and depleted in high-abundance 7mers, (Supplementary Figure S1B), most likely reflecting biased synthesis of the linear library. Nevertheless, a total of 15 382 different 7mers (out of 16 384 possible) were still represented in the library with abundances ranging from a lower cutoff of 1 read per million reads (RMR) up to 467 RMR.
Figure 2.
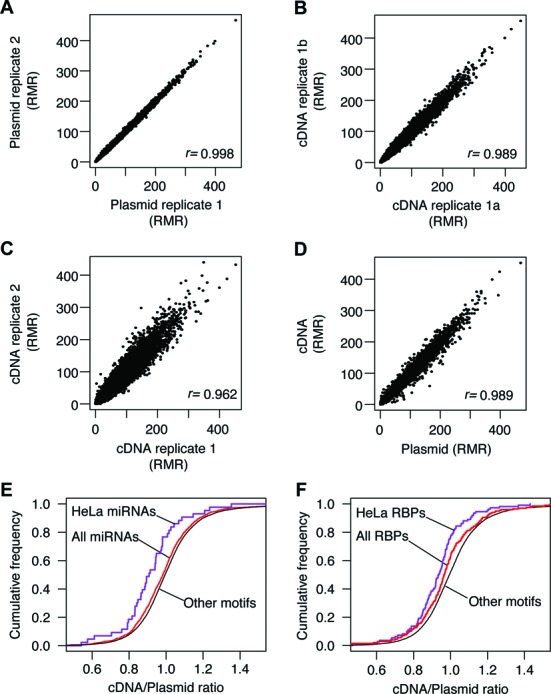
Validation of LibSeq strategy. (A) Reproducibility of 7mer reads per million reads (RMR) from sequencing libraries prepared directly from the LibSeq plasmid pool. The Pearson correlation of RMRs from the two libraries is indicated on the figure. (B) Technical replicate of library preparation using RNA from a LibSeq transfection experiment. Pearson correlation for the 7mer RMR abundances observed in the two libraries is shown on the figure. (C) Comparison of biological replicates of LibSeq RMR abundances prepared from RNA isolated from HeLa cells that were transfected with the LibSeq plasmids on two separate occasions. The Pearson correlation is indicated on the figure. (D) Comparison of LibSeq 7mer RMR abundances obtained from libraries prepared directly from the LibSeq plasmid pool and from RNA isolated from transfected HeLa cells. The Pearson correlation of RMRs from the two libraries is indicated on the figure. (E) Cumulative distribution plot of the ratio of 7mer RMR obtained from libraries prepared from the HeLa transfected RNA and from LibSeq plasmid pool. The curves showing the ratios obtained for 7mers that correspond to miRNA seed complementary sequences and the ratios obtained for 7mers that correspond to miRNA seed complementary sequences of miRNAs highly expressed in HeLa are indicated. (F) Cumulative distribution plots of the ratios of 7mer RMRs obtained from libraries prepared from the HeLa transfected RNA and from LibSeq plasmid pool. The curves showing the ratios obtained for 7mers that correspond to binding motifs for RNA binding proteins and the subset of RNA binding protein known to be expressed in HeLa cells are indicated.
To test the reproducibility of the LibSeq library preparation procedure, a technical replicate was performed on RNA derived from a LibSeq transfection of HeLa cells. After sequencing, a strong correlation of 7mer RMRs was observed (Figure 2B). Next, sequencing libraries were prepared from LibSeq transfections of two different populations of HeLa cells. The correlation between 7mer RMRs from these two biological replicates remains strong (Figure 2C), showing that results from LibSeq experiments are highly reproducible.
Profiling endogenous miRNA and RBP regulation with LibSeq
By comparing 7mer abundances obtained from plasmids to 7mer abundances obtained from cDNA originating from LibSeq transfected cells, it is possible to determine endogenous regulatory effects (Figure 1). In principle, the differences observed could be caused by the particular 7mer having an effect either on transcription of the reporter mRNA or post-transcriptional effects, such as altered mRNA processing or stability. Since the LibSeq library contains an active promoter sequence, (30) we expected the regulatory effects observed (Figure 2D) to be predominantly caused by post-transcriptional effects. To validate this hypothesis, we focused on two groups of 7mers that are known to be targets of regulatory molecules.
The first group consists of 7mers that are complementary to miRNA seed region. Binding of miRNAs repress gene expression and indeed we found that 7mers complementary to seed sequences of miRBase annotated miRNAs (31) was significantly downregulated compared to other 7mers (P < 0.001; Mann–Whitney U test) (Figure 2E). This effect is more pronounced when the analysis is restricted to miRNAs expressed in HeLa cells (21) (P < 0.001).
The second group we tested were 7mers that have previously been found to be strong binders of RNA-BP (22). We found this group of 7mers to be downregulated (P < 0.01) compared to other 7mers (Figure 2F) and that the observed signal is increased when the analysis is restricted to proteins known to be highly expressed in HeLa cells (23) (P < 0.0001). These findings indicate that the LibSeq library can be used to investigate posttranscriptional effects mediated by more than 15 000 different 7mers in parallel.
