


Abstract
Facioscapulohumeral dystrophy (FSHD) is an epi/genetic satellite disease associated with at least two satellite sequences in 4q35: (i) D4Z4 macrosatellite and (ii) β-satellite repeats (BSR), a prevalent part of the 4qA allele. Most of the recent FSHD studies have been focused on a DUX4 transcript inside D4Z4 and its tandem contraction in FSHD patients. However, the D4Z4-contraction alone is not pathological, which would also require the 4qA allele. Since little is known about BSR, we investigated the 4qA BSR functional role in the transcriptional control of the FSHD region 4q35. We have shown that an individual BSR possesses enhancer activity leading to activation of the Adenine Nucleotide Translocator 1 gene (ANT1), a major FSHD candidate gene. We have identified ZNF555, a previously uncharacterized protein, as a putative transcriptional factor highly expressed in human primary myoblasts that interacts with the BSR enhancer site and impacts the ANT1 promoter activity in FSHD myoblasts. The discovery of the functional role of the 4qA allele and ZNF555 in the transcriptional control of ANT1 advances our understanding of FSHD pathogenesis and provides potential therapeutic targets.
INTRODUCTION
Facioscapulohumeral Dystrophy (FSHD) is an autosomal dominant muscular dystrophy with an incidence of 3.2–4.6 per 100 000 throughout the world (1). FSHD is clinically characterized by a slowly advancing muscular weakness in an up-to-down manner involving face, pectoral girdle, upper limbs, lower limbs and hips. Most often the disease begins in childhood and adolescence, between the ages of 10 and 20, and affects both sexes (2,3). Loss of muscular strength limits both personal and occupational activities and leads to the inability to walk in 20% of FSHD patients. In general, the earlier FSHD develops, the more severe it is.
FSHD has been described in 1884 by L. Landouzy and J. Dejerine (4). Since then, its molecular basis has been partially elucidated. More than 95% of the patients have a deletion in the subtelomeric region of the long arm of chromosome 4 (4q35 locus) (5). This region includes a repeated tandem sequence of 3.3 kb, D4Z4. The number of D4Z4 repeats varies in the general population between 11 and 150, whereas it is between 1 and 10 in case of FSHD. The disease is transmitted in an autosomal dominant manner. However, 30% of cases are sporadic resulting from de novo mutation.
There is still no treatment for this disorder that can halt or reverse the symptoms including muscle weakness.
One ORF has been identified in the double homeobox (DUX4) present for each D4Z4 pattern (6). It encodes a transcription factor that is expressed in FSHD myoblasts. It has been shown recently that the FSHD patients carry a specific nucleotide sequence creating a canonical DUX4 poly(A) signal, which stabilizes the D4Z4 transcripts and induces a toxic gain of function of the last DUX4 transcript (7). The expression of DUX4 mRNA is influenced by the shortening of the telomere (8). DUX4 is currently believed to be the main candidate target for FSHD therapy and is the main focus of research (6,9). However, the molecular basis of FSHD and the exact mechanisms of this disease remain poorly understood.
Other genes in 4q35 locus, FRG2, DUX4c, FRG1, FAT1 and ANT1/SLC25A4, have been also considered responsible for the observed pathology (10,11) (Figure 1). The function of FRG2 (FSH Region Gene 2) remains unknown. However, it has been shown that the induction of myoblast differentiation causes strong FRG2 overexpression (12). A truncated and inverted unit of D4Z4 expressing DUX4c can inhibit myoblast differentiation (13). DUX4c protein upregulation has been reported in myoblasts of FSHD patients and in some FSHD biopsies (14). FRG1 (FSH Region Gene 1) is a highly conserved ubiquitous protein possibly involved in RNA biogenesis and actin-binding (15). Transgenic mice overexpressing FRG1 manifest the muscle degeneration identical to human FSHD (16). FAT1 (Atypical cadherin 1) plays a role in cellular polarization, directed cell migration and cell-cell contact. FAT1-deficient mice developed an FSHD-like phenotype (17). The FAT1 defective splice variants were found in FSHD patients (a contraction-independent FSHD variant) (18). Finally, ANT1 (Adenine Nucleotide Translocator 1) is expressed primarily in the heart and skeletal muscles and encodes a carrier of ADP/ATP of the mitochondrial inner membrane. The RNA and protein expression studies have shown an increase of the ANT1 level in FSHD muscles suggesting an early role of the protein in the development of the disease (10,11).
Figure 1.
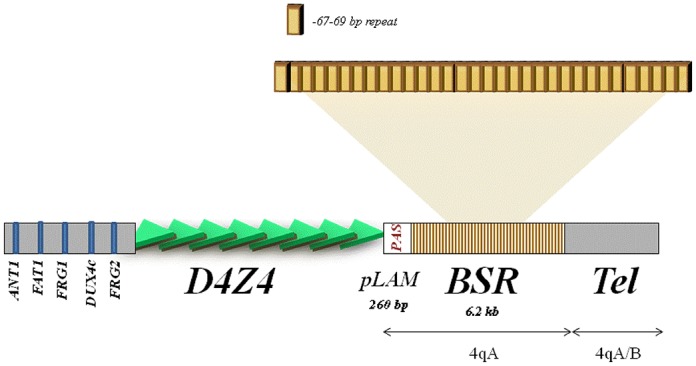
Schematic presentation of the 4qA allele and gene candidates in the 4q35 locus.
It has been also shown that epigenetic changes in the myoblasts of FSHD patients could play a crucial role in the FSHD development (7,19–22). The D4Z4 shortening correlates with hypomethylation of the D4Z4 repeat array and destabilizes the structure of chromatin that could lead to changes in the expression pattern of the neighboring genes.
Another type of FSHD, FSHD-like or phenotypic FSHD, represents 5% of FSHD cases and is characterized by the high frequency of sporadic cases (70% of FSHD-like) and the absence of macrosatellite contraction in 4q (23). This type of FSHD is associated with strong hypomethylation of the D4Z4 macrosatellites on chromosomes 4q and 10q (24,25) and could be related to the haploinsufficiency of SMCHD1 gene, which is involved in a pathway mediating the methylation of CpG islands (26) and probably required for DUX4 silencing in somatic cells. (25,27).
It has been shown that the canonical and phenotypic types of FSHD are associated with a permissive haplotype of the 4q subtelomere (7,23,27,28). It is a necessary condition for the FSHD manifestation, which consists of polymorphisms surrounding the D4Z4 tandem: the 4qA allele in 3′ from D4Z4 (Figure 1) is represented by (i) a pLAM sequence containing a polyadenylation sequence (PAS) of a most distal copy of DUX4 (29,30) and (ii) a β-satellite repeats (BSR) tandem of 6.2 kb length (31), along with a 161 SNP in 5′ from D4Z4 (7). Despite the recent findings demonstrating the heterogeneity of the 5′ haplotype, the conservative presence of the 4qA allele in the 98.7% cases strongly supports the importance of 4qA in FSHD (32).
Although 4qA has been characterized structurally and the role of pLAM sequence has been determined, little is known about the functional role of its BSR part. Previously, using the 3C methodology, which allows the analysis of the chromosome's organization in a cell (33), we demonstrated a transcriptional regulation activity of 4qA and its physical proximity to the promoters of the ANT1 and FRG1 genes in the nuclei of the FSHD primary myoblasts (19). This FSHD-associated chromatin conformation predisposes the 4qA allele as a key player of the transcription module that may influence the expression of genes potentially linked to FSHD. So far, there has been no other data concerning the 4qA-BSR function.
The present study reports the functional role of 4qA BSR in the muscular genetic disease FSHD. We demonstrate that an individual BSR could act as an active unit participating in the transcriptional regulation of 4q35 genes. Moreover, we show that the intrinsic sequence differences between BSR units reveal variable transcriptional activity and could interact with different protein partners. This might have implications for understanding the differences in the severity of the disease. Furthermore, we demonstrate that the BSR 4qAe enhancer can regulate the transcriptional activity of the ANT1 promoter. In order to characterize the molecular mechanism of the BSR action, we have found ZNF555, a trans-acting partner interacting with the 4qAe enhancer. This is an uncharacterized protein containing 15 zinc fingers and a KRAB domain, which could possess the property of transcriptional factor. We investigated the expression level of ZNF555 and found that it was high in committed myogenic cells in comparison with their undifferentiated precursors and non-myogenic origin cells. The depletion of ZNF555 strongly influences the ANT1 gene transcription in FSHD myoblasts possibly via the 4qAe enhancer separated by 5 Mb distance in the genome that was demonstrated by the reporter assay. This could be explained by the precedent looping model based on direct interaction of 4qA and the ANT1 promoter in 4q35 locus (19). Accordingly, the effect of the 4qA allele through the ZNF555 protein opens a new perspective for the understanding of the FSHD disease development at the molecular level, alternative (or complementary) to the DUX4-dependent mechanism. We believe that our findings could be significant for understanding the FSHD pathogenesis and the search for new therapeutic targets.
MATERIALS AND METHODS
Cell culture
We performed experiments on primary human myoblasts from three healthy individuals: Myo_N1, Myo_N2, Myo_N3 and three patients living with FSHD: Myo_F1 (D4Z44), Myo_F2 (D4Z47) and Myo_F3 (D4Z42) (Myobank-AFM, Institute of Myology, Paris and CHU A. de Villeneuve, Montpellier, France) (34), the induced pluripotent stem (iPS) cells were generated from myoblasts and mesenchymal stem cells (MSC) were differentiated from the iPS cells (35).
CD56-positive myoblasts were cultured in a growth medium consisting of 199 medium and Dulbecco's modifiedEagle's medium (DMEM) (Invitrogen) in a 1:4 ratio, supplemented with 20% Fetal Calf serum (FCS) (Invitrogen), 2.5 ng/ml hepatocyte growth factor (Invitrogen), 0.1 μmol/l dexamethasone (Sigma-Aldrich) and 50 μg/ml gentamycin (Invitrogen) at 37°C with 5% CO2. Myoblasts were differentiated in myotubes in DMEM medium with 2% FCS for 6 days. The multinucleated myotubes formation was controlled by haematoxylin staining (Sigma-Aldrich). Immortalized myobalsts were generated at the Institute of Myology (Paris, France) (36).