RNase H recruiting oligonucleotides efficiently repress expression from LibSeq plasmids with complementary sequences
Next, we wanted to use LibSeq to investigate the sequence-specificity and regulatory effects of therapeutic oligonucleotides designed to recruit and activate RNase H upon binding to complementary RNA. We used two different RNase H recruiting oligonucleotides, A and B, which were modified in the flanks with locked nucleic acid (LNA) (32,33) to increase binding affinity and stability (34) (see Table 1). The oligonucleotides have an often-used gapmer design with 3 nt LNA-flanks and a 7 nt DNA gap. The flanks are perfectly complementary to the 3 nt flanking regions up- and downstream of the degenerate 7mer sequence in the LibSeq mRNA reporters, but each of the oligonucleotides have a different sequence in the 7nt DNA-only region (see Table 1).
Each of the oligonucleotides A and B were cotransfected into HeLa cells together with a non-degenerate LibSeq plasmid containing the 7mer perfectly complementary to the respective oligonucleotide. Both oligonucleotides lead to a marked reduction in expression of the luciferase reporter encoded in the LibSeq plasmid, with oligonucleotide B having a slightly more pronounced effect than oligonucleotide A (Figure 3A and B (left side)). This demonstrates that LibSeq plasmids can respond to regulation by RNase H recruiting oligonucleotides.
Figure 3.
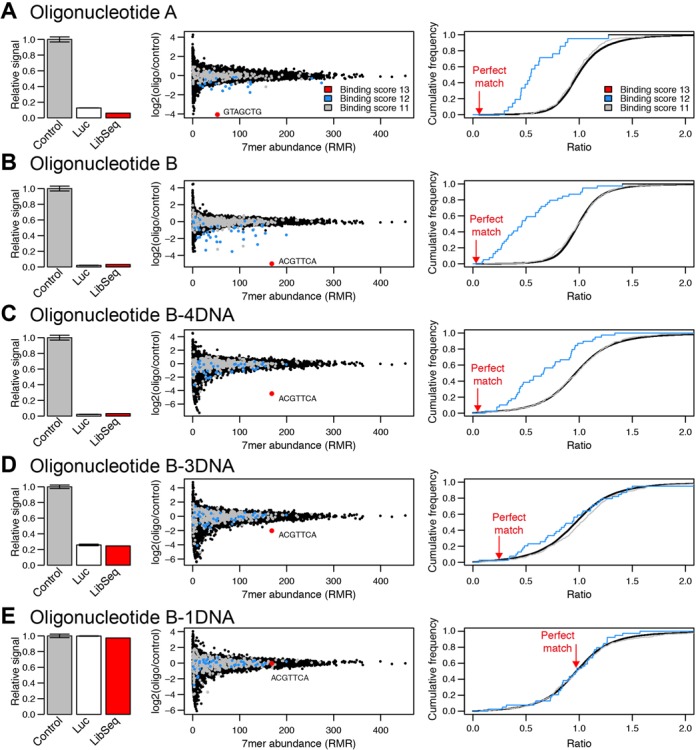
Impact of LNA modified oligonucleotides on 7mer abundances. The five oligonucleotides, (A) oligonucleotide A, (B) oligonucleotide B (C) oligonucleotide B-4DNA (D) oligonucleotide B-3DNA and (E) oligonucleotide B-1DNA were each co-transfected with a luciferase expression vector containing the perfect match target site or with the entire pool of LibSeq reporters. The bar plots on the left shows, for each of the oligonucleotides, the luciferase readout of a cotransfection experiment of the oligonucleotides and the perfect match (binding score 13) reporter vector (Luc) compared with the non-oligo transfection control (Control). For comparison the red bar shows the regulation of the perfect match 7mer in the LibSeq experiment relative to the average RMR in the experiment. The center figures show the result of the LibSeq experiment for each of the oligonucleotides. For each 7mer, the Log2 ratio of the abundance in the co-transfection experiment, divided by abundances from a control no oligonucleotide experiment as a function of the 7mer abundance in the experiment is plotted. The color coding indicates 7mers with binding score 13 (red), 12 (blue), and 11 (grey). The plots on the right shows cumulative distribution for binding score 12 (blue line) and 11 (gray line) 7mers, with the detected down-regulation of the binding score 13 7mer indicated by red arrow.
Importantly, the regulatory effect of oligonucleotide A and B on the perfect matching target was recapitulated when the oligonucleotides were co-transfected with the entire plasmid library and the change of each mRNA reporter level, relative to the mRNA reporter levels in control samples, was determined by sequencing (Figure 3A and B, left and center). This indicates that LibSeq faithfully monitors the regulatory effects mediated by RNase H recruiting oligonucleotides.
LibSeq detects off-targeting by RNase H recruiting oligonucleotides
The major strength of LibSeq is the ability to evaluate both the intended as well as unintended RNA targets. We therefore evaluated the regulatory effects mediated by 7mers having 12 Watson–Crick basepairs out of 13 possible between the RNA target site and the oligonucleotide. Of this group of 7mers, which we assign a binding score of 12 (see ‘Materials and Methods’ section), some but not all of the mRNA reporters are affected by the oligonucleotides (blue dots in Figure 3A and B, center). In contrast, for the mRNA reporters with binding score 11 (gray dots in Figure 3A and B) only a few are affected. Taken as a group, 7mers with binding score 12 are significantly repressed compared to 7mers with binding score <11 (Figure 3A and B, right side). In contrast, the group of 7mers with binding score 11 is not significantly repressed (Figure 3A and B, right). Thus, the LibSeq analysis demonstrates that most off-targets for RNase H recruiting oligonucleotides with the 3 LNA, 7 DNA and 3 LNA design used here have at most one position that is either mismatched or bulged (Supplementary Tables S3 and S4). Moreover, the analysis reveals that RNase H can accommodate many different types of mismatches and bulges, while still retaining the ability to repress the RNA target by the tested oligonucleotides.
Thermodynamic stability of the target oligonucleotide duplex affects RNase H efficiency
Differences in the standard free energy of binding, ΔG, between the oligonucleotide and mRNA target sequence could potentially explain the differential effects of 7mers with the same binding score. We calculated an estimate of ΔG for oligonucleotide-RNA target duplexes using parameters for RNA–RNA interactions (26) (Supplementary Table S1), because it is known that LNA-modifications induce an RNA-like conformation of the surrounding DNA (27). We found a relatively strong correlation between LibSeq fold changes and ΔG for 7mers with binding score 12 and a weaker, but still significant correlation for 7mers with binding score 11 (Supplementary Figure S2B and E). This indicates that the free energy of binding between RNase H recruiting oligonucleotides and RNA target affects the efficiency of gene repression.
7mer accessibility has a negligible effect on LibSeq fold change
The sequence context of the LibSeq 7mers is generally quite accessible (Supplementary Figure S1A), but differences in accessibility could potentially influence the efficiency of the different 7mer and therefore affect our analysis. We therefore tested the correlation between the predicted accessibility and the fold change observed in the LibSeq experiment for all 7mers with a binding score of 13, 12 or 11. This analysis showed no correlation for oligonucleotide A (Supplementary Figure S2C) and a weak, non-significant correlation for oligonucleotide B (Supplementary Figure S2F), indicating that 7mer accessibility has a negligible effect on our results.
LibSeq analysis allow the specificity of RNase H recruiting oligonucleotides to be evaluated
An advantage of our LibSeq analysis of RNase H recruiting oligonucleotides is the ability to obtain data for both the intended and all unintended targets, thereby providing the opportunity to calculate a specificity measure s’ for the tested oligonucleotide, as described in ‘Materials and Methods’ section (Equation 3). Using this method, we calculate the sequence-specificity of oligonucleotide A to be s’ = 0.96, which is twice as high as the sequence-specificity of oligonucleotide B, s’ = 0.48, indicating that oligonucleotide A would be the better choice of the two.
To further confirm the utility of LibSeq in evaluating specificity, and explore whether the sequence-specificity of oligonucleotide B can be improved by changing the modification pattern, we tested three modified versions of oligonucleotide B, denoted B-4DNA, B-3DNA and B-1DNA (Table 1). In these oligonucleotides, we reduced the size of the central DNA gap from seven to 4, 3 or 1 DNA monomer, respectively. It has previously been shown that shortening of the central DNA gap reduces the efficiency of RNase H mediated repression (15), and it is known that an oligonucleotide with only single DNAs between LNAs does not support RNase H cleavage (35). As expected the oligonucleotide B-1DNA did not have any effect on either intended or unintended 7mers (Figure 3E). In contrast, the oligonucleotide B-4DNA had a sequence-specificity of s’ = 2.01 (Figure 3C), whereas. oligonucleotide B-3DNA had a reduced sequence-specificity of s’ = 0.29, mainly because of reduced efficiency on the intended target (Figure 3D). Thus, using LibSeq we find that oligonucleotide B-4DNA has a sequence-specificity four times higher than oligonucleotide B. This demonstrates that reduced length of the central DNA gap in RNase H recruiting oligonucleotides at least in this case can increase the specificity of gene repression.
LibSeq reveals mismatch restrictions for RNase H binding
The oligonucleotides B and B-4DNA have identical sequences, but different location of their LNA modifications (Figure 4A). To gain insight into the mechanisms responsible for the increased specificity of the B-4DNA oligonucleotide, we focused on the 7mers with a binding score of 12 for the B sequence, RMR > 25 and a log2 fold change of at least −0.5 for either oligonucleotide B or B-4DNA (Supplementary Table S5). The non-perfectly matched oligonucleotide–RNA duplexes fall into two categories: mismatches (Figure 4B) and RNA bulges (Figure 4C). For the B oligonucleotide, mismatches and bulges are distributed across all 7 positions in the DNA gap (Figure 4D). This suggests that a DNA gap of 7nt will allow RNase H to accommodate mismatches and bulges in many different locations and still retain the ability to cleave the duplex. In contrast, for 7mers showing repressed expression upon treatment with oligonucleotide B-4DNA, we find a positional pattern of mismatches being accommodated in positions 1, 2 and 3, while RNA bulges are accommodated between positions 5–6, 6–7 and 7–8 (P < 0.05 by binomial test). This striking pattern strongly suggests that the LNA modification located centrally in the B-4DNA oligonucleotide restricts the ability of RNase H to bind to the oligonucleotide-RNA duplex in a manner that supports enzymatic cleavage.
Figure 4.