The iPS cells were maintained on human foreskin fibroblast (BJ1) feeder cells (provided by i-STEM), which were mitomycin-C growth-arrested (Invitrogen). The hES culture medium was KO/DMEM (Invitrogen) supplemented with a 20% knockout serum replacement (KSR) (Invitrogen), 0.1 mM nonessential amino acids (Invitrogen), 2 mM glutamax (Invitrogen), 50 μM β-mercaptoethanol (Invitrogen) and 100 UI/ml penicillin/streptomycin (Invitrogen). The iPS cells were passaged every 7 days.
The MSC were cultured in MSC medium containing KO/DMEM (Invitrogen) supplemented with 20% FCS, 0.1 mM nonessential amino acids (NEAA) (Invitrogen), 2 mM glutamax, 50 μM β-mercaptoethanol and 100 UI/ml penicillin/streptomycin (Invitrogen). The medium was changed every 2–3 days.
The HeLa cell line has been purchased from the ATCC Bioresource centre. The rhabdomyosarcoma (RMS) cells (TE671, Sigma-Aldrich) were maintained in a standard DMEM (Gibco, Life Technologies)/10% FBS (PAA Laboratories BmbH) medium condition with 100 UI/ml penicillin/streptomycin (Invitrogen).
Plasmids and constructs
The constructs on the base of pGL3 vectors (Promega) were used for transient transfection studies (Supplementary Table S1). The pGL3-SV40Pro (pPro) vector contains an SV40 promoter upstream of the luciferase gene in contrast to pGL3 (pBasic) without the promoter. The pGL3-SV40Pro-SV40enh (pControl) vector containing the sequences of the SV40 promoter and the SV40 enhancer was used as a positive control to identify a putative enhancer within the BSR sequence.
The p4qA plasmid containing the BSR fragment corresponding to the 4qA allelic variant was designed earlier (19). We have designed a series of new constructions p4qA_1, p4qA_2 and p4qA_3 first cutting the BSR fragment on three fragments by BglII enzyme: Fr1 (564 bp), Fr2 (435 bp) and Fr (489 bp). Each of these fragments was subcloned into the pPro vector' upstream of the SV40 promoter. In the second series of experiments, we amplified the smaller fragments Fr4-Fr7 from Fr1, Fr2 and Fr3 and inserted them in pPro via BglII sites to find out a minimal activator inside the BSR sequence. The size of fragments varies from 63 to 203 bps. All constructs were analyzed by sequencing.
To construct the pANT1 and p4qAe_ANT1 plasmids expressing the endogenous luciferase under the control of the ANT1 promoter, a DNA fragment of 715 bp containing a wild-type of human ANT1 promoter was amplified by polymerase chain reaction (PCR) using the following primers: FW: 5′-GCTAGATCTGAATTCACCTAGTGGCCC-3′ and RV: 5′-TCTAAGCTTCGCGCAGTCCCCGA-3′; the fragment was inserted into the pBasic and p4qAe vector backbones via BglII and HindIII sites.
The construct pFRG1 (Supplementary Table S1) with the human FRG1 promoter was designed earlier (37) and the 4qAe PCR fragment was inserted in the MluI opened pFRG1 vector upstream from the FRG1 promoter (p4qAe_FRG1).
The sequences of the fragments used in these experiments are presented in Supplemental Materials (Supplementary Table S1).
Luciferase assay
The transfection procedure and the preparation of the complexes were performed using the JetPEI™ reagent and 0.5 μg of DNA according to the manufacturer's recommended protocol (Polyplus transfection Inc). HeLa and TE671 cells were seeded on 48-well culture vessels, 2.5 × 104 cells per well the day before the transfection. After a 48-h incubation period, the gene activity was assessed by reagents and supplied protocol with the Luciferase Assay System Kit (Promega). Luminescence detection was performed by a Microlumat LB96p device (EGG Berthold, Bad Wildbad, Germany). Protein concentration in the extracts was measured by QuantiPro BSA Assay Kit (Sigma-Aldrich).
Nuclear extract preparation and olygonucleotide labeling
Nuclear extracts from TE671 were prepared using high salt lysis/extraction buffer: 10 mM NaCl, 20 mM HEPES [pH 7.6], 1.5 mM MgCl2, 1 mM ZnSO4, 20% glycerol, 0.1% Triton X-100, 1 mM dithiothreitol, 1 mM phenylmethylsulfonyl fluoride (38). The oligonucleotides corresponding to the BSR-fragments were annealed (sense and antisense) and DIG-end-labeled with terminal transferase (Roche) and DIG-ddUTP (Roche).
DIG electrophoresis mobility shift assay (DIG-EMSA)
We based on the protocol from the DIG Gel Shift Kit manual (Roche Applied Science) adding our modifications. Briefly, binding assay was carried out in 25 mM HEPES pH7.6 containing 1 mM EDTA, 10 mM (NH4)2SO4, 1 mM DTT, 0.2% (w/v) Tween20, 30 mM KCl, 0.5 mg poly(dI-dC) as a non-specific nucleotide competitor, 0.2 pmoles of DIG-labeled ds-oligo and 0.25 mg nuclear protein extract in a final volume of 5.5 μl. All samples were incubated for 15 min at room temperature. After the PAAG electrophoresis the DIG-labeled oligo was transferred to the positively charged nylon membrane (Hybond N+, Amersham) equilibrated in 0.5× Tris/Borate/EDTA buffer (Euromedex) by electro-blotting. The transfer was performed at 300mA, 30V for 1 h. Next, the membrane was rinsed in 2× SSC (Euromedex), air-dried during 15 min and baked at 80°C for 30 min without UV-cross-linking. The blot was incubated with the anti-DIG antibodies (Roche Applied Science) and revealed using the ECLplus kit (Amersham). The films were scanned and developed using Kodak X-OMAT 2000 device. The sequences of oligonucleotides corresponding to Fr 4 (4qAe), Fr 6 and Fr 7 are presented in Supplementary Table S1. The DIG-EMSA experiment was performed in triplicate and the results were treated using the ImageJ software (NIH, USA).
Detection of 4qA enhancer-binding proteins
The PCR product corresponding to the strongest enhancer from BSR region was incubated with the nuclear extracts (2.5 mg protein) from TE671 cells and was shifted by the DIG-EMSA procedure. The samples were performed in double. One part of the gel was separated and subjected to DIG-EMSA in order to check for the presence of DNA–protein bands. The other part of the gel was visualized using the Coomassie Brilliant Blue R250 (Thermo Scientific Pierce). The bands of interest were determined by the superposition of the film from DIG-EMSA and excised.
NanoLC-ESI-MS/MS for peptide mass fingerprint analysis
The protein bands corresponding to the proteins bound to the 4qAe oligo (Fr 4) were excised from the gel and processed as in (39). The gel slices were dehydrated with 300 μl of 50% acetonitrile followed by 300 μl of 100% acetonitrile and then re-hydrated with 300 μl of 50 mM ammonium bicarbonate. A final dehydration was performed with two washes of 300 μl of 50% acetonitrile followed by two washes of 300 μl of 100% acetonitrile. Each wash was carried out for 10 min at 25°C with shaking at 1400 rpm. The gel slices were dried in a SpeedVac at 35°C for 10 min. For trypsin digestion, the slices were pre-incubated with 7 ml of 15 ng/ml trypsin (Promega # V5280) at room temperature for 10 min. Afterward, 25 μl of 50 mM ammonium bicarbonate was added, and the gel slices were incubated at 37°C for 16 h. The peptide-containing supernatants were dried at 56°C by SpeedVac for 30 min and then resuspended in 20 μl of solution containing 0.05% formic acid and 3% acetonitrile for mass spectrometry experiments. The resulting peptides were analyzed with a nano-HPLC (Agilent Technologies 1200) directly coupled to an ion-trap mass spectrometer (Bruker 6300 series) equipped with a nano-electrospray source. The separation gradient was 7 min from 5 to 90% acetonitrile. The fragmentation voltage was 1.3 V. The ion trap acquired successive sets of four scan modes consisting of the full scan MS over the ranges of 200–2000 m/z followed by three data-dependent MS/MS scans on the three most abundant ions in the full scan. The analysis of the spectra was performed with the DataAnalysis for the 6300 Series Ion Trap LC/MS Version 3.4 software package and the proteins were identified with Spectrum Mill Software package (Agilent).
The sequences obtained were screened against the SWISS-PROT database using a MASCOT search engine. The searches were carried out with a peptide mass accuracy tolerance of 100 ppm for external calibration.
Total RNA extraction and quantitative RT-PCR analysis
Total RNA was extracted using the Illustra Triple Prep Kit (GE Healthcare Life Sciences). The cDNA was synthesized from 500 ng of total RNA using the RevertAid H Minus First Strand cDNA Synthesis Kit (Fermentas) for RT-PCR analysis according to the manufacturer's instructions. The qPCR was performed on a real-time PCR detection system Step One Plus (Applied Biosystems) using the KiCqStart SYBR Green qPCR Ready Mix (Sigma-Aldrich). The sequences of the oligonucleotides used in this study are listed in the Supplementary Table S2.
Protein isolation and immunoblotting
The sample pellets were diluted in 150–300 μl of 1× phosphate buffered saline with a Protease inhibitor cocktail (Sigma-Aldrich) and sonicated. A NuPAGE® LDS Sample Buffer (Life Technologies) and 5 mM DTT were added and the samples were subjected to the 30 min incubation at 50°C. Alternatively, total proteins were extracted using the Illustra Triple Prep Kit (GE Healthcare Life Sciences). The western blot was carried out according to a standard procedure using pre-cast gel cassettes (Life Technologies). We used anti-ZNF555 (Ab4) (Sigma-Aldrich), anti-Actin (Sigma-Aldrich) and anti-Lamin A/C (Abcam) antibodies in 1/1000 dilution and anti-rabbit secondary antibodies (Sigma-Aldrich) in 1/1000 dilution. The SeeBlue Plus2 Protein Standard (Life Technologies) was used to detect the size of analyzed protein. The detection solutions were the SuperSignal West Pico Chemiluminescent Substrate (Pierce Thermo Scientific) or Chemicon (Millipore).
ZNF555 knockdown
Cultured human TE671 cells, myoblasts from FSHD patients (Myo_F3) and healthy individuals (Myo_N3), were transfected with an esiRNA-targeting ZNF555 (Sigma-Aldrich) as described previously (40) using a Lipofectamine RNAiMAX transfection reagent (Invitrogen, Life Technologies). Briefly, before 24 h of transfection, the cells were trypsinized, collected, diluted with fresh medium and seeded in 6-well plates. Initial transfections for identifying effective esiRNAs were carried out in 24-well cell culture plates. Each sample was assayed in triplicate 72 h post-transfection. A non-specific esiRNA-targeting firefly luciferase was included as a negative control (Sigma-Aldrich). The cotransfection experiments were performed with the luciferase reporter pGL3 plasmids and esiRNA-targeting ZNF555 using a DNA (μg) to Lipofectamine™ 2000 (μl) transfection reagent (Invitrogen, Life Technologies) ratio of 1:2, followed by luciferase assay 72 h post-transfection. The esiRNA were used at a final concentration of 50 nM in all experiments.
ChIP assay
A ChIP assay was performed in primary myoblasts (Myo_N3 and Myo_F3) using anti-ZNF555 (Sigma-Aldrich), antibodies against acetylated H3K9 histone (Abcam) and mouse IgG (Sigma-Aldrich). One microgram of each antibody was used per each precipitation well. We used an Imprint Chromatin Immunoprecipitation Kit (Sigma-Aldrich) and followed the manufacture's instruction. The GAPDH (Sigma-Aldrich) and BSR, 4qAe-f (5′-TCCCCTGTAGGCAGAGA-3′) and 4qAe-r (5′-CACTGATAACCCAGGTGA-3′), primers were used in these experiments. The sequences of all oligonucleotides used in this study are listed in the Supplementary Table S2.
RESULTS
The β-satellite repeat (BSR) from the 4qA allele possess an enhancer activity
The 4qA sequence is represented by 6.2 kb of BSR region that is not present in the 4qB allele (Figure 1). An individual BSR is a 67–69 bp repeat that includes a Sau3AI restriction site and exhibits a nucleotide polymorphism (41) that could influence the activation capability of each repeat (Supplementary Figure S1). All our constructs were based on the pGL3 vector containing the SV40 promoter (pPro). The construct p4qA (19) contains ∼20 BSR (total 1474 bp, Figure 2) from the 4qA allele. First, the 4qA fragment was divided in three 4qA fragments, Fr 1, Fr 2 and Fr 3 in the size range 430–560 bp and subcloned each of them in the pPro vector upstream from the SV40 promoter (Figure 2, Supplementary Table S1). The corresponding constructs, p4qA_1, p4qA_2 and p4qA_3, respectively, were transiently transfected into RMS cells. It was observed that each fragment increased to the same extent of the reporter gene activity relative to the pPro control. Thus, we hypothesized that the presence of any BSR could induce the activation of transcription.
Figure 2.

A BSR repeat from the 4qA allele holds a transcriptional activator property. The transcriptional effect of the fragments from the 4qA allele was tested 48 h after transfection in RMS cells. The enhancer strength was quantified relative to the luciferase activity generated by the pGL3 plasmid with the SV40 promoter (pPro). The equal amounts of the plasmids were transfected. Luciferase signals were normalized to the total protein content in the extracts. All our constructs were on the bases of pGL3 enhancerless vector (pPro) carrying an SV40 promoter. All inserts from the 4qA region were placed upstream of the SV40 promoter. The initial 4qA fragment of the 20 BSR was divided into small fragments, which were sub-cloned in pGL3. Error bars correspond to a standard deviation of the values of three independent experiments.
In order to verify the minimal size of activation unit necessary for the activation of reporter gene expression, the additional constructs derived from the previous vectors were generated by shortening again the fragments in the size range 63–203 bp: Fr 4, 5, 6 and 7 ( p4qA_4–7). The maximum activation (36×) of luciferase expression was achieved with the p4qA_4 construct that contained an insertion fragment of 67 bp. It was at least twice as high comparing to the constructions with other sizes of the 4qA repeat fragments. Interestingly, the 63 bp fragment (less than one BSR) maintained the enhancer activity that was 14× higher than a control vector (pPro).
These results show that the highest enhancer activity corresponds to one BSR from 4qA region. The fragment Fr4 of 67 bp, which we named 4qAe has been used in our further experiments.
The 4qAe enhancer regulates the gene expression of 4q35 locus ex vivo
As we postulated previously from our 3C data, the 4qA region may be involved in the regulation of the transcriptional activity of the FRG1 and ANT1 genes (19). In order to confirm the hypothesis, we proceeded to investigate if the enhancer from 4qA (4qAe) would be able to activate the promoters of the ANT1 and FRG1 genes.
To this end, we designed several constructs on the basis of pBasic vector backbone placing 4qAe together with the promoters of the ANT1 or FRG1 genes (715 and 577 bp, respectively; Supplementary Table S1) and compared them with the constructs missing the enhancer. As shown in Figure 3, the transient transfection of the vector containing the 4qAe enhancer and the ANT1 promoter (p4qAe_ANT1) caused, on average, five-fold augmentation of reporter gene expression as compared with the corresponding enhancerless vectors (pANT1). The result of this experiment shows that the 4qAe sequence may contribute to the ANT1 gene activation, if it is put in a physical proximity to its promoter.
Figure 3.

A BSR repeat from the 4qA allele can regulate the promoter activity of ANT1 gene. The transcriptional effect of the 4qAe was tested 48 h after transfection in RMS cells. The enhancer strength was quantified relative to the luciferase activity generated by the pGL3 plasmid with the SV40 promoter (pPro) referenced as 1. The equal amounts of the plasmids were transfected. Luciferase signals were normalized to the total protein content in the extracts. All our constructs were on the bases of pGL3 vector. Bars indicate the luciferase reporter gene from pGL3, ovals indicate the promoter regions (ANT1 pro, FRG1 pro and SV40 pro) that were placed upstream from the luciferase sequence and arches indicate an enhancer (4qAe, SV40 enh). Error bars correspond to a standard deviation of the values of three independent experiments.
Intriguingly, we did not observe the same effect for 4qAe on the FRG1 promoter (p4qAe_FRG1) (Figure 3, Supplementary Table S1). There was no significant difference in luciferase expression between p4qAe_FRG1 and the corresponding enhancerless construct pFRG1.
The formation of 4qA-enhancer specific protein complex
In order to better understand the mechanism of the transcriptional activation of the genes situated in the 4q35 region associated with FSHD, we decided to test if there are nuclear proteins specifically binding to the 4qAe sequence. The DIG-EMSA essay with nuclear extracts from RMS revealed several slowly migrating complexes. One complex (BS1) appeared to be specific for the 4qA-enhancer fragment (Lane 3, Figure 4A). Its formation was 80% inhibited by the excess copies of the unlabeled 4qAe fragment, a cold competitor (Lane 4, Figure 4A). This loss of the DIG-labeled 4qA fragment from the specific protein complex was compensated by its equivalent enrichment in the free-oligo fraction FO (Lane 4, Figure 4A).
Figure 4.

Identification of 4qAe binding proteins. (A) DIG-EMSA indicates specific binding of 4qA enhancer (4qAe) with proteins from the nuclear extract of RMS cells. Lane 1 corresponds to the DIG-labelled 4qAe free oligo (FO), which was reacted with the nuclear extract (Lane 2) in the presence of poly(dI-dC) (Lane 3). The binding specificity of 4qAe was examined by the competition in binding reaction of nuclear proteins and the DIG-labeled 4qAe probe adding 200-fold molar excess of the unlabeled homologous 4qAe probe (cold competitor, Lane 4) or heterologous probes Fr 6 (Lane 5) or Fr 7 (Lane 6). The band shift 1 (BS 1) was specific for 4qAe. The relative quantity of 4qAe oligo was calculated as a percentage of total quantity (100%) of the oligo per lane. Error bars correspond to a standard deviation of the values of three independent experiments. (B) Schematic description of DIG-EMSA-MS approach. The gel shift experiment was run in double. One set of the samples (Exp 1, Ctl 1) was performed with the DIG labeled EMSA probe and the specific band was detected by anti-DIG antibodies. The resulting from the first set of the EMSA experiment film was superposed with the second set of samples (Exp 2, Ctl 2) and the corresponding bands were excised and analyzed by Mass spectrometry (MS). The nuclear extracts of control (Ctl 2) samples were run without an addition of the DNA probe as it was in experimental probe (Exp 2).
Next, we used different BSR sequences in competition assays to understand whether the intrinsic differences in BSR could influence the binding capacity of the 4qA-enhancer. The inclusion of the excess of heterologous competitors corresponding to Fr 6 and Fr 7 (Supplementary Table S1) partially diminished (about 35% in comparison with the experiment without competitors) the amount of DIG-4qAe-protein complex formation in the band shift 1 (Lane 5 and 6, Figure 4A). These results indicate the presence of 4qAe-specific protein(s) that do not interact with other BSR sequences.
ZNF555 protein specifically binds to the 4qA-enhancer
In the next part of our work, we decided to identify the protein candidates that could be involved in the 4qAe-binding and thus in the 4qA allele function.
For this purpose, we combined the DIG-EMSA and mass-spectrometry approaches, which would allow us to ‘fish out’ such proteins (Figure 4B). Two sets of probes were run in parallel in the gel shift experiment. In both cases, the cold or DIG-labeled 4qAe oligo (Fr 4), and an unspecific competitor poly(dI-dC) were used. In order to render the location of the EMSA bands detectable by chemiluminescence, the samples were spiked with a small amount of the DIG-labeled 4qAe probe in the first set of probes (Exp 1, Ctl 1). The control ‘cold’ EMSA (Ctl 1) was performed with the addition of excess 4qAe in order to detect the specific displaced band. The revealed DIG-EMSA film was superposed with the Coomassie stained gel of second set of probes. The band corresponding to the shift with 4qAe oligo (Exp 1) was excised from the second set of probes performed in triplicate and analyzed by NanoLC-ESI-MS/MS. The second set of experiments (Exp 2) was performed using ‘cold’ 4qA. Importantly, in parallel, the nuclear extracts of control samples (Ctl 2) were run without an addition of the DNA probe and the control band of the size of the specific band was also excised and analyzed. Thus, the protein composition between the experiment (Exp 2) and the control (Ctl 2) was compared.