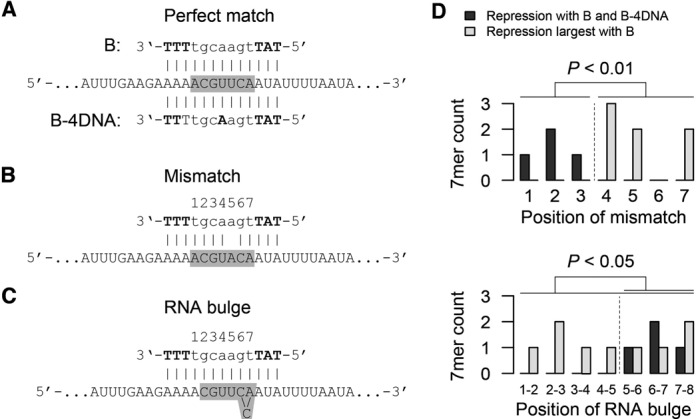
Effective RNA-oligonucleotide duplexes. (A) Perfect match binding of the B and B-4DNA oligonucleotide to the LibSeq RNA sequence. LNA nucleotides are indicated in bold capital letters and the gray box shows the location of the degenerate sequence in the LibSeq library. (B) Specific example of a duplex between the B oligonucleotide and the LibSeq RNA, which affects expression despite a mismatch in position 5. Positions are indicated above the oligonucleotide, LNA nucleotides are indicated in bold capital letters and the gray box shows the location of the degenerate sequence in the LibSeq library. (C) Specific example of a duplex between the B oligonucleotide and the LibSeq RNA, which affects expression despite an RNA bulge between positions 6 and 7. Positions are indicated above the oligonucleotide, LNA nucleotides are indicated in bold capital letters and the gray box shows the location of the degererate sequence in the LibSeq library. (D) Positions of mismatches (top) and RNA bulges (bottom) in LibSeq RNA–oligonucleotide duplexes, which repress expression both for the B and the B-4DNA oligonucleotide (black bars) or only for the B oligonucleotide (gray bars). Both for mismatches and and RNA bulges the positions are significantly different for the two oligonucleotides.
DISCUSSION
In this study, we describe the LibSeq method, which allows exhaustive information on the regulatory potential of a short sequence in a particular context to be obtained. This is similar to a study by Chasin and coworkers, who used a LibSeq-like library-based method to characterize the post-transcriptional effects of 6mer sequences on splicing (36). In these studies post-transcriptional regulation is investigated using reporter constructs with active promoters, which are likely to drive transcription independently of additional enhancer elements. The LibSeq strategy is also related to previous strategies used for unbiased expression analysis of promoters and enhancers. Shendure et al. analyzed the activity of T7 promoter variants using a library of array-synthesized DNA oligos containing a coupled barcode in the transcribed RNA sequence (37). Subsequently, similar strategies have been used for mutational analysis of different human enhancers in cell culture expression experiments (38,39). Enhancer libraries have also been used to evaluate the in vivo regulatory effect of thousands of variants in mouse livers (40,41) and mouse retina (42). Moreover, library-sequencing strategies have been used to dissect the function of promoter elements in yeast cells (43,44) and recently to study the regulatory effects of thousands of 200mer sequences obtained from human 3′ UTR sequences (45).
We validated our LibSeq method by comparing LibSeq counts from plasmid DNA with counts obtained from cDNA produced from plasmid-transfected HeLa cells. In this way, we find that binding motifs for RNA-BPs and miRNA seed matches affect expression of the LibSeq reporter mRNA (Figure 2E and F). For miRNAs it is known that endogenous levels typically have a modest effect on an isolated miRNA seed target sequence (46), which explains why our observed fold changes are relatively modest and not significant for the individual seed target. RNA-BPs are involved in many different regulatory functions and their RNA binding can have both positive and negative effects on target mRNA levels (47). Here, we find that RNA-BPs as a group inhibit the expression of the LibSeq reporter. We speculate that this downregulation could be caused by competition between the RNA-BPs and a stabilizing factor binding to the region or by the RNA-BPs affecting the local RNA structure leading to destabilisation of the RNA.
Having validated LibSeq for the analysis of endogenous regulators, we applied the LibSeq strategy to investigate the sequence requirement of RNase H recruiting oligonucleotides based on the often-used ‘gapmer’-design: 3LNA-7DNA-3LNA. We found that the LibSeq experiment recapitulates the efficient repression of the perfectly complementary sequence observed in a luciferase reporter assay (Figure 3). Importantly, LibSeq also provides detailed information on the regulatory effects on other target sequences, which are affected by the oligonucleotides. Similar information cannot be obtained using conventional transcriptomics, both because it may be very difficult to pinpoint the exact sequence that mediates the effect of the RNase H recruiting oligonucleotide and because the target sequences will be located in different sequence contexts. Therefore, the comprehensive and comparable data obtained with LibSeq has the potential to improve the mechanistic understanding of RNase H recruiting oligonucleotides. For the two tested oligonucleotides, we find that the large majority of unintended targets have only one mismatch or one bulge in either the RNA target or the oligonucleotide strand (Figure 3), suggesting that the catalytic site of the RNase H enzyme can accommodate substrates with a relatively high degree of structural diversity, but not with more than one mismatch or one bulge.