We identified several proteins presented in the experiment (Supplementary Table S3) and control (Supplementary Table S4) bands. Among these proteins some of them were identified in both bands, whereas others were found only in the experiment band, and thus, were presumably associated with the DNA probe. From the set of proteins, a zinc finger protein 555 (ZNF555-Q8NEP9) was the most prominent candidate corresponding to search criteria. An MRM analysis confirmed the absence of ZNF555 in the control bands (Supplementary Figure S2A), whereas the non-specific control protein HSP71 (P11142) was present in both samples (Supplementary Figure S2B).
Thus, by the combination of DIG-EMSA and mass-spectrometry approach we identified a zinc finger protein ZNF555, which specifically binds to the 4qAe sequence.
mRNA and protein expression of ZNF555 in myoblasts
We analyzed the transcriptional profile of the ZNF555 gene during myogenesis by RT-PCR. All experiments were performed on four samples from iPS cells derived from human primary myobalsts, 4 samples from MSC differentiated from the iPS cells and 4 samples from myoblast primary cell lines (35).
We observed an augmentation (200 and 240%) of ZNF555 mRNA expression level in muscle-committed unipotent cells (myoblasts), as compared with pluripotent (iPS) and multipotent precursors (MSC), respectively (Figure 5, A). We did not find any difference in the expression of ZNF555 in FSHD myoblast cells as compared with the cells from healthy individuals (data not shown).
Figure 5.

ZNF555 expression is specific for myoblast cells. (A) Total mRNA from myogenic lineage cells: myoblasts, miPS (myoblast-derived iPS) cells and mMSC (miPS-derived mesenchymal stem cells) and non-myogenic: fibroblasts (fibro) and HeLa cells was tested for the ZNF555 expression by means of RT-PCR (n = 4 − 8). Data are normalized against GAPDH mRNA level and the value for GAPDH was equated to10. t-test for paired differences with HeLa cells was realized; ***P < .001, *P < 0.05. (B) ZNF555 protein expression analysis by Western blotting in FSHD (Myo_F), normal (Myo_N) and immortalized (Myo-i) myoblasts, RMS and HeLa cells. B-actin was used as a control. The data present the results from two independent experiments.
Furthermore, we analyzed the transcriptional level of ZNF555 in non-myogenic cells, fibroblasts and HeLa. The level of ZNF555 mRNA in HeLa cells was statistically lower as compared with all cells of myogenic lineage. The ZNF555 expression in fibroblasts showed no statistically significant difference from that in HeLa cells.
We investigated the ZNF555 protein expression level in order to confirm the data of transcriptome analysis. The ZNF555 protein has not yet been characterized; the antibodies against ZNF555 have been validated only in few cell types including HeLa cells. The purpose of this study was to analyze the level of ZNF555 in myoblast cells from normal and FSHD patients (Figure 5B). Consistent with the ZNF555 mRNA expression data, we did not find any difference in the ZNF555 expression between the cells obtained from healthy and affected individuals on the stage of myoblasts.
Further, we compared the ZNF555 protein level between tumor RMS and HeLa cells. We observed high ZNF555 protein expression in the tumor cells of myogenic origin as compared with HeLa cells, which are of epithelial origin. Surprisingly, unlike myoblasts, which did not undergo immortalization, the immortalized myoblasts cells showed a trace level of the ZNF555 protein expression.
Thus, the data of transcriptional and protein level analysis were consistent with each other. This part of our results suggests a possible myogenic specificity of ZNF555 expression.
ZNF555 is a transcriptional regulator of 4qAe
ZNF555 is an uncharacterized protein with a predicted function of transcriptional regulator according to its structure, which has not been experimentally validated. Therefore, we performed the ZNF555 knockdown in order to (i) experimentally determine the size of protein and (ii) provide the mechanistic insights into the ZNF555 contribution to functional region related to the FSHD disease. RMS cells were transfected with enzymatically prepared siRNA, as described previously (40) and analyzed for the gene and protein expression. An esiRNA-targeting firefly luciferase was used as a negative control. As shown in Figure 6A, we observed that the esiRNA against ZNF555 efficiently knocked down the ZNF555 transcription. Next, we performed a western blot analysis and confirmed the knockdown effect on the level of protein expression. We observed a strong band reduction, which corresponds to the size of 60 kDa, approximately (Figure 6B). In this experiment, we used a polyclonal antibody against ZNF555 provided by Sigma-Aldrich.
Figure 6.
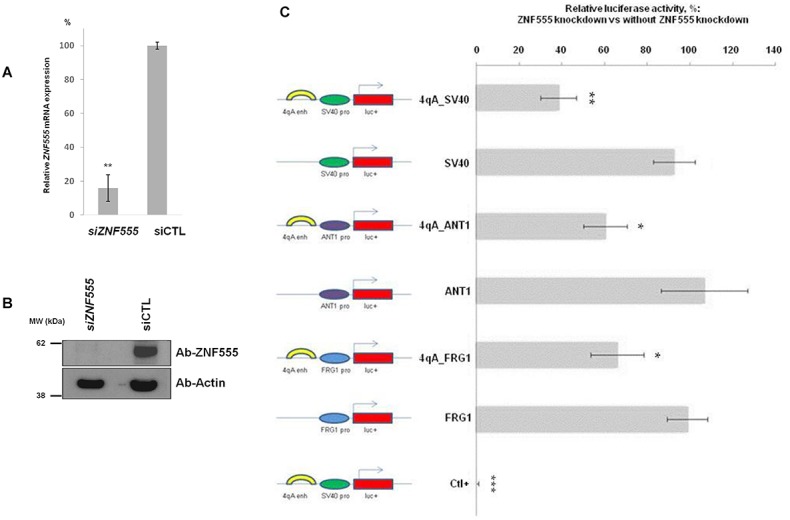
ZNF555 knockdown promotes the changes of luciferase expression in constructs with 4qAe enhancer. (A) Effect of 50 nM esiRNA-mediated knockdown of ZNF555 gene in RMS cells. mRNA quantification by qRT-PCR was done 72 h post transfection. siZNF555: cells transfected with esiRNA targeting ZNF555. Negative control siCTL: transfection of esiRNA targeting luciferase was used as a reference and set to 100%. The results represent three independent experiments measured in triplicate. (B) Western blotting validation of esiRNA-induced downregulation of ZNF555 72 h post transfection. Cells transfected with esiRNA targeting ZNF555 (siZNF555) or luciferase (siCTL). (C) Effect of ZNF555 knockdown using 50 nM esiRNA on 4qAe enhancer activity in RMS cells. The ZNF555 esiRNA was co-transfected with the set of luciferase reporter pGL3-based constructs, which contain the 4qAe enhancer or/and the promoters of ANT1 and FRG1 genes. The luciferase expression was analyzed 72 h post transfection. The data shown represent results of two experiments performed in triplicate and normalized against the luciferase expression of corresponding constructs without ZNF555 knockdown (set to 100%). The positive control (Ctl) performed with siRNA against luciferase gene. Data in A and C are shown as mean ± SD. t-test for paired differences with corresponding esiRNALUC Ctl- (A) and with experiments without ZNF555 knockdown (C) was realized; ***P < 0.001, **P < 0.01, *P < 0.05.
Next, we addressed whether the siRNA-mediated depletion of ZNF555 affected the activity of 4qAe enhancer. For this purpose, we performed the luciferase reporter assay using the same set of constructs as in previous experiments (Supplementary Table S1), i.e. vectors containing 4qAe enhancer and corresponding enhancerless vectors. Each reporter vector was transfected alone or co-transfected with esiRNA targeting ZNF555 in RMS cells. We found a significant reduction of luciferase gene expression upon ZNF555 inhibition in the case of constructs containing the 4qAe enhancer: p4qAe_SV40, p4qAe_FRG1 and p4qAe_ANT1 (Figure 6C). We did not observe the changes of luciferase expression in the corresponding enhancerless constructs containing the promoters of SV40, FRG1 and ANT1 genes (pSV40, pFRG1 and pANT1). These results are consistent with our EMSA-MS analysis regarding the specific interaction of ZNF555 with 4qAe. Together, these data established ZNF555 as a transcriptional regulator of 4qAe enhancer, which could play an important role in the transcriptional control of the activity of ANT1 and FRG1 genes through 4qAe.
ZNF555 knockdown changes the transcription level of ANT1 and FRG1
We next determined whether the suppression of ZNF555 expression could influence the activity of gene candidates ANT1 and FRG1. We performed the ZNF555 knockdown in FSHD and control primary myoblasts and RMS cells and analyzed the transcriptional level of ANT1 and FRG1 (Figures 6A and 7A). We found that the ZNF555 depletion inhibited the mRNA level of the ANT1 gene in both control and disease cases (Figure 7B). However, the ANT1 gene was more sensitive to ZNF555 knockdown in FSHD myoblasts in comparison with normal myoblasts, as the inhibition of ANT1 was considerably stronger (P < 0.01) in FSHD cells, while the changes of ANT1 expression were not statistically significant in normal myoblasts. Additionally, we observed a drastic reduction of FRG1 activity (P < 0.001) in both FSHD and normal myoblasts. Although the effect of ZNF555 knockdown impacted the FRG1 and ANT1 genes, in RMS cells the expression level of these genes stayed practically unmodified, as can be seen from Figure 7B. Therefore, these results provide the first evidence of a functional role of ZNF555 in 4q35 locus via the possible implication in the transcriptional gene regulation.
Figure 7.

ZNF555 knockdown promotes the changes of ANT1 and FRG1 expression. (A) Representative knockdown of ZNF555 in primary normal and FSHD myoblasts using esiRNA. Negative control (Ctl-): esiRNA-targeting luciferase. (B) Effect of 50 nM esiRNAZNF555 on ANT1 and FRG1 gene expression in the primary myoblasts of FSHD patients and healthy individuals and RMS. The extracted mRNA was analyzed by qPCR. Negative control (Ctl-): esiRNA-targeting luciferase. (A and B) Results represent two independent RT-qPCR experiments performed in triplicate and normalized against GAPDH and esiRNALUC Ctl- referenced as 100%. t-test for paired differences with corresponding esiRNALUC Ctl- was realized; ***P < 0.001, **P < 0.01. Myo-N: normal myoblasts, Myo-F: FSHD myoblasts, RMS: rhabdomyosarcoma cells.
ZNF555 association with 4qA BCR in FSHD and normal individuals
Our finding of the ZNF555 interaction with the 4qA BSR region relied on in vitro binding during EMSA-MS assay, and thus needed to be confirmed by experiments in living cells. Accordingly, we performed the ZNF555-specific ChIP assay in human myoblasts from control and FSHD patients. Using sets of specific primers, we analyzed a relative presence of different relevant parts of the 4q35 region in the ZN555 precipitates in FSHD myoblasts and control myoblasts (Supplementary Table S2).
First of all, we analyzed the ZNF555 precipitation in BSR region containing the 4qAe enhancer in FSHD and control myoblasts. As we can see from Figure 8A and B, the enrichment of ZNF555 was remarkably high in both samples. Further, we were interested in examining the transcriptionally functional regions corresponding to the known gene-candidates from 4q35 locus. Intriguingly, we found that ZNF555 binds the ANT1 promoter in both normal and FSHD individuals, but with different level of association (Figure 8). Regarding the FRG1 gene, we did not detect significant association of ZNF555 with the FRG1 promoter region in myoblasts neither from FSHD patients nor from healthy individuals. This is consistent with the observation that the FRG1 promoter was not activated by the 4qAe presence in our reporter assay data. We did not find the ZNF555 binding to the FRG2 promoter neither in normal nor in FSHD myoblasts.
Figure 8.

ZNF555 is associated with the FSHD region 4q35 in vivo. (A) Chromatin immuneprecipitation (ChIP) performed in control (Ct) and facioscapulohumeral muscular dystrophy (FSHD) myoblasts using antibodies against ZNF555 and H3K9Ac. Input (positive control) DNA represents total chromatin and negative control represents the chromatin precipitated by normal rabbit IgG. (B) ChIP analysis of control (Ct) and FSHD myoblasts. ChIP results were normalized against Input = 10 and the IgG negative control was subtracted. GAPDH was amplified as a positive control for H3K9Ac. All PCR experiments were performed in a linear range of amplification and the band intensities were measured by ImageJ software. Student deviation was calculated from the data of two to three independent experiments.
Next, we analyzed the association of ZNF555 to D4Z4 repeats. Interestingly, we observed a strong interaction of ZNF555 with DUX4-containing canonical D4Z4 repeats, the reduction of which is associated with the disease. However, we must note that, because of the extensive amplification of the D4Z4 repeats in the human genome, in addition to 4q, our primers can also recognize the repeat sequences from 10q and a single sequence from 3q. Accordingly, these results reflect the ZNF555 binding to any D4Z4 repeat and we cannot be certain if there is a specificity of the ZNF555 interaction with D4Z4 from the 4q35 region. Then, we tested a short version of D4Z4 that contains a homologous DUX4c gene, located in the truncated and inversed D4Z4 unit. We did not find the presence of ZNF555 in DUX4c in both normal and diseased myoblasts.
In addition to the ZNF555 binding to the relevant regions of the 4q35 locus, we were also interested in the association of these regions with active state of chromatin. For this purpose, we analyzed the enrichment in H3K9 acetylated (H3K9Ac) histone, a mark of transcriptionally active chromatin. We observed that the presence of ZNF555 in the 4q35 region was constantly associated with this modified histone (Figure 8).
Together, our results demonstrate the association of ZNF555 with the FSHD region. Moreover, we confirmed the possibility of interaction of the 4qAe enhancer from the 4qA BSR region and the ZNF555 protein in myoblasts from FSHD patients and normal individuals.
DISCUSSION
FSHD is an epi/genetic satellite DNA related disease associated with at least two satellite repeats in 4q35: (i) macrosatellite (D4Z4) and (ii) β-satellite (a part of the 4qA allele). The slow progress in FSHD research could be explained by the lack of a functional model. The straightforward analysis using an in vivo animal model of the disease cannot be used since linked D4Z4-BSR clusters from the FSHD region are found only in chimpanzees and even this primate never suffers from FSHD (42).
Exacerbating the difficulties in the FSHD studies, recent publications emphasize the complexity of FSHD (29,32,43–45), pointing out that no gene candidate proposed so far is sufficient to explain FSHD development. Most of the current FSHD studies have been focused on the DUX4 transcript inside D4Z4 and its contraction (8,46). However, the inappropriate expression of the DUX4 stable transcript was observed in FSHD fibroblasts (29), which are not affected, as well as in muscle cells of some unaffected individuals without D4Z4 deletion (45). These somewhat controversial observations suggest that the role of DUX4 expression in FSHD and its potential role as a therapeutic target require a more thorough exploration. An additional complexity has been demonstrated by a recent study that identified healthy individuals (2.1% of healthy population) carrying the contracted D4Z4 with 4qA-PAS sequence (32). Accordingly, the role of the genetic regions other than 4q35 cannot be ruled out, as has been shown for the phenotypic form of FSHD (25,27).
However, the contribution of BSR in the 4q35 region has not been explored so far. It is known that the reduction of the macrosatellite tandem is associated with the presence of BSR downstream from the macrosatellite in patients with canonical FSHD or FSHD1. Nevertheless, without the BSR-containing 4qA allele the D4Z4-contraction alone is not pathological. It is important to know that the presence of BSR is associated with phenotypic FSHD or FSHD2/3, which is not related to the D4Z4 contraction (23). These observations strongly suggest the significance of the BSR presence in FSHD development. Therefore, in the present study, we have focused on the second satellite related to FSHD and studied (i) its functional role, (ii) its molecular targets and (iii) the mechanism of its transcriptional control in the FSHD-related 4q35 region.
β-satellite repeat is a transcriptional regulator
The β-satellite DNA is presented by the tandem arrays of divergent Sau3A 67–69 pb monomer repeat units (53% G + C) (41,47). BSR are adjacent to LSau 3.3 kb macrosatellites (D4Z4-like repeats) and show a predominant heterochromatic distribution, which includes all acrocentric chromosomes and chromosomes 1, 3, 4, 9, 10 and Y (7,28,48,49). Recently, the presence of BSR next to a newly created telomere has been shown to retard its replication timing (50). Intriguingly, our experiments demonstrate a transcriptional activity inside the BSR tandem of different length, from one up to several repeats and suggest that each BSR could have this property. Among those repeats, we have identified one, 4qAe, which exhibited particularly strong activation property.
Additionally, the reporter assays show that if 4qAe is put in physical proximity to the promoter of the ANT1 gene (gene located 5 Mbp distal to the 4qA region in the genome), it could strongly activate this promoter. Importantly, ANT1 is the only muscle-specific gene out of several known FSHD gene candidates and is overexpressed in FSHD cells (10,11). Also the ANT1 protein is involved in the redox system stability (51) and related to the hypersensitivity of FSHD muscles to oxidative stress (34).
It appears from this evidence that the role of BSR in the transcriptional control of the 4q35 locus might be other than heterochromatinization. Taking into account the GC-reach structure of BSR, their methylation state remains to be identified.
BSR polymorphism leads to the polymorphism of transcriptional activity
The comparison between 12 BSR sequences reveals a highly conserved Sau3A-containing part and a low homology part (Supplementary Figure S1). The presence of the conserved part might explain why all analyzed BSR fragments could similarly act as transcriptional enhancers in our experiments. One might expect, however, that the sequence differences between individual BSR could influence the degree of enhancer activity, either by changing the affinity to transcriptional activators or else by binding of transcriptional repressors. In addition, comparing to the individual repeats the whole 4qA has a lower transcription activation potential that could be explained by the tendency of repeats to form heterochromatin, inhibitory for transcription (52).
Our further experiments (EMSA data) provided additional evidence that an individual BSR has specific protein partners. At the same time, we do not rule out the possibility of common transcriptional factors shared by all repeats. Therefore, we suggest that a variety of transcription factors could be involved in transcriptional regulation by the 4qA allele. Further work is needed to clarify the role of sequence variability in the 4qA allele function, for example, by identifying the proteins that bind to different BSR sequences.
Overall, our results provide the first experimental evidence that BRS from the 4qA allele could act as transcription regulatory elements with different degree of enhancer activity. This could be a starting point for the future study concerning the BSR polymorphism in FSHD patients and healthy individuals. A cloning of additional 4qA alleles from FSHD patients and their analysis could be envisaged.
ZNF555 is a trans-acting element for BSR
In order to understand the mechanism of transcriptional activation by 4qAe and the role of BSR in the FSHD genesis, we searched for the proteins binding to 4qAe (the strongest BSR enhancer that we could find) using a combination of two technologies: DIG-EMSA and Mass-Spectrometry. This approach was developed by our team and enabled us to identify ZNF555, a protein that specifically interacts with BSR (4qAe). The knockdown of this protein leads to a significant reduction in the 4qAe enhancer activity for the ANT1 promoter in the reporter assay. Moreover, we observed a similar reduction effect with other promoters (SV40 and FRG1) when 4qAe was present. Given that the SV40 enhancer is not sensitive to the ZNF555 depletion, our results indicate a specific interaction between ZNF555 and the 4qAe enhancer. The ChIP data verify that ZNF555 is associated with the 4qAe region.
ZNF555 is an uncharacterized protein. The analysis of the ZNF555 expression profile provides evidence of its strong presence in myoblasts as compared with the cells of non-muscular origin, fibroblasts and HeLa cells. Moreover, we demonstrate that its expression is related to the muscle committed cells as compared with their pluripotent and multipotent precursors. Thus, we hypothesize that ZNF555 could play a role in myogenesis. Supporting this suggestion the transcriptome Meta-Analysis (53) reveals significant transcriptional changes in the ZNF555 level in musculoskeletal diseases (Nextbio.com, Illumina) (Supplementary Figure S3). Interestingly, a predicted functional partner available for the ZNF555 protein is ADAM12, a muscle specific cell surface protein (54,55) (String-db.org (56), Supplementary Figure S4).