Given the relatively high propensity of oligonucleotide A and B to affect target sequences that contain mismatches, we wondered if it would be possible to improve the sequence-specificity by changing the locations of the LNA modifications in the oligonucleotides. As expected, the oligonucleotide with no contiguous DNA monomers did not result in reduction of the target RNA. This oligonucleotide also serves as a control for a possible effect of the LNA oligonucleotides on the efficiency of reverse transcription and PCR. Furthermore, the fact that the luciferase experiments replicate the repression observed in the LibSeq experiments, supports that LibSeq results are dependent on RNase H activity and not the oligonucleotides affecting reverse transcription or PCR. Interestingly, we find that the oligonucleotide with a 4 DNA monomer gap represses expression nearly as efficiently as the oligonucleotide with the seven DNA monomer gap, whereas the oligonucleotide with a three monomer DNA gap was much less efficient. When calculating the specificity measure s’ based on the LibSeq experiment, we find that the oligonucleotide with the 4 DNA monomer gap is four times as specific for the intended target sequence as the original 7 DNA monomer gap oligonucleotide. This finding indicates that reduction of the length of the DNA-gap in therapeutic RNase H recruiting oligonucleotides can be a way of increasing specificity.
The high specificity of the B-4DNA that we observe may result from little or no flexibility in the way it needs to bind to RNA to achieve a duplex structure that can be efficiently cleaved by RNase H. From the crystal structure of the human RNase H1 catalytic domain in complex with an 18 bp DNA/RNA hybrid, it is apparent that the domain specifically recognizes the DNA strand of the duplex by two major determinants (48), as summarized in Figure 5. First, a backbone phosphate of the DNA strand needs to dock into a phosphate-binding pocket. This interaction positions the RNA strand of the duplex for backbone cleavage two nts away from the pocket. The binding of the phosphate in the pocket requires the backbone to adopt a conformation away from the ideal torsion angles, which can only be adopted by DNA (48). Secondly, the three DNA positions 5′ to the phosphate binding pocket fit into a DNA binding channel, which is present on the surface of the catalytic domain and cannot accommodate RNA (48), see Figure 5.
Figure 5.
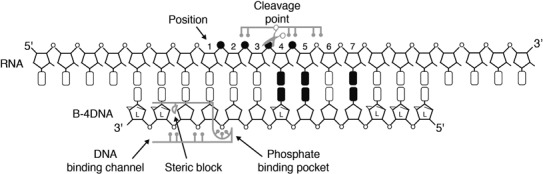
Model for the binding of the RNase H catalytic domain to the RNA B-4DNA oligonucleotide duplex. The duplex is shown with the RNA strand on the top and the oligonucleotide below. Positions are indicated above the RNA strand and LNA nucleotides are indicated with Ls on the backbone ribose rings. In gray selected interactions (as observed in the published crystal structure (46)) between the RNase H enzyme and the duplex is outlined, including the phosphate binding pocket, the cleavage point and the steric block in the DNA binding channel. The indicated locations of the RNase H enzyme interactions on the duplex correspond to the binding mode, which is most likely to support cleavage of the RNA strand. The bases colored black show the positions found to retain the ability to repress expression with mismatches in B, but not B-4DNA oligonucleotide RNA duplexes. Likewise the black backbone positions show the positions found to retain the ability to repress expression with RNA bulges in B, but not B-4DNA oligonucleotide RNA duplexes.
The RNase H specificity determinants (Figure 5) may explain why the B-3DNA oligonucleotide has decreased efficiency on the perfect match target compared to the B-4DNA oligonucleotide. The three DNA monomer gap is likely to be too short to efficiently bind to both the DNA binding channel and the phosphate binding pocket, whereas the four DNA monomer gap may be just sufficient. We speculate that this results in the B-4DNA oligonucleotide RNA duplex being restricted to bind in a specific manner to the RNase H catalytic domain in order to support cleavage of the RNA (Figure 5).
To look further into the mechanistic details of RNase H mediated cleavage, we analysed the position of mismatches and bulges in the duplexes formed by the 7mers that mediated efficient repression for oligonucleotide B, but not oligonucleotide B-4DNA. First, we find that the oligonucleotide B-4DNA compared to oligonucleotide B cannot accommodate mismatches in the positions 4, 5 and 7 of the duplex (Figure 4C). The restricted modes of RNase H binding to the B-4DNA RNA duplex places these mismatches close to the RNase H catalytic site, which most likely distorts the duplex helix enough to interfere with cleavage of the RNA strand (Figure 5, black bases). Second, it is known that RNA bulges in some cases can be accommodated without major distortion of the helical structure (49). This may be the case for the B oligonucleotide-RNA duplexes that have bulges in the RNA, but still effectively repress expression (Figure 4D). However, we find that B-4DNA oligonucleotide RNA duplexes with bulges in positions 2, 3 and 4 do not repress expression, suggesting that bulges in these positions (Figure 5, black colored backbone) cannot be accommodated when the modes of RNase H binding to the duplex are restricted. These rules for accommodation of mismatches and bulges could be similar for all RNase H recruiting oligonucleotides and may only become apparent when the binding mode of the duplex is restricted and an exhaustive method such as LibSeq is used. Further studies with more oligonucleotides and different sequence contexts of the target site will be needed to establish whether these rules are general.