With consideration of the potential molecular functions of ZNF555, this protein contains DNA-binding domains and thus could have a wide variety of functions, most of which encompass some form of transcriptional activation or repression. It could be a transcriptional activator due to the presence of 15 ‘zinc fingers’ of the Cys2-His2 type and a transcriptional repressor due to the presence of the KRAB (Krüppel associated box) domain. In our study, the ZNF555 presence in the 4q35 locus was associated with active chromatin marker and behaved rather as a transcriptional activator in luciferase assay.
Intriguingly, we did not observe a difference in the ZNF555 expression between FSHD and normal individuals, and thus we hypothesized that the permissive chromatin structure could play a role in the interaction of ZNF555 in the 4q35 region. Our data provide additional information consistent with other studies of histone modifications of 4q35 locus associated with repressed and active chromatin (57–59). Furthermore, in our experiments the BSR region is remarkably enriched with H3K9Ac in FSHD myoblasts in comparison with the control. This is in line with the model of epigenetic activation in the 4q35 locus in FSHD (20,24,60).
It should be noted that the 4q35.2 region presents high homology with chromosome 10, and the 4qA-like allele is also present in 10q region (28,61). However, taking together our proteomic and transcriptomic data, as well as the absence of potential myogenic genes in 10q26 and the low level of pairing between the homologous regions of 4q and 10q in the interphase nuclei (19,62), our data results implicate ZNF555 in the function of the 4q35 locus. It remains to be explored, however, whether the BSR and ZNF555 interaction could have broader implications, not restricted to 4q35.
Distant regulation in 4q35 locus
Our current study reveals the first evidence of 4qA BSR implication in the transcriptional regulation of the 4q35 locus and precisely, in ANT1 gene transcriptional activity. On the one hand, BSR can interact with the ZNF555 transcription factor, which impacts the activity of the ANT1 promoter in FSHD myoblasts. On the other hand, ZNF555 influences the promoter activity of the ANT1 gene via the 4qAe enhancer. Our results are in general agreement with the study of abnormal chromatin conformation changes in FSHD myoblasts (20,22,63) leading to the direct interactions between the 4qA region and the long distance genes in 4q35 (19). They are also consistent with the current model of interaction between distantly located enhancer and promoter through formation of the DNA loop enabling their physical interaction (64,65).
We would like to mention that the ZNF555 knockdown provoked the strong reduction of the FRG1 expression. Accordingly, ZNF555 can be involved in the FRG1 transcriptional control, but neither via direct interaction since it was not found (trace level) in the FRG1 promoter by ChIP, nor via BSR since the FRG1 promoter is not sensitive to the presence of 4qAe.
We therefore propose a working model (Figure 9) of the functional role of the 4qA allele in the transcriptional control of the ANT1 gene in FSHD patients, where the cooperative BSR binding with the ZNF555 transcriptional factor could be a critical step in the formation of a transcriptionally productive complex.
Figure 9.

Transcriptional control model of BSR from the 4qA allele through ZNF555 in the 4q35 locus.
Originally considered ‘junk’ DNA, satellite DNA have more recently been reconsidered as having various functions implicating it in different ‘satellite diseases’, including neurological, developmental disorders and cancers (66–70). Satellite repeats could be over-represented in transcription regulatory modules and may have an impact on replication, epigenetic regulation and genomic instability. A potential role of BSR in pathology has been demonstrated (71,72). Therefore, the manipulation of the BSR activity might be beneficial in the treatment of FSHD, although further investigations regarding the therapeutic potential of searching the transcriptional factors associated with 4qA are necessary.
In conclusion, the effect of BSR as a part of 4qA through the ZNF555 protein opens a new perspective for the understanding of the FSHD disease development at the molecular level, alternative (or complementary) to the DUX4-dependent mechanism, which has been considered as a major candidate for FSHD. We believe that our findings could be significant in the search for new therapeutic targets and, in general, for the research strategies for FSHD and other satellite DNA diseases.
Supplementary Material
Acknowledgments
We are grateful to Ac Dr W. Vainchenker (Inserm UMR1009, Villejuif, France) and Dr I. Petrache (Indiana University School of Medicine, Indianapolis, IN, USA) for the critical reading of the manuscript. We thank Dr Y. Vassetzky, Dr M. Lipinski (CNRS UMR8126, Villejuif, France) and A. Vayl, (B.Sc., IUSM, Indianapolis, IN, USA) for the helpful advice. We thank Dr D. Laoudj-Chenivesse and Dr G. Carnac (Inserm U1046, Université Montpellier 1, France) for providing the myoblast cells. We would like to acknowledge the iSTEM (Evry, France) for the help in the iPS cell generation and Dr V. Mouly (Myobank-AFM, Myology Institute, Paris, France) for providing the immortalized human myoblasts and primary myoblasts of patients. We thank Dr N. Droin for providing the EGR2-antibody. We thank the Proteomic Platform for the qualified validation of proteomic data (Gustave Roussy, Villejuif, France) and Mr R. Willett (CNRS) for the copy editing.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Association pour la Recherche sur le Cancer subvention n° [SFI20121205936 to V.O.]; Taxe d'Apprentissage (Gustave Roussy, Villejuif, France) for AK/VO/IP; Erasmus program (to A.K.). Funding for open access charge: CNRS Annual Funding [UMR8126].
Conflict of interest statement. None declared.
REFERENCES
- 1.Theadom A., Rodrigues M., Roxburgh R., Balalla S., Higgins C., Bhattacharjee R., Jones K., Krishnamurthi R., Feigin V. Prevalence of muscular dystrophies: a systematic literature review. Neuroepidemiology. 2014;43:259–268. doi: 10.1159/000369343. [DOI] [PubMed] [Google Scholar]
- 2.Padberg G.W., Lunt P.W., Koch M., Fardeau M. Diagnostic criteria for facioscapulohumeral muscular dystrophy. Neuromuscul. Disord. 1991;1:231–234. doi: 10.1016/0960-8966(91)90094-9. [DOI] [PubMed] [Google Scholar]
- 3.Tawil R., Figlewicz D.A., Griggs R.C., Weiffenbach B. Facioscapulohumeral dystrophy: a distinct regional myopathy with a novel molecular pathogenesis. FSH Consortium. Ann. Neurol. 1998;43:279–282. doi: 10.1002/ana.410430303. [DOI] [PubMed] [Google Scholar]
- 4.Landoyzy L., Dejerine J. De la myopathie atrophique progressive (myopathie héréditaire, débutant dans l'enfance par la face, sans altération du système nerveux) Comptes rendus de l'Académie des sciences, Paris. 1884;98:53–55. [Google Scholar]
- 5.Lemmers R.J., O'Shea S., Padberg G.W., Lunt P.W., van der Maarel S.M. Best practice guidelines on genetic diagnostics of Facioscapulohumeral muscular dystrophy: workshop 9th June 2010, LUMC, Leiden, The Netherlands. Neuromuscul. Disord. 2012;22:463–470. doi: 10.1016/j.nmd.2011.09.004. [DOI] [PubMed] [Google Scholar]
- 6.Tassin A., Laoudj-Chenivesse D., Vanderplanck C., Barro M., Charron S., Ansseau E., Chen Y.W., Mercier J., Coppee F., Belayew A. DUX4 expression in FSHD muscle cells: how could such a rare protein cause a myopathy. J. Cell. Mol. Med. 2013;17:76–89. doi: 10.1111/j.1582-4934.2012.01647.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lemmers R.J., van der Vliet P.J., Klooster R., Sacconi S., Camano P., Dauwerse J.G., Snider L., Straasheijm K.R., van Ommen G.J., Padberg G.W., et al. A unifying genetic model for facioscapulohumeral muscular dystrophy. Science. 2010;329:1650–1653. doi: 10.1126/science.1189044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stadler G., Rahimov F., King O.D., Chen J.C., Robin J.D., Wagner K.R., Shay J.W., Emerson C.P. Jr, Wright W.E. Telomere position effect regulates DUX4 in human facioscapulohumeral muscular dystrophy. Nat. Struct. Mol. Biol. 2013;20:671–678. doi: 10.1038/nsmb.2571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.van der Maarel S.M., Tawil R., Tapscott S.J. Facioscapulohumeral muscular dystrophy and DUX4: breaking the silence. Trends Mol. Med. 2011;17:252–258. doi: 10.1016/j.molmed.2011.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gabellini D., Green M.R., Tupler R. Inappropriate gene activation in FSHD: a repressor complex binds a chromosomal repeat deleted in dystrophic muscle. Cell. 2002;110:339–348. doi: 10.1016/s0092-8674(02)00826-7. [DOI] [PubMed] [Google Scholar]
- 11.Laoudj-Chenivesse D., Carnac G., Bisbal C., Hugon G., Bouillot S., Desnuelle C., Vassetzky Y., Fernandez A. Increased levels of adenine nucleotide translocator 1 protein and response to oxidative stress are early events in facioscapulohumeral muscular dystrophy muscle. J. Mol. Med. 2005;83:216–224. doi: 10.1007/s00109-004-0583-7. [DOI] [PubMed] [Google Scholar]