In conclusion, we present a method for analyzing the regulatory potential of more than 15000 short RNA motifs in parallel. Compared to conventional transcriptomics, LibSeq experiments have the advantage that they allow a direct comparison of many different target sequences in the same context, which potentially can be used to investigate many different types of RNA regulation. Here, we applied the method to gain insights into the mechanistic basis for gene regulation by RNase H recruiting oligonucleotides. We find that RNase H recruiting oligonucleotides with short central DNA gaps can have increased sequence specificity, which may facilitate the design of more specific RNase H recruiting oligonucleotides.
Supplementary Material
Acknowledgments
We would like to thank Marianne B. Mogensen and Michael M. Meldgaard for synthesis and formulation of LNA-modified oligonucleotides. Sequencing was performed at Danish National DNA Sequencing Center and the Section for Computational and RNA Biology provided the computational infrastructure. We also thank Peter Brodersen, Jan Christiansen and Manuel Irimia for proofreading and providing input for the manuscript.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Danish Council for Strategic Research (Center for Computational and Applied Transcriptomics) [DSF-10-092320]. Funding for open access charge: The Danish Council for Strategic Research.
Conflict of interest statement. P.H.H. and M.L. are employees of Roche Innovation Center Copenhagen A/S, a company that is developing LNA-modified oligonucleotidesfor therapeutic purposes.
REFERENCES
- 1.Sharp P.A. The centrality of RNA. Cell. 2009;136:577–580. doi: 10.1016/j.cell.2009.02.007. [DOI] [PubMed] [Google Scholar]
- 2.Vogel C., Marcotte E.M. Insights into the regulation of protein abundance from proteomic and transcriptomic analyses. Nat. Rev. Genet. 2012;13:227–232. doi: 10.1038/nrg3185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Xie X., Lu J., Kulbokas E.J., Golub T.R., Mootha V., Lindblad-Toh K., Lander E.S., Kellis M. Systematic discovery of regulatory motifs in human promoters and 3′ UTRs by comparison of several mammals. Nature. 2005;434:338–345. doi: 10.1038/nature03441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lim L.P., Lau N.C., Garrett-Engele P., Grimson A., Schelter J.M., Castle J., Bartel D.P., Linsley P.S., Johnson J.M. Microarray analysis shows that some microRNAs downregulate large numbers of target mRNAs. Nature. 2005;433:769–773. doi: 10.1038/nature03315. [DOI] [PubMed] [Google Scholar]
- 5.Kenan D.J., Keene J.D. In vitro selection of aptamers from RNA libraries. Methods Mol. Biol. 1999;118:217–231. doi: 10.1385/1-59259-676-2:217. [DOI] [PubMed] [Google Scholar]
- 6.Ray D., Kazan H., Chan E.T., Pena Castillo L., Chaudhry S., Talukder S., Blencowe B.J., Morris Q., Hughes T.R. Rapid and systematic analysis of the RNA recognition specificities of RNA-binding proteins. Nat. Biotechnol. 2009;27:667–670. doi: 10.1038/nbt.1550. [DOI] [PubMed] [Google Scholar]
- 7.Ule J., Jensen K.B., Ruggiu M., Mele A., Ule A., Darnell R.B. CLIP identifies Nova-regulated RNA networks in the brain. Science. 2003;302:1212–1215. doi: 10.1126/science.1090095. [DOI] [PubMed] [Google Scholar]
- 8.Walder R.Y., Walder J.A. Role of RNase H in hybrid-arrested translation by antisense oligonucleotides. Proc. Natl. Acad. Sci. U.S.A. 1988;85:5011–5015. doi: 10.1073/pnas.85.14.5011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stein H., Hausen P. Enzyme from calf thymus degrading the RNA moiety of DNA-RNA Hybrids: effect on DNA-dependent RNA polymerase. Science. 1969;166:393–395. doi: 10.1126/science.166.3903.393. [DOI] [PubMed] [Google Scholar]
- 10.Sachs A.B. Messenger-Rna Degradation in Eukaryotes. Cell. 1993;74:413–421. doi: 10.1016/0092-8674(93)80043-e. [DOI] [PubMed] [Google Scholar]
- 11.Hovingh K., Besseling J., Kastelein J. Efficacy and safety of mipomersen sodium (Kynamro) Expert Opin. Drug Saf. 2013;12:569–579. doi: 10.1517/14740338.2013.793670. [DOI] [PubMed] [Google Scholar]
- 12.Bennett C.F., Swayze E.E. RNA targeting therapeutics: molecular mechanisms of antisense oligonucleotides as a therapeutic platform. Annu. Rev. Pharmacol. Toxicol. 2010;50:259–293. doi: 10.1146/annurev.pharmtox.010909.105654. [DOI] [PubMed] [Google Scholar]
- 13.Woolf T.M., Melton D.A., Jennings C.G. Specificity of antisense oligonucleotides in vivo. Proc. Natl. Acad. Sci. U.S.A. 1992;89:7305–7309. doi: 10.1073/pnas.89.16.7305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lennox K.A., Sabel J.L., Johnson M.J., Moreira B.G., Fletcher C.A., Rose S.D., Behlke M.A., Laikhter A.L., Walder J.A., Dagle J.M. Characterization of modified antisense oligonucleotides in Xenopus laevis embryos. Oligonucleotides. 2006;16:26–42. doi: 10.1089/oli.2006.16.26. [DOI] [PubMed] [Google Scholar]
- 15.Lima W.F., Rose J.B., Nichols J.G., Wu H., Migawa M.T., Wyrzykiewicz T.K., Siwkowski A.M., Crooke S.T. Human RNase H1 discriminates between subtle variations in the structure of the heteroduplex substrate. Mol. Pharmacol. 2007;71:83–91. doi: 10.1124/mol.106.025015. [DOI] [PubMed] [Google Scholar]
- 16.Lindow M., Vornlocher H.P., Riley D., Kornbrust D.J., Burchard J., Whiteley L.O., Kamens J., Thompson J.D., Nochur S., Younis H., et al. Assessing unintended hybridization-induced biological effects of oligonucleotides. Nat. Biotechnol. 2012;30:920–923. doi: 10.1038/nbt.2376. [DOI] [PubMed] [Google Scholar]
- 17.Wiedmann M., Wilson W.J., Czajka J., Luo J., Barany F., Batt C.A. Ligase chain reaction (LCR)–overview and applications. PCR Methods Appl. 1994;3:S51–64. doi: 10.1101/gr.3.4.s51. [DOI] [PubMed] [Google Scholar]
- 18.Vinther J., Hedegaard M.M., Gardner P.P., Andersen J.S., Arctander P. Identification of miRNA targets with stable isotope labeling by amino acids in cell culture. Nucleic Acids Res. 2006;34:e107. doi: 10.1093/nar/gkl590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Goecks J., Nekrutenko A., Taylor J., Galaxy T. Galaxy: a comprehensive approach for supporting accessible, reproducible, and transparent computational research in the life sciences. Genome Biol. 2010;11:R86. doi: 10.1186/gb-2010-11-8-r86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lewis B.P., Burge C.B., Bartel D.P. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;120:15–20. doi: 10.1016/j.cell.2004.12.035. [DOI] [PubMed] [Google Scholar]
- 21.Landgraf P., Rusu M., Sheridan R., Sewer A., Iovino N., Aravin A., Pfeffer S., Rice A., Kamphorst A.O., Landthaler M., et al. A mammalian microRNA expression atlas based on small RNA library sequencing. Cell. 2007;129:1401–1414. doi: 10.1016/j.cell.2007.04.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ray D., Kazan H., Cook K.B., Weirauch M.T., Najafabadi H.S., Li X., Gueroussov S., Albu M., Zheng H., Yang A., et al. A compendium of RNA-binding motifs for decoding gene regulation. Nature. 2013;499:172–177. doi: 10.1038/nature12311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nagaraj N., Wisniewski J.R., Geiger T., Cox J., Kircher M., Kelso J., Paabo S., Mann M. Deep proteome and transcriptome mapping of a human cancer cell line. Mol. Syst. Biol. 2011;7:548. doi: 10.1038/msb.2011.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hofacker I.L., Fontana W., Stadler P.F., Bonhoeffer L.S., Tacker M., Schuster P. Fast folding and comparison of RNA secondary structures. Monatsh Chem. 1994;125:167–188. [Google Scholar]
- 25.SantaLucia J. Jr, Hicks D. The thermodynamics of DNA structural motifs. Annu. Rev. Biophys. Biomol. Struct. 2004;33:415–440. doi: 10.1146/annurev.biophys.32.110601.141800. [DOI] [PubMed] [Google Scholar]
- 26.Rehmsmeier M., Steffen P., Hochsmann M., Giegerich R. Fast and effective prediction of microRNA/target duplexes. RNA. 2004;10:1507–1517. doi: 10.1261/rna.5248604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bondensgaard K., Petersen M., Singh S.K., Rajwanshi V.K., Kumar R., Wengel J., Jacobsen J.P. Structural studies of LNA:RNA duplexes by NMR: conformations and implications for RNase H activity. Chemistry. 2000;6:2687–2695. doi: 10.1002/1521-3765(20000804)6:15<2687::aid-chem2687>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 28.Durbin R., Eddy S.R., Krogh A., Mitchison G. Biological Sequence Analysis: Probabilistic Models of Proteins and Nucleic Acids. Cambridge University Press; 1998. [Google Scholar]
- 29.Herschlag D. Implications of ribozyme kinetics for targeting the cleavage of specific RNA molecules in vivo: more isn't always better. Proc. Natl. Acad. Sci. U.S.A. 1991;88:6921–6925. doi: 10.1073/pnas.88.16.6921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shifera A.S., Hardin J.A. Factors modulating expression of Renilla luciferase from control plasmids used in luciferase reporter gene assays. Anal. Biochem. 2010;396:167–172. doi: 10.1016/j.ab.2009.09.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kozomara A., Griffiths-Jones S. miRBase: integrating microRNA annotation and deep-sequencing data. Nucleic Acids Res. 2011;39:D152–D157. doi: 10.1093/nar/gkq1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Obika S., Nanbu D., Hari Y., Morio K., In Y., Ishida T., Imanishi T. Synthesis of 2′-O,4′-C-methyleneuridine and -cytidine. Novel bicyclic nucleosides having a fixed C-3,-endo sugar puckering. Tetrahedron Lett. 1997;38:8735–8738. [Google Scholar]