- 12.Rijkers T., Deidda G., van Koningsbruggen S., van Geel M., Lemmers R.J., van Deutekom J.C., Figlewicz D., Hewitt J.E., Padberg G.W., Frants R.R., et al. FRG2, an FSHD candidate gene, is transcriptionally upregulated in differentiating primary myoblast cultures of FSHD patients. J. Med. Genet. 2004;41:826–836. doi: 10.1136/jmg.2004.019364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bosnakovski D., Lamb S., Simsek T., Xu Z., Belayew A., Perlingeiro R., Kyba M. DUX4c, an FSHD candidate gene, interferes with myogenic regulators and abolishes myoblast differentiation. Exp. Neurol. 2008;214:87–96. doi: 10.1016/j.expneurol.2008.07.022. [DOI] [PubMed] [Google Scholar]
- 14.Ansseau E., Laoudj-Chenivesse D., Marcowycz A., Tassin A., Vanderplanck C., Sauvage S., Barro M., Mahieu I., Leroy A., Leclercq I., et al. DUX4c is up-regulated in FSHD. It induces the MYF5 protein and human myoblast proliferation. PLoS One. 2009;4:e7482. doi: 10.1371/journal.pone.0007482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu Q., Jones T.I., Tang V.W., Brieher W.M., Jones P.L. Facioscapulohumeral muscular dystrophy region gene-1 (FRG-1) is an actin-bundling protein associated with muscle-attachment sites. J. Cell Sci. 2010;123:1116–1123. doi: 10.1242/jcs.058958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gabellini D., D'Antona G., Moggio M., Prelle A., Zecca C., Adami R., Angeletti B., Ciscato P., Pellegrino M.A., Bottinelli R., et al. Facioscapulohumeral muscular dystrophy in mice overexpressing FRG1. Nature. 2006;439:973–977. doi: 10.1038/nature04422. [DOI] [PubMed] [Google Scholar]
- 17.Caruso N., Herberth B., Bartoli M., Puppo F., Dumonceaux J., Zimmermann A., Denadai S., Lebosse M., Roche S., Geng L., et al. Deregulation of the protocadherin gene FAT1 alters muscle shapes: implications for the pathogenesis of facioscapulohumeral dystrophy. PLoS Genet. 2013;9:e1003550. doi: 10.1371/journal.pgen.1003550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Puppo F., Dionnet E., Gaillard M.C., Gaildrat P., Castro C., Vovan C., Karine B., Bernard R., Attarian S., Goto K., et al. Identification of variants in the 4q35 gene FAT1 in patients with a Facioscapulohumeral dystrophy (FSHD)-like phenotype. Hum. Mutat. 2015;36:443–453. doi: 10.1002/humu.22760. [DOI] [PubMed] [Google Scholar]
- 19.Pirozhkova I., Petrov A., Dmitriev P., Laoudj D., Lipinski M., Vassetzky Y. A functional role for 4qA/B in the structural rearrangement of the 4q35 region and in the regulation of FRG1 and ANT1 in facioscapulohumeral dystrophy. PLoS One. 2008;3:e3389. doi: 10.1371/journal.pone.0003389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Petrov A., Pirozhkova I., Carnac G., Laoudj D., Lipinski M., Vassetzky Y.S. Chromatin loop domain organization within the 4q35 locus in facioscapulohumeral dystrophy patients versus normal human myoblasts. Proc. Natl. Acad. Sci. U.S.A. 2006;103:6982–6987. doi: 10.1073/pnas.0511235103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Krom Y.D., Thijssen P.E., Young J.M., den Hamer B., Balog J., Yao Z., Maves L., Snider L., Knopp P., Zammit P.S., et al. Intrinsic epigenetic regulation of the D4Z4 macrosatellite repeat in a transgenic mouse model for FSHD. PLoS Genet. 2013;9:e1003415. doi: 10.1371/journal.pgen.1003415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xu X., Tsumagari K., Sowden J., Tawil R., Boyle A.P., Song L., Furey T.S., Crawford G.E., Ehrlich M. DNaseI hypersensitivity at gene-poor, FSH dystrophy-linked 4q35.2. Nucleic Acids Res. 2009;37:7381–7393. doi: 10.1093/nar/gkp833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.de Greef J.C., Lemmers R.J., Camano P., Day J.W., Sacconi S., Dunand M., van Engelen B.G., Kiuru-Enari S., Padberg G.W., Rosa A.L., et al. Clinical features of facioscapulohumeral muscular dystrophy 2. Neurology. 2010;75:1548–1554. doi: 10.1212/WNL.0b013e3181f96175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.van Overveld P.G., Lemmers R.J., Sandkuijl L.A., Enthoven L., Winokur S.T., Bakels F., Padberg G.W., van Ommen G.J., Frants R.R., van der Maarel S.M. Hypomethylation of D4Z4 in 4q-linked and non-4q-linked facioscapulohumeral muscular dystrophy. Nat. Genet. 2003;35:315–317. doi: 10.1038/ng1262. [DOI] [PubMed] [Google Scholar]
- 25.Larsen M., Rost S., El Hajj N., Ferbert A., Deschauer M., Walter M.C., Schoser B., Tacik P., Kress W., Muller C.R. Diagnostic approach for FSHD revisited: SMCHD1 mutations cause FSHD2 and act as modifiers of disease severity in FSHD1. Eur. J. Hum. Genet. 2014;23:808–816. doi: 10.1038/ejhg.2014.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Blewitt M.E., Gendrel A.V., Pang Z., Sparrow D.B., Whitelaw N., Craig J.M., Apedaile A., Hilton D.J., Dunwoodie S.L., Brockdorff N., et al. SmcHD1, containing a structural-maintenance-of-chromosomes hinge domain, has a critical role in X inactivation. Nat. Genet. 2008;40:663–669. doi: 10.1038/ng.142. [DOI] [PubMed] [Google Scholar]
- 27.Lemmers R.J., Tawil R., Petek L.M., Balog J., Block G.J., Santen G.W., Amell A.M., van der Vliet P.J., Almomani R., Straasheijm K.R., et al. Digenic inheritance of an SMCHD1 mutation and an FSHD-permissive D4Z4 allele causes facioscapulohumeral muscular dystrophy type 2. Nat. Genet. 2012;44:1370–1374. doi: 10.1038/ng.2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lemmers R.J., de Kievit P., Sandkuijl L., Padberg G.W., van Ommen G.J., Frants R.R., van der Maarel S.M. Facioscapulohumeral muscular dystrophy is uniquely associated with one of the two variants of the 4q subtelomere. Nat. Genet. 2002;32:235–236. doi: 10.1038/ng999. [DOI] [PubMed] [Google Scholar]
- 29.Snider L., Geng L.N., Lemmers R.J., Kyba M., Ware C.B., Nelson A.M., Tawil R., Filippova G.N., van der Maarel S.M., Tapscott S.J., et al. Facioscapulohumeral dystrophy: incomplete suppression of a retrotransposed gene. PLoS Genet. 2010;6:e1001181. doi: 10.1371/journal.pgen.1001181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dixit M., Ansseau E., Tassin A., Winokur S., Shi R., Qian H., Sauvage S., Matteotti C., van Acker A.M., Leo O., et al. DUX4, a candidate gene of facioscapulohumeral muscular dystrophy, encodes a transcriptional activator of PITX1. Proc. Natl. Acad. Sci. U.S.A. 2007;104:18157–18162. doi: 10.1073/pnas.0708659104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.van Geel M., Dickson M.C., Beck A.F., Bolland D.J., Frants R.R., van der Maarel S.M., de Jong P.J., Hewitt J.E. Genomic analysis of human chromosome 10q and 4q telomeres suggests a common origin. Genomics. 2002;79:210–217. doi: 10.1006/geno.2002.6690. [DOI] [PubMed] [Google Scholar]
- 32.Scionti I., Greco F., Ricci G., Govi M., Arashiro P., Vercelli L., Berardinelli A., Angelini C., Antonini G., Cao M., et al. Large-scale population analysis challenges the current criteria for the molecular diagnosis of fascioscapulohumeral muscular dystrophy. Am. J. Hum. Genet. 2012;90:628–635. doi: 10.1016/j.ajhg.2012.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dekker J., Rippe K., Dekker M., Kleckner N. Capturing chromosome conformation. Science. 2002;295:1306–1311. doi: 10.1126/science.1067799. [DOI] [PubMed] [Google Scholar]
- 34.Barro M., Carnac G., Flavier S., Mercier J., Vassetzky Y., Laoudj-Chenivesse D. Myoblasts from affected and non-affected FSHD muscles exhibit morphological differentiation defects. J. Cell Mol. Med. 2010;14:275–289. doi: 10.1111/j.1582-4934.2008.00368.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pirozhkova I., Barat A., Dmitriev P., Kim E., Robert T., Guegan J., Bilhou-Nabera C., Busato F., Tost J., Carnac G., et al. Differences in transcription patterns between induced pluripotent stem cells produced from the same germ layer are erased upon differentiation. PLoS One. 2013;8:e53033. doi: 10.1371/journal.pone.0053033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mamchaoui K., Trollet C., Bigot A., Negroni E., Chaouch S., Wolff A., Kandalla P.K., Marie S., Di Santo J., St Guily J.L., et al. Immortalized pathological human myoblasts: towards a universal tool for the study of neuromuscular disorders. Skelet. Muscle. 2011;1:34. doi: 10.1186/2044-5040-1-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Petrov A., Allinne J., Pirozhkova I., Laoudj D., Lipinski M., Vassetzky Y.S. A nuclear matrix attachment site in the 4q35 locus has an enhancer-blocking activity in vivo: implications for the facio-scapulo-humeral dystrophy. Genome Res. 2008;18:39–45. doi: 10.1101/gr.6620908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Klochkov D., Rincon-Arano H., Ioudinkova E.S., Valadez-Graham V., Gavrilov A., Recillas-Targa F., Razin S.V. A CTCF-dependent silencer located in the differentially methylated area may regulate expression of a housekeeping gene overlapping a tissue-specific gene domain. Mol. Cell. Biol. 2006;26:1589–1597. doi: 10.1128/MCB.26.5.1589-1597.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shevchenko A., Wilm M., Vorm O., Mann M. Mass spectrometric sequencing of proteins silver-stained polyacrylamide gels. Anal. Chem. 1996;68:850–858. doi: 10.1021/ac950914h. [DOI] [PubMed] [Google Scholar]
- 40.Kolas N.K., Chapman J.R., Nakada S., Ylanko J., Chahwan R., Sweeney F.D., Panier S., Mendez M., Wildenhain J., Thomson T.M., et al. Orchestration of the DNA-damage response by the RNF8 ubiquitin ligase. Science. 2007;318:1637–1640. doi: 10.1126/science.1150034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Meneveri R., Agresti A., Della Valle G., Talarico D., Siccardi A.G., Ginelli E. Identification of a human clustered G + C-rich DNA family of repeats (Sau3A family) J. Mol. Biol. 1985;186:483–489. doi: 10.1016/0022-2836(85)90123-8. [DOI] [PubMed] [Google Scholar]