- 33.Koshkin A.A., Singh S.K., Nielsen P., Rajwanshi V.K., Kumar R., Meldgaard M., Olsen C.E., Wengel J. LNA (Locked Nucleic Acids): synthesis of the adenine, cytosine, guanine, 5-methylcytosine, thymine and uracil bicyclonucleoside monomers, oligomerisation, and unprecedented nucleic acid recognition. Tetrahedron. 1998;54:3607–3630. [Google Scholar]
- 34.Wahlestedt C., Salmi P., Good L., Kela J., Johnsson T., Hokfelt T., Broberger C., Porreca F., Lai J., Ren K.K., et al. Potent and nontoxic antisense oligonucleotides containing locked nucleic acids. Proc. Natl. Acad. Sci. U.S.A. 2000;97:5633–5638. doi: 10.1073/pnas.97.10.5633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cerritelli S.M., Crouch R.J. Ribonuclease H: the enzymes in eukaryotes. FEBS J. 2009;276:1494–1505. doi: 10.1111/j.1742-4658.2009.06908.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ke S.D., Shang S.L., Kalachikov S.M., Morozova I., Yu L., Russo J.J., Ju J.Y., Chasin L.A. Quantitative evaluation of all hexamers as exonic splicing elements. Genome Res. 2011;21:1360–1374. doi: 10.1101/gr.119628.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Patwardhan R.P., Lee C., Litvin O., Young D.L., Pe'er D., Shendure J. High-resolution analysis of DNA regulatory elements by synthetic saturation mutagenesis. Nat. Biotechnol. 2009;27:1173–1175. doi: 10.1038/nbt.1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Melnikov A., Murugan A., Zhang X., Tesileanu T., Wang L., Rogov P., Feizi S., Gnirke A., Callan C.G. Jr, Kinney J.B., et al. Systematic dissection and optimization of inducible enhancers in human cells using a massively parallel reporter assay. Nat. Biotechnol. 2012;30:271–277. doi: 10.1038/nbt.2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kheradpour P., Ernst J., Melnikov A., Rogov P., Wang L., Zhang X.L., Alston J., Mikkelsen T.S., Kellis M. Systematic dissection of regulatory motifs in 2000 predicted human enhancers using a massively parallel reporter assay. Genome Res. 2013;23:800–811. doi: 10.1101/gr.144899.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Smith R.P., Taher L., Patwardhan R.P., Kim M.J., Inoue F., Shendure J., Ovcharenko I., Ahituv N. Massively parallel decoding of mammalian regulatory sequences supports a flexible organizational model. Nat. Genet. 2013;45:1021–1028. doi: 10.1038/ng.2713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Patwardhan R.P., Hiatt J.B., Witten D.M., Kim M.J., Smith R.P., May D., Lee C., Andrie J.M., Lee S.I., Cooper G.M., et al. Massively parallel functional dissection of mammalian enhancers in vivo. Nat. Biotechnol. 2012;30:265–270. doi: 10.1038/nbt.2136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kwasnieski J.C., Mogno I., Myers C.A., Corbo J.C., Cohen B.A. Complex effects of nucleotide variants in a mammalian cis-regulatory element. Proc. Natl. Acad. Sci. U.S.A. 2012;109:19498–19503. doi: 10.1073/pnas.1210678109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mogno I., Kwasnieski J.C., Cohen B.A. Massively parallel synthetic promoter assays reveal the in vivo effects of binding site variants. Genome Res. 2013;23:1908–1915. doi: 10.1101/gr.157891.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sharon E., Kalma Y., Sharp A., Raveh-Sadka T., Levo M., Zeevi D., Keren L., Yakhini Z., Weinberger A., Segal E. Inferring gene regulatory logic from high-throughput measurements of thousands of systematically designed promoters. Nat. Biotechnol. 2012;30:521–530. doi: 10.1038/nbt.2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhao W., Pollack J.L., Blagev D.P., Zaitlen N., McManus M.T., Erle D.J. Massively parallel functional annotation of 3′ untranslated regions. Nat. Biotechnol. 2014;32:387–391. doi: 10.1038/nbt.2851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Baek D., Villen J., Shin C., Camargo F.D., Gygi S.P., Bartel D.P. The impact of microRNAs on protein output. Nature. 2008;455:64–71. doi: 10.1038/nature07242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mitchell S.F., Parker R. Principles and properties of eukaryotic mRNPs. Mol. Cell. 2014;54:547–558. doi: 10.1016/j.molcel.2014.04.033. [DOI] [PubMed] [Google Scholar]
- 48.Nowotny M., Gaidamakov S.A., Ghirlando R., Cerritelli S.M., Crouch R.J., Yang W. Structure of human RNase h1 complexed with an RNA/DNA hybrid: Insight into HIV reverse transcription. Mol. Cell. 2007;28:264–276. doi: 10.1016/j.molcel.2007.08.015. [DOI] [PubMed] [Google Scholar]
- 49.Hermann T., Patel D.J. RNA bulges as architectural and recognition motifs. Struct. Fold. Des. 2000;8:R47–R54. doi: 10.1016/s0969-2126(00)00110-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.