- 42.Kramer H., Han C., Post W., Goff D., Diez-Roux A., Cooper R., Jinagouda S., Shea S. Racial/ethnic differences in hypertension and hypertension treatment and control in the multi-ethnic study of atherosclerosis (MESA) Am J. Hypertens. 2004;17:963–970. doi: 10.1016/j.amjhyper.2004.06.001. [DOI] [PubMed] [Google Scholar]
- 43.Ricci G., Zatz M., Tupler R. Facioscapulohumeral muscular dystrophy: more complex than it appears. Curr. Mol. Med. 2014 doi: 10.2174/1566524014666141010155054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ricci G., Scionti I., Sera F., Govi M., D'Amico R., Frambolli I., Mele F., Filosto M., Vercelli L., Ruggiero L., et al. Large scale genotype-phenotype analyses indicate that novel prognostic tools are required for families with facioscapulohumeral muscular dystrophy. Brain. 2013;136:3408–3417. doi: 10.1093/brain/awt226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jones T.I., Chen J.C., Rahimov F., Homma S., Arashiro P., Beermann M.L., King O.D., Miller J.B., Kunkel L.M., Emerson C.P. Jr, et al. Facioscapulohumeral muscular dystrophy family studies of DUX4 expression: evidence for disease modifiers and a quantitative model of pathogenesis. Hum. Mol. Genet. 2012;21:4419–4430. doi: 10.1093/hmg/dds284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kowaljow V., Marcowycz A., Ansseau E., Conde C.B., Sauvage S., Matteotti C., Arias C., Corona E.D., Nunez N.G., Leo O., et al. The DUX4 gene at the FSHD1A locus encodes a pro-apoptotic protein. Neuromuscul. Disord. 2007;17:611–623. doi: 10.1016/j.nmd.2007.04.002. [DOI] [PubMed] [Google Scholar]
- 47.Waye J.S., Willard H.F. Human beta satellite DNA: genomic organization and sequence definition of a class of highly repetitive tandem DNA. Proc. Natl. Acad. Sci. U.S.A. 1989;86:6250–6254. doi: 10.1073/pnas.86.16.6250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Meneveri R., Agresti A., Marozzi A., Saccone S., Rocchi M., Archidiacono N., Corneo G., Della Valle G., Ginelli E. Molecular organization and chromosomal location of human GC-rich heterochromatic blocks. Gene. 1993;123:227–234. doi: 10.1016/0378-1119(93)90128-p. [DOI] [PubMed] [Google Scholar]
- 49.Cardone M.F., Ballarati L., Ventura M., Rocchi M., Marozzi A., Ginelli E., Meneveri R. Evolution of beta satellite DNA sequences: evidence for duplication-mediated repeat amplification and spreading. Mol. Biol. Evol. 2004;21:1792–1799. doi: 10.1093/molbev/msh190. [DOI] [PubMed] [Google Scholar]
- 50.Arnoult N., Schluth-Bolard C., Letessier A., Drascovic I., Bouarich-Bourimi R., Campisi J., Kim S.H., Boussouar A., Ottaviani A., Magdinier F., et al. Replication timing of human telomeres is chromosome arm-specific, influenced by subtelomeric structures and connected to nuclear localization. PLoS Genet. 2010;6:e1000920. doi: 10.1371/journal.pgen.1000920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lena A., Rechichi M., Salvetti A., Vecchio D., Evangelista M., Rainaldi G., Gremigni V., Rossi L. The silencing of adenine nucleotide translocase isoform 1 induces oxidative stress and programmed cell death in ADF human glioblastoma cells. FEBS J. 2010;277:2853–2867. doi: 10.1111/j.1742-4658.2010.07702.x. [DOI] [PubMed] [Google Scholar]
- 52.Khobta A., Anderhub S., Kitsera N., Epe B. Gene silencing induced by oxidative DNA base damage: association with local decrease of histone H4 acetylation in the promoter region. Nucleic Acids Res. 2010;38:4285–4295. doi: 10.1093/nar/gkq170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kupershmidt I., Su Q.J., Grewal A., Sundaresh S., Halperin I., Flynn J., Shekar M., Wang H., Park J., Cui W., et al. Ontology-based meta-analysis of global collections of high-throughput public data. PLoS One. 2010;5:e13066. doi: 10.1371/journal.pone.0013066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Galliano M.F., Huet C., Frygelius J., Polgren A., Wewer U.M., Engvall E. Binding of ADAM12, a marker of skeletal muscle regeneration, to the muscle-specific actin-binding protein, alpha -actinin-2, is required for myoblast fusion. J. Biol. Chem. 2000;275:13933–13939. doi: 10.1074/jbc.275.18.13933. [DOI] [PubMed] [Google Scholar]
- 55.Kronqvist P., Kawaguchi N., Albrechtsen R., Xu X., Schroder H.D., Moghadaszadeh B., Nielsen F.C., Frohlich C., Engvall E., Wewer U.M. ADAM12 alleviates the skeletal muscle pathology in mdx dystrophic mice. Am. J. Pathol. 2002;161:1535–1540. doi: 10.1016/S0002-9440(10)64431-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Franceschini A., Szklarczyk D., Frankild S., Kuhn M., Simonovic M., Roth A., Lin J., Minguez P., Bork P., von Mering C., et al. STRING v9.1: protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Res. 2013;41:D808–D815. doi: 10.1093/nar/gks1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bodega B., Ramirez G.D., Grasser F., Cheli S., Brunelli S., Mora M., Meneveri R., Marozzi A., Mueller S., Battaglioli E., et al. Remodeling of the chromatin structure of the facioscapulohumeral muscular dystrophy (FSHD) locus and upregulation of FSHD-related gene 1 (FRG1) expression during human myogenic differentiation. BMC Biol. 2009;7:41. doi: 10.1186/1741-7007-7-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jiang G., Yang F., van Overveld P.G., Vedanarayanan V., van der Maarel S., Ehrlich M. Testing the position-effect variegation hypothesis for facioscapulohumeral muscular dystrophy by analysis of histone modification and gene expression in subtelomeric 4q. Hum. Mol. Genet. 2003;12:2909–2921. doi: 10.1093/hmg/ddg323. [DOI] [PubMed] [Google Scholar]
- 59.Yang F., Shao C., Vedanarayanan V., Ehrlich M. Cytogenetic and immuno-FISH analysis of the 4q subtelomeric region, which is associated with facioscapulohumeral muscular dystrophy. Chromosoma. 2004;112:350–359. doi: 10.1007/s00412-004-0280-x. [DOI] [PubMed] [Google Scholar]
- 60.Cabianca D.S., Casa V., Bodega B., Xynos A., Ginelli E., Tanaka Y., Gabellini D. A long ncRNA links copy number variation to a polycomb/trithorax epigenetic switch in FSHD muscular dystrophy. Cell. 2012;149:819–831. doi: 10.1016/j.cell.2012.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Giussani M., Cardone M.F., Bodega B., Ginelli E., Meneveri R. Evolutionary history of linked D4Z4 and Beta satellite clusters at the FSHD locus (4q35) Genomics. 2012;100:289–296. doi: 10.1016/j.ygeno.2012.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Stout K., van der Maarel S., Frants R.R., Padberg G.W., Ropers H.H., Haaf T. Somatic pairing between subtelomeric chromosome regions: implications for human genetic disease. Chromosome Res. 1999;7:323–329. doi: 10.1023/a:1009287111661. [DOI] [PubMed] [Google Scholar]
- 63.Tsumagari K., Qi L., Jackson K., Shao C., Lacey M., Sowden J., Tawil R., Vedanarayanan V., Ehrlich M. Epigenetics of a tandem DNA repeat: chromatin DNaseI sensitivity and opposite methylation changes in cancers. Nucleic Acids Res. 2008;36:2196–2207. doi: 10.1093/nar/gkn055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kulaeva O.I., Nizovtseva E.V., Polikanov Y.S., Ulianov S.V., Studitsky V.M. Distant activation of transcription: mechanisms of enhancer action. Mol. Cell. Biol. 2012;32:4892–4897. doi: 10.1128/MCB.01127-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Petrascheck M., Escher D., Mahmoudi T., Verrijzer C.P., Schaffner W., Barberis A. DNA looping induced by a transcriptional enhancer in vivo. Nucleic Acids Res. 2005;33:3743–3750. doi: 10.1093/nar/gki689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Erliandri I., Fu H., Nakano M., Kim J.H., Miga K.H., Liskovykh M., Earnshaw W.C., Masumoto H., Kouprina N., Aladjem M.I., et al. Replication of alpha-satellite DNA arrays in endogenous human centromeric regions and in human artificial chromosome. Nucleic Acids Res. 2015;42:11502–11516. doi: 10.1093/nar/gku835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Jurka J., Kapitonov V.V., Kohany O., Jurka M.V. Repetitive sequences in complex genomes: structure and evolution. Annu. Rev. Genomics Hum. Genet. 2007;8:241–259. doi: 10.1146/annurev.genom.8.080706.092416. [DOI] [PubMed] [Google Scholar]
- 68.Zhu Q., Pao G.M., Huynh A.M., Suh H., Tonnu N., Nederlof P.M., Gage F.H., Verma I.M. BRCA1 tumour suppression occurs via heterochromatin-mediated silencing. Nature. 2011;477:179–184. doi: 10.1038/nature10371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Li Y., Miyanari Y., Shirane K., Nitta H., Kubota T., Ohashi H., Okamoto A., Sasaki H. Sequence-specific microscopic visualization of DNA methylation status at satellite repeats in individual cell nuclei and chromosomes. Nucleic Acids Res. 2013;41:e186. doi: 10.1093/nar/gkt766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Rich J., Ogryzko V., Pirozhkova I. Satellite DNA and related diseases. Biopolym. Cell. 2014;30:249–259. [Google Scholar]
- 71.Ting D.T., Lipson D., Paul S., Brannigan B.W., Akhavanfard S., Coffman E.J., Contino G., Deshpande V., Iafrate A.J., Letovsky S., et al. Aberrant overexpression of satellite repeats in pancreatic and other epithelial cancers. Science. 2011;331:593–596. doi: 10.1126/science.1200801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Scott H.S., Kudoh J., Wattenhofer M., Shibuya K., Berry A., Chrast R., Guipponi M., Wang J., Kawasaki K., Asakawa S., et al. Insertion of beta-satellite repeats identifies a transmembrane protease causing both congenital and childhood onset autosomal recessive deafness. Nat. Genet. 2001;27:59–63. doi: 10.1038/83768. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.